

Abstract
The exposure of Staphylococcus aureus to a broad range of cell wall-damaging agents triggers the induction of a cell wall stress stimulon (CWSS) controlled by the VraSR two-component system. The vraSR genes form part of the four-cistron autoregulatory operon orf1-yvqF-vraS-vraR. The markerless inactivation of each of the genes within this operon revealed that orf1 played no observable role in CWSS induction and had no influence on resistance phenotypes for any of the cell envelope stress-inducing agents tested. The remaining three genes were all essential for the induction of the CWSS, and mutants showed various degrees of increased susceptibility to cell wall-active antibiotics. Therefore, the role of YvqF in S. aureus appears to be opposite that in other Gram-positive bacteria, where YvqF homologs have all been shown to inhibit signal transduction. This role, as an activator rather than repressor of signal transduction, corresponds well with resistance phenotypes of ΔYvqF mutants, which were similar to those of ΔVraR mutants in which CWSS induction also was completely abolished. Resistance profiles of ΔVraS mutants differed phenotypically from those of ΔYvqF and ΔVraR mutants on many non-ß-lactam antibiotics. ΔVraS mutants still became more susceptible than wild-type strains at low antibiotic concentrations, but they retained larger subpopulations that were able to grow on higher antibiotic concentrations than ΔYvqF and ΔVraR mutants. Subpopulations of ΔVraS mutants could grow on even higher glycopeptide concentrations than wild-type strains. The expression of a highly sensitive CWSS-luciferase reporter gene fusion was up to 2.6-fold higher in a ΔVraS than a ΔVraR mutant, which could be linked to differences in their respective antibiotic resistance phenotypes. Bacterial two-hybrid analysis indicated that the integral membrane protein YvqF interacted directly with VraS but not VraR, suggesting that it plays an essential role in sensing the as-yet unknown trigger of CWSS induction.
The bacterial cell envelope is a major target for antimicrobial agents, most of which act by blocking or disrupting peptidoglycan synthesis (7). The integrity of the cell envelope is essential in Staphylococcus aureus for survival and pathogenicity, as it not only protects cells against environmental stresses but also modulates colonization, virulence, and antimicrobial resistance. The structural rigidity of the staphylococcal cell envelope is provided by a multilayered and highly cross-linked peptidoglycan cell wall surrounding the cytoplasmic membrane. Accessory features governing physicochemical properties of the cell surface include wall and lipoteichoic acids, membrane-associated and peptidoglycan-anchored proteins, and extracellular polysaccharide matrices. Bacteria rely on sensory elements within their cell envelope to trigger adaptive responses to wide-ranging environmental conditions. A large family of such signal transducers is the two-component systems (TCS), consisting of membrane-anchored sensor kinases that respond to environmental signals and activate cognate response regulators to induce or repress specific sets of target genes.
Many Gram-positive bacteria contain TCS regulatory systems that respond to cell envelope damage or the disruption of cell wall synthesis and trigger protective cell envelope stress responses (reviewed in reference 19). The exposure of S. aureus to diverse cell wall-targeting antibiotics, or the depletion of essential cell wall synthesis enzymes, have been shown to induce the VraSR TCS, which controls a large VraSR-dependent cell wall stress stimulon (CWSS) (14, 27, 43, 47). The induction of the VraSR-dependent CWSS in S. aureus is thought to protect against cell envelope damage by enhancing peptidoglycan synthesis through the induction of genes, including pbp2, the only bifunctional staphylococcal penicillin binding protein (PBP) with both transglycosylase and transpeptidase activity (39), murZ (a redundant MurA isozyme [5]), sgtB (a soluble transglycosylase [50]), and fmtA (an accessory PBP with low affinity for beta-lactams [13]) (27, 30, 47). The CWSS of S. aureus also contains several genes of currently unknown function or unknown significance to the stress response. The upregulation of the VraSR-dependent CWSS has been linked to increased ß-lactam and glycopeptide resistance phenotypes in several clinical S. aureus isolates (23, 24, 28, 30).
The VraSR-TCS shares homology with other well-studied Gram-positive TCS modulators of cell wall stress, such as LiaRS from Bacillus subtilis, Streptococcus pneumoniae, and Streptococcus mutans and CesSR from Lactococcus species (12, 20, 29, 45). Although these homologous systems have all been shown to respond to similar signals and function in similar ways, there is little conservation in the size of their regulons and the types of genes they control, indicating that the respective stress responses mounted are highly genus or species specific, probably in response to niche-specific stresses and required adaptive responses. The exact signal sensed by these TCS is unknown, and while there is significant overlap in the types of antibiotics or enzymes able to induce signal transduction, there also are organism-specific differences, suggesting that TCS are activated by a signal resulting from general cell wall damage and/or the inhibition of cell wall synthesis (19).
TCS genes belonging to this family are all cotranscribed with a third gene, homologous to liaF in B. subtilis, on a single autoregulatory transcript (19). In B. subtilis and S. mutans, the membrane-anchored LiaF protein was shown to repress LiaSR-dependent signal transduction under normal growth conditions (20, 45). Because of the integral part LiaF plays in LiaSR signal transduction and the identical resistance phenotypes of liaF and liaR mutants in S. mutans, it was proposed that these loci are in fact three-component systems (e.g., LiaFSR). In S. aureus, little is known about the liaF homolog, yvqF, which is located directly upstream of vraS in the four-gene operon orf1-yvqF-vraS-vraR. However, the step selection of a methicillin-resistant S. aureus (MRSA) isolate first on inhibitory concentrations of imipenem and then teicoplanin was shown to select for point mutations in either yvqF or vraS at a high frequency, and some clinical glycopeptide intermediate-resistant S. aureus (GISA) isolates from diverse geographical locations were found to have different point mutations in either yvqF or vraS. However, no strains analyzed had mutations in both loci, indicating that the mutation of both genes would not confer a further resistance advantage (23).
The inactivation of VraSR decreases resistance to most VraSR-inducing cell wall-damaging agents, indicating that the CWSS controlled by this TCS is essential for the expression of several different resistance phenotypes. Conversely, a specific point mutation in VraS, activating signal transduction in the absence of antibiotic induction, was shown to contribute to increased teicoplanin resistance in some clinical GISA (24). No specific relationship has been found yet between any of the yvqF point mutations found in clinical S. aureus or generated by in vitro selection on imipenem/teicoplanin and the resistance phenotypes of the strains analyzed (23).
In this study, we introduced nonpolar mutations into each of the four open reading frames (ORFs) of the orf1-yvqF-vraS-vraR operon and compared their impacts on the induction of the CWSS and on resistance phenotypes to cell wall-active antibiotics, targeting several different steps of the cell wall synthesis pathway in both an MRSA and a near-isogenic methicillin-susceptible S. aureus (MSSA) strain background. We also analyzed protein-protein interactions between YvqF, VraS, and VraR using a bacterial two-hybrid (BTH) system developed by Karimova et al. (21, 22), which was used previously to successfully show protein-protein interactions in S. aureus (40) and between members of TCS (46).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Strains and plasmids used in this study are listed in Table 1. Bacteria were grown on sheep blood or Luria-Bertani (LB) agar plates, and liquid cultures were grown in LB with shaking at 180 rpm at a medium/flask volume ratio of 1:4. All optical density (OD) measurements given were taken at 600 nm. Media were supplemented with the following antibiotics when appropriate: 25 or 50 μg/ml of kanamycin, 50 or 100 μg/ml of ampicillin, 10 μg/ml of tetracycline, or 10 μg/ml of chloramphenicol. Phage 80α was used for transduction.
TABLE 1.
Strains and plasmids
| Strain/plasmid | Relevant genotype/phenotypea | Reference/source |
|---|---|---|
| S. aureus | ||
| RN4220 | Restriction-negative derivative of NCTC8325-4 | 26 |
| BB270 | MRSA derivative of NCTC8325 containing an SCCmec type I | 4 |
| COL | Early clinical MRSA isolate, CC8/ST250, SCCmec type I | 15 |
| RN4220Δorf1 | RN4220 containing markerless deletion of orf1 (sa1703) | This study (Fig. 1) |
| RN4220ΔYvqF | RN4220 containing yvqF mutation, truncating YvqF at the 5th aa | This study (Fig. 1) |
| RN4220ΔVraS | RN4220 containing vraS mutation, truncating VraS at the 2nd aa | This study (Fig. 1) |
| RN4220ΔVraR | RN4220 containing vraR mutation, truncating VraR at the 2nd aa | This study (Fig. 1) |
| BB270Δorf1 | BB270 containing markerless deletion of orf1 (sa1703) | This study (Fig. 1) |
| BB270ΔYvqF | BB270 containing yvqF mutation, truncating YvqF at the 5th aa | This study (Fig. 1) |
| BB270ΔVraS | BB270 containing vraS mutation, truncating VraS at the 2nd aa | This study (Fig. 1) |
| BB270ΔVraR | BB270 containing vraR mutation, truncating VraR at the 2nd aa | This study (Fig. 1) |
| E. coli | ||
| DH5α | F− f80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK+) phoA supE44 thi-1 gyrA96 relA1 λ− | Invitrogen |
| BTH101 | F−cya-99 araD139 galE15 galK16 rpsL1 (Strr) hsdR2 mcrA1 mcrB1 | 22 |
| Plasmids | ||
| pKOR1 | E. coli-S. aureus shuttle plasmid for creating markerless deletions; repF (ts), cat, attP, ccdB, ori ColE1, bla, Pxyl/tetO, secY570; Apr, Cmr | 1 |
| pAW17 | E. coli-S. aureus shuttle plasmid, ori ColE1, ori pAMα1, aac-aph; Kmr | 41 |
| pvra | pAW17 containing entire vra operon of S. aureus COL; Kmr | This study |
| pvraΔYvqF | pAW17 containing entire vra operon of BB270ΔYvqF; Kmr | This study |
| pvraΔVraR | pAW17 containing entire vra operon of BB270ΔVraR; Kmr | This study |
| pBUS1 | S. aureus-E. coli shuttle vector, tetL; Tcr | 41 |
| pSP-luc+ | Luciferase fusion plasmid, ori ColE1, bla, luc+; Apr | Promega |
| psas016p-luc+ | pBUS1 containing sas016 promoter-luciferase reporter gene fusion; Tcr | This study |
| pKT25 | BTH vector encoding the T25 fragment of CyaA upstream of the multiple cloning site; ori p15A; Kmr | 22 |
| pKNT25 | BTH vector encoding the T25 fragment of CyaA downstream of the multiple cloning site, ori p15A; Kmr | 21 |
| pUT18C | BTH vector encoding the T18 fragment of CyaA upstream of the multiple cloning site; ori ColE1; Apr | 22 |
| pUT18 | BTH vector encoding the T18 fragment of CyaA downstream of the multiple cloning site; ori ColE1; Apr | 22 |
Abbreviations: Strr, streptomycin resistance; Apr, ampicillin resistance; Cmr, chloramphenicol resistance; Kmr, kanamycin resistance; Tcr, tetracycline resistance.
Construction of pKOR1 markerless mutations.
The pKOR1 system created by Bae et al. (1) was used to construct a markerless deletion of orf1 and to truncate YvqF, VraS, and VraR proteins by inserting two stop codons in frame into the beginning of their respective ORFs (Fig. 1). Primers used for plasmid construction are listed in Table 2. For orf1 deletion, upstream and downstream genomic regions flanking orf1 were amplified using primer pairs attB1-yvqF.upF/BamHI-orf1.upR and BamHI-orf1.downF/attB2-yvqF.downR, respectively. Flanking regions were digested with BamHI, ligated together, and recombined into pKOR1 using Gateway BP Clonase II enzyme mix (Invitrogen). To create pKOR1 constructs that would insert an XhoI site and two in-frame stop codons into the beginning of yvqF, vraS, and vraR ORFs, regions directly upstream and containing the first 2 to 5 codons of the ORFs, followed by an XhoI site, were amplified using the primer pairs attB1-yvqF.upF/XhoI-yvqF.upR, attB1-yvqF-upF/XhoI-vraS.upR, and attB1-vraR.upF/XhoI-vraR.upR, respectively. Adjacent regions, containing the remainder of the yvqF, vraS, or vraR ORFs, preceded by two stop codons and an XhoI site, were amplified using primer pairs XhoI-stop-yvqF.downF/attB2-yvqF.downR, XhoI-stop-vraS.downF/attB2-yvqF.downR, and XhoI-stop-vraR.downF/attB2-vraR.downR, respectively. Amplicon pairs were digested with XhoI, ligated together, and recombined into pKOR1. Mutations were introduced into the genomes of S. aureus strains RN4220 and BB270 using the inducible counterselection protocol described by Bae et al. (1) and confirmed by PCR and sequencing across the deleted or genetically manipulated region. The sequence analysis of the entire vra operon region of the yvqF mutant was also performed to ensure that no additional mutations had been introduced. All mutants were also screened by pulsed-field gel electrophoresis (PFGE) (48) to confirm that no additional major genomic rearrangements had occurred (data not shown).
FIG. 1.

Map of the orf1-yvqF-vraS-vraR operon and construction of mutants. (A) Wild-type operon structure, with the single autoregulatory promoter element indicated by an arrow (3) and the predicted transcriptional terminator (http://cmr.jcvi.org/) indicated by a stem-loop symbol. (B) Markerless deletion of orf1, leaving only the first 3 amino acids (aa) fused in frame to the last 8 aa. (C) YvqF was truncated by markerless insertion of an XhoI site and two in-frame stop codons after the 5th aa. (D) VraS mutant truncated by stop codons inserted after the 2nd aa. (E) VraR mutant truncated by stop codons inserted after the 2nd aa.
TABLE 2.
Primers used in this study
| Primer name and function | Nucleotide sequencea (5′-3′) | Source |
|---|---|---|
| Construction of pKOR1 mutagenesis plasmids | ||
| attB1-yvqF.upF | GGGGACAAGTTTGTACAAAAAAGCAGGCTGATTCCAAGTAAGCGTGTCAT | This study |
| attB2-yvqF.downR | GGGGACCACTTTGTACAAGAAAGCTGGGTCATCCATGTCATCCATAAGTA | This study |
| yvqF-XhoI.upR | ATTTACTCGAGATATTTGTGTGTCATGTTTCG | This study |
| XhoI-stop-yvqF.downF | AATTTCTCGAGTGATAGATATCAACGCAAATGTTGATC | This study |
| BamHI-orf1.upR | AATTTGGATCC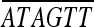 CATAACTATCACCTT CATAACTATCACCTT |
This study |
| BamHI-orf1.downF | AATTTGGATCCAATTATATGAACGCTGTGGCA | This study |
| XhoI-vraS.upR | ATTTACTCGAGGTTCATCGATAAATCACCTCTA | This study |
| XhoI-stop-VraS.downF | AATTTCTCGAGTGATAGCACTACATTAGAACAATTGGTT | This study |
| attB1-vraR.upF |
GGGGACAAGTT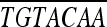 AAAAGCAGGCTGCTATTAATTGCATTCACATTAT AAAAGCAGGCTGCTATTAATTGCATTCACATTAT |
This study |
| XhoI-vraR.upR | ATTTACTCGAGCGTCATACGAATCCTCCTTAT | This study |
| XhoI-stop-vraR.downF | AATTTCTCGAGTGATAGATTAAAGTATTGTTTGTGGAT | This study |
| attB2-vraR.downR |
GGGGACCACTT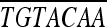 GAAAGCTGGGTAAACGAAGCTTAAGTCAGTATTA GAAAGCTGGGTAAACGAAGCTTAAGTCAGTATTA |
This study |
| Construction of complementation plasmids | ||
| vra.compF | AATTTGGATCCGCACATGTACTTAATTACTT | This study |
| vra.compR | AATTTGGTACCCGAATATGATGAAGATAGTA | This study |
| Construction of luciferase-fusion plasmid | ||
| SAS016.lucF | AATTA GGTACC TGGATCACGGTGCATACAAC | This study |
| SAS016.lucR | AATTA CCATGG CCTATATTACCTCCTTTGCT | This study |
| Amplification of DIG-labeled probes | ||
| vraR.For | ATGACGATTAAAGTATTGTT | This study |
| vraR.Rev | TTGAATTAAATTATGTTGGA | This study |
| SAS016.For | TCATACGTTCTATGTCTGAT | This study |
| SAS016.Rev | GATCTATATCGTCTTGTAAT | This study |
| Construction of BTH plasmids | ||
| yvqF.pKT25F | ATTAACTGCAGTGACACACAAATATATATCAA | This study |
| yvqF.pUT18CF | ATTAACTGCAGGACACACAAATATATATCAAC | This study |
| yvqF.BTHR | ATTAAGGTACCCGATAAATCACCTCTACGTC | This study |
| vraS.pKT25F | ATTAACTGCAGTGAACCACTACATTAGAACAA | This study |
| vraS.pUT18CF | ATTAACTGCAGGAACCACTACATTAGAACAAT | This study |
| vraS.BTHR | ATTAAGGTACCTCGTCATACGAATCCTCCTT | This study |
| vraR.pKT25F | ATTAACTGCAGTGACGATTAAAGTATTGTTT | This study |
| vraR.pUT18CF | ATTAACTGCAGGACGATTAAAGTATTGTTTG | This study |
| vraR.BTHR | ATTAAGGTACCTGAATTAAATTATGTTGGAA | This study |
| sigB.pKT25F | ATTAACTGCAGTGGCGAAAGAGTCGAAATCA | This study |
| sigB.pKT25R | ATTAAGGATCCTATTGATGTGCTCGTTCTTG | This study |
| rsbW.pUT18CF | ATTAACTGCAGGCAATCTAAAGAAGATTTTA | This study |
| rsbW.pUT18CR | ATTAAGGATCCTAGCTGATTTCGACTCTTTC | This study |
Restriction sites are underlined, attB sites are in boldface, and introduced stop codons are double underlined.
vra operon complementation.
The entire orf1-yvqF-vraS-vraR operon, including the published promoter region (3, 52) and predicted transcriptional terminator (http://cmr.jcvi.org/), was amplified from S. aureus COL using primers vra.compF and vra.compR (Table 2) and cloned into pAW17 to create plasmid pvra. To demonstrate that the mutation of yvqF introduced no polar effects on the downstream vraS and vraR genes, complementation plasmids pvraΔYvqF and pvraΔVraR were also constructed. These plasmids contained vra operon inserts from BB270ΔYvqF and BB270ΔVraR and were amplified using primers vra.compF and vra.compR.
Antibiotic resistance tests.
Several different techniques were employed to quantitatively and qualitatively compare resistance phenotypes to 15 different antibiotics, targeting various different cell wall structures or stages of cell wall synthesis (Fig. 2). MICs of antibiotics were determined by Etest (AB-Biodisk) according to the manufacturer's instructions. Glycopeptide MICs also were determined using the macro-Etest method, which is recommended for its high specificity and sensitivity in identifying differences in glycopeptide resistance levels (49). Macro-Etests were performed on brain heart infusion (BHI) (BBL) agar swabbed with 2.0 McFarland suspensions and incubated at 35°C for 48 h. Broth microdilutions were performed according to CLSI guidelines (9).
FIG. 2.

S. aureus peptidoglycan synthesis and targets of cell wall-active antibiotics. The inhibition of enzymatic reactions is indicated by blocked arrows; the inhibition of cell wall synthesis by the binding of antibiotics to peptidoglycan precursors is indicated by half-moon symbols; pentaglycine bridge cleavage by lysostaphin and membrane disruption/depolarization by daptomycin are indicated by arrows. (Adapted from reference 31 with the permission of the publisher.)
Antibiotic resistance profiles were compared by population analysis profile (PAP), whereby appropriate dilutions of an overnight culture, ranging from 100 to 10−8, were plated on increasing concentrations of oxacillin (0.5 to 512 μg/ml) (InfectoPharm), teicoplanin (0.0625 to 8 μg/ml) (Hoechst Marion Roussel), vancomycin (0 to 8 μg/ml) (Eli Lilly & Company), flavomycin (0 to 8 μg/ml) (BC Biochemie GmbH), or daptomycin (0 to 2 μg/ml) (Cubist Pharmaceuticals, Inc.) and incubated at 35°C. CFU per ml were determined after 48 h. Daptomycin plates were supplemented with 50 μg/ml of CaCl2, corresponding to ∼27 μg/ml Ca2+, which, in addition to the ∼20-μg/ml Ca2+ from the Difco agar (BD Biosciences), present in the plates gave a final concentration of ∼47 μg/ml Ca2+.
Gradient plates were used to compare resistance phenotypes for lysostaphin (Ambi), ramoplanin (Sigma), ceftobiprole (Johnson & Johnson Pharmaceutical Research & Development), d-cycloserine (Sigma), and Triton X-100 (Fluka). Bacterial suspensions of 2.0 McFarland were used to enhance the visualization of population heterogeneity. The suspensions were swabbed across agar plates containing appropriate antibiotic concentration gradients, and plates were incubated at 35°C for 24 to 48 h.
Relative resistance levels of strains containing the empty plasmid pAW17 and the complementing plasmids pvra, pvraΔYvqF, or pvraΔVraR were also compared by gradient plating, in which bacterial suspensions of 0.5 McFarland were swabbed across agar plates containing a teicoplanin gradient of 0 to 4 μg/ml. Plates for complementation comparisons were supplemented with kanamycin (50 μg/ml) to ensure plasmid maintenance.
Northern blotting.
Northern blot analyses were performed as previously described (32). Overnight cultures were diluted to an OD of 0.05 in fresh prewarmed LB containing kanamycin (50 μg/ml) and grown to an OD of 0.5. Uninduced samples were harvested, and the remaining culture was induced with vancomycin (10 μg/ml) for 30 min before induced samples were collected. Total RNA was extracted as described by Cheung et al. (8). RNA samples (8 μg) were separated in a 1.5% agarose-20 mM guanidine thiocyanate gel in 1× Tris-borate-EDTA (TBE) buffer (16). Digoxigenin (DIG)-labeled probes were amplified using the PCR DIG probe synthesis kit (Roche) with primer pairs listed in Table 2. All Northern blottings were repeated at least two times using independently isolated RNA samples.
Luciferase reporter gene fusion assay.
The promoter region of sas016, which corresponds to ORF SACOL0625 from S. aureus COL (accession number NC_002951), was amplified using primers SAS016.lucF and SAS016.lucR (Table 2), digested with Asp718 and NcoI, and ligated directly upstream of the promoterless luciferase (luc+) gene in vector pSP-luc+ (Promega). A fragment containing the sas016 promoter-luc+ translational fusion was then excised with Asp718 and EcoRI and cloned into the Escherichia coli-S. aureus shuttle vector pBUS1. The fusion plasmid, psas016p-luc+, was transformed into S. aureus RN4220 and then transduced into further strains.
For induction assays, two identical culture broths were inoculated with an overnight culture to an OD of 0.05. Cultures were grown to between OD 0.4 and 0.5 and then split into two flasks; one was left uninduced and the other induced with vancomycin (10 μg/ml). After 30 min, samples were collected from both uninduced and induced cultures. To compare luciferase activity throughout different growth stages, three separate culture broths for each mutant were inoculated with an overnight culture to an OD of 0.05 and grown for 9 h. Samples were collected at 1.5-, 3-, 4.5-, 6-, 7.5-, and 9-h time points, and ODs were recorded. Samples were harvested by centrifugation, and pellets were frozen at −20°C. For luciferase activity measurements, pellets were resuspended in phosphate-buffered saline (PBS) to an OD of 10, and aliquots were mixed with equal amounts of Luciferase assay system substrate (Promega). Luminescence was measured for 15 s after a delay of 3 s on a Turner Designs TD-20/20 luminometer (Promega).
Bacterial two-hybrid (BTH) plasmid construction and interaction screening.
The yvqF, vraS, and vraR gene sequences were amplified from S. aureus COL genomic DNA and cloned into four BTH vectors to create N-terminal (pKT25 and pUT18C) and C-terminal (pKNT25 and pUT18) protein fusions to the T25 or T18 domain of Bordetella pertussis CyaA (21, 22). To create in-frame fusions, two different inserts were amplified for each gene. Inserts amplified with x.pKT25F and x.BTHR primers were cloned into pKT25, whereas inserts amplified with x.pUT18CF and x.BTHR primers were cloned into pUT18C, pUT18, and pKNT25 vectors.
Combinations of constructs then were transformed into the reporter strain E. coli BTH101 and cotransformants were selected on LB agar containing ampicillin (100 μg/ml), kanamycin (50 μg/ml), 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) (40 μg/ml), and isopropyl-β-d-thiogalactopyranoside (IPTG) (24 μg/ml) and on M63 minimal agar containing lactose as a sole carbon source (22) and supplemented with ampicillin (50 μg/ml), kanamycin (25 μg/ml), X-Gal (40 μg/ml), and IPTG (24 μg/ml). Cotransformants then were spotted onto LB or M63 agar containing X-Gal, IPTG, ampicillin, and kanamycin. Interactions also were quantified by performing ONPG (o-nitrophenyl-ß-d-galactopyranoside) (Sigma) cleavage assays to measure ß-galactosidase activity according to the standard protocol (34).
Cotransformants containing the empty vectors pKT25 and pUT18C were used as a negative control. As a positive control, plasmids pKT25-sigB and pUT18C-rsbW, containing the alternate sigma factor SigB and its anti-sigma factor RsbW, from S. aureus COL, were constructed and cotransformed into E. coli BTH101. These two proteins were chosen because they have been shown to interact strongly in S. aureus (35).
RESULTS
Antibiotic resistance profiles of orf1, yvqF, vraS, and vraR mutants. (i) MIC comparisons.
MICs were measured to determine the impact of orf1, yvqF, vraS, and vraR mutations on antibiotic resistance levels in the near-isogenic MSSA (RN4220) and MRSA (BB270) strains (Table 3). The deletion of orf1 had no noticeable effect on antibiotic resistance levels in either strain background. The inactivation of yvqF, vraS, and vraR generally decreased resistance levels, although ß-lactam resistances (oxacillin, cefoxitin, and imipenem) were affected only in the MRSA background.
TABLE 3.
MICs for vra operon mutants
| Strain | MICc (μg/ml) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| OX | FX | IP | TPa | VAa | BA | FM | DPC | TMb | |
| RN4220 | 0.25 | 2 | 0.032 | 1 (3) | 2 (4) | 64 | 0.38 | 0.19 | 32 |
| RN4220Δorf1 | 0.25 | 2 | 0.032 | 1 (2) | 2 (4) | 64 | 0.5 | 0.19 | 32 |
| RN4220ΔYvqF | 0.25 | 2 | 0.032 | 0.38 (1) | 1.5 (4) | 16 | 0.125 | 0.25 | 16 |
| RN4220ΔVraS | 0.25 | 2 | 0.032 | 0.75 (3) | 2 (6) | 16 | 0.38 | 0.38 | 8 |
| RN4220ΔVraR | 0.25 | 2 | 0.032 | 0.125 (0.25) | 1.5 (2) | 12 | 0.125 | 0.19 | 16 |
| BB270 | >256 | >256 | >32 | 1 (3) | 2 (4) | >256 | 2 | 0.25 | >32 |
| BB270Δorf1 | >256 | >256 | >32 | 1 (3) | 2 (4) | >256 | 2 | 0.25 | >32 |
| BB270ΔYvqF | 96 | 128 | 8 | 0.094 (0.125) | 1 (2) | 48 | 0.38 | 0.19 | 32 |
| BB270ΔVraS | 96 | 96 | 16 | 0.5 (4) | 3 (8) | 48 | 4 | 0.25 | 32 |
| BB270ΔVraR | 96 | 96 | 8 | 0.094 (0.125) | 1 (2) | 48 | 0.38 | 0.19 | 32 |
MICs in parentheses were determined by macro-Etest (49).
MICs were determined by microdilution.
Abbreviations: OX, oxacillin; FX, cefoxitin; IP, imipenem; TP, teicoplanin; VA, vancomycin; BA, bacitracin; FM, fosfomycin; DPC, daptomycin; TM, tunicamycin.
Strain-dependent differences between RN4220 and BB270 mutants also were seen in levels of resistance to some non-ß-lactam antibiotics, including fosfomycin, daptomycin, tunicamycin, and bacitracin. ΔYvqF, ΔVraS, and ΔVraR mutations influenced resistance levels to various degrees, with huge MIC decreases for teicoplanin (up to 30-fold), more moderate decreases (2- to 6-fold) for bacitracin, fosfomycin, and the ß-lactam antibiotics in the BB270 background, and only marginal effects (≤2-fold) on daptomycin and vancomycin resistance levels.
In general, yvqF and vraR mutants gave highly comparable MIC readings, indicating that YvqF was as essential as VraR for protecting against cell wall damage. MICs for ΔVraS mutants were similar to those for ΔYvqF and ΔVraR mutants for some antibiotics (oxacillin, cefoxitin, bacitracin, and tunicamycin), but they were up to severalfold higher for others (teicoplanin, vancomycin, fosfomycin, and daptomycin).
(ii) PAP.
For more in-depth comparisons of resistance profiles, BB270 and its orf1, yvqF, vraS, and vraR mutants were also analyzed by PAP on selected antibiotics (Fig. 3A). Profiles of BB270Δorf1 were identical to those of wild-type BB270 for all antibiotics tested. Teicoplanin PAPs showed severe changes in resistance profiles in ΔYvqF, ΔVraS, and ΔVraR mutants, with an immediate drop of 2 to 4 log in CFU in all mutants at 0.0625 μg/ml of teicoplanin, while wild-type and orf1 mutants grew on concentrations of up to 0.5 μg/ml before CFU numbers began to decrease. Profiles of ΔYvqF, ΔVraS, and ΔVraR mutants also showed various levels of resistance heterogeneity. ΔYvqF and ΔVraR mutant curves were almost identical and showed that both strains had become highly susceptible, retaining only a small subpopulation able to grow at concentrations between 0.0625 and 8 μg/ml. The ΔVraS mutant showed a markedly different phenotype; even though CFU/ml dropped immediately at 0.0625 μg/ml of teicoplanin, the ΔVraS mutant retained a much larger subpopulation that was able to grow on higher teicoplanin concentrations. Between 1 and 8 μg/ml, the surviving subpopulation of the ΔVraS mutant was even higher, by up to 2 log, than that of the wild type.
FIG. 3.

Antibiotic resistance profiles of vra operon mutants. (A) Population analysis profiles of BB270 and its Δorf1, ΔYvqF, ΔVraS, and ΔVraR mutants. (B) Growth phenotypes of RN4220 and BB270 compared to those of their respective vra operon mutants on antibiotic gradient plates. Antibiotic and Triton X-100 concentration gradients are indicated.
Resistance profiles of all strains displayed the same trends on vancomycin, although overall differences in resistance were much smaller and were seen within a much narrower range of vancomycin concentrations (0.5 to 4 μg/ml). The resistant subpopulation of the VraS mutant once again was larger than that of the wild type at concentrations above 1 μg/ml. The ΔVraS mutant also retained a much larger resistant subpopulation than the ΔYvqF and ΔVraR mutants on flavomycin, although it never became more resistant than the wild type.
Conversely, ΔYvqF, ΔVraS, and ΔVraR mutants all had identical resistance phenotypes on oxacillin, with all three mutants becoming much more heterogeneous at oxacillin concentrations of >16 μg/ml, while the wild-type and orf1 mutants remained homogeneously resistant until they were grown at concentrations of >128 μg/ml.
Daptomycin PAPs showed only small differences, but they gave a clearer picture of resistance phenotypes than the Etest readings. ΔYvqF and ΔVraR mutants were more susceptible than the other three strains at daptomycin concentrations of >0.25 μg/ml. The ΔVraS mutant once again appeared to be more resistant than all other strains at higher daptomycin concentrations, although only marginally so.
(iii) Gradient plate comparisons.
Resistance phenotypes of RN4220 and BB270 strain sets were also compared on lysostaphin, ramoplanin, d-cycloserine, ceftobiprole, and Triton X-100 gradient plates (Fig. 3B). Triton X-100 gradient plates showed that yvqF vraS vraR mutants were all much more susceptible than the wild type and all displayed heterogeneous resistance profiles, although the ΔVraS mutant appeared to have a larger subpopulation able to grow on higher concentrations of Triton X-100 than ΔYvqF and ΔVraR. All three yvqF, vraS, and vraR mutants had indistinguishable resistance phenotypes on ramoplanin and d-cycloserine, although ramoplanin resistance was decreased to a much greater extent than d-cycloserine resistance, where observable differences were very small but highly reproducible. BB270 ΔYvqF, ΔVraS, and ΔVraR mutants were also slightly more susceptible to ceftobiprole, but as with other ß-lactam-family antibiotics tested, none of the mutations affected resistance in RN4220. BB270-derived strains were also more intrinsically resistant than their RN4220 counterparts to Triton X-100 and d-cycloserine but not to ramoplanin or lysostaphin. Resistance levels to lysostaphin did not appear to be affected by any of the vra operon mutations in either strain background.
Complementation.
Gradient plates comparing teicoplanin resistance levels of BB270, BB270Δorf1, BB270ΔYvqF, BB270ΔVraS, and BB270ΔVraR, containing either the empty control plasmid pAW17 or plasmid pvra (Fig. 4A), showed that yvqF, vraS, and vraR mutants all could be complemented by introducing the orf1-yvqF-vraS-vraR operon in trans.
FIG. 4.

Transcomplementation of vra operon mutants. (A) Teicoplanin gradient plates comparing the resistance levels of wild-type and mutant strains containing either the empty vector pAW17 or the complementing plasmid pvra. (B) Schematic representation of transcomplementation plasmids pvra, containing the wild-type vra operon, pvraΔYvqF, containing the vra operon from BB270ΔYvqF, and pvraΔVraR, containing the vra operon from BB270ΔVraR. (C) Teicoplanin gradient plates showing the transcomplementation of BB270ΔYvqF with pvraΔVraR, BB270ΔVraS with pvraΔYvqF and pvraΔVraR, and BB270ΔVraR with pvraΔYvqF.
To exclude the possibility that polar effects were introduced during mutant construction, a further two complementing plasmids, containing the vra operons amplified from BB270ΔYvqF or BB270ΔVraR, were created (Fig. 4B). BB270ΔVraS and BB270ΔVraR could both be complemented by pvraΔYvqF, indicating that vraS and vraR genes were both still functional in BB270ΔYvqF. The visual comparison of growth on gradient plates indicated that pvra and pvraΔVraR both complemented the growth of BB270ΔVraS to wild-type levels. However, when complemented with pvraΔYvqF, the growth of BB270ΔVraS appeared fainter, indicating that although resistance was clearly complemented, wild-type growth was not fully restored. Both BB270ΔYvqF and BB270ΔVraS could also be complemented by pvraΔVraR, further suggesting that the introduced mutations had not created polar effects on other genes within this operon (Fig. 4C).
CWSS induction in orf1, yvqF, vraS, and vraR mutants.
The induction of CWSS genes in BB270, BB270Δorf1, BB270ΔYvqF, BB270ΔVraS, and BB270ΔVraR was analyzed by Northern blotting. Genes used as probes included sas016, because it is highly induced by cell wall-active antibiotics (33), and vraR, because the orf1-yvqF-vraS-vraR transcript is autoregulated and transcription should not be disrupted in the ΔVraR mutant. Both transcripts were highly induced in wild-type BB270 and the orf1 mutant, but no induction was visible in the ΔYvqF, ΔVraS, or ΔVraR mutants (Fig. 5A).
FIG. 5.

Induction of CWSS transcripts in vra operon mutants. (A) Transcriptional profiles of sas016 and the vra operon in BB270 and its four vra operon mutants. RNA was harvested from strains containing either the empty plasmid pAW17 or the transcomplementation plasmid pvra both before (−) and after (+) vancomycin induction. Ethidium bromide-stained 16S rRNA bands are shown below Northern blots as an indication of RNA loading. (B) Levels of luciferase activity from psas016p-luc+ in both uninduced and vancomycin-induced cultures of BB270 and its Δorf1, ΔYvqF, ΔVraS, and ΔVraR mutants. All values shown represent the means ± standard deviations (SD) obtained from two independent cultures. (C) Profiles of luciferase activity over growth in BB270ΔYvqF, BB270ΔVraS, and BB270ΔVraR, which contain psas016p-luc+. All values shown represent the means ± SD obtained from three independent cultures.
Luciferase activity from plasmid psas016p-luc+ was also measured in both uninduced and vancomycin-induced cultures of BB270 and its four mutants (Fig. 5B). Results confirmed that there was no induction in ΔYvqF, ΔVraS, and ΔVraR mutants and showed that expression levels in the absence of induction were much lower in these three strains than in the wild type. ODs both before and after induction were very similar for all five strains, indicating that there were no significant differences in growth phenotypes (data not shown).
Luciferase activity measurements over growth showed that all three mutants had subtle but highly reproducible differences in sas016 promoter activity (Fig. 5C). Luciferase activity was lowest in the ΔVraR mutant throughout all sampling points. Activity in the ΔYvqF mutant ranged between 1.1- and 1.7-fold higher than that in the ΔVraR mutant, while in the ΔVraS mutant it ranged from 1.5- to 2.6-fold higher.
BTH protein-protein interactions.
The BTH system was used to analyze protein-protein interactions between YvqF, VraS, and VraR (Fig. 6A and B). Genes yvqF, vraS, and vraR were cloned into all four BTH vectors; however, restriction digest profiles of pKNT25(X-T25) constructs containing yvqF or vraS suggested that these clones were unstable and prone to frequent rearrangements (data not shown), and subsequently they were omitted from the study. Restriction profiles of the remaining 10 constructs appeared stable, and pairs of these plasmids were cotransformed into the reporter strain E. coli BTH101.
FIG. 6.

BTH analysis YvqF, VraS, and VraR. (A) Scheme of VraSR signal transduction. Cell wall stress triggers VraS to activate VraR by phosphotransfer. Activated VraR controls a large regulon in addition to autoregulating its own expression. VraR also is deactivated by VraS-specific dephosphorylation (2, 19). (Reprinted from reference 31 with the permission of the publisher.). (B) Potential protein-protein interactions between the transmembrane protein YvqF and VraS/VraR. (C) Phenotypes of cotransformants containing combinations of BTH fusion protein pairs on minimal medium containing lactose as a sole carbon source and X-Gal. Positive interactions are indicated by growth and blue pigmentation. Three clones were tested from each cotransformation, and negative and positive controls were included for phenotypic comparison. (D) ß-Galactosidase activity of the BTH clones shown above as measured by ONPG cleavage assays. Values given indicate the mean expression ± standard deviations of the three clones.
All cotransformations were repeated at least four times using independently isolated plasmid constructs. Figure 6C shows the phenotypes of three representative clones from each cotransformation, replica plated onto minimal media containing lactose and X-Gal. Levels of ß-galactosidase activity in each of the cotransformants were also measured to compare relative interaction strengths to those of negative and positive controls (Fig. 6D).
Positive interactions between T25-VraS and both T18-VraS and VraS-T18 fusion clones were consistent with the dimeric structure of sensor histidine kinases (2, 46) and indicated that all plasmids containing vraS functioned correctly in this system. Positive interactions resulted from three of four combinations of the YvqF and VraS plasmids, suggesting a positive interaction between these two proteins. The pairing of T25-VraS and YvqF-T18 clones consistently gave negative results, which could be due to a genuine lack of interaction or could arise as a false negative due to the orientation of fusion protein domains sterically hindering the interaction.
All interactions between YvqF and VraR were negative, with ß-galactosidase levels in the same range as that of the negative control. The positive interaction between T25-YvqF and YvqF-T18 clones suggested that YvqF could dimerize, although this was not supported for cotransformants containing T25-YvqF and T18-YvqF fusions.
Remaining combinations, including VraR dimerization and VraS-VraR interaction, gave heterogenous results, with interactions arising from some plasmid combinations and not others. However, interactions between these proteins, including the signal transduction cascade between VraS and VraR and the dimerization of VraR when phosphorylated by VraS or by acetyl phosphate in vitro, have already been thoroughly described (2, 3).
DISCUSSION
VraSR-controlled CWSS induction is the major defense mounted by S. aureus in response to cell wall damage and provides various levels of intrinsic resistance/tolerance to cell wall-targeting antibiotics. The importance of this stress response in S. aureus appears to be reflected in the relatively large size of the CWSS and its induction by all classes of cell wall-active antibiotics (27, 36, 38, 42, 44, 47). Homologous, LiaFSR-controlled CWSS from diverse species, including B. subtilis and the naturally competent S. pneumoniae, tend to be much smaller and induced only by antibiotics interacting directly with lipid II and/or specific murein hydrolases (12, 20, 45). The CWSS are perhaps more specialized in these bacteria due to the presence of additional regulatory mechanisms, such as the multiple-cell envelope stress-responsive ECF sigma factors in B. subtilis and the production of the ComM immunity protein during competence in S. pneumoniae (12, 19). These extra layers of cell envelope stress response systems may have diminished the importance and/or narrowed the scope of the LiaFSR-dependent CWSS in these bacteria (19, 51).
This study investigated the roles of the individual genes within the of1-yvqF-vraS-vraR operon on CWSS induction and resistance to cell wall-active antibiotics. orf1 mutants were phenotypically identical to their respective wild-type parent strains, indicating that Orf1 plays no role in CWSS induction. Analyses of the remaining mutants identified similarities but also key differences between the VraSR signal transduction system in S. aureus and the corresponding LiaFSR systems of B. subtilis, S. pneumoniae, and S. mutans and the CesRS system of lactococci.
The most striking difference was that YvqF was essential for both CWSS induction and CWSS-mediated antibiotic tolerance in S. aureus. The homologous LiaF protein from B. subtilis and S. mutans represses LiaSR signal transduction, with liaF deletion leading to the constitutive expression of CWSS genes in the absence of an inducing signal (12, 20, 45). The mutation of yvqF, however, abolished CWSS induction in S. aureus.
The inactivation of yvqF and vraR had almost identical effects on resistance phenotypes to all antibiotics tested. MICs for vraS mutants, however, were often higher than those for yvqF/vraR mutants. These phenotypes correlated with those published for S. mutans, whereby the deletion of liaF and liaR both decreased resistance to lipid II-interacting antibiotics to comparable levels, while liaS mutants were generally slightly more antibiotic tolerant (45). Disparate results regarding the effects of LiaFSR inactivation on resistance phenotypes in S. mutans have been reported (45, 53) and were attributed to the methods used for resistance determination, with some methods, such as disk tests, giving less sensitive results than comparisons of MICs and minimum bactericidal concentrations (MBCs) by broth microdilution (45).
In this study, resistance phenotypes were compared using several different methods. MIC determination enabled the quantification of the impact of vra operon mutants on resistance levels, while PAP analysis and gradient plate comparisons enabled a more in-depth comparison of population resistance heterogeneity and the identification of subtle differences in resistance levels. PAP results confirmed that ΔVraS mutants were less susceptible to glycopeptides than ΔYvqF/ΔVraR mutants. Resistance profiles also showed that while ΔVraS mutants were much more susceptible than wild-type strains at low antibiotic concentrations, they contained a subpopulation that was able to grow on higher glycopeptide concentrations than the wild type, indicating that vraS inactivation may actually increase tolerance to glycopeptides. ΔVraS mutants also showed greater tolerance than ΔYvqF/ΔVraR mutants to several other antibiotics, including flavomycin, fosfomycin, daptomycin, and the detergent Triton X-100. However, with the exception of daptomycin, ΔVraS mutants were not able to grow on higher antibiotic concentrations than the wild type for any of these antibiotics. No vraS mutant-specific phenotype was detected on ß-lactams, ramoplanin, or d-cycloserine, as ΔYvqF/ΔVraS/ΔVraR mutants all displayed near-identical resistance phenotypes by PAP or gradient plate comparison.
Decreased resistance to ß-lactams, including ceftobiprole, was only detected in BB270 (MRSA) mutants and not in RN4220 (MSSA) mutants, indicating that the CWSS only contributes to mecA-mediated resistance and not intrinsic ß-lactam tolerance. In BB270, oxacillin resistance levels decreased 8-fold, which is consistent with reported fold decreases for vraS or vraSR mutants of other MRSA (6, 27). In some clinical, community-acquired MRSA, VraSR inactivation decreases MICs to below susceptible breakpoint levels (6); however, in highly resistant strains, such as BB270, the MICs of yvqF/vraSR mutants are still classified as resistant (9).
Despite being closely related, the RN4220 and BB270 strain sets also showed different resistance phenotypes on several non-ß-lactam antibiotics, including fosfomycin, tunicamycin, d-cycloserine, and bacitracin, which should not be influenced by the presence of the staphylococcal cassette chromosome mec element (SCCmec) in BB270. Reasons for these differences in both intrinsic resistance profiles of the wild-type strains and on the impacts of ΔYvqF/ΔVraS/ΔVraR mutations are currently unknown. RN4220 is known to differ from other NCTC8325-derived strains in its general global regulation (17), which may contribute to altered susceptibly profiles to some antimicrobial agents.
Resistance profiling by gradient plate analysis showed that ΔYvqF/ΔVraS/ΔVraR mutation in S. aureus severely increased susceptibility to the detergent Triton X-100. Detergents are extremely potent inducers of the S. aureus CWSS (unpublished data), and although they are known to directly damage cell membranes, their induction of the CWSS is probably indirect, as Triton X-100 is known to alter autolytic enzyme activities (10). In staphylococci and other Gram-positive bacteria, Triton X-100 has been shown to induce the release of lipoteichoic acids from the cell wall, which subsequently stimulates autolysin activities (18, 25, 37) and could thereby indirectly trigger CWSS induction. Detergent susceptibility was also shown to increase in S. mutans when liaFSR genes were deleted (45).
Lysostaphin was the only inducing agent tested for which the inactivation of YvqF/VraSR had no observable impact on resistance levels in either strain; some other resistance levels, including those for daptomycin, d-cycloserine, and ceftobiprole, were also only marginally affected, although all four of these agents are inducers of the CWSS (36, 47, and unpublished data). No apparent links could be drawn between the types of antibiotics or their specific targets and the impact of yvqF or vraSR inactivation on respective resistance/tolerance levels.
Reasons for enhanced tolerance or increased heterogeneous resistance patterns in vraS mutants could be, as hypothesized for liaS mutants in S. mutans, that VraR is being activated to some extent in the absence of VraS. VraR was shown to be autophosphorylated by acetyl phosphate in vitro and could therefore be activated by phosphotransfer independently of VraS in vivo and upregulate CWSS genes to a certain extent (2, 45). VraS is also responsible for terminating the stress response signal by acting as a VraR-specific phosphatase (2). It is therefore possible that VraR-controlled promoters are affected differently in strains with no VraR compared to strains with functional VraR but no VraS or YvqF. The expression of psas016p-luc+ was substantially lower in ΔYvqF, ΔVraS, and ΔVraR mutants than in the wild type; however, there were small but highly reproducible differences in expression between these three mutant backgrounds. Reporter gene activity was ∼2-fold higher throughout growth in BB270ΔVraS than in BB270ΔVraR, indicating that CWSS expression is somewhat different in these two mutants. However, the reasons for these subtle changes in CWSS gene expression and their relevance to the corresponding resistance phenotypes of ΔYvqF, ΔVraS, and ΔVraR mutants requires further investigation.
The role of YvqF as a positive rather than negative modulator of VraSR signal transduction corresponds well with the resistance phenotypes of yvqF mutants. Increased CWSS expression has been linked to increased glycopeptide resistance in GISA clinical isolates and has been shown to contribute to in vitro-selected heterogeneous teicoplanin resistance (11, 23, 24). Therefore, if YvqF inactivation had led to constitutive CWSS expression, as liaF deletion does in B. subtilis and S. mutans (20, 45), resistance to teicoplanin would have increased rather than drastically decreasing. Following this logic, the point mutations found in clinical GISA and selected by imipenem/teicoplanin step selection (23) are likely to enhance signal transduction rather than leading to YvqF loss of function.
BTH results indicated that the transmembrane protein YvqF interacted directly with VraS but not VraR and was therefore likely to be involved in sensing the unknown cell envelope stress signal responsible for triggering signal transduction, as previously suggested by Jordan et al. (20).
Acknowledgments
This study was carried out with financial support from the Commission of the European Communities, specifically the infectious diseases research domain of the Health Theme of the 7th Framework Programme, contract number 241446, “The effects of antibiotic administration on the emergence and persistence of antibiotic-resistant bacteria in humans and on the composition of the indigenous microbiotas at various body sites,” from the Forschungskredit der Universität Zürich, no. 54232501, to N.M., and the Swiss National Science Foundation grant 31-117707 to B.B.-B.
We are grateful to T. Bae (Department of Microbiology, University of Chicago) for providing the plasmid pKOR1 and to D. Ladant (Unité de Biochimie Cellulaire, Institut Pasteur) for providing the BTH plasmids and host strain. We also thank Cubist Pharmaceuticals for providing daptomycin and Johnson & Johnson Pharmaceutical Research and Development for providing ceftobiprole.
Footnotes
Published ahead of print on 10 January 2011.
REFERENCES
- 1.Bae, T., and O. Schneewind. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58-63. [DOI] [PubMed] [Google Scholar]
- 2.Belcheva, A., and D. Golemi-Kotra. 2008. A close-up view of the VraSR two-component system: a mediator of Staphylococcus aureus response to cell wall damage. J. Biol. Chem. 283:12354-12364. [DOI] [PubMed] [Google Scholar]
- 3.Belcheva, A., V. Verma, and D. Golemi-Kotra. 2009. DNA-binding activity of the vancomycin resistance associated regulator protein VraR and the role of phosphorylation in transcriptional regulation of the vraSR operon. Biochemistry 48:5592-5601. [DOI] [PubMed] [Google Scholar]
- 4.Berger-Bächi, B., and M. L. Kohler. 1983. A novel site on the chromosome of Staphylococcus aureus influencing the level of methicillin resistance: genetic mapping. FEMS Microbiol. Lett. 20:305-309. [Google Scholar]
- 5.Blake, K. L., et al. 2009. The nature of Staphylococcus aureus MurA and MurZ and approaches for detection of peptidoglycan biosynthesis inhibitors. Mol. Microbiol. 72:335-343. [DOI] [PubMed] [Google Scholar]
- 6.Boyle-Vavra, S., S. Yin, and R. S. Daum. 2006. The VraS/VraR two-component regulatory system required for oxacillin resistance in community-acquired methicillin-resistant Staphylococcus aureus. FEMS Microbiol. Lett. 262:163-171. [DOI] [PubMed] [Google Scholar]
- 7.Bugg, T. D. H. 1999. Bacterial peptidoglycan biosynthesis and its inhibition, p. 241-294. In M. Pinto (ed.), Comprehensive natural products chemistry, vol. 3. Elsevier, Oxford, United Kingdom. [Google Scholar]
- 8.Cheung, A. L., K. J. Eberhardt, and V. A. Fischetti. 1994. A method to isolate RNA from Gram-positive bacteria and mycobacteria. Anal. Biochem. 222:511-514. [DOI] [PubMed] [Google Scholar]
- 9.Clinical and Laboratory Standards Institute. 2010. Performance standards for antimicrobial susceptibility testing. M100-S20, vol. 30, no. 1. Clinical and Laboratory Standards Institute, Wayne, PA.
- 10.Cornett, J. B., and G. D. Shockman. 1978. Cellular lysis of Streptococcus faecalis induced with Triton X-100. J. Bacteriol. 135:153-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui, L., H. M. Neoh, M. Shoji, and K. Hiramatsu. 2009. Contribution of vraSR and graSR point mutations to vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob. Agents Chemother. 53:1231-1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eldholm, V., et al. 2010. The pneumococcal cell envelope stress-sensing system LiaFSR is activated by murein hydrolases and lipid II-interacting antibiotics. J. Bacteriol. 192:1761-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan, X., et al. 2007. Diversity of penicillin-binding proteins. Resistance factor FmtA of Staphylococcus aureus. J. Biol. Chem. 282:35143-35152. [DOI] [PubMed] [Google Scholar]
- 14.Gardete, S., S. W. Wu, S. Gill, and A. Tomasz. 2006. Role of VraSR in antibiotic resistance and antibiotic-induced stress response in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:3424-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gill, S. R., et al. 2005. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187:2426-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goda, S. K., and N. P. Minton. 1995. A simple procedure for gel electrophoresis and northern blotting of RNA. Nucleic Acids Res. 23:3357-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbert, S., et al. 2010. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 78:2877-2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Höltje, J. V., and A. Tomasz. 1975. Lipoteichoic acid: a specific inhibitor of autolysin activity in Pneumococcus. Proc. Natl. Acad. Sci. U. S. A. 72:1690-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordan, S., M. I. Hutchings, and T. Mascher. 2008. Cell envelope stress response in Gram-positive bacteria. FEMS Microbiol. Rev. 32:107-146. [DOI] [PubMed] [Google Scholar]
- 20.Jordan, S., A. Junker, J. D. Helmann, and T. Mascher. 2006. Regulation of LiaRS-dependent gene expression in Bacillus subtilis: identification of inhibitor proteins, regulator binding sites, and target genes of a conserved cell envelope stress-sensing two-component system. J. Bacteriol. 188:5153-5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karimova, G., N. Dautin, and D. Ladant. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 187:2233-2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karimova, G., J. Pidoux, A. Ullmann, and D. Ladant. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U. S. A. 95:5752-5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato, Y., T. Suzuki, T. Ida, and K. Maebashi. 2010. Genetic changes associated with glycopeptide resistance in Staphylococcus aureus: predominance of amino acid substitutions in YvqF/VraSR. J. Antimicrob. Chemother. 65:37-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato, Y., et al. 2008. Microbiological and clinical study of methicillin-resistant Staphylococcus aureus (MRSA) carrying VraS mutation: changes in susceptibility to glycopeptides and clinical significance. Int. J. Antimicrob. Agents 31:64-70. [DOI] [PubMed] [Google Scholar]
- 25.Komatsuzawa, H., J. Suzuki, M. Sugai, Y. Miyake, and H. Suginaka. 1994. The effect of Triton X-100 on the in-vitro susceptibility of methicillin-resistant Staphylococcus aureus to oxacillin. J. Antimicrob. Chemother. 34:885-897. [DOI] [PubMed] [Google Scholar]
- 26.Kreiswirth, B. N., et al. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709-712. [DOI] [PubMed] [Google Scholar]
- 27.Kuroda, M., et al. 2003. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 49:807-821. [DOI] [PubMed] [Google Scholar]
- 28.Kuroda, M., K. Kuwahara-Arai, and K. Hiramatsu. 2000. Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strains Mu3 and Mu50 by cDNA differential hybridization method. Biochem. Biophys. Res. Commun. 269:485-490. [DOI] [PubMed] [Google Scholar]
- 29.Martínez, B., A. L. Zomer, A. Rodríguez, J. Kok, and O. P. Kuipers. 2007. Cell envelope stress induced by the bacteriocin Lcn972 is sensed by the lactococcal two-component system CesSR. Mol. Microbiol. 64:473-486. [DOI] [PubMed] [Google Scholar]
- 30.McAleese, F., et al. 2006. Overexpression of genes of the cell wall stimulon in clinical isolates of Staphylococcus aureus exhibiting vancomycin-intermediate-S. aureus-type resistance to vancomycin. J. Bacteriol. 188:1120-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCallum, N., B. Berger-Bächi, and M. M. Senn. 2010. Regulation of antibiotic resistance in Staphylococcus aureus. Int. J. Med. Microbiol. 300:118-129. [DOI] [PubMed] [Google Scholar]
- 32.McCallum, N., A. K. Brassinga, C. D. Sifri, and B. Berger-Bächi. 2007. Functional characterization of TcaA: minimal requirement for teicoplanin susceptibility and role in Caenorhabditis elegans virulence. Antimicrob. Agents Chemother. 51:3836-3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCallum, N., G. Spehar, M. Bischoff, and B. Berger-Bächi. 2006. Strain dependence of the cell wall-damage induced stimulon in Staphylococcus aureus. Biochim. Biophys. Acta 1760:1475-1481. [DOI] [PubMed] [Google Scholar]
- 34.Miller, J. H. 1972. Experiments in molecular genetics, p. 352-355. Cold Spring Harbor Laboratories, Cold Spring Harbor, NY.
- 35.Miyazaki, E., J. M. Chen, C. Ko, and W. R. Bishai. 1999. The Staphylococcus aureus rsbW (orf159) gene encodes an anti-sigma factor of SigB. J. Bacteriol. 181:2846-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muthaiyan, A., J. A. Silverman, R. K. Jayaswal, and B. J. Wilkinson. 2008. Transcriptional profiling reveals that daptomycin induces the Staphylococcus aureus cell wall stress stimulon and genes responsive to membrane depolarization. Antimicrob. Agents Chemother. 52:980-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohta, K., H. Komatsuzawa, M. Sugai, and H. Suginaka. 2000. Triton X-100-induced lipoteichoic acid release is correlated with the methicillin resistance in Staphylococcus aureus. FEMS Microbiol. Lett. 182:77-79. [DOI] [PubMed] [Google Scholar]
- 38.Pietiäinen, M., et al. 2009. Transcriptome analysis of the responses of Staphylococcus aureus to antimicrobial peptides and characterization of the roles of vraDE and vraSR in antimicrobial resistance. BMC Genomics 10:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinho, M. G., H. de Lencastre, and A. Tomasz. 2001. An acquired and a native penicillin-binding protein cooperate in building the cell wall of drug-resistant staphylococci. Proc. Natl. Acad. Sci. U. S. A. 98:10886-10891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohrer, S., and B. Berger-Bächi. 2003. Application of a bacterial two-hybrid system for the analysis of protein-protein interactions between FemABX family proteins. Microbiology 149:2733-2738. [DOI] [PubMed] [Google Scholar]
- 41.Rossi, J., M. Bischoff, A. Wada, and B. Berger-Bachi. 2003. MsrR, a putative cell envelope-associated element involved in Staphylococcus aureus sarA attenuation. Antimicrob. Agents Chemother. 47:2558-2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sass, P., et al. 2008. The lantibiotic mersacidin is a strong inducer of the cell wall stress response of Staphylococcus aureus. BMC Microbiol. 8:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sobral, R. G., et al. 2007. Extensive and genome-wide changes in the transcription profile of Staphylococcus aureus induced by modulating the transcription of the cell wall synthesis gene murF. J. Bacteriol. 189:2376-2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steidl, R., et al. 2008. Staphylococcus aureus cell wall stress stimulon gene-lacZ fusion strains: potential for use in screening for cell wall-active antimicrobials. Antimicrob. Agents Chemother. 52:2923-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suntharalingam, P., M. D. Senadheera, R. W. Mair, C. M. Lévesque, and D. G. Cvitkovitch. 2009. The LiaFSR system regulates the cell envelope stress response in Streptococcus mutans. J. Bacteriol. 191:2973-2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szurmant, H., M. A. Mohan, P. M. Imus, and J. A. Hoch. 2007. YycH and YycI interact to regulate the essential YycFG two-component system in Bacillus subtilis. J. Bacteriol. 189:3280-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Utaida, S., et al. 2003. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 149:2719-2732. [DOI] [PubMed] [Google Scholar]
- 48.Wada, A., Y. Katayama, K. Hiramatsu, and T. Yokota. 1991. Southern hybridization analysis of the mecA deletion from methicillin-resistant Staphylococcus aureus. Biochem. Biophys. Res. Commun. 176:1319-1325. [DOI] [PubMed] [Google Scholar]
- 49.Walsh, T. R., et al. 2001. Evaluation of current methods for detection of staphylococci with reduced susceptibility to glycopeptides. J. Clin. Microbiol. 39:2439-2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang, Q. M., et al. 2001. Identification and characterization of a monofunctional glycosyltransferase from Staphylococcus aureus. J. Bacteriol. 183:4779-4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolf, D., et al. 2010. In-depth profiling of the LiaR response of Bacillus subtilis. J. Bacteriol. 192:4680-4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin, S., R. S. Daum, and S. Boyle-Vavra. 2006. VraSR two-component regulatory system and its role in induction of pbp2 and vraSR expression by cell wall antimicrobials in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:336-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang, J., and I. Biswas. 2009. A phenotypic microarray analysis of Streptococcus mutans liaS mutant. Microbiology 155:61-68. [DOI] [PMC free article] [PubMed] [Google Scholar]