

Abstract
Mycobacterium goodii strain 12523 is an actinomycete that is able to oxidize phenol regioselectively at the para position to produce hydroquinone. In this study, we investigated the genes responsible for this unique regioselective oxidation. On the basis of the fact that the oxidation activity of M. goodii strain 12523 toward phenol is induced in the presence of acetone, we first identified acetone-induced proteins in this microorganism by two-dimensional electrophoretic analysis. The N-terminal amino acid sequence of one of these acetone-induced proteins shares 100% identity with that of the protein encoded by the open reading frame Msmeg_1971 in Mycobacterium smegmatis strain mc2155, whose genome sequence has been determined. Since Msmeg_1971, Msmeg_1972, Msmeg_1973, and Msmeg_1974 constitute a putative binuclear iron monooxygenase gene cluster, we cloned this gene cluster of M. smegmatis strain mc2155 and its homologous gene cluster found in M. goodii strain 12523. Sequence analysis of these binuclear iron monooxygenase gene clusters revealed the presence of four genes designated mimABCD, which encode an oxygenase large subunit, a reductase, an oxygenase small subunit, and a coupling protein, respectively. When the mimA gene (Msmeg_1971) of M. smegmatis strain mc2155, which was also found to be able to oxidize phenol to hydroquinone, was deleted, this mutant lost the oxidation ability. This ability was restored by introduction of the mimA gene of M. smegmatis strain mc2155 or of M. goodii strain 12523 into this mutant. Interestingly, we found that these gene clusters also play essential roles in propane and acetone metabolism in these mycobacteria.
Mycobacterium goodii strain 12523 is a unique actinomycete that is able to oxidize phenol regioselectively at the para position to produce hydroquinone (20). This microorganism was discovered for application in biocatalysis: chemical synthesis of hydroquinone is accompanied by side reactions leading to undesired by-products such as catechol, whereas M. goodii strain 12523 enabled gram-scale production of hydroquinone without by-products (21). Although M. goodii strain 12523 cannot utilize phenol, this microorganism can use acetone and methylethylketone as sources of carbon and energy. Interestingly, the oxidation activity of M. goodii strain 12523 toward phenol is induced in the presence of acetone and methylethylketone (20). To date, there have been only a few reports concerning monooxygenases that convert phenol to hydroquinone. P450BM-3, a cytochrome P450 monooxygenase from Bacillus megaterium, exhibits such an activity, although this activity is very low (23). A P450BM-3 mutant was then created, which exhibited 16.5 times higher activity than the wild type (23). A strain with a mutation of toluene-o-xylene monooxygenase, a binuclear iron monooxygenase from Pseudomonas stutzeri, also converts phenol to hydroquinone and catechol in a proportion of 80:20, although the wild type produces only catechol (27).
In this study, we identified a monooxygenase gene cluster that is responsible for the conversion of phenol to hydroquinone in M. goodii strain 12523. On the basis of the fact that the oxidation activity of M. goodii strain 12523 toward phenol is induced in the presence of acetone, we first identified acetone-induced proteins in this microorganism by two-dimensional electrophoretic analysis. N-terminal amino acid sequencing of these acetone-induced proteins, combined with analysis of the genome sequence of Mycobacterium smegmatis strain mc2155, subsequently led to identification of the monooxygenase gene cluster by deletion and complementation analyses. We found that this gene cluster encodes a multicomponent monooxygenase that belongs to the binuclear iron monooxygenase family and that its homologous gene cluster found in M. smegmatis strain mc2155 also encodes monooxygenase activity toward phenol. Furthermore, we found that these gene clusters were also involved in propane and acetone metabolism in these mycobacteria.
MATERIALS AND METHODS
Bacterial strains, plasmids, and cultivation media.
The bacterial strains and plasmids that were used or constructed in this study are listed in Table 1. Strain 12523, which had previously been identified as a Mycobacterium sp. (20), was recently identified as M. goodii based on 16S rRNA gene sequence analysis (1). The bacteria were grown in Luria-Bertani (LB) medium, which contained (per liter) Bacto tryptone (10 g), Bacto yeast extract (5 g), and NaCl (10 g) (pH 7.0), or in KG medium, which contained (per liter) (NH4)2SO4 (3 g), KH2PO4 (1.4 g), Na2HPO4 (2.1 g), MgSO4·7H2O (0.2 g), FeCl2·5H2O (10.6 mg), CaCl2·2H2O (8 mg), ZnSO4·7H2O (4 mg), MnCl2·4H2O (2 mg), CuSO4·5H2O (0.02 mg), KI (0.2 mg), Na2MoO4·2H2O (0.2 mg), CoCl2·6H2O (0.2 mg), H3BO3 (0.4 mg), and NaCl (10 mg) (pH 7.2).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Characteristics | Reference or source |
|---|---|---|
| Strains | ||
| M. goodii 12523 | Wild type | 20 |
| M. smegmatis mc2155 | Wild type, ATCC 700084 | ATCC |
| M. smegmatis mc2155 ΔmimA | M. smegmatis strain mc2155 with mimA deletion | This study |
| Gordonia sp. strain TY-5 | Wild type | 7 |
| Gordonia sp. strain TY-5 prmB::Kanr | Gordonia sp. strain TY-5 with prmB disruption | 7 |
| E. coli DH5α | Host used for cloning | Takara Bio |
| Plasmids | ||
| pET21a | Vector used for cloning | Novagen |
| pETmimABCDgo | pET21a containing mimABCDgo of M. goodii 12523 | This study |
| pETmimABCDsm | pET21a containing mimABCDsm of M. smegmatis mc2155 | This study |
| pK18mobsacB | Vector used for deletion mutagenesis, aph sacB | NBRP (NIG, Japan) |
| pKΔmimA | pK18mobsacB containing a deleted mimA gene of M. smegmatis mc2155 | This study |
| pRHK1 | Rhodococcus (Mycobacterium)-E. coli shuttle vector, Kanr | 6 |
| pUCkap1 | pUC18 containing the kap1 promoter region from R. erythropolis | 13 |
| pRHKkap1 | pRHK1 containing the kap1 promoter region | This study |
| pRHKkap1mimAgo | pRHKkap1 containing the mimAgo gene under the control of the kap1 promoter | This study |
| pRHKkap1mimAsm | pRHKkap1 containing the mimAsm gene under the control of the kap1 promoter | This study |
Two-dimensional electrophoretic analysis.
M. goodii strain 12523 cells were cultivated for 6 days in LB medium (2 ml) at 30°C. After centrifugation at 10,000 × g for 10 min at 4°C, the cells were suspended in KG medium (2 ml) with or without acetone (1%, vol/vol), and were incubated for 24 h at 30°C. M. goodii strain 12523 cells with and without acetone induction were harvested by centrifugation, washed with potassium phosphate buffer (50 mM, pH 7.5) containing glycerol (10%, vol/vol), and stored at −80°C until use.
The frozen cells were suspended in a solution containing Tris (60 mM), urea (5 M), thiourea (1 M), Complete Mini EDTA-free (one tablet per 10 ml; Roche, Mannheim, Germany), 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonic acid (CHAPS) (10 g/liter), Triton X-100 (1% vol/vol), and dithiothreitol (10 g/liter) (pH 8.8 to 9.0) and were disrupted with an ultraoscillator. After centrifugation at 100,000 × g for 20 min at 4°C, the resulting supernatant was supplemented with acrylamide (0.1 M) and was subjected to two-dimensional electrophoretic analysis. Isoelectric focusing was performed for the first dimension. The supernatant (75 μg protein) was loaded onto an agar gel (pH 3 to 10; ATTO, Tokyo, Japan) and was analyzed using the AE-6540 electrophoresis unit (ATTO). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed for the second dimension. The agar gel after isoelectric focusing was loaded onto a polyacrylamide gel (12.5%) and was analyzed using the AE-6541 electrophoresis unit (ATTO). Proteins in the gel after SDS-PAGE were electroblotted onto a polyvinylidene difluoride (PVDF) membrane, and their N-terminal amino acid sequences were determined by APRO Life Science (Tokushima, Japan) using Edman degradation.
Cloning and sequence analysis.
The genomic DNAs of M. goodii strain 12523 and M. smegmatis strain mc2155 were prepared using a DNeasy tissue kit (Qiagen, Germantown, MD). Two oligonucleotide primers, mimABCD-F and mimABCD-R (Table 2), were designed to amplify the binuclear iron monooxygenase gene clusters of M. goodii strain 12523 and M. smegmatis strain mc2155, based on the N-terminal amino acid sequences of two acetone-induced proteins from M. goodii strain 12523 and the genome sequence of M. smegmatis strain mc2155 (GenBank accession number, NC_008596). The region between the two oligonucleotide primers was amplified from the genomic DNAs of M. goodii strain 12523 and M. smegmatis strain mc2155 using PCR. These amplified DNA fragments were digested with NdeI and EcoRI and were inserted into the pET21a vector (Table 1). The resulting plasmids, pETmimABCDgo for M. goodii strain 12523 and pETmimABCDsm for M. smegmatis strain mc2155, were amplified in Escherichia coli DH5α cells, and the inserts were sequenced using an ABI PRISM 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA).
TABLE 2.
Oligonucleotide primers used in this study
| Primer | Sequence (5′ to 3′)a | Restriction site |
|---|---|---|
| mimABCD-F | GAATTCCATATGAGCAGACAAAGCCTGACCAAG | NdeI |
| mimABCD-R | CCGGAATTCCTGTTGGCCGTCTTTTTCGTACAT | EcoRI |
| mimA5′-F | CCCAAGCTTGTGATCCAGCAGCACCGTCTGATC | HindIII |
| mimA5′-R | GCTCTAGACGGCTCCCACGTGAGTTCGCTTATC | XbaI |
| mimA3′-F | GCTCTAGAATGACCGACGACGAGCGCGAC | XbaI |
| mimA3′-R | CGGAATTCTCTTTGATTCCCTTGCCCAGTTCG | EcoRI |
| kap1-F | AACTGCAGGCCGCGACGTGGCTGTTGCTGA | PstI |
| kap1-R | GCTCTAGAACGAATTGATCTTCTGGCGCGT | XbaI |
| mimA-F | GCTCTAGACCAACTATCAGGAGGCTCACGTTG | XbaI |
| mimA-R | CCGGAATTCTCAGGCCGGGACCCCGCCGGCGCG | EcoRI |
Restriction sites are underlined.
Deletion of the mimA gene in M. smegmatis strain mc2155.
The mimA gene in M. smegmatis strain mc2155 was deleted in frame using the pK18mobsacB vector (19) (Table 1). Two oligonucleotide primers, mimA5′-F and mimA5′-R (Table 2), were designed to amplify the 5′-terminal region of the mimA gene based on the genome sequence of M. smegmatis strain mc2155. The region between the two oligonucleotide primers was amplified from the genomic DNA of M. smegmatis strain mc2155 using PCR. These amplified DNA fragments were digested with HindIII and XbaI and were inserted into the pK18mobsacB vector. Two other oligonucleotide primers, mimA3′-F and mimA3′-R (Table 2), were designed to amplify the 3′-terminal region of the mimA gene. These amplified DNA fragments were digested with XbaI and EcoRI and were subsequently inserted into the pK18mobsacB vector containing the 5′-terminal region of the mimA gene. The resulting plasmid, pKΔmimA, contained a deleted mimA gene encoding a 42-amino-acid protein instead of a full-length mimA gene encoding a 542-amino-acid protein.
A two-step recombination was performed to delete the mimA gene from the chromosome of M. smegmatis strain mc2155 as described previously (11, 22, 26). The pKΔmimA plasmid was introduced into M. smegmatis strain mc2155 cells by electroporation. Single-crossover mutants, into which the plasmid was integrated, were selected on an LB plate containing kanamycin (50 μg/ml). Kanamycin-resistant strains were then subjected to repeated cultivation in LB medium without kanamycin. Finally, double-crossover mutants, which had lost the vector backbone and were sensitive to kanamycin, were selected on LB plates with or without kanamycin. Deletion of the mimA gene was confirmed by PCR using the two oligonucleotide primers mimA5′-F and mimA3′-R (Table 2). This procedure resulted in the deletion mutant M. smegmatis mc2155 ΔmimA (Table 1).
Construction of the mimA expression plasmids.
The plasmids used for expression of the mimA genes of M. goodii strain 12523 and M. smegmatis strain mc2155 were constructed using the pRHK1 vector (6) (Table 1) and the kap1 promoter (13) (GenBank accession number, AB083090) from Rhodococcus erythropolis. Two oligonucleotide primers, kap1-F and kap1-R (Table 2), were designed to amplify the kap1 promoter region. The region between the two oligonucleotide primers was amplified from the pUCkap1 plasmid (13) (Table 1) using PCR. These amplified DNA fragments were digested with PstI and XbaI and were inserted into the pRHK1 vector to construct pRHKkap1. Two other oligonucleotide primers, mimA-F and mimA-R (Table 2), were designed to amplify the mimA genes and their putative ribosome binding sites, based on the determined sequences of these genes and the sequence upstream from the mimA gene in the genome sequence of M. smegmatis strain mc2155. The region between the two oligonucleotide primers was amplified from the genomic DNAs of M. goodii strain 12523 and M. smegmatis strain mc2155 using PCR. These amplified DNA fragments were digested with XbaI and EcoRI and were subsequently inserted into the pRHKkap1 vector. The resulting plasmids, pRHKkap1mimAgo for M. goodii strain 12523 and pRHKkap1mimAsm for M. smegmatis strain mc2155, were introduced into M. smegmatis strain mc2155 ΔmimA cells by electroporation.
Assays of monooxygenase activity and growth substrates.
Monooxygenase activities of mycobacterial strains toward phenol were examined using growing cells. Wild-type and mutant strains were inoculated into KG medium (2 ml) containing Tween 80 (0.05%, vol/vol) and pyruvate (1 g/liter), which was used as a source of carbon and energy, in glass vials (14 ml) sealed with screw caps. The headspace volume was sufficient to prevent any oxygen limitation during growth and reaction. The strains were cultivated at 37°C with reciprocal shaking at a speed of 240 strokes per min. After cultivation for 24 h, acetone (1%, vol/vol) and phenol (500 μM) were added to the medium as an inducer and a substrate, respectively, and the growing cells were reacted with phenol for 5 days. Following the reaction, high-pressure liquid chromatography (HPLC) analysis was performed using an HPLC system (1100 series; Agilent, Palo Alto, CA) with a Cosmosil 5C18-PAQ column (4.6 mm by 250 mm; particle size, 5 μm; Nacalai Tesque, Kyoto, Japan). The reaction mixture was acidified by the addition of HCl (pH 2 to 3) and was then extracted with ethyl acetate (2 ml). The extract (1 ml) was evaporated, and the resulting residue was dissolved in a water-methanol mixture at a ratio of 50:50 (200 μl). After filtration through a 0.20-μm-pore-size polytetrafluoroethylene membrane (Advantec, Tokyo, Japan), the sample (10 μl) was injected into the HPLC system. Mobile phases A and B were composed of potassium phosphate buffer (10 mM, pH 6.9) and methanol, respectively. The samples were eluted with a linear gradient of 0% to 20% B for 10 min, followed by a linear gradient of 20% to 80% B for 5 min at a flow rate of 1 ml/min. Hydroquinone produced from phenol was spectrophotometrically detected at 290 nm.
We also examined the monooxygenase activities of Gordonia sp. strain TY-5 and its mutant strain (7) (Table 1) toward phenol using resting cells. The strains were cultivated for 2 days in LB medium (2 ml) at 30°C. After centrifugation at 10,000 × g for 10 min at 4°C, the cells were suspended in KG medium (2 ml) with acetone (1%, vol/vol) and were incubated for 24 h at 30°C. The cells of Gordonia sp. strain TY-5 and its mutant strain with acetone induction were harvested by centrifugation, washed with potassium phosphate buffer (50 mM, pH 7.5) containing glycerol (10%, vol/vol), and suspended in the same buffer (200 μl). The resting cells were reacted with phenol (2 mM) at 30°C with vigorous shaking for 5 h. Following the reaction, HPLC analysis was performed as described above.
Propane, 1-propanol, 2-propanol, acetone, methylethylketone, and acetol were examined as potential growth substrates of mycobacterial strains. Wild-type and mutant strains were inoculated into KG medium (2 ml) containing Tween 80 (0.05%, vol/vol) and individual compounds as a source of carbon and energy in glass vials (14 ml) sealed with screw caps. The strains were cultivated at 37°C with reciprocal shaking at a speed of 240 strokes per min. Propane was added to the headspace at a concentration of 20% (vol/vol). 1-Propanol, acetone, methylethylketone, and acetol were added to the medium at a concentration of 1% (vol/vol), whereas 2-propanol was added at a concentration of 0.5% (vol/vol). Cell growth was measured turbidimetrically at 660 nm and is expressed as cell dry weight.
Nucleotide sequence accession number.
The nucleotide sequence of the monooxygenase gene cluster of M. goodii strain 12523 has been submitted to GenBank, and the assigned accession number is AB568291.
RESULTS
Identification of acetone-induced proteins by two-dimensional electrophoretic analysis.
On the basis of the fact that the oxidation activity of M. goodii strain 12523 toward phenol is induced in the presence of acetone (20), we first examined acetone-induced proteins in this microorganism by two-dimensional electrophoretic analysis. Comparison of the protein profiles of extracts from M. goodii strain 12523 cells with and without acetone induction revealed the presence of several spots that appeared only with acetone induction (Fig. 1). We chose two major spots corresponding to proteins A (ca. 57 kDa) and B (ca. 43 kDa) on the gel shown in Fig. 1 and determined the N-terminal amino acid sequences of these proteins. The N-terminal sequences of proteins A and B were SRQSLTKAHAKISELTWE and MYEKDGQQYFIVDSHVHL, respectively.
FIG. 1.

Identification of acetone-induced proteins. Two-dimensional electrophoretic protein profiles of extracts from M. goodii strain 12523 cells in the absence and presence of acetone (left and right panels, respectively) are shown. The two acetone-induced proteins whose N-terminal sequences were determined (A and B) are indicated by arrows.
These N-terminal sequences were analyzed using the BLAST program at the National Center for Biotechnology Information (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The sequence of protein A shares 100% identity with that of the protein encoded by the open reading frame (ORF) Msmeg_1971 in M. smegmatis strain mc2155, whose genome sequence has been determined. Similarly, the sequence of protein B shares 100% identity with that of the protein encoded by Msmeg_1975. Although the functions of the Msmeg_1971 and Msmeg_1975 proteins have not been experimentally determined, these proteins exhibit amino acid similarities with oxygenase large subunits of binuclear iron monooxygenases and amidohydrolases, respectively. Furthermore, the Msmeg_1972, Msmeg_1973, and Msmeg_1974 proteins, which are encoded by the ORFs between Msmeg_1971 and Msmeg_1975, exhibit amino acid similarities with reductases, oxygenase small subunits, and coupling proteins, respectively, of binuclear iron monooxygenases.
Cloning and sequence analysis of monooxygenase gene clusters in M. goodii strain 12523 and M. smegmatis strain mc2155.
Assuming that the binuclear iron monooxygenase gene cluster consisting of Msmeg_1971, Msmeg_1972, Msmeg_1973, and Msmeg_1974 in M. smegmatis strain mc2155 and its homologous gene cluster in M. goodii strain 12523 are involved in the conversion of phenol to hydroquinone, we cloned these gene clusters. Two oligonucleotide primers were designed based on the N-terminal amino acid sequences of the two acetone-induced proteins from M. goodii strain 12523 and the genome sequence of M. smegmatis strain mc2155. The region between the two oligonucleotide primers was amplified from the genomic DNAs of M. goodii strain 12523 and M. smegmatis strain mc2155 using PCR and was cloned into the pET21a vector. Sequence analysis revealed the presence of four ORFs, Msmeg_1971, Msmeg_1972, Msmeg_1973, and Msmeg_1974, in the PCR product from M. smegmatis strain mc2155 and their homologous four ORFs in that from M. goodii strain 12523. These four ORFs were carried on the same strand and were designated mimABCD for the mycobacterial binuclear iron monooxygenase gene clusters. For convenience, we named the gene cluster from M. goodii strain 12523 mimABCDgo and that from M. smegmatis strain mc2155 mimABCDsm.
We confirmed that the nucleotide sequence of mimABCDsm (4,238 bp) from M. smegmatis strain mc2155 coincided with that determined by the genome sequencing project and further sequence analysis (3). The nucleotide sequence of mimABCDgo (4,231 bp) from M. goodii strain 12523 shares 92% overall identity with that of mimABCDsm from M. smegmatis strain mc2155 based on a BLAST search. Intergenic distances between the stop codons and the start codons were 91 bp for mimAsm and mimBsm and 23 bp for mimBsm and mimCsm, whereas the 3′-terminal nucleotide of mimCsm and the 5′-terminal nucleotide of mimDsm overlapped by 4 bp. Similarly, intergenic distances between the stop codons and the start codons were 83 bp for mimAgo and mimBgo and 24 bp for mimBgo and mimCgo, whereas the 3′-terminal nucleotide of mimCgo and the 5′-terminal nucleotide of mimDgo overlapped by 4 bp. Putative ribosome binding sites were found in the upstream regions of mimBsm, mimCsm, and mimDsm and mimBgo, mimCgo, and mimDgo. We also confirmed that a putative ribosome binding site and a putative promoter exist in the upstream region of the mimAsm gene in the genome sequence of M. smegmatis strain mc2155. These observations suggest that the gene clusters are transcribed polycistronically.
Unexpectedly, we found that the multicomponent monooxygenases encoded by the mimABCD gene clusters exhibit high amino acid similarities with two propane monooxygenases (Prm) from Rhodococcus sp. strain RHA1 (22) and Gordonia sp. strain TY-5 (7) (Table 3). Furthermore, these monooxygenases also exhibit appreciable amino acid similarities with a tetrahydrofuran monooxygenase (Thm) from Pseudonocardia sp. strain K1 (25) and a propene monooxygenase (Pmo) from Mycobacterium sp. strain M156 (2) (Table 3). The order of the mimABCD gene clusters is an oxygenase large subunit, a reductase, an oxygenase small subunit, and a coupling protein, which is identical to that of the prm and thm gene clusters but is different from that of the pmo gene cluster. The mimAgo and mimAsm genes each encode 542-amino-acid proteins. The MimAgo protein of strain 12523 shares 99, 94, and 93% amino acid identity with MimAsm of strain mc2155, PrmA of strain RHA1, and PrmA of strain TY-5, respectively (Table 3). We confirmed that a pair of motif sequences (Glu-X-X-His) that coordinate to the binuclear iron center at the active site (16) are conserved in MimAgo and MimAsm. Similarly, the MimBgo, MimCgo, and MimDgo proteins of strain 12523 exhibit amino acid similarities with their corresponding components of strains mc2155, RHA1, and TY-5 (Table 3). Compared with MimAgo and MimDgo, MimBgo and MimCgo exhibit relatively low similarities (64 to 81%) with the propane monooxygenase components.
TABLE 3.
Amino acid identity between mimABCDgo-encoded proteins and other binuclear iron monooxygenases
| mimABCDgo gene and comparison gene | No. of amino acid residues | % Identity (no. of identical amino acids/total no. of amino acids)a | Organism | Accession no. |
|---|---|---|---|---|
| mimAgo | 542 | M. goodii 12523 | AB568291 | |
| mimAsm | 542 | 99 (537/542) | M. smegmatis mc2155 | YP_886336 |
| prmA | 544 | 94 (516/544) | Rhodococcus sp. strain RHA1 | YP_700435 |
| prmA | 545 | 93 (501/537) | Gordonia sp. strain TY-5 | BAD03956 |
| thmA | 545 | 41 (217/520) | Pseudonocardia sp. strain K1 | CAC10506 |
| pmoC | 501 | 38 (202/524) | Mycobacterium sp. strain M156 | AAS19484 |
| mimBgo | 348 | M. goodii 12523 | AB568291 | |
| mimBsm | 348 | 93 (327/348) | M. smegmatis mc2155 | YP_886337 |
| prmB | 370 | 76 (267/347) | Rhodococcus sp. strain RHA1 | YP_700436 |
| prmB | 346 | 64 (229/355) | Gordonia sp. strain TY-5 | BAD03957 |
| thmD | 360 | 47 (164/347) | Pseudonocardia sp. strain K1 | CAC10508 |
| pmoD | 340 | 36 (123/341) | Mycobacterium sp. strain M156 | AAS19485 |
| mimCgo | 368 | M. goodii 12523 | AB568291 | |
| mimCsm | 368 | 94 (346/368) | M. smegmatis mc2155 | ABJ96310 |
| prmC | 368 | 81 (299/368) | Rhodococcus sp. strain RHA1 | YP_700437 |
| prmC | 368 | 74 (273/368) | Gordonia sp. strain TY-5 | BAD03958 |
| thmB | 346 | 32 (96/296) | Pseudonocardia sp. strain K1 | CAC10509 |
| pmoA | 346 | 29 (90/304) | Mycobacterium sp. strain M156 | AAS19482 |
| mimDgo | 114 | M. goodii 12523 | AB568291 | |
| mimDsm | 114 | 96 (110/114) | M. smegmatis mc2155 | YP_886338 |
| prmD | 113 | 91 (102/112) | Rhodococcus sp. strain RHA1 | YP_700438 |
| prmD | 111 | 90 (100/110) | Gordonia sp. strain TY-5 | BAD03959 |
| thmC | 117 | 30 (32/106) | Pseudonocardia sp. strain K1 | CAC10510 |
| pmoB | 111 | 28 (29/102) | Mycobacterium sp. strain M156 | AAS19483 |
Amino acid identity was determined using the BLAST program.
Deletion and complementation analyses of the monooxygenase gene clusters.
To confirm that the binuclear iron monooxygenase gene clusters in M. goodii strain 12523 and M. smegmatis strain mc2155 are involved in the conversion of phenol to hydroquinone, we performed deletion and complementation analyses of the mimA genes. We first confirmed that M. smegmatis strain mc2155 was also able to oxidize phenol to hydroquinone (Fig. 2). M. smegmatis strain mc2155 was used for construction of the deletion mutant because this strain can be easily transformed, whereas attempts to introduce heterologous genes into M. goodii strain 12523 cells were unsuccessful. We examined the monooxygenase activities of the wild-type and mutant M. goodii 12523 and M. smegmatis mc2155 strains toward phenol using growing cells in the presence of pyruvate and acetone, which were used as a source of carbon and energy and an inducer, respectively. M. goodii strain 12523, M. smegmatis strain mc2155, and the deletion mutant mc2155 ΔmimA were all able to grow on a pyruvate carbon source. The growing cells of M. goodii strain 12523 and M. smegmatis strain mc2155 produced 114 μM and 104 μM hydroquinone, respectively, from 500 μM phenol in 5 days, whereas the deletion mutant mc2155 ΔmimA did not produce any hydroquinone (Fig. 2). To confirm that the loss of monooxygenase activity toward phenol in this mutant was due only to deletion of the mimA gene, we determined if complementation of this gene in the deletion mutant mc2155 ΔmimA would restore hydroquinone production. The mimAsm gene was constitutively expressed under the control of the kap1 promoter derived from R. erythropolis using the pRHK1 vector (Table 1). The growing cells of M. smegmatis strain mc2155 ΔmimA that was complemented with the mimAsm gene produced 61.1 μM hydroquinone from phenol (Fig. 2). Furthermore, when we complemented the deletion mutant mc2155 ΔmimA with the mimAgo gene from M. goodii strain 12523, the growing cells produced 63.9 μM hydroquinone from phenol (Fig. 2). These results indicate that the mimA genes are essential for the oxidation of phenol to hydroquinone.
FIG. 2.

Monooxygenase activities of wild-type and mutant strains toward phenol. Growing cells of wild-type 12523 and mc2155, the deletion mutant mc2155 ΔmimA, and the complemented strains mc2155 ΔmimA (mimAsm) and mc2155 ΔmimA (mimAgo), which were induced with acetone, were reacted with phenol, and the monooxygenation product hydroquinone was quantified using HPLC. Bars represent the averages from three independent experiments, and error bars represent the standard deviations from the means.
We also examined the monooxygenase activity of Gordonia sp. strain TY-5 and its mutant strain with a prmB disruption in the prmABCD propane monooxygenase gene cluster (7) (Table 1) using resting cells with acetone induction. The resting cells of Gordonia sp. strain TY-5 regioselectively produced 1.1 mM hydroquinone from 2 mM phenol in 5 h, whereas the disruption mutant TY-5 prmB::Kanr did not produce any hydroquinone (data not shown).
Growth on propane and acetone.
Since the multicomponent monooxygenases encoded by the mimABCD gene clusters exhibit high amino acid similarities with propane monooxygenases from Rhodococcus sp. strain RHA1 (22) and Gordonia sp. strain TY-5 (7), we examined the growth of the wild-type and mutant M. goodii 12523 and M. smegmatis mc2155 strains on propane. We first confirmed that the wild-type M. goodii strain 12523 and M. smegmatis strain mc2155 were able to grow on propane as a source of carbon and energy (Fig. 3). We then found that the deletion mutant mc2155 ΔmimA had lost the ability to grow on propane, whereas this ability was restored by introduction of the mimAsm gene or the mimAgo gene into this mutant (Fig. 3). These results suggest that the gene clusters encode monooxygenase activity toward propane as well as phenol. We also confirmed that the wild-type M. goodii 12523 and M. smegmatis mc2155 strains were able to grow on 1-propanol and 2-propanol (Fig. 3). Furthermore, we found that growth on 2-propanol was dependent on the presence of the mimAsm gene or the mimAgo gene, whereas growth on 1-propanol was not (Fig. 3).
FIG. 3.

Growth of wild-type and mutant strains on propane and related compounds. Cells of wild-type 12523 and mc2155, the deletion mutant mc2155 ΔmimA, and the complemented strains mc2155 ΔmimA (mimAsm) and mc2155 ΔmimA (mimAgo) were cultivated on individual compounds as a source of carbon and energy for 8 days. Cell growth is expressed as cell dry weight per liter of medium. Bars represent the averages from two independent experiments, and error bars represent the standard deviations from the means.
We next examined the growth of the wild-type and mutant M. goodii 12523 and M. smegmatis mc2155 strains on acetone and methylethylketone, since the monooxygenase activity of these strains toward phenol is known to be induced in the presence of acetone and methylethylketone (20). The wild-type M. goodii 12523 and M. smegmatis mc2155 strains were able to grow on acetone and methylethylketone as a source of carbon and energy (Fig. 3 and 4). Surprisingly, the deletion mutant mc2155 ΔmimA had lost the ability to grow on acetone and methylethylketone, whereas this ability was restored by introduction of the mimAsm gene or the mimAgo gene into this mutant (Fig. 3 and 4). These results indicate that the monooxygenase gene clusters are involved in acetone and methylethylketone metabolism. Although the wild-type M. goodii 12523 and M. smegmatis mc2155 strains were able to grow on acetol, the deletion mutant mc2155 ΔmimA did not lose the ability to grow on this substrate (Fig. 3).
FIG. 4.
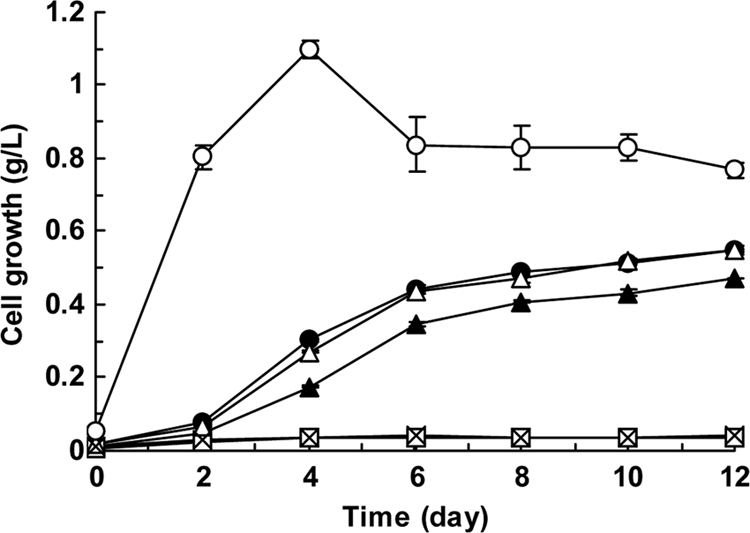
Growth profiles of wild-type and mutant strains on acetone. Cells of wild-type 12523 (white circles) and mc2155 (white triangles), the deletion mutant mc2155 ΔmimA (squares), and the complemented strains mc2155 ΔmimA (mimAsm) (black triangles) and mc2155 ΔmimA (mimAgo) (black circles) were cultivated on acetone as a source of carbon and energy. The growth of the deletion mutant mc2155 ΔmimA in the absence of acetone is indicated by crosses. Cell growth is expressed as cell dry weight per liter of medium. Plots represent the averages from two independent experiments, and error bars represent the standard deviations from the means.
DISCUSSION
Binuclear iron monooxygenases are a family of proteins that contain a binuclear iron center at the active site and introduce one oxygen atom derived from molecular oxygen into organic molecules, including alkanes, alkenes, and aromatics (9). These monooxygenases are widely distributed throughout prokaryotes such as actinomycetes, methanotrophs, and pseudomonads. The monooxygenases in actinomycetes have only recently been reported and constitute a new subfamily. The first monooxygenase to be reported was the alkene monooxygenase (Amo) of Rhodococcus corallines strain B-276 (18). This enzyme was able to produce optically active epoxides from their corresponding alkenes. A second alkene monooxygenase, propene monooxygenase (Pmo), was discovered in Mycobacterium sp. strain M156 (2). Propane monooxygenase (Prm) gene clusters were identified in Gordonia sp. strain TY-5 (7) and Rhodococcus sp. strain RHA1 (22) by deletion analysis, although enzymatic characterization or heterologous expression of these monooxygenases has not been reported, probably due to technical difficulties. The report regarding Gordonia sp. strain TY-5 suggested that this monooxygenase oxidized propane to 2-propanol (7). All of the reported monooxygenase gene clusters of this actinomycete subfamily consist of four genes that encode an oxygenase large subunit, an oxygenase small subunit, a reductase, and a coupling protein. The oxygenase component, consisting of the catalytic large subunit and the structural small subunit, activates molecular oxygen using electrons, which are transferred from NAD(P)H by the reductase component. The coupling protein is believed to be involved in electron transfer and substrate oxidation (9).
In this study, we succeeded in identifying binuclear iron monooxygenase gene clusters in M. goodii strain 12523 and M. smegmatis strain mc2155. The gene clusters consist of four genes, mimA, mimB, mimC, and mimD, which encode an oxygenase large subunit, a reductase, an oxygenase small subunit, and a coupling protein, respectively. We demonstrated that the mimABCD gene clusters were responsible for the conversion of phenol to hydroquinone (Fig. 2). These are the first binuclear iron monooxygenases that have been shown to oxidize phenol regioselectively at the para position, although several monooxygenases in this family have been reported to convert phenol to catechol (4, 12, 14), and many mutants of binuclear iron monooxygenases have been created for their application in biocatalysis (15, 17, 27). Furthermore, we confirmed that the prmABCD propane monooxygenase gene cluster in Gordonia sp. strain TY-5, which exhibits high sequence similarity with mimABCD, also encodes the same regioselective oxidation activity toward phenol. It will be of interest to study the factors that control the unique regioselectivity.
We found that M. smegmatis strain mc2155, which is a typical strain of mycobacteria that has been well studied, was able to not only oxidize phenol to hydroquinone but also grow on propane and acetone as a source of carbon and energy. M. smegmatis and M. goodii are closely related phylogenetically, and the 16S rRNA gene sequence of M. smegmatis strain mc2155 shows 99% identity with that of M. goodii strain 12523. We demonstrated that the mimABCD gene clusters in M. goodii strain 12523 and M. smegmatis strain mc2155 were also responsible for propane oxidation (Fig. 3). Although M. smegmatis strain mc2155 has often been used as a surrogate for Mycobacterium tuberculosis, this human pathogen does not have a homolog of the mimABCD gene cluster. In contrast, homologs of this gene cluster were found in the sequenced genomes of several Rhodococcus and Gordonia strains in addition to Rhodococcus sp. strain RHA1 and Gordonia sp. strain TY-5 (data not shown). It is conceivable that the mim and prm gene clusters and their homologs might play important roles in hydrocarbon degradation by actinomycetes in polluted environments. Recently, it was reported that the prm gene cluster in Rhodococcus sp. strain RHA1 was also responsible for N-nitrosodimethylamine degradation (22).
We also showed that the deletion mutant mc2155 ΔmimA had lost the ability to grow on acetone as a source of carbon and energy and that this ability was restored by introduction of the mimA gene of M. smegmatis strain mc2155 or of M. goodii strain 12523 into this mutant. Although acetone metabolism in bacteria has been well studied (5), this study led to the first identification of binuclear iron monooxygenases that are involved in acetone metabolism. Several Gram-positive bacteria, including mycobacteria, have been reported to metabolize acetone via acetol (10, 24, 28). We confirmed that the deletion mutant mc2155 ΔmimA did not lose the ability to grow on acetol as a source of carbon and energy. These results suggest that the mimABCD gene clusters might be responsible for the conversion of acetone to acetol. Elucidation of the catalytic function in acetone metabolism awaits detailed characterization. Gordonia sp. strain TY-5 was reported to oxidize acetone to methyl acetate by a Baeyer-Villiger monooxygenase (AcmA) during acetone metabolism (8). We confirmed that no homolog of the acmA gene exists in the genome sequence of M. smegmatis strain mc2155 (data not shown). There is a possibility that M. smegmatis strain mc2155 metabolizes acetone through a pathway that is different from that in Gordonia sp. strain TY-5.
In conclusion, we identified binuclear iron monooxygenase gene clusters that are responsible for the regioselective oxidation of phenol to hydroquinone in M. goodii strain 12523 and M. smegmatis strain mc2155. Furthermore, this study fortunately provided new insights into propane and acetone metabolism in mycobacteria. In other words, it would be reasonable to conclude that the acetone-induced monooxygenases that play physiologically essential roles in propane and acetone metabolism fortuitously have the ability to oxidize phenol regioselectively at the para position. This unique fortuitous reaction is of biochemical interest and biotechnological importance. Further investigations will focus on heterologous expression of the monooxygenases for their enzymatic characterization and also for their practical application in biocatalysis.
Acknowledgments
We thank Yasuyoshi Sakai (Kyoto University) for the gift of Gordonia sp. strain TY-5 and the disruption mutant TY-5 prmB::Kanr. We thank Yoshikazu Izumi (Tottori University) for the gift of pRHK1. We also thank Ken-ichi Noda (Hiroshima University) for the gift of pUCkap1.
Footnotes
Published ahead of print on 23 December 2010.
REFERENCES
- 1.Brown, B. A., et al. 1999. Mycobacterium wolinskyi sp. nov. and Mycobacterium goodii sp. nov., two new rapidly growing species related to Mycobacterium smegmatis and associated with human wound infections: a cooperative study from the International Working Group on Mycobacterial Taxonomy. Int. J. Syst. Bacteriol. 49:1493-1511. [DOI] [PubMed] [Google Scholar]
- 2.Chan Kwo Chion, C. K., S. E. Askew, and D. J. Leak. 2005. Cloning, expression, and site-directed mutagenesis of the propene monooxygenase genes from Mycobacterium sp. strain M156. Appl. Environ. Microbiol. 71:1909-1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deshayes, C., et al. 2007. Interrupted coding sequences in Mycobacterium smegmatis: authentic mutations or sequencing errors? Genome Biol. 8:R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ehrt, S., F. Schirmer, and W. Hillen. 1995. Genetic organization, nucleotide sequence and regulation of expression of genes encoding phenol hydroxylase and catechol 1,2-dioxygenase in Acinetobacter calcoaceticus NCIB8250. Mol. Microbiol. 18:13-20. [DOI] [PubMed] [Google Scholar]
- 5.Hausinger, R. P. 2007. New insights into acetone metabolism. J. Bacteriol. 189:671-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirasawa, K., Y. Ishii, M. Kobayashi, K. Koizumi, and K. Maruhashi. 2001. Improvement of desulfurization activity in Rhodococcus erythropolis KA2-5-1 by genetic engineering. Biosci. Biotechnol. Biochem. 65:239-246. [DOI] [PubMed] [Google Scholar]
- 7.Kotani, T., T. Yamamoto, H. Yurimoto, Y. Sakai, and N. Kato. 2003. Propane monooxygenase and NAD+-dependent secondary alcohol dehydrogenase in propane metabolism by Gordonia sp. strain TY-5. J. Bacteriol. 185:7120-7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kotani, T., H. Yurimoto, N. Kato, and Y. Sakai. 2007. Novel acetone metabolism in a propane-utilizing bacterium, Gordonia sp. strain TY-5. J. Bacteriol. 189:886-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leahy, J. G., P. J. Batchelor, and S. M. Morcomb. 2003. Evolution of the soluble diiron monooxygenases. FEMS Microbiol. Rev. 27:449-479. [DOI] [PubMed] [Google Scholar]
- 10.Lukins, H. B., and J. W. Foster. 1963. Methyl ketone metabolism in hydrocarbon-utilizing mycobacteria. J. Bacteriol. 85:1074-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyamoto, Y., et al. 2004. Aggregation of mycobacteria caused by disruption of fibronectin-attachment protein-encoding gene. FEMS Microbiol. Lett. 236:227-234. [DOI] [PubMed] [Google Scholar]
- 12.Ng, L. C., V. Shingler, C. C. Sze, and C. L. Poh. 1994. Cloning and sequences of the first eight genes of the chromosomally encoded (methyl) phenol degradation pathway from Pseudomonas putida P35X. Gene 151:29-36. [DOI] [PubMed] [Google Scholar]
- 13.Noda, K., K. Watanabe, and K. Maruhashi. 2002. Cloning of a rhodococcal promoter using a transposon for dibenzothiophene biodesulfurization. Biotechnol. Lett. 24:1875-1882. [DOI] [PubMed] [Google Scholar]
- 14.Nordlund, I., J. Powlowski, and V. Shingler. 1990. Complete nucleotide sequence and polypeptide analysis of multicomponent phenol hydroxylase from Pseudomonas sp. strain CF600. J. Bacteriol. 172:6826-6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pikus, J. D., J. M. Studts, K. McClay, R. J. Steffan, and B. G. Fox. 1997. Changes in the regiospecificity of aromatic hydroxylation produced by active site engineering in the diiron enzyme toluene 4-monooxygenase. Biochemistry 36:9283-9289. [DOI] [PubMed] [Google Scholar]
- 16.Rosenzweig, A. C., C. A. Frederick, S. J. Lippard, and P. Nordlund. 1993. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature 366:537-543. [DOI] [PubMed] [Google Scholar]
- 17.Rui, L., Y. M. Kwon, A. Fishman, K. F. Reardon, and T. K. Wood. 2004. Saturation mutagenesis of toluene ortho-monooxygenase of Burkholderia cepacia G4 for enhanced 1-naphthol synthesis and chloroform degradation. Appl. Environ. Microbiol. 70:3246-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saeki, H., and K. Furuhashi. 1994. Cloning and characterization of a Nocardia corallina B-276 gene cluster encoding alkene monooxygenase. J. Ferment. Bioeng. 78:399-406. [Google Scholar]
- 19.Schäfer, A., A. Tauch, W. Jäger, J. Kalinowski, G. Thierbach, and A. Pühler. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69-73. [DOI] [PubMed] [Google Scholar]
- 20.Semba, H., M. Mukouyama, and K. Sakano. 1996. A para-site-specific hydroxylation of various aromatic compounds by Mycobacterium sp. strain 12523. Appl. Microbiol. Biotechnol. 46:432-437. [DOI] [PubMed] [Google Scholar]
- 21.Semba, H., and K. Sakano. 1997. A para-site-specific hydroxylation of aromatic compounds by Mycobacterium sp. strain 12523: stabilization of the hydroxylation activity. Appl. Microbiol. Biotechnol. 48:256-260. [DOI] [PubMed] [Google Scholar]
- 22.Sharp, J. O., et al. 2007. An inducible propane monooxygenase is responsible for N-nitrosodimethylamine degradation by Rhodococcus sp. strain RHA1. Appl. Environ. Microbiol. 73:6930-6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sulistyaningdyah, W. T., et al. 2005. Hydroxylation activity of P450 BM-3 mutant F87V towards aromatic compounds and its application to the synthesis of hydroquinone derivatives from phenolic compounds. Appl. Microbiol. Biotechnol. 67:556-562. [DOI] [PubMed] [Google Scholar]
- 24.Taylor, D. G., P. W. Trudgill, R. E. Cripps, and P. R. Harris. 1980. The microbial metabolism of acetone. J. Gen. Microbiol. 118:159-170. [Google Scholar]
- 25.Thiemer, B., J. R. Andreesen, and T. Schräder. 2003. Cloning and characterization of a gene cluster involved in tetrahydrofuran degradation in Pseudonocardia sp. strain K1. Arch. Microbiol. 179:266-277. [DOI] [PubMed] [Google Scholar]
- 26.van der Geize, R., G. I. Hessels, R. van Gerwen, P. van der Meijden, and L. Dijkhuizen. 2001. Unmarked gene deletion mutagenesis of kstD, encoding 3-ketosteroid Δ1-dehydrogenase, in Rhodococcus erythropolis SQ1 using sacB as counter-selectable marker. FEMS Microbiol. Lett. 205:197-202. [DOI] [PubMed] [Google Scholar]
- 27.Vardar, G., and T. K. Wood. 2004. Protein engineering of toluene-o-xylene monooxygenase from Pseudomonas stutzeri OX1 for synthesizing 4-methylresorcinol, methylhydroquinone, and pyrogallol. Appl. Environ. Microbiol. 70:3253-3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vestal, J. R., and J. J. Perry. 1969. Divergent metabolic pathways for propane and propionate utilization by a soil isolate. J. Bacteriol. 99:216-221. [DOI] [PMC free article] [PubMed] [Google Scholar]