

Abstract
The Gram-negative facultative chemolithoautotrophic bacterium Ralstonia eutropha strain H16 is known for its narrow carbohydrate utilization range, which limits its use for biotechnological production of polyhydroxyalkanoates and possibly other products from renewable resources. To broaden its substrate utilization range, which is for carbohydrates and related compounds limited to fructose, N-acetylglucosamine, and gluconate, strain H16 was engineered to use mannose and glucose as sole carbon sources for growth. The genes for a facilitated diffusion protein (glf) from Zymomonas mobilis and for a glucokinase (glk), mannofructokinase (mak), and phosphomannose isomerase (pmi) from Escherichia coli were alone or in combination constitutively expressed in R. eutropha strain H16 under the control of the neokanamycin or lac promoter, respectively, using an episomal broad-host-range vector. Recombinant strains harboring pBBR1MCS-3::glf::mak::pmi or pBBR1MCS-3::glf::pmi grew on mannose, whereas pBBR1MCS-3::glf::mak and pBBR1MCS-3::glf did not confer the ability to utilize mannose as a carbon source to R. eutropha. The recombinant strain harboring pBBR1MCS-3::glf::pmi exhibited slower growth on mannose than the recombinant strain harboring pBBR1MCS-3::glf::mak::pmi. These data indicated that phosphomannose isomerase is required to convert mannose-6-phosphate into fructose-6-phosphate for subsequent catabolism via the Entner-Doudoroff pathway. In addition, all plasmids also conferred to R. eutropha the ability to grow in the presence of glucose. The best growth was observed with a recombinant R. eutropha strain harboring plasmid pBBR1MCS-2::Pnk::glk::glf. In addition, expression of the respective enzymes was demonstrated at the transcriptional and protein levels and by measuring the activities of mannofructokinase (0.622 ± 0.063 U mg−1), phosphomannose isomerase (0.251 ± 0.017 U mg−1), and glucokinase (0.518 ± 0.040 U mg−1). Cells of recombinant strains of R. eutropha synthesized poly(3-hydroxybutyrate) to ca. 65 to 67% (wt/wt) of the cell dry mass in the presence of 1% (wt/vol) glucose or mannose as the sole carbon sources.
The facultative chemolithoautotrophic Gram-negative Ralstonia eutropha strain H16, formerly known as Alcaligenes eutrophus, serves as model organism for studying the metabolism of poly(3-hydroxybuyrate) (PHB) and hydrogen-based chemolithoautotrophy. If grown autotrophically, strain H16 assimilates CO2 via the Calvin-Benson-Bassham cycle. It uses also various organic compounds as a carbon and energy source if H2 is absent. Under unbalanced growth conditions, PHB is accumulated in granules as a storage compound for carbon and energy (8). This is the case when a carbon source is abundant and if another macroelement such as nitrogen is lacking. The well-understood physiology of this “Knallgas” bacterium, its ability to be cultivated to high cell densities also at large scale, and the enormous polyhydroxyalkanoate (PHA) content of the cells led the industry consider to producing PHAs by fermentation of R. eutropha (7, 26, 35). The biodegradability of PHAs made these polyesters attractive for numerous applications. One prominent example is the commercial production of the thermoplastic Biopol (14).
Although R. eutropha H16 is capable of utilizing various organic compounds such as organic acids, aromatic compounds, and gluconate, its ability to metabolize sugars is restricted to fructose and the amino sugar N-acetylglucosamine; in addition, gluconate is used (30, 36). Fructose is presumably transported into the cell by an ABC-type transporter, and the transfer of N-acetylglucosamine is probably mediated by a phosphotransferase-type transport system (30). Further catabolism of these sugars occurs via the Entner-Doudoroff (KDPG) pathway. The key enzymes of the Embden-Meyerhoff-Parnas (EMP) and the oxidative pentose phosphate pathways, fructose-1,6-bisphosphate aldolase (FBPA) and 6-phosphogluconate dehydrogenase (6PGDH), respectively, are lacking in R. eutropha (16, 30). Interestingly, genes for an anabolic EMB pathway (gluconeogenesis) exist (30). A catabolic EMP pathway was established in R. eutropha by heterologous expression of the pfkA gene from Escherichia coli encoding an FBPA (41).
Efforts to extend the substrate utilization range of R. eutropha H16 were frequently made in the past. Schlegel and Gottschalk (36) isolated several glucose-utilizing mutants of R. eutropha strain H16. All mutants capable of glucose utilization exhibited elevated levels of a constitutively expressed glucose-6-phosphate dehydrogenase. It was assumed that glucose is taken up via a facilitative diffusion process. Despite further efforts, no mannose-metabolizing mutants were isolated (23). Pries et al. (31) established lactose utilization in R. eutropha by inserting the β-galactosidase gene fused with the PHB biosynthesis gene promoter into the genome of R. eutropha strain H16. Subsequently, the cells utilized the residual glucose for growth, whereas galactose was secreted into the medium. Only after the gal operon of E. coli was cloned on a separate vector and also introduced to R. eutropha was the complete utilization of lactose achieved (31). There were also numerous attempts to extend the substrate utilization range for cheap and abundant carbon sources in other microorganisms such as the utilization of unfermentable pentoses like xylose and arabinose occurring in lignocellulosic biomass. These studies often aimed at the extension of carbohydrate metabolism in ethanol producers such as Zymomonas mobilis and Saccharomyces cerevisiae (12, 20, 28, 44, 50).
The aim of the present study was to further extend the substrate utilization range of R. eutropha and to establish utilization of the carbohydrates glucose and mannose in strain H16. Glucose is the most abundant sugar in nature and is primarily found in sucrose, vegetable starch, and cellulose (27). Mannose is a wood sugar and is present in a polymerized form in the so-called hemicelluloses of plant cell walls. Predominantly, it is abundant in the lignocellulosic biomass of soft woods. Lignocellulose is the major renewable resource in several applications (18). We report here on the development of a mannose- and glucose-utilizing R. eutropha strain by metabolic engineering. The introduction of different genes necessary for the utilization of glucose and mannose in R. eutropha strain H16 complemented the lacking parts of glucose and mannose catabolism.
MATERIALS AND METHODS
Bacterial strains, plasmids, oligonucleotides, and cultivation conditions.
All of the bacteria, plasmids, and primers used in the present study are listed in Table 1 . Cells of R. eutropha strain H16 were cultivated in mineral salts medium (MSM) as described by Schlegel et al. (34). Carbon sources were added to liquid MSM as indicated in the text. Liquid cultures in Erlenmeyer flasks were incubated on a horizontal rotary shaker at an agitation of 110 rpm. Solid media were prepared by the addition of 1.5% (wt/vol) agar/agar. E. coli cells were cultivated at 37°C in lysogeny broth (LB [33]). Z. mobilis cells were cultivated in a medium containing 10 g of Bacto peptone, 10 g of yeast extract, and 20 g of glucose/liter. Antibiotics were applied according to the method of Sambrook et al. (33) and as indicated in the text.
TABLE 1.
Bacterial strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Relevant characteristics or sequence (5′-3′)a | Source or reference |
|---|---|---|
| Strains | ||
| E. coli K-12 | Wild type | DSM 426 |
| E. coli TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC)φ80dlacZΔM15 ΔlacX74 deoR recA1 araD139 Δ(ara-leu)7697 galU galK rpsL endA1 nupG | Invitrogen |
| E. coli S17-1 | recA1 thi-1 hsdR17(rK− mK+) proA tra genes of RP4 plasmid chromosomally integrated (mobilization strain) | 40 |
| E. coli JE5511 | Hfr; manA4 lpp-1 pps-6 | 19; E. coli Genetic Stock Center |
| R. eutropha strain H16 | Wild type | DSM 428 |
| R. eutropha strain G+1 | Glucose-utilizing mutant of R. eutropha strain H16 | 36 |
| Z. mobilis subsp. mobilis | Wild type | DSM 424, ATCC 10988 |
| Z. mobilis subsp. mobilis | Wild type | DSM 3580, ATCC 29191 |
| Plasmids | ||
| pJET1.2/blunt | Apr | Fermentas |
| pCR2.1 | Apr Kmr; LacZ-α | Invitrogen |
| pJET1.2::glf | pJET1.2 with glf as a blunt-end PCR product | This study |
| pJET1.2::mak | pJET1.2 with mak as a blunt-end PCR product | This study |
| pJET1.2::pmi | pJET1.2 with pmi as a blunt-end PCR product | This study |
| pCR2.1::Pnk | pCR2.1 with neokanamycin promoter as a XhoI/NdeI fragment | This study |
| pCR2.1::Pnk::glk | pCR2.1 with neokanamycin promoter and glk as a XhoI/EcoRI fragment | This study |
| pBBR1MCS-3 | Tcr; LacZ-α mob rep | 24 |
| pBBR1MCS-2::Pnk::glf | pBBR1MCS-2 with neokanamycin promoter and glf as a XhoI/SacI fragment | This study |
| pBBR1MCS-2::Pnk::glk::glf | pBBR1MCS-2 with neokanamycin promoter, glk, and glf as a XhoI/SacI fragment | This study |
| pBBR1MCS-3::glf | pBBR1MCS-3 with glf as a KpnI/PstI fragment | This study |
| pBBR1MCS-3::glf::mak | pBBR1MCS-3 with glf and mak as a KpnI/XbaI fragment | This study |
| pBBR1MCS-3::glf::pmi | pBBR1MCS-3 with glf as a KpnI/PstI fragment and a pmi XbaI/SacI fragment | This study |
| pBBR1MCS-3::glf::mak::pmi | pBBR1MCS-3 with glf, mak, and pmi as KpnI/SacI fragment | This study |
| Oligonucleotides | ||
| Pglf_KpnI_f | GGTACCAAGGAAGGACTGATCATGAGTTCTGAAAGTAGTCAGGGTCTAGTC3 | |
| Pglf_PstI_r | CTGCAGCTACTTCTGGGAGCGCCACATC | |
| Pmak_PstI_f | CTGCAGAAGGAAGGACTGATCGTGCGTATAGGTATCGATTTAGGCG | |
| Pmak_XbaI_r | TCTAGATTACTCTTGTGGCCATAACCACGC | |
| Ppmi_XbaI_f | TCTAGAAAGGAAGGTCGACTCATGCAAAAACTCATTAACTCAGTGCAAAAC | |
| Ppmi_SacI_r | GAGCTCTTACAGCTTGTTGTAAACACGCGCTAAAC | |
| glf_RT_f | GTTCTATCGATTGGGTTAATGCCAGTGG | |
| glf_RT_r | GGAACATCTGCGGTGCATAATACAGC | |
| mak_RT_f | GCGAGGTTGCAGCGGGAAGTG | |
| mak_RT_r | AATTTCACTGCCTTTCAGCGCATGTCC | |
| pmi_RT_f | CGCTGACGCCTTTCCTTGCGAT | |
| pmi_RT_r | GCGGTGTTTCAGCGAACAGGAACA | |
| pJET1.2 forward sequencing primer, 23-mer | CGACTCACTATAGGGAGAGCGGC | Fermentas |
| pJET1.2 reverse sequencing primer, 24-mer | AAGAACATCGATTTTCCATGGCAG | Fermentas |
| M13/pUC-forward, 17-mer | GTAAAACGACGGCCAGT | Jena Bioscience |
| F_Pnk_XhoI | CTCGAGCCGGAATTGCCAGCTGGGG | |
| R_Pnk_NdeI | CATATGAAACGATCCTCATCCTGTCTCTTG | |
| F_glk_NdeI | CATATGACAAAGTATGCATTAGTCGGTGATGTGGG | |
| R_glk_SalI | GTCGACTTACAGAATGTGACCTAAGGTCTGGCGTAAATGTGC | |
| F_glf_BamHI | GGATCCATGAGGATCGTTTCGCATGAGTTCTGAAAGTAGTCAGGGTCTAGTC | |
| R_glf_SacI | GAGCTCCTACTTCTGGGAGCGCCACATCTCCTCG |
Tcr, tetracycline resistance; Apr, ampicillin resistance; Kmr, kanamycin resistance. In the oligonucleotide sequences, restriction sites are indicated by underlining.
Isolation, analysis, and modification of DNA.
Plasmid DNA was prepared from crude lysates by the alkaline extraction method (4). The total DNA of Z. mobilis DSM 424 or DSM 3580 and of E. coli strain K-12 was prepared using a DNeasy blood and tissue kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Restriction endonucleases (Gibco-BRL, Gaithersburg, MD) were applied under conditions recommended by the manufacturer. All other genetic procedures and manipulations were conducted as described by Sambrook et al. (33).
Constructions of plasmids and transfer into R. eutropha.
The coding regions of glf from Z. mobilis DSM 424 and DSM 3580 and of glk, mak, and pmi from E. coli, respectively, were amplified by PCR using the oligonucleotides Pglf_KpnI_f, Pglf_PstI_r, F_glf_BamHI, and R_glf_SacI for glf; F_Pnk_XhoI and R_Pnk_NdeI for Pnk; F_glk_NdeI and R_glk_SalI for glk; Pmak_PstI_f and Pmak_XbaI_r for mak; or Ppmi_XbaI_f and Ppmi_SacI_r for pmi (Table 1). For PCR, KOD Hot Start DNA polymerase (Merck, Darmstadt, Germany) was used according to the manufacturer's instructions. PCR products were then cloned into the pJET1.2/blunt-cloning vector (Fermentas, Germany) using T4 DNA ligase (Gibco-BRL) or into cloning vector pCR2.1 (Invitrogen, Carlsbad, Germany) and transferred into E. coli strain TOP10. Plasmids were isolated from ampicillin-resistant clones, and the cloned fragments were excised by using restriction enzymes appropriate for further cloning and extracted from the gel after separation using an EZNA gel extraction kit (Omega Bio-Tec, Bangalore, India). For expression experiments in R. eutropha, the broad-host-range vectors pBBR1MCS-2 and pBBR1MCS-3 (24) were used for cloning of glf, glk, mak, and pmi. Whereas pBBR1MCS-2 conferred kanamycin resistance (50 μg/ml for E. coli and 300 μg/ml for R. eutropha strain H16), pBBR1MCS-3 conferred tetracycline (12.5 μg/ml) resistance for selection to E. coli and R. eutropha strain H16. The coding region of glf was excised by restriction with KpnI and PstI and ligated to KpnI/PstI-linearized plasmid pBBR1MCS-3, yielding plasmid pBBR1MCS-3::glf (Fig. 2). The coding sequence of the neokanamycin promoter Pnk was amplified using the primers F_Pnk_XhoI and R_Pnk_NdeI with isolated vector pBBR1MCS-2 and further ligated to vector pCR2.1, yielding pCR2.1::Pnk. The glk gene was amplified by tailored PCR from total DNA of E. coli K-12 by using the oligonucleotides F_glk_NdeI and R_glk_SalI as primers. The resulting PCR product was cloned as an NdeI/SalI fragment into pCR2.1::Pnk, yielding pCR2.1::Pnk::glk. The glf gene was amplified by tailored PCR from the total DNA of Z. mobilis DSM 3580 using the oligonucleotides F_glf_BamHI and R_glf_SacI as primers. The resulting PCR product was cloned as BamHI/SacI fragment into pBBR1MCS-2, yielding pBBR1MCS-2::glf. To obtain plasmid pBBR1MCS-2::Pnk::glk::glf, the coding region of Pnk::glk was excised by restriction with XhoI and EcoRI from plasmid pCR2.1::Pnk::glk and ligated into XhoI/EcoRI-linearized pBBR1MCS-2::glf. The coding sequence of glk was further excised by restriction with NdeI/SalI from plasmid pBBR1MCS-2::Pnk::glk::glf; the linearized plasmid was treated with T4 polymerase (Gibco-BRL), yielding blunt ends, and religated using T4 DNA ligase (Gibco-BRL), yielding plasmid pBBR1MCS-2::Pnk::glf. To obtain the plasmids pBBR1MCS-3::glf::mak and pBBR1MCS-3::glf::pmi, the coding regions of mak and pmi were excised by restriction with PstI/XbaI and XbaI/SacI, respectively, and ligated to PstI/XbaI- and XbaI/SacI-linearized plasmid pBBR1MCS-3::glf, respectively (Fig. 2). The excised coding regions of mak and pmi were also ligated together to PstI/SacI-linearized plasmid pBBR1MCS-3::glf, yielding pBBR1MCS-3::glf::mak::pmi (Fig. 2). All plasmids were transferred to E. coli strain S17-1 by transformation (17).
Transfer of DNA by conjugation.
Plasmids pBBR1MCS-2, pBBR1MCS-2:: Pnk::glf, pBBR1MCS-2::Pnk::glk::glf, pBBR1MCS-3, pBBR1MCS-3::glf, pBBR1MCS-3::glf::mak, pBBR1MCS-3::glf::pmi, and pBBR1MCS-3::glf::mak::pmi (Table 1) were transferred by conjugation, applying a previously described protocol (15), using E. coli S17-1 as the donor and R. eutropha strains as recipients.
DNA sequence analysis.
DNA was sequenced in a 3730 DNA Analyzer electrophoresis system (Applied Biosystems, Foster City, CA). For sequencing of inserts in the pJET1.2/blunt vector (Fermentas, Germany), the pJET1.2 forward sequencing primer, 23-mer, and pJET1.2 reverse sequencing primer, 24-mer, were used. Universal M13/pUC-forward and M13/pUC-reverse primers were used for sequencing inserts in plasmids pBBR1MCS-2 and pBBR1MCS-3 (24).
RT-PCR analysis of total RNA isolated from R. eutropha.
DNA-free total RNA of R. eutropha strain H16 was prepared by using an RNeasy RNA purification kit (Qiagen) according to the manufacturer's protocol. For identification of glf-, mak-, and pmi-derived mRNA, reverse transcription-PCR (RT-PCR) was applied using oligonucleotides glf_RT_f, glf_RT_r, mak_RT_f, mak_RT_r, pmi_RT_f, and pmi_RT_r (Table 1). RT-PCR was carried out with a Qiagen OneStep RT-PCR kit (Qiagen) and 0.5 ng of RNA as a template. To exclude any DNA contamination that could serve as a template for PCR, template RNA was added in a control experiment, after inactivation of reverse transcriptase for 15 min at 95°C in the presence of Taq polymerase. The absence of PCR products indicated that the RT-PCR products were not derived from contaminating DNA.
Preparation of soluble protein fractions.
Cells of recombinant R. eutropha strains were cultivated as described above in the presence of 1% (wt/vol) sodium gluconate as the sole carbon source. Cells were harvested by 15 min of centrifugation at 4°C and 3,500 × g, washed with 100 mM morpholinepropanesulfonic acid (MOPS) buffer (pH 7.0) and resuspended in 2 volumes of the same buffer. Disruption was done by sonication in a Sonifier 250 (Branson Sonic Power Company) with an amplitude of 16 μm (1 min/ml; 50% output control), followed by cooling with an ice-NaCl mixture. Soluble membrane-free protein fractions were prepared by 60 min of ultracentrifugation of the crude extracts at 100,000 × g and 4°C.
Solubilization of membrane proteins.
Cells of R. eutropha were cultivated as described above in the presence of 1% (wt/vol) glucose as the sole carbon source. After 48 h the cells were harvested by 15 min of centrifugation at 3,500 × g and 4°C, washed once with 0.9% (wt/vol) NaCl, and resuspended in 1 volume of 100 mM Tris-HCl buffer (pH 7.0). Cell disruption was carried out by a 3-fold passage through a precooled French pressure cell at 1,000 MPa. The obtained lysates were centrifuged as before to remove residual cells. Membrane fractions were prepared by 1 h centrifugation of the supernatants at 100,000 × g and 4°C, resuspension in 2 ml of 20 mM Tris-HCl buffer (pH 7.4) containing 200 mM NaCl and 2% (vol/vol) Triton X-114, and 12 h of incubation on ice on a VV3 rotary shaker (VWR GmbH, Darmstadt, Germany). The lysate was centrifuged for 20 min at 3,500 × g and 4°C. The supernatant was incubated for 5 min at 37°C, centrifuged for 20 min for at 3,500 × g and 37°C to remove the Triton X-114 (5), and then further diluted with 2 volumes of the same buffer without Triton X-114 to decrease the detergent concentration.
One-dimensional SDS-PAGE.
Protein samples were resuspended in gel loading buffer and separated in 12.5% (wt/vol) SDS-polyacrylamide gels as described by Laemmli (25). The proteins were stained with Coomassie brilliant blue R-250 (45). Samples of crude extracts and solubilized membrane proteins were analyzed by this method.
MAK, PMI, and GLK activity assays.
Mannofructokinase (MAK), phosphomannose isomerase (PMI), and glucokinase (GLK) activity measurements were done in soluble protein fractions of recombinant R. eutropha strains at 30°C using a Nicolet Evolution 100 UV/VIS spectrophotometer (Thermo Electron Corp., Cambridge, United Kingdom). The activity of MAK was assayed in an NADH-coupled system as described by Sebastian and Asensio (38) with some modifications, using pyruvate kinase and lactate dehydrogenase as auxiliary enzymes. The buffer (100 mM MOPS [pH 7.0]) contained 2 mM MgCl2, 2 mM ATP, 0.2 mM NADH, 5 mM mannose, 0.2 mM phosphoenolpyruvate (PEP), 5 U of pyruvate kinase ml−1, 5 U of lactate dehydrogenase ml−1, and 5 to 100 μl of soluble extract. The PMI activity was measured in an NADP-coupled assay (21) with some modifications, using phosphoglucose isomerase and glucose-6-phosphate dehydrogenase as auxiliary enzymes. The buffer (100 mM MOPS [pH 7.0]) contained 5 mM MgCl2, 1 mM NADP, 3 mM mannose-6-phosphate, 1 U of phosphoglucose isomerase ml−1, 1 U of glucose-6-phosphate dehydrogenase ml−1, and 10 to 100 μl of soluble extract. The GLK activity was measured in an NADPH-coupled system according to the method of Gottschalk et al. (16) using a modified buffer. The buffer mixture (100 mM Tris-HCl [pH 7.6]) contained 7 mM MgCl2, 0.9 mM NADP, 460 mM d-glucose, 0.7 mM ATP, 0.35 U of glucose-6-phosphate-dehydrogenase ml−1, and 10 to 50 μl of enzyme preparation.
Analysis of PHB content of recombinant R. eutropha cells by gas chromatography.
Determination of the PHB content was performed as described in detail elsewhere (9, 42).
RESULTS
Search for genes encoding enzymes catabolizing mannose and glucose and transporters of these sugars in the genome of R. eutropha strain H16.
Previous studies (16) concluded that all enzymes for the catabolism of glucose are present in this bacterium and that its cryptic behavior was due to the lack of a glucose transporter. An in silico analysis using the blastp algorithm (1) and a protein database for R. eutropha strain H16 (30) investigated the genome of R. eutropha strain H16 for the presence of genes coding for functional enzymes required for catabolism of mannose and glucose. Consistent with the previous conclusions, no genes encoding a PEP-dependent phosphotransferase system (PTS) or a transporter specific for glucose or mannose could be identified in the genome of strain H16. Although strain H16 possesses transport proteins belonging to the major facilitator superfamily (MFS), these proteins exhibit only a few similarities to other energy-independent glucose transporters. Most hexokinases occurring in prokaryotes are specific for their substrates (10), and strain H16 possesses genes for a fructokinase and a glucokinase (30). Previous studies have demonstrated that fructose, glucose, and mannose were phosphorylated by crude extracts of wild-type R. eutropha strain H16 (16). Although our in silico analyses did not detect an unspecific hexokinase in strain H16, a side activity of one of these kinases with the other substrate could not be excluded. Furthermore, a pmi gene putatively encoding a phosphomannose isomerase (PMI) or mannose-6-phosphate isomerase, which catalyzes the reversible isomerization of the phosphorylated forms of mannose and fructose, was annotated in R. eutropha strain H16; however, it is unknown whether the protein exhibits any activity with mannose-6-phosphate. Such an enzyme in combination with a phosphoglucose isomerase present in R. eutropha strain H16 could then convert mannose-6-phosphate into glucose-6-phosphate. All other genes encoding enzymes of the KDPG pathway for further catabolism of glucose-6-phosphate are present in R. eutropha strain H16.
Strategy to establish mannose and glucose utilization in R. eutropha.
Because the in silico analysis did not reveal a glucose and mannose transporter in strain H16 (30), the transport of these sugars had to be established first. Due to the complexity of PEP-dependent PTS, the energy-independent glucose-facilitated diffusion transporter (GLF) from Z. mobilis was chosen for heterologous expression in R. eutropha strain H16. GLF not only has a high affinity for glucose but also uses mannose as a substrate (29, 47). GLF was repeatedly heterologously expressed in E. coli to complement mutants defective in glucose-specific PTS components (22, 29, 47) and, since GLF is primarily a glucose transporter, it was assumed that it will also confer glucose utilization to R. eutropha.
Since crude extracts of R. eutropha H16 were shown to phosphorylate not only glucose but also fructose and mannose (16), an episomally introduced glucokinase should not be mandatory. Since the specific activity of this kinase was relatively low in comparison to the subsequent glucose-6-phosphate dehydrogenase and the Entner-Doudoroff pathway (23), we also introduced the E. coli glk gene encoding a glucokinase to prevent a possible metabolite bottleneck.
The mak gene encoding MAK in E. coli K-12 should phosphorylate mannose after its uptake into the cell and was chosen for heterologous expression in R. eutropha strain H16. MAK from E. coli is known to phosphorylate mannose and fructose with almost the same efficiency (37). Subsequent conversion of mannose-6-phosphate to fructose-6-phosphate by PMI should enable its further catabolism via the KDPG pathway. PMI from E. coli was already successfully expressed in Z. mobilis, enabling this bacterium to use mannose as a sole carbon source (47). Therefore, we episomally introduced and expressed the Z. mobilis glf gene plus the E. coli mak and pmi genes in R. eutropha and characterized the recombinant strains. The engineered pathways of glucose and mannose catabolism in the recombinant R. eutropha strain are depicted in Fig. 1.
FIG. 1.

Pathways of mannose and glucose utilization established in R. eutropha strain H16. GLF, glucose-facilitated diffusion transporter; KDPG, Entner-Doudoroff pathway; MAK, mannofructokinase; PMI, phosphomannose isomerase, GLK, glucokinase; PGI, phosphoglucose isomerase.
Construction of different pBBR1 expression vectors for R. eutropha strain H16.
The PCR products of the Z. mobilis gene glf and of the E. coli genes mak, pmi, and glk comprising suitable ribosome-binding sites for R. eutropha were cloned into the broad-host-range vectors pBBR1MCS-2 and pBBR1MCS-3 under the control of the neokanamycin or lac promoter, respectively, which allow constitutive expression of the cloned genes in R. eutropha H16 (39), yielding the plasmids pBBR1MCS-2::Pnk::glf, pBBR1MCS-2::Pnk::glk::glf, pBBR1MCS-3::glf, pBBR1MCS-3::glf::mak, pBBR1MCS-3::glf::pmi, and pBBR1MCS-3::glf::mak::pmi (Fig. 2). In addition, the functionality of the pmi gene in pBBR1MCS-3::glf::pmi and pBBR1MCS-3::glf::mak::pmi was confirmed by complementation of the pmi (manA)-deficient E. coli mutant JE5511 (19). The recombinant E. coli manA mutants exhibited good growth on solid M9 medium containing 0.5% (wt/vol) mannose as a sole carbon source after 2 days of incubation at 37°C. In contrast, the pBBR1MCS-3 vector did not confer growth to the manA mutant.
FIG. 2.

Physical maps of the constructed plasmids pBBR1MCS-3::glf, pBBR1MCS-3::glf::mak, pBBR1MCS-3::glf::pmi, pBBR1MCS-3::glf::mak::pmi, pBBR1MCS-2::Pnk::glf, and pBBR1MCS-2::Pnk::glk::glf. Relevant cleavage sites and structural genes are indicated. Kmr, kanamycin resistance cassette, Tcr, tetracycline resistance cassette; mob, mobilization site; rep, origin of replication; glf, gene encoding the glucose-facilitated diffusion transporter (GLF) from Z. mobilis (accession no. P21906); glk, gene encoding the glucokinase (GLK) from E. coli (accession no. NP_416889); mak, gene encoding a mannofructokinase (MAK) from E. coli (accession no. P23917); pmi, gene encoding a phosphomannose isomerase (PMI) from E. coli (accession no. P00946).
Transfer of plasmids to R. eutropha strain H16 and establishment of glucose and mannose utilization.
Both plasmids pBBR1MCS-2::Pnk::glf and pBBR1MCS-2::Pnk::glk::glf were transferred to R. eutropha strain H16. Only recombinant strains harboring either pBBR1MCS-2::Pnk::glf or pBBR1MCS-2::Pnk::glk::glf exhibited significant growth after 5 days of incubation.
To investigate whether all three genes are necessary for mannose utilization, all constructed plasmids and plasmid pBBR1MCS-3 were mobilized to R. eutropha strain H16. The cells were then transferred to MSM agar plates containing mannose as the sole carbon source at various concentrations (0.2, 0.5, 1.0, 2.0, and 4.0% [wt/vol]). Only recombinant strains harboring pBBR1MCS-3::glf::pmi or pBBR1MCS-3::glf::mak::pmi exhibited significant growth in the presence of any tested mannose concentration. Whereas cells harboring plasmid pBBR1MCS-3::glf::mak::pmi exhibited distinct growth on mannose after 3 to 4 days of incubation, cells with pBBR1MCS-3::glf::pmi only showed growth after an incubation time of 5 to 6 days. In contrast, recombinant strains harboring plasmid pBBR1MCS-3::glf or pBBR1MCS-3::glf::mak, respectively, exhibited barely no growth even after 30 days of incubation. Only after prior exposure of the latter recombinant strains on MSM plates containing fructose instead of gluconate did slight growth occur after a prolonged incubation time.
Control of expression of glf, glk, mak, and pmi.
To confirm the presence of glf-, mak-, and pmi-derived mRNA, transcriptional analyses of the recombinant strains were performed. Since Plac is a constitutive promoter in R. eutropha strain H16 (39), all three genes should be expressed constitutively without further induction. PCR products of the expected size of 351 bp for glf, mak, and pmi, respectively, were obtained if RNA isolated from recombinant pBBR1MCS-3::glf::mak::pmi harboring cells of strain H16 was analyzed. This clearly demonstrated that glf, mak, and pmi were only transcribed in cells of the recombinant strain of R. eutropha strain H16 harboring pBBR1MCS-3::glf::mak::pmi but not in the negative control harboring only the vector pBBR1MCS-3. The absence of PCR products in the control without any activity of reverse transcriptase indicated that the RT-PCR product was not derived from contaminating DNA (Fig. 3).
FIG. 3.
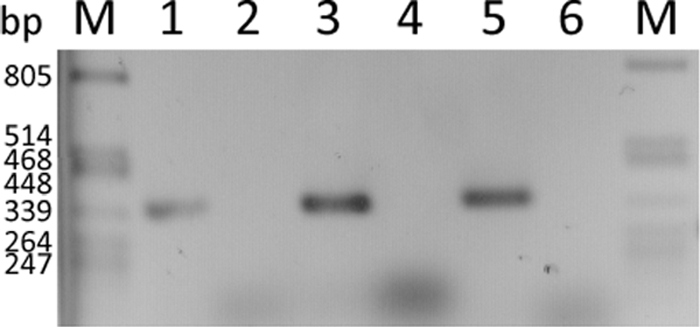
Transcription analysis of glf, mak, and pmi in R. eutropha strain H16 harboring pBBR1MCS-3::glf::mak::pmi and in the negative control harboring only the vector pBBR1MCS-3. Lanes 1, 3, and 5 represent the RT-PCR assays for glf, mak, and pmi, respectively, for R. eutropha strain H16 harboring pBBR1MCS-3::glf::mak::pmi, whereas lanes 2, 4, and 6 represent the negative controls to detect whether there is any expression of glf, mak, and pmi in the strain harboring only the vector pBBR1MCS-3. A λ/PstI DNA marker (lanes M; Fermentas, Germany) served for size comparison.
To further confirm the transcription of glk and glf in strains harboring plasmid pBBR1MCS-2::Pnk::glk::glf at the protein level, an analysis by SDS-PAGE was performed. Since Glf is a transmembrane protein (29), membrane proteins of both R. eutropha strain H16 harboring vector pBBR1MCS-2 and R. eutropha strain H16 harboring plasmid pBBR1MCS-2::Pnk::glk::glf were solubilized and separated by SDS-PAGE. The electropherogram showed a distinct protein band with an apparent mass of 50 kDa representing Glf that was absent in the control. Similarly, analysis of the soluble cell fraction showed a distinct protein band of 35 kDa, representing GLK (Fig. 4).
FIG. 4.
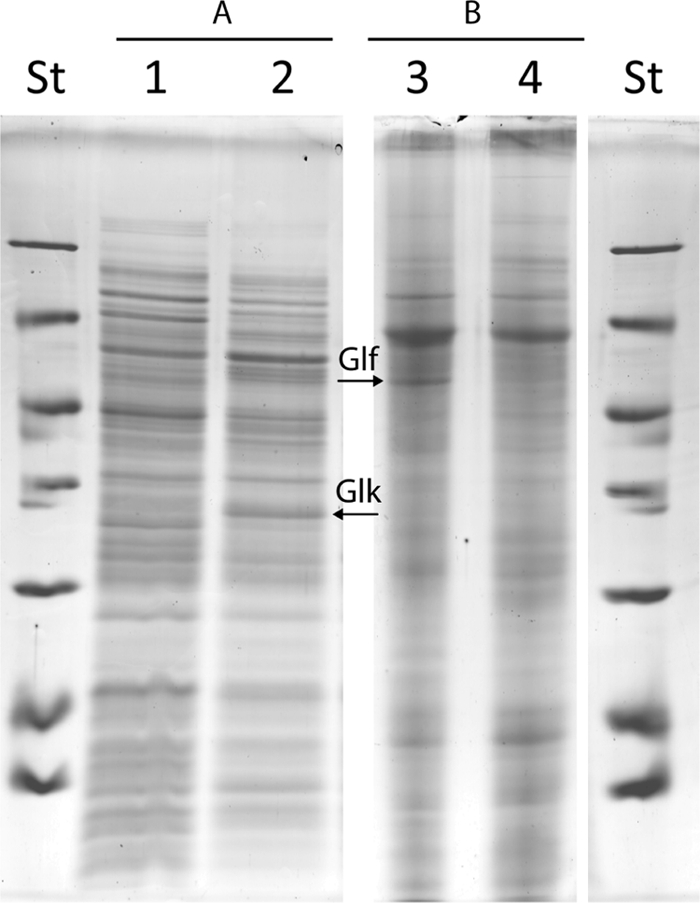
SDS-PAGE analysis of crude extracts and solubilized membrane proteins of recombinant R. eutropha strain H16 harboring pBBR1MCS-2 or pBBR1MCS-2::Pnk::glk::glf. (A) Crude extracts. Lane 1, R. eutropha strain H16 pBBR1MCS-2; lane 2, R. eutropha strain H16 pBBR1MCS-2::Pnk::glk::glf. (B) Solubilized membrane proteins. Lane 3, R. eutropha strain H16 pBBR1MCS-2::Pnk::glk::glf; lane 4, R. eutropha strain H16 pBBR1MCS-2. St, molecular weight standard. Arrows indicate detected recombinant proteins glucokinase (Glk) and glucose transporter (Glf).
The presence of functional active MAK, PMI, and GLK in the recombinant strains was confirmed by enzymatic analyses. Coupled enzyme assays using the soluble protein fractions obtained from cells of the recombinant strain of H16 harboring pBBR1MCS-3::glf::mak::pmi or pBBR1MCS-2::Pnk::glk::glf demonstrated the presence of active PMI (0.251 ± 0.017 U mg−1), MAK (0.622 ± 0.063 U mg−1), and also GLK (0.518 ± 0.040 U mg−1), respectively, whereas activities were distinctly diminished for PMI (0.019 ± 0.005 U mg−1), MAK (0.021 ± 0.006 U mg−1), and GLK (0.052 ± 0.001 U mg−1) in the soluble protein fractions of the negative control of strain H16 harboring plasmids pBBR1MCS-2 or pBBR1MCS-3, respectively. For comparison and to give evidence for enzyme expression in R. eutropha, the respective enzyme assays were also performed with soluble protein fractions from E. coli harboring pBBR1MCS-2::Pnk::glk::glf and pBBR1MCS-3::glf::mak::pmi. The enzyme activities were as follows: PMI, 2.010 ± 0.048 U mg−1; MAK, 1.230 ± 0.059 U mg−1; and GLK, 0.708 ± 0.050 U mg−1. E. coli strains harboring the empty vector exhibited diminished activity levels for PMI (0.220 ± 0.035 U mg−1), MAK (0.160 ± 0.013 U mg−1), and GLK (0.004 ± 0.001 U mg−1).
Investigations on utilization of various carbon sources by the recombinant strains of R. eutropha H16.
In comparison to the other recombinant strains, R. eutropha strain H16 harboring the plasmids pBBR1MCS-2::Pnk::glk::glf or pBBR1MCS-3::glf::mak::pmi exhibited the fastest growth on glucose or mannose of all tested substrates, respectively, as a sole carbon source. These strains were therefore further investigated with regard to the utilization of several other carbon sources using MSM agar plates each containing 1% (wt/vol) l-arabinose, d-fructose, d-galactose, d-gluconate, d-glucose, glycerol, maltose, d-mannitol, d-mannose, N-acetyl-d-glucosamine, pyruvate, d-ribose, trehalose, or d-xylose as the sole carbon source. Wild-type R. eutropha served as a negative control. Both the wild-type strain and the recombinant strains were able to grow in the presence of d-fructose, d-gluconate, glycerol, N-acetylglucosamine, and pyruvate. All other carbon sources mentioned above were not utilized for growth, except mannose by the recombinant strains harboring pBBR1MCS-3::glf::mak::pmi and glucose by the recombinant strains harboring pBBR1MCS-2::Pnk::glk::glf or pBBR1MCS-3::glf::mak::pmi.
Growth of the recombinant R. eutropha with glucose as the sole carbon source.
Recombinant strains of R. eutropha strain H16 were cultivated in liquid MSM containing 1% (wt/vol) glucose. Although all strains exhibited growth on glucose, the recombinant strain harboring pBBR1MCS-2::Pnk::glk::glf showed the fastest growth (μ = 0.32 h−1) compared to the recombinant strain harboring pBBR1MCS-3::glf::mak::pmi (μ = 0.12 h−1) (Fig. 5 A). This was in the same order as the positive control strain G+1 (μ = 0.30 h−1) (36). Interestingly, recombinant cells harboring plasmids carrying only the glf gene either under the control of the neokanamycin or lac promoter exhibited a strongly diminished growth (μ = 0.085 and 0.073, respectively) compared to recombinant cells harboring plasmids with also the other genes (Fig. 5A).
FIG. 5.

Growth of recombinant R. eutropha strain H16. (A) Recombinant strains of R. eutropha harboring different plasmids. The cells were cultivated in liquid MSM containing 1% (wt/vol) glucose as sole carbon source. ▪, pBBR1MCS-3::glf; □, pBBR1MCS-2::Pnk::glf; •, pBBR1MCS-2::Pnk::glk::glf; ▴, pBBR1MCS-3::glf::mak::pmi; ▵, control R. eutropha H16 strain G+1. (B) Recombinant strains of R. eutropha harboring plasmid pBBR1MCS-3::glf::mak::pmi. Cells were cultivated in liquid MSM containing tetracycline (12.5 μg/ml) and either 0.5% (wt/vol) sodium gluconate (▵), 0.5% (wt/vol) mannose (▴), 1% (wt/vol) mannose (□), 2% (wt/vol) mannose (•), or 4% (wt/vol) mannose (○) as the sole carbon source.
Growth of the recombinant R. eutropha with mannose or gluconate as the sole carbon source.
Recombinant cells of R. eutropha strain H16 harboring plasmid pBBR1MCS-3::glf::mak::pmi were cultivated in liquid MSM containing different concentrations of mannose as the sole carbon source to evaluate the optimal concentration for maximal growth. Furthermore, we examined whether the introduced genes were affecting the growth of the recombinant strain in comparison to the wild-type strain H16 in MSM containing 0.5% (wt/vol) sodium gluconate as the sole carbon source. According to our expectations, the recombinant strain H16 harboring pBBR1MCS-3::glf::mak::pmi exhibited faster growth on gluconate (μ = 0.33 h−1) than on mannose (μ = 0.078 h−1) (Fig. 5B). With gluconate as the sole carbon source, the growth of the recombinant strain was only slightly diminished compared to the wild type. Although strain H16 pBBR1MCS-3::glf::mak::pmi was engineered for mannose utilization, it exhibited a shorter lag phase on glucose than on mannose (Fig. 5B).
Investigations of PHB accumulation by recombinant R. eutropha strains.
To investigate whether the recombinant R. eutropha strains can be used for PHB production from mannose or glucose, cells were cultivated under conditions permissive for PHB accumulation. The PHB contents of the cells were analyzed gas chromatographically as described in Materials and Methods. Cultures containing gluconate as a sole carbon source served as a reference for the recombinant strain, and samples were taken in the early stationary growth phase. Gas chromatographic analysis of the cells revealed a PHB content of 63% (wt/wt) of cell dry mass. Cells grown in the presence of 1% (wt/vol) glucose or mannose were also examined in the stationary growth phase, and PHB contents of 65 and 67% (wt/wt) of cell dry mass, respectively, were determined.
DISCUSSION
In this study, recombinants of R. eutropha strain H16 were generated which, in contrast to the wild type, were able to utilize mannose and/or glucose as sole carbon sources. Up to four genes (glf, glk, mak, and pmi) were introduced episomally in R. eutropha strain H16 under the control of a constitutive neokanamycin or lac promoter, enabling uptake and conversion of glucose and mannose into intermediates of the KDPG pathway. Interestingly, only after prior cultivation on fructose instead of gluconate did recombinant strains containing pBBR1MCS-3::glf and pBBR1MCS-3::glf::mak show very slight growth on mannose as the sole carbon source. Possibly, prior cultivation on fructose resulted in the induction of enzymes of the KDPG pathway and/or in the expression of the PMI present in R. eutropha strain H16. Consistent with this, genes encoding enzymes of the KDPG pathway leading to gluconate were reported to be fully induced in the presence of fructose in R. eutropha strain H16 (6). All three episomally introduced genes are necessary for an efficient utilization of mannose. Since GLF is known to be a low-affinity, high-velocity, and energy-independent facilitated diffusion transport system for glucose and fructose in Z. mobilis (3, 13), the observation that recombinant cells of R. eutropha strain H16 harboring glf gained the ability to utilize glucose is not surprising. However, in agreement with previous observations, we found that for stable establishment of glucose utilization the constitutive expression of only a glucose transporter is not sufficient since recombinant strains needed a prolonged initial incubation period to grow on MSM plates containing glucose. After this, glucose utilization was stably maintained. Therefore, a pleiotropic effect has also to be considered for the recombinant strains of the present study, as was done by others (23) for a spontaneous mutant of R. eutropha strain H16 that is able to utilize glucose (36) and was used as control for experiments on utilization of glucose (Fig. 5A). The transport kinetics and substrate specificity of Z. mobilis GLF have been investigated in detail (29, 46, 47).
Initial experiments using the glucokinase-enzyme assay, as well as the Glf solubilization experiments, suggested that Pnk is a stronger constitutive promoter in R. eutropha strain H16 than Plac (data not shown). However, the use of different promoters for control of glf to test different expression levels exerted only minor effects on cell growth, indicating that glucose uptake was only to a certain extent the limiting step. In contrast, additional expression of E. coli glucokinase resulted in a strongly elevated specific enzyme activity (∼9-fold in comparison to the wild type) and in an enhanced growth of recombinant strains on glucose (Fig. 5A). This provides further evidence that the sugar kinase naturally present in R. eutropha is a bottleneck for the efficient utilization of glucose. This hypothesis is supported by the observation that R. eutropha strain H16 harboring the pBBR1MCS-3::glf::mak::pmi also exhibited better growth rates than recombinant strains harboring only the glf gene, as MAK was shown to have also an activity for glucose (37). Interestingly, the activity of the glucose-6-phosphate dehydrogenase (G6PDH) was found to be not elevated in the recombinant strain harboring plasmid pBBR1MCS-2::Pnk::glk::glf in comparison to the spontaneous mutant G+1 (data not shown). Therefore, the G6PDH activity is unlikely to be the bottleneck for a more efficient glucose utilization. However, since all engineered strains still grow more slowly on glucose than on gluconate, and taking into account the initial incubation phase on glucose needed by all recombinant strains, it is likely that enzymatic adaptations beside an elevated G6PDH activity are needed. This aspect requires further investigation in order to improve the strains for a more efficient utilization of this sugar.
Utilization of mannose as the sole carbon source for growth was only established in R. eutropha after the pmi gene from E. coli was additionally expressed. In Z. mobilis, the accumulation of mannose-6-phosphate occurred after uptake of mannose by GLF and phosphorylation by the chromosomally encoded fructokinase, thus indicating the lack of PMI activity. After heterologous expression of the pmi gene, Z. mobilis converted mannose-6-phosphate into the KDPG pathway intermediate fructose-6-phosphate, and hence enabled the utilization of this sugar (47). Unlike recombinant strains harboring pBBR1MCS-3::glf::mak, recombinant cells containing plasmid pBBR1MCS-3::glf::pmi exhibited distinct growth with mannose as sole carbon source, thus indicating that PMI activity is essential for mannose utilization in R. eutropha and that the chromosomally encoded PMI is neither constitutively expressed nor induced by mannose, since no PMI enzyme activity was detectable in the control strain, and the recombinant strain harboring plasmid pBBR1MCS-3::glf::mak showed no growth on mannose without initial incubation on fructose. This may also be the reason why mannose-utilizing mutants of R. eutropha have not been isolated thus far (23, 30; for a review, see reference 32. In R. eutropha strain H16 phosphorylation of mannose to mannose-6-phosphate is mediated by the chromosomally encoded kinase (16). However, analogously to glucose, a more efficient mannose utilization was achieved in R. eutropha after expression of an additional sugar kinase: E. coli MAK. Mannose is not the primary substrate of hexokinase; the enzyme exhibits just a side activity toward this sugar and is heterologously expressed. Transcriptional and activity analyses of the two enzymes confirmed the presence of either transcriptional or translational products, respectively, of the heterologously expressed genes that are necessary for efficient mannose utilization. Consistent with these results, R. eutropha strain H16 containing pBBR1MCS-3::glf::mak::pmi exhibited the fastest growth on mannose compared to the other recombinant strains harboring only one or two of these genes.
Interestingly, cells of the recombinant strain harboring pBBR1MCS-3::glf::mak::pmi exhibited a shorter lag phase on glucose than on mannose. This may be due to the higher affinity of GLF for glucose (29, 46) resulting in a faster transport of glucose into the cells. Recombinant strains of R. eutropha with the extended substrate utilization range synthesized PHB from mannose and glucose in similar amounts as from gluconate. Under optimized conditions, also higher PHB contents of the recombinant strains, as described in literature for the wild type (48, 49; for a review, see reference 32), will most likely be possible. The extension of the substrate utilization range is only a first step toward the use of R. eutropha for industrial production of PHAs or cyanophycin (43) from cheap carbohydrates such as glucose and mannose, which are available in agricultural residues and later also in abundant lignocellulosic biomass. In contrast to previous chemically induced mutants of R. eutropha, which are, for example, capable of utilizing glucose as a sole carbon source for growth, the strategy in the present study provides the advantage that the extension of the substrate range can be achieved with the existing plasmids in any strain of this bacterium and that possible additional mutations that might affect its productiveness and/or may lead to unfavorable side effects can be excluded.
Further investigations should primarily focus on the improvement of an appropriate expression of genes and on sufficient sugar uptake and conversion rates. Hexokinases from other prokaryotes such as Leuconostoc mesenteroides or Streptomyces sp., which also exhibit an activity for mannose (2, 11), may be candidates for expression in R. eutropha to improve sugar conversion in R. eutropha strain H16. An overexpression of the chromosomally encoded hexokinase with its side activity for mannose may provide another possibility to enhance mannose utilization in R. eutropha. Since pBBR is a low-copy-number vector (24) and, since the encoded Plac is a weak constitutive promoter for R. eutropha (24, 39), other vectors may be more suitable for expression of the desired genes for mannose utilization. Improvements might also aim at replacing the lac promoter by a stronger constitutive promoter such as, for example, the neokanamycin promoter, which was also used in the present study and which resulted in elevated expression levels. An integration of the genes into the genome of R. eutropha may also provide advantages.
Acknowledgments
We thank Fred B. Oppermann-Sanio (Institut für Molekulare Mikrobiologie und Biotechnologie, Westfälische Wilhelms-Universität Münster, Münster, Germany) for helpful instructions and discussions concerning the enzyme assays.
Footnotes
Published ahead of print on 17 December 2010.
REFERENCES
- 1.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, R. L., and V. L. Sapico. 1975. d-fructose (d-mannose) kinase. Methods Enzymol. 42:39-43. [DOI] [PubMed] [Google Scholar]
- 3.Barnell, W. O., K. C. Yi, and T. Conway. 1990. Sequence and genetic organization of a Zymomonas mobilis gene cluster that encodes several enzymes of glucose metabolism. J. Bacteriol. 172:7227-7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birnboim, H. C., and J. Doly. 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7:1513-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bordier, C. 1981. Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 256:1604-1607. [PubMed] [Google Scholar]
- 6.Bowien, B., and H. G. Schlegel. 1972. Isolation and characterization of mutants of Hydrogenomonas eutropha strain H16 defective in catabolism. II. Mutants defective in 2-keto-3-deoxy-6-phosphogluconate aldolase. Arch. Mikrobiol. 87:221-234. [PubMed] [Google Scholar]
- 7.Bowien, B., and H. G. Schlegel. 1981. Physiology and biochemistry of aerobic hydrogen-oxidizing bacteria. Annu. Rev. Microbiol. 35:405-452. [DOI] [PubMed] [Google Scholar]
- 8.Bowien, B., and B. Kusian. 2002. Genetics and control of CO2 assimilation in the chemoautotroph Ralstonia eutropha. Arch. Microbiol. 178:85-93. [DOI] [PubMed] [Google Scholar]
- 9.Brandl, H., R. A. Gross, R. W. Lenz, and R. C. Fuller. 1988. Pseudomonas oleovorans as a source of poly(β-hydroxyalkanoates) for potential applications as biodegradable polyesters. Appl. Environ. Microbiol. 54:1977-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cárdenas, M. L., A. Cornish-Bowden, and T. Ureta. 1998. Evolution and regulatory role of the hexokinases. Biochim. Biophys. Acta Mol. Cell Res. 1401:242-264. [DOI] [PubMed] [Google Scholar]
- 11.Coulombel, C., M. J. Foglietti, and F. Percheron. 1982. Identification and kinetic studies of an inducible mannokinase from a Streptomyces strain. Biochim. Biophys. Acta 706:117-122. [DOI] [PubMed] [Google Scholar]
- 12.Deanda, K., M. Zhang, C. Eddy, and S. Picataggio. 1996. Development of an arabinose-fermenting Zymomonas mobilis strain by metabolic pathway engineering. Appl. Environ. Microbiol. 62:4465-4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiMarco, A. A., and A. H. Romano. 1985. d-Glucose transport system of Zymomonas mobilis. Appl. Environ. Microbiol. 49:151-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans, J. 2010. Bioplastics get growing. Plastics Eng. 66:14-20. [Google Scholar]
- 15.Friedrich, B., C. Hogrefe, and H. G. Schlegel. 1981. Naturally occurring genetic transfer of hydrogen-oxidizing ability between strains of Alcaligenes eutrophus. J. Bacteriol. 147:198-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottschalk, G., U. Eberhardt, and H. G. Schlegel. 1964. Verwertung von Fructose durch Hydrogenomonas H16. Arch. Mikrobiol. 48:95-108. [PubMed] [Google Scholar]
- 17.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 18.Hatti-Kaul, R., U. Tornvall, L. Gustafsson, and P. Borjesson. 2007. Industrial biotechnology for the production of bio-based chemicals: a cradle-to-grave perspective. Trends Biotechnol. 25:119-124. [DOI] [PubMed] [Google Scholar]
- 19.Hirota, Y., H. Suzuki, Y. Nishimura, and S. Yasuda. 1977. On the process of cellular division in Escherichia coli: a mutant of E. coli lacking a murein-lipoprotein. Proc. Natl. Acad. Sci. U. S. A. 74:1417-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeffries, T. W., and Y. S. Jin. 2004. Metabolic engineering for improved fermentation of pentoses by yeasts. Appl. Microbiol. Biotechnol. 63:495-509. [DOI] [PubMed] [Google Scholar]
- 21.Kang, S., and A. Markovitz. 1967. Induction of capsular polysaccharide synthesis by rho-fluorophenylalanine in Escherichia coli wild type and strains with altered phenylalanyl soluble ribonucleic acid synthetase. J. Bacteriol. 93:584-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaup, B., S. Bringer-Meyer, and H. Sahm. 2003. Metabolic engineering of Escherichia coli: construction of an efficient biocatalyst for d-mannitol formation in a whole-cell biotransformation. Commun. Agric. Appl. Biol. Sci. 68:235-240. [PubMed] [Google Scholar]
- 23.König, C., I. Sammler, E. Wilde, and H. G. Schlegel. 1969. Constitutive glucose-6-phosphate dehydrogenase in mutants utilizing glucose, which are derived from cryptic wild-type strains. Arch. Mikrobiol. 67:51-57. [PubMed] [Google Scholar]
- 24.Kovach, M. E., et al. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175-176. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 26.Lee, S. Y., and S. J. Park. 2005. Fermentative production of short-chain-length PHAs, p. 207-234. In A. Steinbüchel and Y. Doi (ed.), Biotechnology of biopolymers: from synthesis to patents. Wiley-VCH, Weinheim, Germany.
- 27.Linde, M., M. Galbe, and G. Zacchi. 2008. Bioethanol production from non-starch carbohydrate residues in process streams from a dry-mill ethanol plant. Bioresour. Technol. 99:6505-6511. [DOI] [PubMed] [Google Scholar]
- 28.McMillan, J. D., and B. L. Boynton. 1994. Arabinose utilization by xylose-fermenting yeasts and fungi. Appl. Biochem. Biotechnol. 46:569-584. [DOI] [PubMed] [Google Scholar]
- 29.Parker, C., W. O. Barnell, J. L. Snoep, L. O. Ingram, and T. Conway. 1995. Characterization of the Zymomonas mobilis glucose facilitator gene product (glf) in recombinant Escherichia coli: examination of transport mechanism, kinetics and the role of glucokinase in glucose transport. Mol. Microbiol. 15:795-802. [DOI] [PubMed] [Google Scholar]
- 30.Pohlmann, A., et al. 2006. Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat. Biotechnol. 24:1257-1262. [DOI] [PubMed] [Google Scholar]
- 31.Pries, A., A. Steinbüchel, and H. G. Schlegel. 1990. Lactose and galactose utilizing strains of poly(hydroxyalkanoic acid) accumulating Alcaligenes eutrophus and Pseudomonas saccharophila obtained by recombinant DNA technology. Appl. Microbiol. Biotechnol. 33:410-417. [Google Scholar]
- 32.Reinecke, F., and A. Steinbüchel. 2009. Ralstonia eutropha strain H16 as model organism for PHA metabolism and for biotechnological production of technically interesting biopolymers. J. Mol. Microbiol. Biotechnol. 16:91-108. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 34.Schlegel, H. G., H. Kaltwasser, and G. Gottschalk. 1961. Ein Submersverfahren zur Kultur wasserstoffoxydierender Bakterien: Wachstumsphysiologische Untersuchungen. Arch. Mikrobiol. 38:209-222. [PubMed] [Google Scholar]
- 35.Schlegel, H. G., and R. Lafferty. 1965. Growth of Knallgas bacteria (Hydrogenomonas) using direct electrolysis of culture medium. Nature 205:308-309. [Google Scholar]
- 36.Schlegel, H. G., and G. Gottschalk. 1965. Verwertung von Glucose durch eine Mutante von Hydrogenomonas H16. Biochem. Z. 341:249-259. [PubMed] [Google Scholar]
- 37.Sebastian, J., and C. Asensio. 1967. Identification of mannokinase in Escherichia coli. Biochem. Biophys. Res. Commun. 28:197-202. [DOI] [PubMed] [Google Scholar]
- 38.Sebastian, J., and C. Asensio. 1972. Purification and properties of the mannokinase from Escherichia coli. Arch. Biochem. Biophys. 151:227-233. [DOI] [PubMed] [Google Scholar]
- 39.Siedow, A., R. Cramm, R. A. Siddiqui, and B. Friedrich. 1999. A megaplasmid-borne anaerobic ribonucleotide reductase in Alcaligenes eutrophus H16. J. Bacteriol. 181:4919-4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1:784-791. [Google Scholar]
- 41.Steinbüchel, A. 1986. Expression of the Escherichia coli pfkA gene in Alcaligenes eutrophus and in other Gram-negative bacteria. J. Bacteriol. 166:319-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Timm, A., D. Byrom, and A. Steinbüchel. 1990. Formation of blends of various poly(3-hydroxyalkanoic acids) by a recombinant strain of Pseudomonas oleovorans. Appl. Microbiol. Biotechnol. 33:296-301. [Google Scholar]
- 43.Voss, I., S. C. Diniz, E. Aboulmagd, and A. Steinbüchel. 2004. Identification of the Anabaena sp. strain PCC7120 cyanophycin synthetase as suitable enzyme for production of cyanophycin in Gram-negative bacteria like Pseudomonas putida and Ralstonia eutropha. Biomacromolecules 5:1588-1595. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe, S., et al. 2007. Ethanol production from xylose by recombinant Saccharomyces cerevisiae expressing protein-engineered NADH-preferring xylose reductase from Pichia stipitis. Microbiology 153:3044-3054. [DOI] [PubMed] [Google Scholar]
- 45.Weber, K., and M. Osborn. 1969. The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J. Biol. Chem. 244:4406-4412. [PubMed] [Google Scholar]
- 46.Weisser, P., R. Krämer, H. Sahm, and G. A. Sprenger. 1995. Functional expression of the glucose transporter of Zymomonas mobilis leads to restoration of glucose and fructose uptake in Escherichia coli mutants and provides evidence for its facilitator action. J. Bacteriol. 177:3351-3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weisser, P., R. Krämer, and G. A. Sprenger. 1996. Expression of the Escherichia coli pmi gene, encoding phosphomannose-isomerase in Zymomonas mobilis, leads to utilization of mannose as a novel growth substrate, which can be used as a selective marker. Appl. Environ. Microbiol. 62:4155-4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.York, G. M., B. H. Junker, J. A. Stubbe, and A. J. Sinskey. 2001. Accumulation of the PhaP phasin of Ralstonia eutropha is dependent on production of polyhydroxybutyrate in cells. J. Bacteriol. 183:4217-4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.York, G. M., et al. 2003. Ralstonia eutropha H16 encodes two and possibly three intracellular poly[d-(−)-3-hydroxybutyrate] depolymerase genes. J. Bacteriol. 185:3788-3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang, M., C. Eddy, K. Deanda, M. Finkelstein, and S. Picataggio. 1995. Metabolic engineering of a pentose metabolism pathway in ethanologenic Zymomonas mobilis. Science 267:240-243. [DOI] [PubMed] [Google Scholar]