

Abstract
Ribosome-targeting antibiotics exert their antimalarial activity on the apicoplast of the malaria parasite, an organelle of prokaryote origin having essential metabolic functions. These antibiotics typically cause a delayed-death phenotype, which manifests in parasite killing during the second replication cycle following administration. As an exception, treatment with the antibiotic thiostrepton results in an immediate killing. We recently demonstrated that thiostrepton and its derivatives interfere with the eukaryotic proteasome, a multimeric protease complex that is important for the degradation of ubiquitinated proteins. Here, we report that the thiostrepton-based compounds are active against chloroquine-sensitive and -resistant Plasmodium falciparum, where they rapidly eliminate parasites before DNA replication. The minor parasite fraction that escapes the fast killing of the first replication cycle is arrested in the schizont stage of the following cycle, displaying a delayed-death phenotype. Thiostrepton further exhibits gametocytocidal activity by eliminating gametocytes, the sexual precursor cells that are crucial for parasite transmission to the mosquito. Compound treatment results in an accumulation of ubiquitinated proteins in the blood stages, indicating an effect on the parasite proteasome. In accordance with these findings, expression profiling revealed that the proteasome is present in the nucleus and cytoplasm of trophozoites, schizonts, and gametocytes. In conclusion, thiostrepton derivatives represent promising candidates for malaria therapy by dually acting on two independent targets, the parasite proteasome and the apicoplast, with the capacity to eliminate both intraerythrocytic asexual and transmission stages of the parasite.
Proteasomes are highly conserved multimeric protein complexes that are essential in all eukaryotes. Their main purpose is the proteolysis and recycling of nonfunctional proteins, which are tagged for degradation by polyubiquitin tails (13). Proteasomes localize to both the cell nucleus and the cytoplasm and are involved in a variety of cellular processes having high protein turnover rates, such as cell cycle control, stress response, and apoptosis (36). Due to these essential roles, the proteasome has emerged as an important drug target. Several proteasome inhibitors currently are used in the clinic, including bortezomib for the treatment of multiple myeloma (14) and the anti-HIV drug ritonavir (43).
Genome sequence annotation of the human malaria parasite Plasmodium falciparum recently revealed 14 predicted proteins homologous to yeast proteasome subunits (32, 35, 54). In addition to homologs of eukaryotic proteasome proteins, a predicted bacteria-like proteasomal predecessor was identified with similarity to ClpQ/hslV threonine peptidase (17, 32). In malaria parasites, protein quality control is of particular importance because of the high replication rate during liver and blood-stage schizogony and due to shifts in temperature that the parasite experiences during transmission back and forth between human and mosquito. It therefore is likely that proteasome inhibitors will show promise as antimalarial agents.
In an initial study on the efficacy of proteasome inhibitors for malaria therapy, lactacystin was shown to inhibit Plasmodium liver and blood-stage growth prior to DNA synthesis (16). A similar effect was reported for MLN-273 (28), and the activity of this compound against the proteasome was confirmed by demonstrating an accumulation of ubiquitinated proteins in treated parasites. Inhibitors MG132, lactacystin, epoxomicin, and bortezomib inhibit the growth of P. falciparum chloroquine (CQ)-sensitive and -resistant strains prior to DNA synthesis (23, 40). Moreover, epoxomicin recently was reported to exhibit gametocytocidal activity and to reduce the transmission of malaria parasites to the mosquito (10). While these data indicated that proteasome inhibition offers attractive alternatives for malaria chemotherapy, the overall low therapeutic index of most current inhibitors remains a challenge (16).
We recently reported that the thiopeptide antibiotic thiostrepton and newly generated derivatives have an impact on the caspase-like activity of the human proteasome in vitro (46). Thiostrepton inhibits bacterial protein biosynthesis by binding to the GTPase-associated center on the 70S ribosome (4, 19, 21) and is predicted to mediate antiplasmodial activity by targeting the translation machinery of the malaria parasite apicoplast (31, 41). This semiautonomous organelle of prokaryotic origin plays a crucial role in a variety of metabolic and housekeeping functions, including fatty acid, isoprenoid, and heme biosynthesis (reviewed in references 26 and 38). Ribosome-targeting antibiotics like azithromycin, telithromycin, or the tetracyclines interfere with apicoplast function by inhibiting the prokaryotic-like translation machinery of the organelle (3, 12, 49). This results in a slow killing of the parasite, termed delayed death, in which parasite growth is arrested at the schizont stage of the second replication cycle following antibiotic treatment. The delayed-death phenotype is defined as the requirement for high initial drug concentrations to achieve moderate growth inhibition after 48 h and an approximate 10-fold-increased activity when the parasites have entered the second replication cycle (reviewed in references 11 and 38). In contrast to these antibiotics, thiostrepton treatment leads to a fast killing of the malaria parasite (18, 49), thus confounding the explanation for the atypical kinetics of parasite killing.
Here, we describe that thiostrepton and its derivatives target the proteasome of the human malaria parasite P. falciparum, resulting in rapid parasite elimination prior to DNA replication. Parasites that escaped the immediate killing then are caught by the interference of the compounds with the apicoplast. In addition to the effect on the asexual blood stages, the compounds exhibit gametocytocidal activity. In conclusion, we show that thiostrepton and derivatives target the 20S proteasome in the parasite with a very beneficial activity profile.
MATERIALS AND METHODS
Gene identities.
The following gene identifiers are assigned to the proteins investigated in this study: Pf39, PF11_0098; PfCCp1, PF14_0723; P. falciparum AMA-1, PF11_0344; P. falciparum MSP-1, PFI1475w; P. falciparum proteasome alpha subunit (α-SU) type 5, PF07_0112; and P. falciparum proteasome beta subunit (β-SU) type 5, PF10_0111.
Compounds.
The following compounds were used in this study: azithromycin (LKT Laboratories), chloroquine (Sigma-Aldrich), doxycycline (Invitrogen); epoxomicin (Sigma-Aldrich), MG132 (Sigma-Aldrich), primaquine diphosphate (Sigma-Aldrich), and thiostrepton (Sigma-Aldrich). The thiostrepton-based compounds were synthesized as described previously (46). The synthesis of the fluorescent thiostrepton probe was described previously (45).
Cell culture.
P. falciparum CQ-sensitive strain 3D7 and CQ-resistant strain Dd2 were cultivated in human erythrocytes with Albumax II medium as described previously (9, 52). RPMI 1640 medium (Gibco) was supplemented with hypoxanthine (Sigma-Aldrich), 0.5% Albumax II (Invitrogen), and gentamicin (Invitrogen), and cultures were maintained at 37°C in an atmosphere of 5% O2, 5% CO2, 90% N2. Cultures were synchronized by repeated sorbitol treatment as described previously (25). Briefly, cultures containing mainly ring-stage parasites were centrifuged at 1,500 rpm, and the pellet was resuspended in 5% sorbitol. After 10 min of incubation at room temperature, the cells were washed once with culture medium, diluted to 5% hematocrit, and cultured as described above. To generate gametocytes, P. falciparum isolate strain NF54 was cultivated in RPMI 1640 medium (Gibco) in vitro in the presence of 10% inactivated human serum (Bavarian Red Cross) as described previously (20). HeLa (human epithelial cervical cancer) cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and l-glutamine at 37°C (Sigma-Aldrich) and a 5% CO2 atmosphere.
Malstat assay.
Compounds were screened for growth inhibition activity against P. falciparum at concentrations of 10 nM to 100 μM using the Malstat assay as described previously (29, 30). Synchronized ring stages of P. falciparum strains 3D7 and Dd2 were plated in triplicate in 96-well plates (200 μl/well) at a parasitemia of 1% (if not indicated otherwise) in the presence of the compounds dissolved in dimethyl sulfoxide (DMSO). Chloroquine (dissolved in double-distilled water), epoxomicin, and MG132 (both dissolved in DMSO) served as positive controls in the experiments. The incubation of parasites with DMSO alone at a concentration of 0.5% was used as a negative control. Parasites were cultivated in vitro for 72 h and resuspended, and aliquots of 20 μl were removed and added to 100 μl of the Malstat reagent in a 96-well microtiter plate. The assessment of pLDH activity was obtained by adding 20 μl of a mixture of NBT (Nitro Blue Tetrazolium)-diaphorase (1:1; 1 mg/ml stock each) to the Malstat reaction, and optical densities were measured at 630 nm (OD630). Each compound was tested two to four times, and the 50% inhibitory concentrations (IC50s) were calculated from variable-slope sigmoidal dose-response curves using the GraphPad Prism program version 4. For the delayed-death test, the Malstat assay was carried out as described above at 48, 72, 96, and 120 h after drug treatment with an initial parasitemia of 0.5%, and the respective IC50s were calculated.
MTT cytotoxicity test.
HeLa cells were plated in 96-well microtiter plates and cultured at 37°C overnight. Compounds were added to the cultures in concentrations of 0.8 to 100 μM in triplicate, and the cells were incubated for 48 h. DMSO at a final concentration of 1% was used as a negative control, and a toxic concentration of 50% DMSO was applied as a positive control. After 48 h of drug treatment, MTT [3-(4, 5-dimethylthiazol-2-yl)-2,5 diphenyltetrazoliumbromid; 5 mg/ml stock] was added to each well at a final concentration of 1 mg/ml and incubated for 3 h at 37°C. The medium subsequently was replaced by 100 μl solubilization solution (5% SDS, 10 mM HCl in 50% DMSO) and incubated for 5 min under rotation at room temperature to solubilize the formazan crystals. The optical density of the solution was measured at 550 nm (Multiscan Ascent). The OD550s of compound-treated cultures were normalized to the OD550s of the DMSO control (set to 100%).
Erythrocyte pretreatment.
A volume of 1 ml of uninfected washed erythrocytes was incubated for 3 h at 37°C in the presence of thiostrepton derivatives (5 μM), MG 132 (250 nM), epoxomicin (125 nM), and CQ (250 nM). After incubation, erythrocytes were washed twice with incomplete medium and once with Albumax II medium. Ring-stage cultures were added to the pretreated erythrocytes in a final concentration of 0.5% parasitemia and cultured for 72 h. Parasite viability subsequently was measured using the Malstat assay (as described above).
Gametocyte toxicity test.
P. falciparum NF54 parasites were grown at high parasitemia to favor gametocyte formation. Upon the appearance of gametocyte stage II, 1 ml of culture was aliquoted in triplicate in a 24-well plate in the presence of compounds at their respective IC50s or IC90s. The gametocytes were cultivated for 7 days, and the medium was replaced daily. For the first 48 h of cultivation, the gametocytes were treated with test compounds at IC50s or IC90s, and subsequently the medium was compound free. After 7 days, Giemsa-stained blood smears were prepared and the gametocytemia was evaluated by counting the numbers of gametocytes of stage IV and V in a total number of 1,000 erythrocytes.
Erythrocyte lysis test.
A volume of 1 ml of washed uninfected erythrocytes was resuspended in Albumax II medium at a final hematocrit of 5% and plated in triplicate in a 96-well plate (200 μl/well) in the presence of compounds at their IC50s. Medium supplemented with 0.15% saponin was use for the lysis control, and 0.5% DMSO was used for the negative control. After 48 h of incubation at 37°C, the plate was centrifuged at 500 × g for 2 min. A volume of 100 μl of the resulting supernatant was carefully transferred to another plate, and the optical density was measured at 550 nm (Multiscan Ascent).
RNA isolation and transcript level analysis.
Synchronized P. falciparum NF54 asexual blood-stage parasites were harvested at the ring, trophozoite, or schizont stage and treated with 0.15% saponin for the lysis of erythrocytes. Gametocytes were enriched from mixed gametocyte cultures by Percoll gradient purification (22). RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. RNA preparations were treated with RNase-free DNase I (Invitrogen) to remove genomic DNA contamination, followed by phenol-chloroform extraction and ethanol precipitation. The synthesis of cDNA was accomplished using a Superscript II cDNA synthesis kit (Invitrogen). Reverse transcription-PCR (RT-PCR) assays were performed with 125 ng cDNA using gene-specific primers by conventional thermocycling (25 cycles) separated by 2% agarose gel electrophoresis. The following primers were used: PfAMA-1 S, 5′-GGA TTA TGG GTC GAT GGA-3′; PfAMA-1 AS, 5′-GAT CAT ACT AGC GTT CTT-3′; PfCCp1 S, 5′-CTG GTA TGG ACC ATT ATG TTG GG-3′; PfCCp1 AS, 5′-CGA AAT TAC AGA AGA ATC AAC ACC ATG-3′; α-SU type 5 S, 5′-AGT TGA ATA TGC CTT AGG-3′; α-SU type 5 AS, 5′-GAT TAC ACT CGA CTC TTG-3′; β-SU type 5, 5′-TTG CAG TAG ATT CCC GAG-3′; and β-SU type 5 AS, 5′-CTA GCT GCA CGT ACT GAT-3′.
Recombinant protein expression and production of mouse antisera.
Recombinant proteins were expressed as fusion proteins with a glutathione S-transferase (GST) tag using the pGEX 4T-1 vector (Amersham Bioscience). The coding sequence of proteasome β-SU type 5 was amplified by PCR from genomic DNA extracted from P. falciparum 3D7 culture using the following primers: β-SUrp1 S, 5′-TAGGATCCATGGTAATAGCAAG-3′; and β-SUrp1 AS, 5′-TAGCGGCCGCTCACATAACATATTG-3′. Cloning was mediated by BamHI and NotI restriction sites (underlined). Recombinant proteins were expressed in BL21(DE3) RIL cells according to the manufacturer's protocol (Stratagene). Inclusion bodies containing recombinant proteins were purified from bacterial extracts as described previously (44). Specific immune sera were generated by immunizing 6-week-old female NMRI mice (Charles River Laboratories) with 100 μg recombinant protein emulsified in Freund's incomplete adjuvant (Sigma-Aldrich), followed by a second immunization after 4 weeks. Sera were collected 10 days after the second immunization. The housing and handling of the animals followed the guidelines of the animal welfare committee of Lower Franconia.
Western blot analysis.
Synchronized P. falciparum 3D7 asexual blood-stage parasites were harvested at the ring, trophozoite, or schizont stage and treated with 0.15% saponin for erythrocyte lysis. Nonactivated NF54 gametocytes were enriched from mixed gametocyte cultures by Percoll gradient purification (22). Parasite pellets were washed with phosphate-buffered saline (PBS), resuspended, and sonicated in lysis buffer (20 mM Tris-HCl, pH 8.0, 10 mM EDTA, pH 8.0, 400 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF], 10 mM β-glycerophosphate, 10 mM NaF, 0.25% Triton X-100) supplemented with a protease inhibitor cocktail (Sigma-Aldrich). Parasite proteins were separated by SDS-PAGE and transferred to Hybond ECL nitrocellulose membrane (Amersham Biosciences) according to the manufacturer's protocol. Membranes were blocked for nonspecific binding by incubation in blocking buffer (1× Tris-buffered saline [TBS], 5% nonfat milk powder, 0.5% bovine serum albumin [BSA]) for 1 h. Blots were washed and incubated for 2 h or overnight with primary antibody, i.e., mouse anti-β SU type 5 (1:50), mouse anti-ubiquitin antibody P4D1 (1:500; Santa Cruz), or mouse anti-Pf39 (1:500) diluted in 1× TBS, 3% milk. The blots subsequently were washed and incubated for 1 h with secondary goat anti-mouse antibody conjugated to alkaline-phosphatase (Sigma-Aldrich) diluted 1:5,000 in 1× TBS, 0.1% Tween 20, 3% milk. Membranes were washed and developed in NBT-5-bromo-4-chloro-3-indolylphosphate (BCIP) solution (Sigma-Aldrich) for 5 to 30 min. Scanned blots were processed using Adobe Photoshop CS software.
Indirect immunofluorescence assay.
Synchronized P. falciparum 3D7 parasite cultures at ring, trophozoite, and schizont stages, as well as nonactivated NF54 gametocyte cultures, were air dried on slides and fixed for 10 min in −80°C methanol. The specimens were incubated in blocking buffer (1% neutral goat serum, 0.01% saponin, and 0.5% BSA in PBS) for 30 min at room temperature. After being blocked, slides were incubated for 2 h at 37°C with mouse anti-β SU type 5 (1:20) or rabbit anti-MSP-1 (1:1,000; ATCC) primary antibody diluted in blocking buffer. Specimens were washed three times with 0.01% saponin in PBS and incubated with fluorochrome-coupled goat anti-mouse or anti-rabbit secondary antibodies (Alexa Fluor 488 and 594; Molecular Probes). The counterstaining of erythrocytes was performed using 0.05% Evans blue in PBS for 1 min (Sigma-Aldrich). The slides were rinsed twice in PBS and then stained with Hoechst 33342 (Invitrogen) according to the manufacturer's protocol. Specimens were mounted on a coverslip with antifade mounting medium (Bio-Rad). Labeled specimens were examined by confocal laser-scanning microscopy using a Leica TCS SP5 (see Fig. 5A) or a Zeiss Axiolab fluorescence microscope in combination with a Zeiss AxioCam ICc1 camera (Fig. 5C). Digital images were processed using Adobe Photoshop CS software.
Immunoelectron microscopy.
Intraerythrocytic asexual-stage parasites of P. falciparum NF54 were fixed in 4% paraformaldehyde in PBS overnight and treated with 50 mM NH4Cl in PBS for 15 min to block aldehyde groups. Specimens were dehydrated in increasing concentrations of ethanol at −25°C and then incubated for 1 h in a 1:1 mixture of 100% ethanol and LR White (Scientific Services) at 4°C. Specimens subsequently were embedded in LR White at 40°C for 3 days. Ultrathin sections of specimens were subjected to postembedding labeling. Samples were blocked in 1% BSA and 0.1% Tween 20 in PBS for 10 min prior to immunolabeling with mouse anti-β SU type 5 antibodies (1:50). The binding of primary antibody was visualized using 12-nm gold-conjugated goat anti-mouse secondary antibodies (Dianova). Ultrathin sections were postfixed with 1% glutaraldehyde in PBS and poststained with 2% uranylacetate. Photographs were taken with a Zeiss EM10 transmission electron microscope, and scanned images were processed using Adobe Photoshop.
RESULTS
Thiostrepton derivatives exhibit antimalarial activity.
We recently generated a number of thiostrepton derivatives by the selective oxidization of the thiazoline residue to improve chemical stability and created a small focused library of candidate compounds via combinations of tail truncation, oxidation, and the addition of lipophilic thiols to the terminal dehydroamino acid (46). A selection of these derivatives, SS231/[14], SS234/[05], SS238/[09], SS257/[13], and SS266/[10] (Fig. 1 ), was tested for their antimalarial activity in the CQ-sensitive strain 3D7 and the CQ-resistant strain Dd2 of P. falciparum. The antimalarial activity of thiostrepton and derivatives was determined by the Malstat viability assay, which measures the activity of the Plasmodium-specific enzyme lactate dehydrogenase, and it was calculated as growth inhibition at a 50% inhibitory concentration (IC50).
FIG. 1.

Chemical structures of thiostrepton and derivatives. Dashed frame, thiazoline residue; gray frame, thiazole; black frame, appendage.
Thiostrepton exhibited a modest antimalarial activity, with an IC50 of 8.9 μM. The thiostrepton-based derivatives revealed an increased activity, with an 8-fold higher activity for derivatives SS231/[14] and SS234/[05] (IC50s of 1.0 μM each) (Table 1). No differences were observed between the activities of compounds against CQ-sensitive and CQ-resistant malaria parasites. Additionally, the proteasome inhibitors epoxomicin and MG132 were tested (Table 1). Among several proteasome inhibitors, epoxomicin and MG132 were shown previously to exhibit the highest antiplasmodial activity of the various laboratory strains (23). We found growth inhibition with IC50s of 0.03 and 0.05 μM, respectively. CQ was used as a positive control in the assay (Table 1).
TABLE 1.
Antimalarial activities of the compounds under studyb
| Compound | IC50 [μM] |
||
|---|---|---|---|
| Antimalarial activity |
Cytotoxicity for HeLa | ||
| 3D7 | Dd2 | ||
| Thiostrepton | 8.9 ± 1.70 | 16.7 ± 3.27 | 27.8 ± 10.89 |
| SS231/[14] | 1.0 ± 0.44 | 1.7 ± 0.27 | >100 |
| SS234/[05] | 1.0 ± 0.10 | 1.3 ± 0.16 | >100 |
| SS238/[09] | 3.0 ± 0.29 | 2.3 ± 1.08 | 21.1 ± 10.32 |
| SS257/[13] | 1.5 ± 0.40 | 0.77 ± 0.097 | >100 |
| SS266/[10] | 3.5 ± 0.44 | 1.3 ± 0.10 | >100 |
| MG132 | 0.05 ± 0.025 | 0.04 ± 0.001 | NA |
| Epoxomicin | 0.03 ± 0.001 | NT | NA |
| Chloroquine | 0.03 ± 0.002 | 0.45a | NA |
Tested once.
NA, not applicable; NT, not tested.
Compound cytotoxicity was tested in HeLa cells by the MTT viability assay, which measures the activity of the human mitochondrial dehydrogenases. Among thiostrepton derivatives, only SS238/[09] showed a moderate cytotoxic effect on HeLa cells (IC50 = 21.1 μM) (Table 1). Moderate cytotoxicity was further observed for thiostrepton (IC50 = 27.8). The cytotoxic effect of thiostrepton on human cancer cell lines has been reported previously (5). Additionally, the possible effect of thiostrepton and derivatives on erythrocyte integrity was investigated. Treatment with compounds did not increase the amount of free hemoglobin in the medium compared to levels for controls treated with medium alone or with 0.5% DMSO (see Fig. S1A in the supplemental material). Erythrocyte lysis with saponin was used as a positive control, and it resulted in a 26-fold increase in OD550s of the supernatant.
Thiostrepton derivatives eliminate malaria parasites in two steps.
To gain phenotypic insight into the mode of action of thiostrepton and derivatives, we investigated the stage of growth inhibition via blood-stage quantification. Compounds were added to synchronized parasites at IC80s either at the ring, trophozoite, or early schizont stage (Fig. 2). Blood smears were taken at seven time points between 0 and 48 h of drug treatment and stained with Giemsa. The proportions of ring stages, trophozoites, early schizonts, and mature schizonts were counted under the microscope, and the parasitemia was calculated. The two most active compounds, SS231/[14] and SS234/[05], as well as thiostrepton and MG132, were chosen for these experiments, and results were compared to those of DMSO treated control parasites.
FIG. 2.

Stage of growth inhibition of P. falciparum during 48 h of compound treatment. Compounds at IC80 or in a 0.5% vol of DMSO were added to synchronized parasites at the ring, trophozoite, or early schizont stage. Giemsa-stained blood smears were prepared at seven time points between 0 and 48 h of compound incubation, and the numbers of ring stages, trophozoites, early schizonts, and mature schizonts were counted. Histograms indicate the percentages of developmental stages present in the respective blood smears. The respective parasitemia (P) is indicated above each column. Fifty parasites were counted for each condition. In samples with low parasite numbers, 20 parasites were counted. RS, ring stage; SZ, schizont; TZ, trophozoite.
While control cultures of P. falciparum completed the typical 48-h replication cycle, the growth of parasites was stopped immediately when MG132 was added to the ring or trophozoite stage prior to DNA replication (Fig. 2, left and center). When added to the early schizont stage, MG132 was not able to eliminate all parasites within the first replication cycle, and a fraction of them escaped the immediate killing and entered the second cycle (Fig. 2, right).
Similarly to MG132, thiostrepton and derivatives were able to eliminate the majority of parasites in the ring and trophozoite stages when the inhibitors were added to these stages (Fig. 2, left and center). A minority of parasites, however, escaped the immediate killing and entered the second replication cycle. When added to the early schizont stage, the compounds were not able to eliminate all parasites within the first cycle, which was particularly obvious for SS231/[14]- and SS234/[05]-treated cultures (Fig. 2, right). Here, the majority of parasites entered the second replication cycle and were present as ring stages at 48 h of drug treatment.
To investigate the fate of the parasite fraction that had escaped the immediate killing and entered the second replication cycle, we applied a delayed-death test and determined parasite growth inhibition at 48, 72, 96, and 120 h of drug treatment. The ribosome-targeting antibiotics doxycycline and azithromycin were used as positive controls. These antibiotics were previously reported to induce the typical delayed-death phenotype (3, 12, 49). As expected, treatment with doxycycline and azithromycin showed a more than 10-fold decrease in IC50s between 48 and 96 h of incubation time (Table 2). The treatment of parasites with thiostrepton, SS231/[14], and SS234/[05] also resulted in a decrease of the respective IC50s during the incubation time. However, only approximately 3-fold decreases in the IC50s were detected between 48 and 96 h of drug treatment. At 120 h of incubation time, compounds SS231/[14] and SS234/[05] exhibited antimalarial activities with IC50s of 0.46 and 0.26 μM, respectively.
TABLE 2.
Antimalarial activities of compounds over timea (delayed-death test)
| Compound | IC50 (μM) at: |
|||
|---|---|---|---|---|
| 48 h | 72 h | 96 h | 120 h | |
| Doxycycline | 17.6 | 5.4 | 3.1 | 0.52 |
| Azithromycin | 18.3 | 2.1 | 4.1 | 0.12 |
| Thiostrepton | 8.2 | 4.7 | 2.8 | 2.1 |
| SS231/[14] | 1.6 | 0.62 | 0.52 | 0.46 |
| SS234/[05] | 2.0 | 0.46 | 0.57 | 0.26 |
All compounds were tested once.
We then quantified the stage of growth inhibition for compound-treated parasites screened in the delayed-death test (Fig. 3A). Parasites were treated with compounds at IC50s. CQ was used as an immediate killing control and azithromycin as a delayed-death control (not shown) in the tests. The DMSO-treated control went through the 48-h replication cycle, and ring stages were observed at 0, 48, and 96 h of culturing (Fig. 3A). Treatment with CQ, which interferes with the biocrystallization of heme following hemoglobin degradation in the parasite (50), resulted in a strong reduction of parasite numbers during each replication cycle, while a minor fraction of parasites continued to grow (Fig. 3A). In parasites treated with thiostrepton, SS231/[14], or SS234/[05], parasite numbers decreased drastically during the first replication cycle. Interestingly, the fraction of parasites that entered the second cycle was arrested in the trophozoite/schizont stage (Fig. 3A), thus displaying the delayed-death phenotype typical for apicoplast-targeting antibiotics like azithromycin, doxycycline, and telithromycin (3).
FIG. 3.

Delayed-death effect and gametocytocidal activity of compounds. (A) For the delayed-death test, compounds at IC50s or a 0.5% volume of DMSO was added to synchronized ring-stage parasites. Giemsa-stained blood smears were made at 0, 48, 72, 96, and 120 h of incubation with inhibitor, and the developmental stages were quantified as described for Fig. 1. The respective parasitemia (P) is indicated above each column. A total number of 50 parasites were counted for each condition. In samples with low parasite numbers, 20 parasites were counted. (B) Compounds at IC50s or IC90s or a 0.5% volume of DMSO were added to stage II gametocyte cultures for 2 days. The numbers of stage IV and V gametocytes were counted after 7 days and correlated to the gametocyte numbers of the DMSO control (set to 100%). An asterisk indicates the significant reduction of gametocyte numbers (P < 0.05, Student's t test). AZ, azithromycin; PQ, primaquine.
Thiostrepton derivatives exhibit gametocytocidal activity.
We also investigated the effect of the thiostrepton-based compounds on gametocyte development. Gametocytes are intraerythrocytic sexual precursor cells that are formed by the parasite in response to stress and that mediate the transition of the parasite from the human to the mosquito (reviewed in references 24 and 37). The gametocytocidal effect of a compound would block the transmission of parasites that escaped the blood-stage killing and thus counteract the propagation of parasite genotypes that confer drug resistance.
Thiostrepton and derivatives SS231/[14] and SS234/[05] were added to young gametocyte cultures of stage II. The numbers of gametocytes of stages IV and V were counted 7 days later and compared to results for DMSO-treated control gametocytes (Fig. 3B). For a positive control, gametocytes were treated with epoxomicin and primaquine, while azithromycin-treated gametocytes were used for the negative control. The gametocyte toxicity assay revealed that epoxomicin and thiostrepton fully eliminated gametocytes in the cultures at IC50s, while primaquine (IC50 activity on asexual blood stages of 3 μM) resulted in a 62% reduction of gametocyte numbers (Fig. 3B). Treatment with MG132, SS231/[14], and SS234/[05] did not result in a significant reduction of gametocytes when added at IC50s, while a significant decrease in gametocyte numbers was observed when these compounds were added at IC90s. Azithromycin treatment had no significant effect on the development of gametocytes (Fig. 3B).
Thiostrepton and derivatives interfere with the parasite proteasome.
The rapid elimination of malaria parasites treated with thiostrepton and derivatives leads to the assumption that the compounds have a second target in addition to the apicoplast. All compounds have been shown previously to inhibit human proteasome in vitro, therefore an effect on the parasite proteasome can be expected.
To exclude the growth inhibition of parasites due to an effect on the erythrocyte proteasome, uninfected erythrocytes were incubated with thiostrepton and derivatives as well as with MG132, epoxomicin, and CQ for 48 h. Pretreated erythrocytes then were used to culture parasites, and growth inhibition was quantified by the Malstat assay. Parasite growth was found to be unaffected for any of the pretreated erythrocyte cultures after 72 h compared to data for untreated controls (see Fig. S1B in the supplemental material), indicating that the pretreatment of erythrocytes with proteasome inhibitors has no effect on parasite viability.
We then investigated the effect of thiostrepton and derivatives on the parasite proteasome by monitoring ubiquitinated proteins. Synchronized parasites at the early trophozoite stage were treated with the compounds for 6 h and harvested, and the lysates were screened by Western blotting using mouse anti-ubiquitin antibody. Treatment with MG132 was used for the positive control and CQ treatment for the negative control in the assays. Western blotting revealed an accumulation of ubiquitinated proteins in parasites treated with MG132, thiostrepton, or derivatives compared to results for the 0.5% DMSO-treated control (Fig. 4, upper). Labeling for ubiquitinated proteins was particularly strong when parasites were treated with thiostrepton, SS234/[05], or SS238/[09]. No accumulation of ubiquitinated proteins was detected in CQ-treated parasites (Fig. 4, upper). The Coomassie staining of protein gels was used to show equal loading (Fig. 4, lower).
FIG. 4.

Accumulation of ubiquitinated blood-stage proteins following compound treatment. Synchronized early trophozoites were incubated for 6 h with compounds at IC80s or with a 0.5% volume of DMSO. Parasite extracts were separated by polyacrylamide gel electrophoresis and screened via Western blot analysis. Ubiquitinated parasite proteins were detected with mouse antibody against ubiquitin (upper). The Coomassie blue staining of protein gels was used to demonstrate the equal loading of parasite protein extract (lower).
Expression and subcellular distribution of the 20S proteasome in blood stages of P. falciparum.
The identification of genes in the P. falciparum genome that are homologous to proteasome subunits of other eukaryotes strongly suggests the presence of a 20S proteasome in the malaria parasite. However, before our study no detailed expression and subcellular localization data were available. To study the proteasome expression in blood-stage parasites, we performed diagnostic RT-PCR analysis using primers corresponding to distinct regions in the α-SU type 5 and the β-SU type 5 gene sequences. Synchronized parasites were harvested in the ring, trophozoite, schizont, or gametocyte stage, and cDNA was generated from isolated parasite RNA. Quality was verified by monitoring transcripts of stage-specific marker proteins, e.g., AMA-1 for asexual blood stages or PfCCp1 for gametocytes (Fig. 5A). No PCR products were amplified from mock-treated RNA samples lacking reverse transcriptase, indicating that genomic DNA was absent (data not shown). The RT-PCR analysis of stage-specific cDNA using α-SU type 5- and β-SU type 5-specific primers revealed corresponding PCR products in all samples, indicating that these putative proteasome subunits are expressed in these stages. However, transcript for both subunits was less abundant in ring-stage parasites than in trophozoites, schizonts, and gametocytes (Fig. 5A).
FIG. 5.
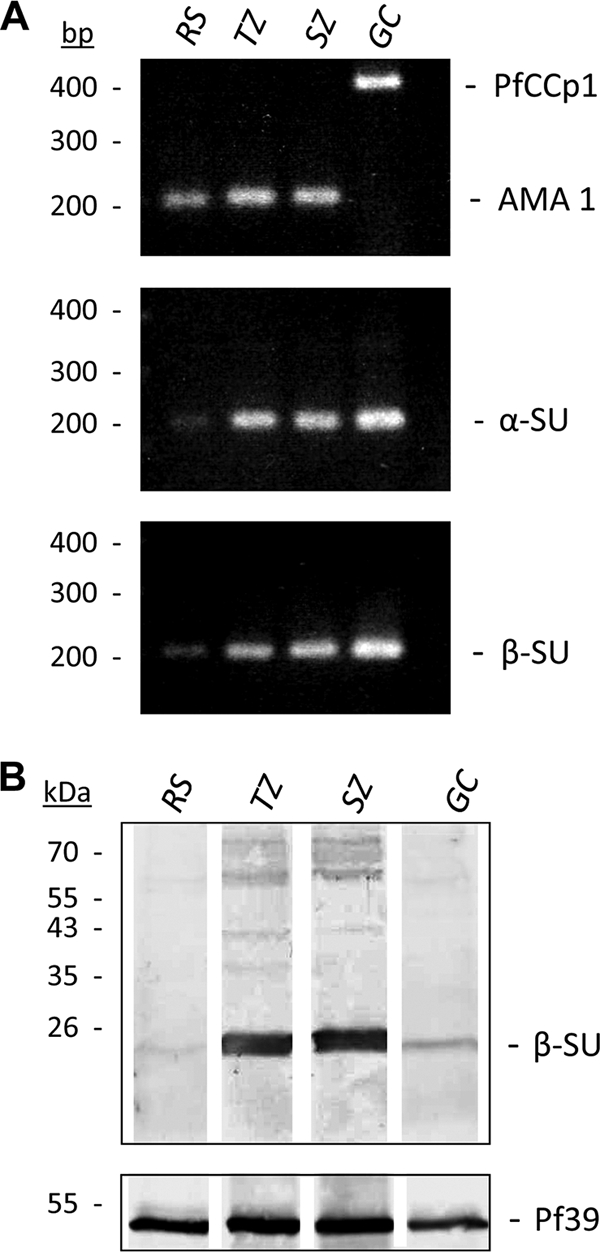
Expression of proteasome subunits in the blood stages of P. falciparum. (A) Diagnostic RT-PCR analysis on cDNA from ring, trophozoite, schizont, or gametocyte stage indicated transcript expression for proteasome subunits α-SU type 5 (224 bp) and β-SU type 5 (200 bp) in all blood stages with a low transcript abundance in ring stages. The detection of transcript for the asexual marker gene AMA-1 (189 bp) and the gametocyte marker gene PfCCp1 (371 bp) were used as quality and equal-loading controls. (B) Protein expression of β-SU type 5 in parasite extracts was demonstrated by Western blot analysis using mouse polyclonal antisera (upper). The detected protein band migrates at an approximate molecular mass of 25 kDa (calculated full-length protein, 31 kDa). Screening with antisera against the endoplasmic reticulum protein Pf39 was used to demonstrate the equal loading of parasite extracts (lower). GC, gametocyte; RS, ring stage; SZ, schizont; TZ, trophozoite.
The presence of proteasomes in blood-stage parasites subsequently was investigated by Western blot analysis. Antisera against recombinant P. falciparum 3D7 β-SU type 5 protein were generated by the immunization of mice and blotted against SDS-PAGE-separated lysates of ring stages, trophozoites, schizonts, and mature gametocytes. Western blotting detected one protein band with a molecular mass of approximately 25 kDa (Fig. 5B, upper). The protein was prominent in the trophozoite and schizont stages but less abundant in ring stages and gametocytes. Blotting against the endoplasmic reticulum-specific protein Pf39 was used as a control for equal protein loading (Fig. 5B, lower).
The gene sequence of P. falciparum proteasome β-SU type 5 indicates a molecular mass of 31 kDa for the proprotein, but most of the proteasome β-SU proteins undergo proteolysis during proteasome maturation (7, 15, 47) to unmask their active-site residue. At the predicted cleavage site, the amino acids Gly(−1) and Thr(1) are conserved in all active proteasome beta subunits, with Gly(−1) being important for the recognition of the cleavage site and Thr(1) being the catalytic nucleophile (42, 48). The P. falciparum proteasome β-SU type 5 possesses the conserved GTTT active domains described for β-SU type 5 of humans (see Fig. S2 in the supplemental material) and other organisms. The cleavage between Gly(−1) and Thr(1) is predicted to release an active protein of 23.5 kDa (size estimated by the ExPASy Proteomics Server), which correlates well with the observed protein band at 25 kDa. The nonprocessed protein was not detected in Western blot analysis (Fig. 5B).
We also investigated the subcellular localization of proteasome β-SU 5 in the parasite blood stages by immunofluorescence assay. While there was minor punctuate labeling detected in the ring stages (Fig. 6A, RS), β-SU type 5 was abundantly expressed in trophozoites and schizonts, where it was present in the cytoplasm and associated with the parasite nucleus (Fig. 6A, TZ and SZ). In gametocytes, β-SU type 5 was localized primarily to the cytoplasm (Fig. 6A, GC). Uninfected erythrocytes did not label for β-SU type 5, and sera of nonimmunized mice did not result in any labeling of the parasite (data not shown). Immunoelectron microscopy subsequently revealed that β-SU type 5 is abundantly present in the nuclei of trophozoites and schizonts but not in the nucleolus (Fig. 6B). The protein additionally is present in the cytoplasm of blood-stage parasites. No gold labeling was detected in the erythrocyte, the parasite rhoptries, or the food vacuole (Fig. 6B).
FIG. 6.

Subcellular localization of β-SU type 5 in the P. falciparum blood stages. (A) Indirect immunofluorescence assays using mouse polyclonal antisera against β-SU type 5 revealed the localization of the protein in the ring, trophozoite, schizont, or gametocyte stage (green; Alexa Fluor 488). Nuclei were highlighted by Hoechst nuclear staining (blue), and erythrocytes were counterstained with Evans blue (red). Bar, 5 μm. (B) Immunoelectron microscopy using anti-β-SU type 5 antisera in combination with gold (12-nm)-coupled secondary antibody indicated the ultrastructural localization of the protein in trophozoites and schizonts. Arrowheads indicate gold particles. Bar, 1 μm. (C) Incubation of asexual blood-stage parasites with a fluorescent thiostrepton probe (termed MP40) revealed the subcellular binding of thiostrepton in a schizont. Parasites were visualized by double labeling with anti-MSP-1 antibody (red, Alexa Fluor 596), and nuclei were highlighted by Hoechst staining (blue). Arrows indicate hemozoin in the parasite food vacuole. Bar, 5 μm. BF, bright field; C, cytoplasm; E, erythrocyte; FV, food vacuole; GC, gametocyte; N, nucleus; No, nucleolus; R, rhoptry; RS, ring stage; SZ, schizont; TZ, trophozoite.
Lastly, we visualized the binding of a fluorescent thiostrepton probe (45) to blood-stage parasites. Parasites were incubated with the probe for 4 h, fixed with methanol, and processed for immunofluorescence assay. The fluorescent probe was localized mainly in schizonts associated with the nuclei in a staining pattern similar to that found with anti-β-SU 5 antisera (Fig. 6C). No labeling was detected in the food vacuole of treated parasites (Fig. 6C, arrow) or when the parasites were incubated with a fluorescent control probe, which lacks thiostrepton (data not shown).
DISCUSSION
The P. falciparum genome possesses genes predicted to encode all 14 subunits of an eukaryote-type 20S proteasome. The genome additionally encodes a predicted homolog of the bacterium-like threonine protease ClpQ/hslV homolog PfhslV (17, 32), a heat shock protein encoded by the heat shock locus V (hslV) that is regarded as a phylogenetic ancestor of the 20S proteasome (6). We recently described that the antiplasmodial activities of the antibiotic thiostrepton and newly obtained semisynthetic thiostrepton derivatives correlate with the in vitro inhibition of the human 20S proteasome, where they preferentially inhibited the caspase-like activity of the proteasome β-SU type 1 (46). Thiostrepton and structurally related thiazole antibiotics were known previously to block protein translation in bacteria by binding tightly to the GTPase-associated region on the 70S ribosome (1, 2, 4, 19, 21). Likewise, thiostrepton also binds to the similar apicoplast ribosome and inhibits plastid protein synthesis, suggesting a first mode of action on P. falciparum culture (8, 31). In contrast to other antibiotics, thiostrepton did not cause a delayed-death phenotype, instead resulting in the immediate killing of the parasites (18, 49).
Here, we show that the immediate killing effect of thiostrepton and its derivatives correlates with the inhibition of the parasite 20S proteasome. The treatment of P. falciparum blood stages with these compounds results in an immediate killing at the trophozoite stage similarly to the killing mechanism of the proteasome inhibitor MG132. Neither MG132 nor thiostrepton and derivatives are able to eliminate the parasites at the schizont stage after the initiation of DNA replication. This is in accordance with previous findings on the antimalarial effect of proteasome inhibitors, which describe parasite growth inhibition prior to DNA synthesis (23, 40). The treatment of blood-stage parasites with MG132 or with the thiostrepton-based compounds led to the accumulation of ubiquitinated proteins, supporting the proteasome as the target of thiostrepton derivatives. Notably, thiostrepton has been reported to be active against cancer cell proliferation (5), which likely can be explained by proteasome inhibition as well.
Compound-treated parasites that have escaped killing during the first replication cycle then were arrested in the schizont stage of the second cycle. Such an arrest was described for parasites treated with apicoplast-targeting antibacterials, consequently termed the delayed-death effect. Hence, the dual killing mechanism of thiostrepton and its derivatives, i.e., immediate killing followed by delayed death, indicates that in P. falciparum the 20S proteasome and the apicoplast ribosomes both are targeted.
In the second part of our study, we provided a detailed characterization of the proteasome expression in blood-stage parasites. We showed that components of α- and β-SU type 5 are transcribed in all blood stages, including gametocytes. β-SU type 5 labeling revealed that the proteasome is present predominantly in trophozoites and schizonts, namely, in stages that are metabolically highly active and have to prepare for DNA replication and cell division. The proteasome is localized in the cytoplasm and in the nucleus of parasites but not in the nucleolus, similarly to the 26S proteasome in human cells (36). The binding of a fluorescently labeled thiostrepton probe correlated with the presence and location of the proteasome in blood stages.
Interestingly, the thiostrepton derivatives also exhibited a gametocytocidal activity in our assays. Gametocyte toxicity was described for other antimalarial drugs such as primaquine (39) and recently was reported for the proteasome inhibitor epoxomicin (10). Since the apicoplast-targeting antibiotic azithromycin did not affect gametocyte differentiation, we must conclude that the gametocytocidal activity of thiostrepton and its derivatives correlates with gametocyte proteasome inhibition. In accordance with our observations, thiostrepton treatment was reported to reduce by 10-fold the parasite transmission from P. berghei-infected mice (51).
The thiostrepton-based derivatives do not exhibit pronounced toxicity against human cell lines (this study and reference 46), suggesting a considerable selectivity for the parasite proteasome. In this context, a dose-response study showed that thiostrepton can completely cure P. berghei-infected mice with no apparent toxicity for up to 500 mg/kg of body weight intraperitoneally (51). It was shown recently that the species-selective inhibition of proteasomes may lead to novel therapeutic options (27). Although thiostrepton has never been advanced to systemic applications due to rather low aqueous solubility and formulation problems (2), it is currently used on large scale as a topical antibiotic in animal health care applications (34). Other thiopeptide antibiotics resulted in systemically applicable antibiotics after derivatization (53). Therefore, it can be expected that compounds with improved properties can be obtained by following this precedent. Likewise, further optimization should lead to higher in vitro potency.
In conclusion, the semisynthetic thiostrepton derivatives described herein provide a highly promising first generation of novel antimalarial drugs for the following reasons. First, thiostrepton and derivatives act immediately on the malaria parasite and therefore do not have to be combined with a fast-acting antimalarial. Second, the thiostrepton-based compounds inhibit two independent targets, the 20S proteasome and the large ribosomal subunit of the prokaryotic apicoplast, in a dual mode of action, which renders thiostrepton derivatives more refractory to resistance than single-target-based inhibitors. Third, the compounds exhibit gametocytocidal activity and hence eliminate all gametocyte stages that have escaped the asexual stage-dependent killing, thus providing a stopgap against the transmission of genotypes conferring drug resistance. It is noteworthy in this context that antibiotics with antiplasmodial effect may be used in antibiotic-based combinations for the treatment of severe malaria (33). Taken together, our present work demonstrates that the proteasome is a viable target for antimalaria drug research. This approach may become especially powerful when combined with a second mode of action, such as being intrinsically embedded in the thiostrepton derivatives characterized herein.
Supplementary Material
Acknowledgments
We thank T. J. Templeton (Weill Cornell Medical College, New York) for reviewing the manuscript, T. Kreuzahler and the EM team of G. Krohne (University of Würzburg) for technical support, G. Harms and A. Mehlitz (University of Würzburg) for assistance with the confocal laser-scanning microscopy, and V. Heussler (BNI Hamburg) for helpful discussions.
G.P. and H.-D.A. gratefully acknowledge the Emmy-Noether Young Investigator awards from the Deutsche Forschungsgemeinschaft (DFG). This work was funded in part by the IRTG1522 of the DFG (to G.P.).
Footnotes
Published ahead of print on 18 January 2011.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Arndt, H. D., S. Schoof, and J. Y. Lu. 2009. Thiopeptide antibiotic biosynthesis. Angew Chem. Int. Ed. Engl. 48:6770-6773. [DOI] [PubMed] [Google Scholar]
- 2.Bagley, M. C., J. W. Dale, E. A. Merritt, and X. Xiong. 2005. Thiopeptide antibiotics. Chem. Rev. 105:685-714. [DOI] [PubMed] [Google Scholar]
- 3.Barthel, D., M. Schlitzer, and G. Pradel. 2008. Telithromycin and quinupristin-dalfopristin induce delayed death in Plasmodium falciparum. Antimicrob. Agents Chemother. 52:774-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumann, S., S. Schoof, S. D. Harkal, and H. D. Arndt. 2008. Mapping the binding site of thiopeptide antibiotics by proximity-induced covalent capture. J. Am. Chem. Soc. 130:5664-5666. [DOI] [PubMed] [Google Scholar]
- 5.Bhat, U. G., M. Halasi, and A. L. Gartel. 2009. Thiazole antibiotics target FoxM1 and induce apoptosis in human cancer cells. PLoS One 4:e5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bochtler, M., L. Ditzel, M. Groll, C. Hartmann, and R. Huber. 1999. The proteasome. Annu. Rev. Biophys. Biomol. Struct. 28:295-317. [DOI] [PubMed] [Google Scholar]
- 7.Chen, P., and M. Hochstrasser. 1995. Biogenesis, structure and function of the yeast 20S proteasome. EMBO J. 14:2620-2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clough, B., M. Strath, P. Preiser, P. Denny, and I. R. Wilson. 1997. Thiostrepton binds to malarial plastid rRNA. FEBS Lett. 406:123-125. [DOI] [PubMed] [Google Scholar]
- 9.Cranmer, S. L., C. Magowan, J. Liang, R. L. Coppel, and B. M. Cooke. 1997. An alternative to serum for cultivation of Plasmodium falciparum in vitro. Trans. R. Soc. Trop. Med. Hyg. 91:363-365. [DOI] [PubMed] [Google Scholar]
- 10.Czesny, B., S. Goshu, J. L. Cook, and K. C. Williamson. 2009. The proteasome inhibitor epoxomicin has potent Plasmodium falciparum gametocytocidal activity. Antimicrob. Agents Chemother. 53:4080-4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dahl, E. L., and P. J. Rosenthal. 2008. Apicoplast translation, transcription and genome replication: targets for antimalarial antibiotics. Trends Parasitol. 24:279-284. [DOI] [PubMed] [Google Scholar]
- 12.Dahl, E. L., et al. 2006. Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob. Agents Chemother. 50:3124-3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Etlinger, J. D., and A. L. Goldberg. 1977. A soluble ATP-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc. Natl. Acad. Sci. U. S. A. 74:54-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher, R. I., et al. 2006. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 24:4867-4874. [DOI] [PubMed] [Google Scholar]
- 15.Frentzel, S., B. Pesold-Hurt, A. Seelig, and P. M. Kloetzel. 1994. 20 S proteasomes are assembled via distinct precursor complexes. Processing of LMP2 and LMP7 proproteins takes place in 13-16 S preproteasome complexes. J. Mol. Biol. 236:975-981. [DOI] [PubMed] [Google Scholar]
- 16.Gantt, S. M., et al. 1998. Proteasome inhibitors block development of Plasmodium spp. Antimicrob. Agents Chemother. 42:2731-2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gille, C., et al. 2003. A comprehensive view on proteasomal sequences: implications for the evolution of the proteasome. J. Mol. Biol. 326:1437-1448. [DOI] [PubMed] [Google Scholar]
- 18.Goodman, C. D., V. Su, and G. I. McFadden. 2007. The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 152:181-191. [DOI] [PubMed] [Google Scholar]
- 19.Harms, J. M., et al. 2008. Translational regulation via L11: molecular switches on the ribosome turned on and off by thiostrepton and micrococcin. Mol. Cell 30:26-38. [DOI] [PubMed] [Google Scholar]
- 20.Ifediba, T., and J. P. Vanderberg. 1981. Complete in vitro maturation of Plasmodium falciparum gametocytes. Nature 294:364-366. [DOI] [PubMed] [Google Scholar]
- 21.Jonker, H. R., S. Ilin, S. K. Grimm, J. Wohnert, and H. Schwalbe. 2007. L11 domain rearrangement upon binding to RNA and thiostrepton studied by NMR spectroscopy. Nucleic Acids Res. 35:441-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kariuki, M. M., et al. 1998. Plasmodium falciparum: purification of the various gametocyte developmental stages from in vitro-cultivated parasites. Am. J. Trop. Med. Hyg. 59:505-508. [DOI] [PubMed] [Google Scholar]
- 23.Kreidenweiss, A., P. G. Kremsner, and B. Mordmuller. 2008. Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malar. J. 7:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuehn, A., and G. Pradel. 2010. The coming-out of malaria gametocytes. J. Biomed. Biotechnol. 2010:976827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambros, C., and J. P. Vanderberg. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65:418-420. [PubMed] [Google Scholar]
- 26.Lim, L., and G. I. McFadden. 2010. The evolution, metabolism and functions of the apicoplast. Philos Trans. R. Soc. Lond. B Biol. Sci. 365:749-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin, G., et al. 2009. Inhibitors selective for mycobacterial versus human proteasomes. Nature 461:621-626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindenthal, C., N. Weich, Y. S. Chia, V. Heussler, and M. Q. Klinkert. 2005. The proteasome inhibitor MLN-273 blocks exoerythrocytic and erythrocytic development of Plasmodium parasites. Parasitology 131:37-44. [DOI] [PubMed] [Google Scholar]
- 29.Makler, M. T., and D. J. Hinrichs. 1993. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am. J. Trop. Med. Hyg. 48:205-210. [DOI] [PubMed] [Google Scholar]
- 30.Makler, M. T., et al. 1993. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg. 48:739-741. [DOI] [PubMed] [Google Scholar]
- 31.McConkey, G. A., M. J. Rogers, and T. F. McCutchan. 1997. Inhibition of Plasmodium falciparum protein synthesis. Targeting the plastid-like organelle with thiostrepton. J. Biol. Chem. 272:2046-2049. [DOI] [PubMed] [Google Scholar]
- 32.Mordmüller, B., et al. 2006. Plasmodia express two threonine-peptidase complexes during asexual development. Mol. Biochem. Parasitol. 148:79-85. [DOI] [PubMed] [Google Scholar]
- 33.Noedl, H. 2009. ABC-antibiotics-based combinations for the treatment of severe malaria? Trends Parasitol. 25:540-544. [DOI] [PubMed] [Google Scholar]
- 34.Papich, M. G., and J. E. Riviere. 2001. Chloramphenicol and derivatives, macrolides, lincosamides, and miscellaneous antimicrobials, p. 868-897. In H. R. Adams (ed.), Veterinary pharmacology and therapeutics. Iowa State University Press, Ames, IA.
- 35.Paugam, A., A. L. Bulteau, J. Dupouy-Camet, C. Creuzet, and B. Friguet. 2003. Characterization and role of protozoan parasite proteasomes. Trends Parasitol. 19:55-59. [DOI] [PubMed] [Google Scholar]
- 36.Peters, J. M., W. W. Franke, and J. A. Kleinschmidt. 1994. Distinct 19 S and 20 S subcomplexes of the 26 S proteasome and their distribution in the nucleus and the cytoplasm. J. Biol. Chem. 269:7709-7718. [PubMed] [Google Scholar]
- 37.Pradel, G. 2007. Proteins of the malaria parasite sexual stages: expression, function and potential for transmission blocking strategies. Parasitology 134:1911-1929. [DOI] [PubMed] [Google Scholar]
- 38.Pradel, G., and M. Schlitzer. 2010. Antibiotics in malaria therapy and their effect on the parasite apicoplast. Curr. Mol. Med. 10:335-349. [DOI] [PubMed] [Google Scholar]
- 39.Pukrittayakamee, S., et al. 2004. Activities of artesunate and primaquine against asexual- and sexual-stage parasites in falciparum malaria. Antimicrob. Agents Chemother. 48:1329-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reynolds, J. M., K. El Bissati, J. Brandenburg, A. Gunzl, and C. B. Mamoun. 2007. Antimalarial activity of the anticancer and proteasome inhibitor bortezomib and its analog ZL3B. BMC Clin. Pharmacol. 7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rogers, M. J., E. Cundliffe, and T. F. McCutchan. 1998. The antibiotic micrococcin is a potent inhibitor of growth and protein synthesis in the malaria parasite. Antimicrob. Agents Chemother. 42:715-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidtke, G., et al. 1996. Analysis of mammalian 20S proteasome biogenesis: the maturation of beta-subunits is an ordered two-step mechanism involving autocatalysis. EMBO J. 15:6887-6898. [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidtke, G., et al. 1999. How an inhibitor of the HIV-I protease modulates proteasome activity. J. Biol. Chem. 274:35734-35740. [DOI] [PubMed] [Google Scholar]
- 44.Scholz, S. M., et al. 2008. PfCCp proteins of Plasmodium falciparum: gametocyte-specific expression and role in complement-mediated inhibition of exflagellation. Int. J. Parasitol. 38:327-340. [DOI] [PubMed] [Google Scholar]
- 45.Schoof, S., S. Baumann, B. Ellinger, and H. D. Arndt. 2009. A fluorescent probe for the 70 S-ribosomal GTPase-associated center. Chembiochem 10:242-245. [DOI] [PubMed] [Google Scholar]
- 46.Schoof, S., et al. 2010. Antiplasmodial thiostrepton derivatives: proteasome inhibitors with a dual mode of action. Angew Chem. Int. ed. Engl. 49:3317-3321. [DOI] [PubMed] [Google Scholar]
- 47.Seemüller, E., et al. 1995. Proteasome from Thermoplasma acidophilum: a threonine protease. Science 268:579-582. [DOI] [PubMed] [Google Scholar]
- 48.Seemuller, E., A. Lupas, and W. Baumeister. 1996. Autocatalytic processing of the 20S proteasome. Nature 382:468-471. [DOI] [PubMed] [Google Scholar]
- 49.Sidhu, A. B., et al. 2007. In vitro efficacy, resistance selection, and structural modeling studies implicate the malarial parasite apicoplast as the target of azithromycin. J. Biol. Chem. 282:2494-2504. [DOI] [PubMed] [Google Scholar]
- 50.Sullivan, D. J., Jr., I. Y. Gluzman, D. G. Russell, and D. E. Goldberg. 1996. On the molecular mechanism of chloroquine's antimalarial action. Proc. Natl. Acad. Sci. U. S. A. 93:11865-11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sullivan, M., J. Li, S. Kumar, M. J. Rogers, and T. F. McCutchan. 2000. Effects of interruption of apicoplast function on malaria infection, development, and transmission. Mol. Biochem. Parasitol. 109:17-23. [DOI] [PubMed] [Google Scholar]
- 52.Trager, W., and J. B. Jensen. 1976. Human malaria parasites in continuous culture. Science 193:673-675. [DOI] [PubMed] [Google Scholar]
- 53.Xu, L.-B., et al. 2009. Nocathiacin analogs: synthesis and antibacterial activity of novel water-soluble amides. Bioorg. Med. Chem. Lett. 19:3531-3535. [DOI] [PubMed] [Google Scholar]
- 54.Young, J. A., et al. 2005. The Plasmodium falciparum sexual development transcriptome: a microarray analysis using ontology-based pattern identification. Mol. Biochem. Parasitol. 143:67-79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.