

Abstract
The formation of stable foam in activated sludge plants is a global problem for which control is difficult. These foams are often stabilized by hydrophobic mycolic acid-synthesizing Actinobacteria, among which are Tsukamurella spp. This paper describes the isolation from activated sludge of the novel double-stranded DNA phage TPA2. This polyvalent Siphoviridae family phage is lytic for most Tsukamurella species. Whole-genome sequencing reveals that the TPA2 genome is circularly permuted (61,440 bp) and that 70% of its sequence is novel. We have identified 78 putative open reading frames, 95 pairs of inverted repeats, and 6 palindromes. The TPA2 genome has a modular gene structure that shares some similarity to those of Mycobacterium phages. A number of the genes display a mosaic architecture, suggesting that the TPA2 genome has evolved at least in part from genetic recombination events. The genome sequence reveals many novel genes that should inform any future discussion on Tsukamurella phage evolution.
A common problem in activated sludge systems is the formation of stable foams on the aerated reactor, leading to major environmental, operational, cosmetic, and health problems (12, 41, 42). These foams are typically stabilized by members of the mycolic acid-containing Actinobacteria (the Mycolata), although other hydrophobic filamentous bacteria, including “Candidatus Microthrix parvicella,” are also important (12, 24, 39). One potentially attractive approach for controlling such foaming events is the use of lytic bacteriophages targeting the problematic foam-stabilizing populations (46, 48). Similar “phage therapy” strategies have been proposed to treat infectious diseases (6) and decrease bacterial contaminants in food (50). Furthermore, Thomas et al. (46) showed that Mycolata lytic bacteriophages could be isolated readily from activated sludge, making the development of a phage-based foam control approach an environmentally safe and attractive option for this worldwide operational problem.
Tsukamurella is in the suborder Corynebacterineae, and on the basis of its mycolic acid-containing hydrophobic cell surface it has been categorized as a Mycolata (18). Tsukamurella spp. have been isolated from activated sludge foams (12, 31), arthropods (44), and soil (18) and more recently in opportunistic clinical infections (23, 43). Although Tsukamurella spp. are a problem in environmental and medical contexts, little attention has been directed toward the isolation and characterization of bacteriophages specific for members of this genus. To our knowledge, only one partially characterized Tsukamurella phage (TPA1, targeting T. paurometabola) has been reported in the literature (46). With the aim of developing a biocontrol approach to manage foaming within activated sludge systems, we have sought to isolate and characterize new lytic phages for members of this genus.
More fundamentally, the ecological role of bacteriophages in activated sludge communities has received relatively little attention. This is surprising given the high numbers of bacteriophages found in this environment (107 to 109 per ml) and the importance of phages in regulating bacterial community biodiversity (33). Characterizing new phages from environments such as activated sludge will provide a greater insight into phage evolution and diversity, as most phage studies have been dominated by the use of a limited range of bacterial hosts (48).
In this paper, we report the isolation and characterization of two Tsukamurella phages (TPA2 and TPA3) from activated sludge plants. These phages, together with the phage TPA1 isolated by Thomas et al. (46), were characterized on the basis of their morphologies, host ranges, phage life cycle, burst sizes, and complete genome sequences. Annotation of these phage genome sequences reveals that TPA1 and TPA2 phages share some similarity with other Corynebacterineae phages (21), but their DNA sequences reveal many novel attributes.
MATERIALS AND METHODS
Bacterial strains used in study.
The bacterial strains used in this study are listed in Table 1. All were grown on peptone-yeast-calcium (PYCa) (0.1% yeast extract [Oxoid, Adelaide, Australia], 1.5% peptone [Oxoid], 0.5% CaCl, 0.1% glucose) broth and agar (PYCa plus 1.4% agar [Oxoid]) at 30°C. All chemicals were obtained from Sigma (Sydney, Australia) unless otherwise noted.
TABLE 1.
Strains used in this study
| Name of organisma | Culture collection number(s) and other synonymsb |
|---|---|
| Tsukmurella inchonensisT | DSMZ44067 |
| T. inchonensis | NCTC10741 |
| T. paurometabolaT | Tpau37, ATCC 25938 |
| T. paurometabola | NCTC107411 |
| T. paurometabola | IMRU1283 |
| T. paurometabola | DSMZ20162 |
| T. paurometabola | IMRU1520, M334, DSMZ44119 |
| T. paurometabola | IMRU1312, M343 |
| T. paurometabola | IMRU1505, M337 |
| T. pseudospumaeT | N1176, DSMZ44118 |
| T. pulmonis | DSM44142 |
| T. spumaeT | N1171, DSMZ44113, NCIMB139647 |
| T. spumae | JC85 |
| T. tyrosinosolvensT | DSMZ44234 |
| Nocardia asteroides | Ben600 |
| N. asteroidesT | Nast23, DSMZ43757 |
| N. asteroides | Noast4, UQCC131 |
| N. brasiliensisT | Nbra42, DSMZ43758 |
| N. carneaT | Ncar30, DSMZ 43397 |
| N. novaT | Nnov47, ATCC 33726 |
| N. otitdidiscariarum | Noti14, AMMRL 19.11 |
| N. otitdidiscariarumT | Noti25, DSMZ43242 |
| N. otitdidiscariarum | Noti15, AMMRL 19.12 |
| N. transvalensisT | Ntra40, DSMZ43405 |
| Rhodococcus coprohilusT | Rcop41, DSMZ43347 |
| R. coprohilus | Rcop18, UQCC1259 |
| R. equi | Requ10, UQCC702 |
| R. equiT | Requ28, UQCC20307, DSMZ20307 |
| R. erythropolis | Rery19, UQCC379 |
| R. erythropolisT | Rery29, DSMZ43066 |
| R. fasciansT | Rlut36, DSMZ43673 |
| R. globerulusT | Rglo35, DSMZ43954 |
| R. rhodniiT | Rrho46, DSMZ43336 |
| R. rhodochrous | Rrho3, UQCC2807 |
| R. rhodochrousT | Rrho39, DSMZ43241 |
| R. rhodochrous | Rrho11S, UQCC2808 |
| R. ruberT | Rrub33, DSMZ43338 |
| R. tritomaeT | DSM44892 |
| Gordonia aichiensisT | Raic22, DSMZ43978 |
| G. alkanivorous | Ben606 |
| G. amaraeT | Gama44, DSMZ43392 |
| G. amarae | Gama9, UQCC2810 |
| G. amictica | Ben607 |
| G. australis | 18F3 M |
| G. defluviiT | J4, DSMZ44981 |
| G. desulfuricansT | 213E, NCIMB40816 |
| G. hydrophobicaT | N1123, DSMZ44015 |
| G. malaquaeT | A554, ATCC 35215 |
| G. malaquae | A448 |
| G. rubropertinctaT | Grub48, DSMZ43197 |
| G. sputi | Gobu31, DSMZ43979 |
| G. sputiT | Gspu49, ATCC2 9627 |
| G. sputi | Gspu48, ATCC 336609 |
| G. terraeT | Gter34, DSMZ43249 |
| G. terrae | Ben601 |
| G. terrae | Ben602 |
| G. terrae | Ben603 |
| G. terrae | Ben604 |
| Dietzia marisT | Dmar27, DSMZ43672 |
| Streptomyces griseus | Sgri05 |
| Millisia brevisT | J82, DSMZ44463 |
| Mycobacterium chlorophenolicusT | Mchl24, DSMZ43826 |
| M. smegmatis | Msme1, UQCC422 |
| M. fortuitum | Mfor21, UQCC120 |
T, type strain.
Culture collection numbers have been obtained from the following organizations: DSMZ, German Collection of Microorganisms and Cell Cultures; ATCC, American Type Culture Collection; UQCC, Australian Collection of Microorganisms (now ACM); AMMRL, Australian National Reference Laboratory in Medical Mycology; and NCIMB, National Collection of Industrial Bacteria. All culture numbers have been derived from the La Trobe University Bendigo culture collection.
Isolation and purification of phages.
Bacteriophages were isolated from mixed liquor samples taken from activated sludge plants in Australia as follows. Samples (20 ml) were centrifuged (3,000 × g for 20 min) before being filtered through cellulose acetate membrane filters (0.2-μm pore size) to remove bacterial cells. Tsukamurella phage enrichment was performed by pooling 1 ml of the six species of Tsukamurella listed in Table 1 (∼106 per ml) in 50 ml of PYCa broth in 300-ml flasks and incubating with 1 ml of filtered activated sludge supernatant. Flasks were left for 1 h at room temperature without shaking to encourage phage infection before a further incubation with shaking at 30°C for 2 days. Following enrichment, the remaining bacterial cells were removed by centrifugation and the supernatants filtered through 0.2-μm cellulose acetate membrane filters. Individual lawn plates of each Tsukamurella species were prepared by swabbing, and 20-μl aliquots of enriched supernatants were drop diluted onto each lawn plate and allowed to dry. Plates were incubated for 48 h to allow visualization of plaques. Single plaques were seen on plates inoculated with filtered mixed liquor from two activated sludge plants in Victoria, Australia. Plaques were purified through four rounds of dilution and reisolation to ensure that each plaque resulted from a single virion before further studies were undertaken. These phages were named TPA2 and TPA3.
Phage DNA isolation.
T. paurometabola (Tpau37) was inoculated in PYCa broth and incubated overnight. One milliliter (∼107 CFU/ml) of T. paurometabola culture was added to 50 ml of PYCa broth and infected with the appropriate TPA phage using a multiplicity of infection (MOI) equal to 0.1 PFU/bacterium and incubated as described previously. The lysate was recovered by centrifugation at 3,000 × g for 20 min, and the supernatant was removed and filtered through a 0.2-μm filter to remove bacterial cells and cell debris. Nonphage nucleic acids were eliminated from the filtered supernatant (the titer was ∼1010 PFU/ml) by nuclease digestion (final concentrations of 10 μg/ml DNase I, 10 μg/ml RNase A, and 5 mM MgCl2; 30 min at room temperature). Phage virions were recovered by polyethylene glycol (PEG) precipitation (final concentrations of 10% [wt/vol] polyethylene glycol 8000 and 1 M NaCl; 4°C overnight) and collected by centrifugation at 13,000 × g for 15 min before being resuspended in 50 μl of TE buffer (10 mM Tris [pH 8.0], 1 mM EDTA).
Phage DNA was extracted from ∼1010 PFU using SDS-proteinase K (final concentrations of 20 mM EDTA, 0.5% SDS, and 50 μg/ml proteinase K; 55°C for 1 h) in a total volume of 1 ml. After incubation, an equal volume of phenol-chloroform-isoamyl alcohol (29:28:1) was added and mixed by vortexing before centrifugation at 10,000 × g for 3 min. The upper aqueous phase was removed to a fresh tube containing 1 volume isopropanol and 0.1 volume 3 M sodium acetate (pH 5.2) and incubated at room temperature for 30 min. The precipitated phage DNA was recovered by centrifugation at 10,000 × g for 10 min. The DNA pellet was washed once with 70% ethanol, air dried, and finally resuspended in 50 μl of TE buffer.
Phage genome sequencing.
Phage DNA for TPA1 and TPA2 was genome sequenced using the Roche GS FLX genome sequencer and titanium chemistry by Genoseq (UCLA, Los Angeles, CA). The pyrosequencing reads were assembled using the gsAssembler (Roche Applied Science, Indianapolis, IN). All resulting single contigs obtained for each phage had a minimum of 50 times read coverage.
DNA annotation.
Geneious 4.0.4 (14) was used initially to identify all open reading frames (ORFs) longer than 30 codons in all six reading frames. The putative proteins encoded by the identified ORFs were screened using the NCBI database and the BLAST P algorithm with a significance cutoff E value of 10−4. The conserved domain database (CDD) (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) and Pfam database (http://pfam.sanger.ac.uk) were used to identify conserved motifs and make predicted protein family allocations (15). The presence of putative tRNA and tmRNA was screened for using ARAGORN (http://www.acgt.se/online.html) (25).
Phage host range determination.
All six Tsukamurella species were screened for lytic infection by TPA1, TPA2, and TPA3 phages. A drop dilution series of each purified phage was spotted onto swabbed lawn plates of each strain and incubated for 2 days. Additional actinobacterial strains (Table 1) were screened for their ability to support TPA2 lytic infection.
Single-step growth curve.
Four replicas of a 1-ml volume of an early-exponential-phase culture of T. paurometabola (Tpau37) was infected with TPA2 at a multiplicity of infection (MOI) of 0.1 (2). After an adsorption period of 5 min at 30°C, the cells were washed twice in PYCa broth and resuspended in 1 ml of broth. This culture was diluted 1 in 100 to minimize the possibility of nonadsorbed phages infecting cells. All further steps were performed as described by Adams (2) except that the incubations were conducted at 30°C.
Electron microscopy.
Virus particles were allowed to adsorb to Formvar-coated 200-mesh copper grids for 5 min. These were washed twice for 1 min in double-distilled water (ddH2O) and negatively stained with 2% (wt/vol) uranyl acetate for 2 min. Excess liquid was absorbed onto filter paper, and the disc was allowed to air dry. Grids were examined under a JEOL JEM-100cx transmission electron microscope (TEM) at an accelerating voltage of 100 kV.
Nucleotide sequence accession number.
The nucleotide sequence for TPA2 has been deposited in GenBank under accession number HM486077.
RESULTS AND DISCUSSION
Activated sludge samples from 10 different wastewater treatment plants in Australia were screened for Tsukamurella phages by enrichment and plaque plating onto lawn plates of six Tsukamurella species (T. paurometabola, T. pulmonis, T. tyrosinosolvens, T. spumae, T. pseudospumae, and T. inchonensis) (see Materials and Methods). Single plaques were observed on lawn plates of T. paurometabola from the activated sludge samples obtained from the Daylesford and Ballarat (Victoria, Australia) wastewater treatment plants. Both phages (TPA2 and TPA3) formed plaques of ∼1 mm in diameter after 2 days incubation at 30°C. Restriction mapping of the TPA2 and TPA3 genomes suggested that these two phages were the same (see below), so only TPA2 was selected for genome sequencing, together with the T. paurometabola phage (TPA1), which had been isolated earlier by Thomas et al. (46). Subsequent sequence analysis showed that TPA1 and TPA2 were identical, and therefore this paper reports the characterization of the TPA2 genome.
TPA2 belongs to the Siphoviridae family, possessing the characteristic long, noncontractile tail (∼340 nm) of members of the Caudovirales with a B1 isometric capsid (∼58 nm) morphotype (1) (Fig. 1). The average burst size was determined to be 155 ± 5 particles per infective center, with a latency period of 4 h in PYCa broth at 30°C. The TPA2 phage displays a broad-host-range phenotype within the genus Tsukamurella. In addition to infecting T. pauomotabola (Tpau37), TPA2 lysed a further six T. paurometabola strains (DSMZ20162, IMRU1520, IMRU1312, IMRU1505, IMRU1283, and NCTC107411), as well as members of four other Tsukamurella species, i.e., T. pulmonis (DSM44142), T. tyrosinosolvens (DSMZ44234), T. pseudospumae (N1176), and two strains of T. spumae (N1171 and JC85). Mock infection controls were performed using each strain to ensure that lysis was not due to the release of prophages from the different hosts. The only Tsukamurella strain tested that was not lysed by TPA2 was T. inchonensis (DSMZ44067). Although clear lytic plaques were not observed, the TPA2 phage did induce a turbid lysis on T. inchonensis at a high MOI. Further investigations revealed that TPA2 was unable to propagate in this strain, suggesting that infected cultures of TPA2 generate a factor that can kill T. inchonensis. TPA2 appears to be restricted in its host range to members of the genus Tsukamurella, since it was unable to lyse any other Mycolata strain, including 20 different Gordonia spp., 15 different Rhodococcus spp., 10 different Nocardia spp., 3 Mycobacterium spp., Dietzia maris, Millisia brevis, and Streptomyces griseus (Table 1). The molecular basis for this host specificity remains to be determined.
FIG. 1.
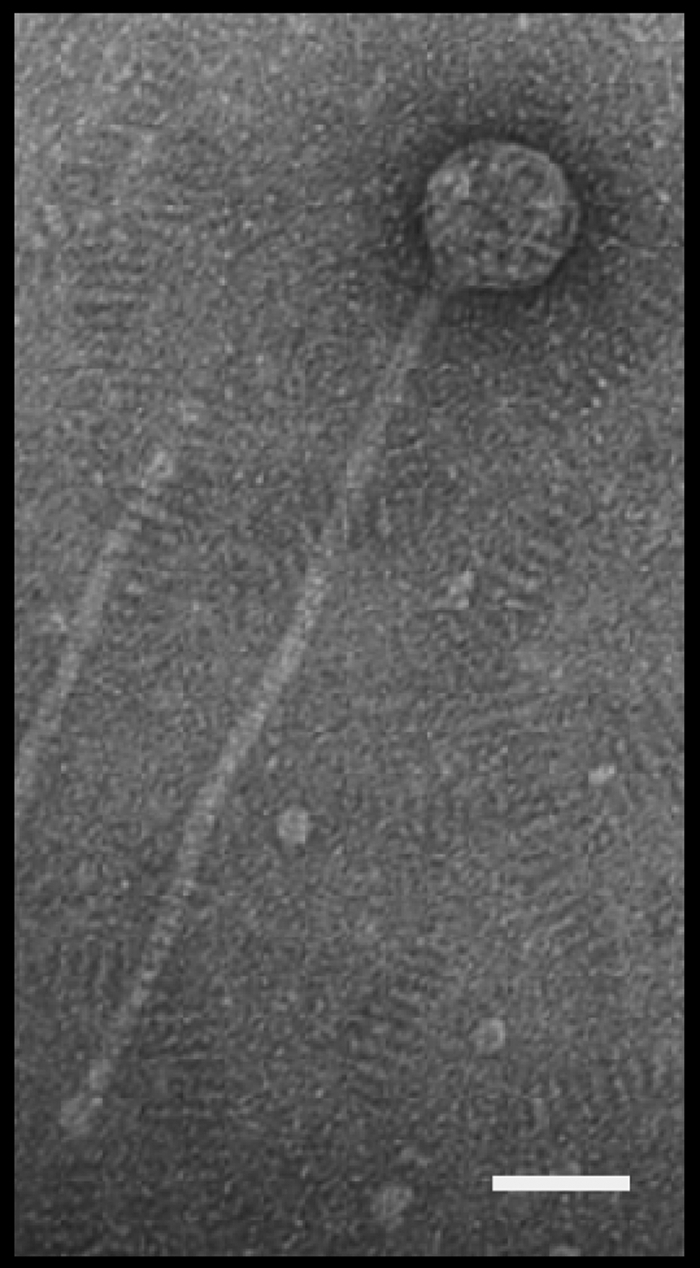
Electron micrograph of TPA1. Scale bar, 50 nm.
Genomic features of TPA2.
The genomic DNAs isolated from the TPA1, TPA2 and TPA3 bacteriophages were digested using a range of restriction enzymes. Several (EcoRI, HindIII, and BamHI) failed to digest DNA from any of these genomes. However, genomic DNA from all three was digestible by BglII, and each gave an identical restriction enzyme digest pattern (data not shown). Consequently, only TPA1 and TPA2 were selected for genome sequencing.
The genome sequences of TPA1 and TPA2 after Roche/454 pyrosequencing generated 30,440 and 27,589 reads, respectively, with a minimum of 50 times sequence coverage. The assembled genomes of TPA1 and TPA2 were identical despite their being isolated from separate activated sludge plants 9 years apart (46). The assembled sequence of TPA1 and TPA2 suggested a circularly permuted genome, which was further confirmed by BglII restriction endonuclease analysis (data not shown). While laboratory cross-contamination is a possibility, it is unlikely in this case given that TPA1 was recovered from a freezer stock only after TPA2 and TPA3 had been isolated and initially characterized. These results suggest not only that TPA-like phages may be widespread within activated sludge systems but that they may persist for many years within this environment. Since the two phages were identical at the DNA sequence level, all further discussion is based on TPA2.
TPA2 has a circularly permutated, double-stranded DNA (dsDNA) genome consisting of 61,440 bp, with a G+C content of 69.1 mol%. This is within the 67 to 74 mol% G+C range for Tsukamurella spp. (18), suggesting that TPA2 is adapted to its host. A comparison between the in silico and the experimentally determined restriction digest patterns indicated that the TPA2 genome was correctly assembled and lacked any EcoRI, HindIII, and BamHI sites (data not shown). At the DNA level, 30% of the TPA2 genome shares identity with a number of sequenced mycobacteriophages (21), while the remaining 70% of the genome sequence appears to be unique so far. Analysis of the TPA2 genome revealed 78 ORFs larger than 90 nucleotides, with no tRNA or tmRNA detected. These ORFs are numbered consecutively except in the case of the highly conserved large terminase (terL), lysin (lysA), and Holliday junction resolvase (ruvC) genes. Thirty-three of the ORFs are located on one strand and 45 on the opposite strand (Fig. 2). A total of 44 ORFs showed significant identity with previously reported ORFs, and only 15 (19%) of these could be assigned predicted functions. Thirty-four ORFs exhibited no significant identity to any hypothetical protein (Table 2).
FIG. 2.

Circular map of the TPA2 genome. The arrows represent the putative genes and the directions in which they are transcribed. Modules are shaded in similar tones, and the outer circle indicates the genes encoded within the modules. Arrows represent the repeat regions R1 to R6.
TABLE 2.
Summary of ORFs and gene products of TPA2
| ORFa | No. of amino acids (% identity)b | Predicted function (conserved motif)c | Matchd | E valuee |
|---|---|---|---|---|
| 1 | 146 | Unknown | ||
| 2 | 193 | Unknown | ||
| 3 | 146 | Unknown | ||
| 4 | 172 | Unknown | ||
| 5 | 79 | Unknown | ||
| 6 | 53 | Unknown | ||
| 7 | 139 | Unknown | ||
| 8 | 140 | Unknown | ||
| terL | 554 (51) | Large terminase (Pfamn03237) | Gp7, Mycobacterium phage Rosebush | 1e−145 |
| 10 | 240 | Unknown | ||
| 11 | 79 | Unknown | ||
| 12 | 118 | Unknown | ||
| 13 | 95 | Unknown | ||
| 14 | 113 | Unknown (Pfam05305) | ||
| ruvC | 192 (40) | RuvC resolvase (cd00529) | Mycobacterium phage Pipefish | 6e−28 |
| 16 | 114 (42) | Unknown (Pfam07098) | Gp6, Mycobacterium phage Nigel | 1e−19 |
| 17 | 611 (52) | Putative structural protein | Gp12, Mycobacterium phage Qyrzula | 4e−164 |
| 18 | 793 (52) | Structural protein (Pfam04233) | Gp12, Mycobacterium phage Pipefish | 0.0 |
| 19 | 64 (53) | Putative structural protein | Gp13, Mycobacterium phage Pipefish | 8e−12 |
| 20 | 72 (0) | Unknown | ||
| 21 | 414 (69) | Unknown | Gp2, Mycobacterium phage Cooper | 6e−167 |
| 22 | 260 (45) | Unknown | Gp13, Mycobacterium phage Orion | 5e−53 |
| 23 | 199 (34) | Unknown | Gp16, Mycobacterium phage Pipefish | 8e−17 |
| 24 | 101 (59) | Unknown | Gp15, Mycobacterium phage Cooper | 1e−21 |
| 25 | 151 (49) | Unknown | GTE5p031, Gordonia terrae phage GTE5 | 5e−34 |
| 26 | 147 | Unknown | ||
| 27 | 268 (67) | Major tail protein | Gp21, Mycobacterium phage Pipefish | 2e−102 |
| 28 | 256 (50) | Putative tail assembly | Gp23, Mycobacterium phage Pipefish | 1e−64 |
| 29 | 234 (48) | Putative tail assembly | Gp23, Mycobacterium phage Qyrzula | 3e−45 |
| 30 | 97 (68) | Unknown | Gp20, Mycobacterium phage Nigel | 2e−06 |
| 31 | 147 (51) | Tail component (Pfam04883) | Gp25, Rhodococcus phage ReqiPine5 | 5e−29 |
| 32 | 145 (49) | Unknown | Gp27, Mycobacterium phage Pipefish | 1e−29 |
| 33 | 2,386 (36) | Tape measure protein (Pfam10145) | TMP, Mycobacterium abscessus | 2e−102 |
| 34 | 331 (49) | Unknown | Gp30, Mycobacterium phage pipefish | 3e−96 |
| 35 | 292 (49) | Unknown | Gp30, Mycobacterium phage Colbert | 3e−69 |
| 36 | 227 | Unknown | ||
| 37 | 171 | Unknown | ||
| 38 | 188 | Unknown | ||
| 39 | 209 (59) | Unknown | Gp36, Mycobacterium phage Rosebush | 3e−59 |
| 40 | 196 (65) | Unknown | Gp32, Mycobacterium phage Nigel | 3e−70 |
| 41 | 184 (45) | Unknown | Gp38, Mycobacterium phage Phlyer | 9e−39 |
| 42 | 133 (37) | Unknown | Gp39, Mycobacterium phage Rosebush | 2e−13 |
| 43 | 58 (44) | Unknown | Gp37, Mycobacterium phage Cooper | 6e−04 |
| 44 | 137 (46) | Putative tail fiber | Gp43, Mycobacterium phage Pipefish | 3e−23 |
| 45 | 88 (50) | Unknown | Gp44, Mycobacterium phage Pipefish | 3e−12 |
| 46 | 364 (36) | Putative tail fiber | Gp6, Mycobacterium phage Jasper | 4e−15 |
| 47 | 218 | Unknown | ||
| 48 | 94 (65) | Putative regulator | Gp45, Mycobacterium phage Pipefish | 2e−18 |
| 49 | 50 (56) | Unknown | Gp46, Mycobacterium phage Pipefish | 6e−04 |
| 50 | 83 | Unknown | ||
| 51 | 113 | Unknown | ||
| 52 | 131 | Unknown | ||
| 53 | 244 (32) | Unknown | Gp41, Mycobacterium phage PBI1 | 3e−18 |
| 54 | 294 (27) | Putative holing | Rhodococcus opacus | 1e−04 |
| lysA | 536 (59) | Lysin (Pfam01510, Pfam08310) | Nocardia facinica | 4e−103 |
| 56 | 169 (46) | Unknown | Gp48, Mycobacterium phage Phlyer | 8e−26 |
| 57 | 435 (39) | Unknown | Gp47, Mycobacterium phage Cooper | 1e−82 |
| 58 | 549 (56) | Helicase (Pfam04851) (Pfam00271) | Gp50, Mycobacterium phage Rosebush | 4e−167 |
| 59 | 161 (40) | Unknown | Gp51, Mycobacterium phage Cooper | 4e−15 |
| 60 | 86 | Unknown | ||
| 61 | 870 (51) | Primase (Pfam09250) | Gp52, Mycobacterium phage Phaedrus | 0.0 |
| 62 | 178 (47) | Unknown | Hypothetical, Jonesia denitrificans | 7e−19 |
| 63 | 626 (52) | DNA polymerase I (Pfam01612, Pfam00476) | Gp54, Mycobacterium phage Cooper | 6e−174 |
| 64 | 259 | Unknown | ||
| 65 | 85 | Unknown | ||
| 66 | 202 | Unknown | ||
| 67 | 136 | Unknown | ||
| 68 | 150 | Unknown | ||
| 69 | 124 | Unknown | ||
| 70 | 99 | Unknown | ||
| 71 | 85 | Unknown | ||
| 72 | 208 (27) | Unknown | Gp65, Mycobacterium phage Rosebush | 2e−07 |
| 73 | 79 | Unknown | ||
| 74 | 151 | Unknown | ||
| 75 | 112 (63) | Putative endonuclease | Gp85, Mycobacterium phage Scoot17C | 7e−22 |
| 76 | 182 (46) | Unknown | Gp72, Mycobacterium phage Phaedrus | 1e−29 |
| 77 | 102 | Unknown | ||
| 78 | 110 (50) | Metallophoesterase (COG4186) | Gp66, Rhodococcus jostii | 1e−17 |
ORFs were numbered consecutively, with the exceptions of terL, ruvC, and lysA.
Percent identity is based on the best match when a BLAST P analysis was performed.
Predicted function is based on amino acid identity, conserved motifs, N-terminal sequencing, and gene location within functional modules.
The most closely related gene (only if named) and the name of the organism.
The probability of obtaining a match by chance as determined by BLAST analysis. Only values of less than 10−4 were considered significant.
The phage genome is modularly organized, consisting of DNA packaging, head and tail morphogenesis, cell lysis, DNA replication, modification, and regulation modules (Fig. 2). In the absence of an obvious origin of replication, the ORFs were ordered from the putative large terminase (terL) gene. However, since orf8 overlaps terL and is part of a putative operon of unknown function transcribed in the opposite direction to terL, the ORFs were numbered consecutively from the large noncoding region located before orf1 in the transcriptional direction of terL.
Sequence repeats.
A large number of repeat structures were observed in the TPA2 genome. Of these one is a near-identical 58-bp or 59-bp repeat (R1 to R6) located throughout the TPA2 genome (Table 3). Five of these repeat sequences (R1 to R5) occur on one strand, and one (R6) occurs on the opposite strand (Fig. 2). Four of the repeat sequences (R2, R4, R5, and R6) overlap the beginnings or ends of putative genes. Repeats R4 and R5 occur in regions overlapping the translational starts of ORFs, while R6 overlaps the end of a putative gene and R2 encompasses the start of one gene and stop of another, overlapping gene. The location of this overlap corresponds to the direction in which the repeat occurs (i.e., R6 overlaps the end of a putative gene and occurs in the opposite orientation to repeats that overlap the start of genes). Such a repeat structure raises the possibility that they may be involved in antisense translation regulation. Expression of the ORF containing R6 would produce an RNA that would bind with high affinity to the RNA transcripts containing R1 to R5. Furthermore, the repeat structures carry a sequence highly similar to the sigma −35 (TTGACA) and −10 (TATAAT) sequences (Table 3). To the best of our knowledge, such structures have not been observed in other bacteriophages and warrant further study.
TABLE 3.
Repeats R1 to R6 located within the TPA2 genome
 |
a Repeats R1 and R6 are 58 bp long, whereas R2 to R5 are 59 bp. Shaded nucleotides indicate differences in comparison to the consensus.
b Overlaps the start of the gene.
c Overlaps the end of the gene.
d Repeat is present in an intergenic region.
e Sequences in boldface indicate the start codons and those underlined indicate the stop codons of a gene. The boxed sequences represent putative −35 and −10 promoter regions.
Analysis of the TPA2 genome also reveals 95 pairs of inverted repeats ranging in size from 16 to 53 bp (see the supplemental material). These sequences occur within both putative genes and intergenic regions. Their large number and wide distribution throughout the genome suggests that they may have a functional role. Interestingly, all the repeats identified are inverted, with none occurring in a direct orientation. Inverted repeat structures are associated with replication origins (30) and transposable elements, neither of which could be identified in the genome of TPA2. The functional role (if any) of these repeats remains to be experimentally determined.
In addition to the inverted repeats, six palindrome sequences were identified. The smallest is 22 bp long, and the largest is 49 bp long (Table 4). Four of the palindromes occur in intergenic regions, suggesting that they may function as rho-independent transcriptional terminators. The downstream flanking sequences do not display the T-rich region typical of rho-independent transcriptional terminators in Escherichia coli (26); however, it has been observed that this T-rich region is not required for transcriptional termination in other bacteria such as the Gram-positive Streptomyces lividans (13, 32).
TABLE 4.
Palindrome sequences found within the TPA2 genome
| Palindrome | Coordinates | Sequence | Melting temp (°C) | Location within TPA2 |
|---|---|---|---|---|
| P1 | 14252-14300 | AGGGCGGGCGGCGTGGACCACCACAGGCCCGGTCTACGCCGCCCGCCCT | 84 | Intergenic (orf21-orf22) |
| P2 | 20738-20777 | GGCCGCGCGCGATGTCGAAGCTCAGATGTCGCGCGCGGCC | 79 | Start of tmp |
| P3 | 42728-42763 | GAGAAGGCCCGTCGCGGGGAAGCGACGGGCCTTCTC | 76 | Intergenic (lysA-orf56) |
| P4 | 47408-47429 | GACGGCGTCGGCCGACGCCGTC | 68 | End of orf61 |
| P5 | 59817-59851 | GAGAACCCCGCCGTCCTGAGTTCGGCGGGGTTCTC | 69 | Intergenic (orf75-orf76) |
| P6 | 60852-60875 | TCGTGCCTACCGCGGTAGACACGA | 57 | Intergenic (orf77-orf78) |
DNA packaging gene region.
The protein encoded by the ninth ORF (terL) is highly similar to the putative large terminase from the Mycobacterium phage Rosebush (21). The terminase enzyme is essential for DNA packing of the phage genome into the phage head (38). It normally functions as a complex with a small terminase subunit (8), which determines the specificity of DNA binding, while the large terminase subunit mediates cleavage of the phage DNA packaged into the prohead (17). Typically, the genes encoding the small terminase are located immediately upstream of those encoding the large terminase subunit and transcribed in the same direction. The putative gene upstream of terL, orf8, is transcribed in the opposite direction to terL and appears to be part of an unrelated operon. Furthermore, an examination of the complete genome reveals no ORF with any identity to known or putative small terminase genes, suggesting that either TPA2 utilizes an alternative DNA packaging mechanism or the small terminase has no similarity to any previously described one.
Structural gene region.
The genes encoding the structural proteins are located from orf17 to orf47. SDS-PAGE analysis of TPA1 phage proteins revealed 10 structural proteins (45). The major protein of ∼35 kDa was previously extracted and N-terminally sequenced (ATFPLVKGTRLRATRINSCG) by Thomas (45). This sequence is identical to that of the protein encoded by orf27 of the TPA2 genome. The N-terminal methionine is absent, a situation seen with other phage proteins and caused by the activity of a host cell methionine aminopeptidase (28).
The phage head morphogenesis module (Fig. 2) appears to be encoded by the genes orf17 to orf20. All four ORFs are expressed in the same orientation and share high levels of amino acid identity with Mycobacterium phages and the Rhodococcus phage ReqiPine5. The gene orf18 contains a pfam04233 motif, which is indicative of proteins involved in viral head morphogenesis of dsDNA phages (4). It is typical for the head protein genes in phage genomes to be clustered together and to precede the tail protein genes (7). This suggests that orf17 and orf19 are likely to be involved in viral head structural formation despite their products lacking identity to any known bacteriophage structural proteins. The function of orf20 is unknown, and it displays no identity to any other gene.
Immediately downstream of the phage head morphogenesis-encoding region is a putative tail morphogenesis region. This region (orf22 to orf49, with the exception of orf36 to orf38) shares a high level of amino acid identity with similar regions from a range of Mycobacterium phages (19), the Rhodococcus phage ReqiPine5 (accession no. GU580943), and the Gordonia terrae phage GTE5 (accession no. AY846870). These genes are transcribed in the same direction and are flanked by putative rho-independent transcriptional terminators (Table 4).
The major tail protein appears to be encoded by the orf27 gene, whose product has been confirmed by N-terminal sequencing to be a structural protein (45). On the basis of amino acid identity, we predict that an additional four structural proteins (encoded by orf31, orf33, orf44, and orf46) are involved in tail assembly. The protein encoded by orf31 (15.8 kDa) contains a pfam04883 domain, which is common to tail component proteins in tailed phages. Furthermore, orf33 encodes the largest protein (249 kDa) within the TPA2 genome and contains both the pfam10145 (found in tape measure proteins [TMPs]) and pfam06737 (lytic transglycosylase) motifs. The lytic transglycosylase domain located at the C terminus of this TMP also contains a peptidoglycan hydrolase domain, a feature seen in the Mycobacterium phage TM4 (37). The genes orf31 and orf33 encode conserved motifs specifically found in phage tail proteins.
The putative genes orf28 and orf29 display no significant identity to any known proteins. These two ORFs are located between the genes encoding the major tail protein (orf27) and the tape measure protein (orf33). Interestingly, between the major tail and tape measure protein genes of bacteriophages λ and Mu is a single ORF that expresses two separate proteins via a programmed translational frameshift (49). A similar phenomenon has been noted in some Mycobacterium phages, such as Mycobacterium phage Tweety (36). No translational frameshift was identified in this region of TPA2; however, orf28 and orf29 overlap by 169 bp, and they have their own initiation codons. This intriguing structure suggests that TPA2 may be using an alternative mechanism to achieve coding overlap in this region.
Lastly, two ORFs (orf44 and orf46) within this putative operon show identity at the amino acid level to genes encoding tail fibers in other bacteriophages (21). In addition to the major tail protein encoded by orf27, Thomas (45) detected nine minor structural proteins. Five of these proteins could be identified in the genome of TPA2 with sizes consistent with the proteins identified by Thomas (45). The identities of the remaining structural genes remain unknown, but it is possible that some are components of larger proteins that have undergone posttranslational cleavage (9).
Host cell lysis gene region.
Phage lysis modules typically consist of endolysin and holin genes that together are responsible for bacterial lysis and release of phage progeny (11). Within the TPA2 genome we could identify only the putative endolysin gene (lysA), with no ORF displaying identity to any known holin protein. At the amino acid level this putative endolysin shares high identity with proteins found in Mycobacterium phages and several hypothetical proteins thought to belong to the esterase and lipase family, encoded in the genomes of Nocardia facinica and Corynebacterium and Rhodococcus species. Close examination of the putative LysA amino acid sequence reveals two overlapping conserved motifs in the protein center with identity to the amidase 2 (Pfam01510) and the peptidoglycan recognition protein (PGRP) superfamily. These motifs suggest that this protein is most probably a zinc amidase involved in cleaving the amide bond between N-acetylmuramyl and l-amino acid residues in the bacterial peptidoglycan. This enzyme may have a structure and biological function similar to those in the T7 phage lysozyme homologue (10, 22). The C terminus of the putative endolysin contains three conserved 54-amino-acid repeat sequences with identity to a repeat structure seen in the Corynebacterium glutamicum and Corynebacterium efficiens genomes. These repeats belong to the pfam08310 family and are hypothesized to anchor proteins into the bacterial cell wall (3).
No lysin B (lysB) gene was identified in TPA2, which suggests that lysA may be sufficient for lysis or that if present its product is significantly different from previously identified lysin B proteins. Some Mycobacterium phages have been shown to encode a mycolylarabinogalactan esterase responsible for the release of the mycolic acid during cell lysis (34).
The identification of a holin-encoding gene remains unresolved. Holin genes are generally found adjacent to the lysin genes in phages (29) and typically contain two transmembrane domains. The putative gene orf54 and lysA overlap and are expressed in the same direction. In silico analysis of the orf54 protein sequence reveals two transmembrane domains, further suggesting that orf54 encodes a novel holin; however, this has not been shown experimentally.
DNA replication, modification, and regulation region.
The majority of genes involved in bacteriophage DNA replication and regulation appear to be located between orf48 and orf1. Some exceptions exist (e.g., ruvC). The gene denoted ruvC (between orf14 and orf16) appears to encode a Holliday junction resolvase with high identity to products of genes observed in Mycobacterium phage genomes. Holliday endonucleases resolve Holliday junction structures formed during homologous recombination events (40). Such enzymes are frequently encountered in bacterial genomes, but similar ruvC genes have also been identified in phage genomes, including Lactococcus lactis phage bIL66 and a number of Mycobacterium phages (5). It has been speculated that the phage ruvC-like gene products are more similar to general branch-cutting endonucleases, such as those encoded by the T4 and T7 phage genomes (47), than to typical bacterial Holliday junction resolvases (40). The genes flanking ruvC, orf14 and orf16, encode products with no known function. However, conserved motifs (pfam05305 and pfam07098, respectively) were detected in their amino acid sequences. Both motifs occur in bacterial proteins of unknown function.
The first gene in this cluster, orf48, encodes a putative regulator that shares identity with gene products in mycobacterium phages that may also have a similar function. The genes following orf48, which include orf49 to orf53 encode products of unknown function. Transcribed in the opposite direction to lysA are eight putative genes (orf56 to orf63) organized into what appear to be two operons, since these appear to contain rho-independent transcriptional terminators located after orf56 and between orf61 and orf62 (Table 4). A further two ORFs (orf64 and orf65) are transcribed in the same direction as orf56 to orf63 but are separated by a 750-bp noncoding region. The putative genes in this cluster share identity with genes from Mycobacterium phages. Presumed functions can be assigned to only three of the gene products (orf58, orf61, and orf63). Genes encoding a putative helicase (orf58), primase (orf61), and DNA polymerase I (orf63) can be identified on the basis of their amino acid sequences. The products of orf60, orf64, and orf65 have no significant similarity to products of any sequenced gene.
The protein encoded by orf58 has two conserved motifs (Pfam04851 and Pfam00271) found in DNA helicases. The N-terminal portion of the protein has a type III restriction enzyme res subunit motif (Pfam04851) that is characteristic of ATP-dependent helicase proteins. The center of this protein possesses a helicase motif (Pfam00271), whose function is to unwind the phage DNA duplex by utilizing energy from nucleoside triphosphate hydrolysis. Two putative ATP-binding sites were also identified within the amino acid sequence.
The N-terminal region of the putative primase (Orf61) possesses three conserved motifs involved in nucleotide binding, primase-nucleotide binding, and polymerase-nucleotide binding. This region also encodes a primase polymerase domain (primpol; Pfam09250) commonly seen in archaeal plasmids and phages. The central region of Orf61 has an additional ATP-binding motif, together with Walker A and Walker B motifs, both of which are related to the 5′-3′ DNA helicase found in plasmids and utilize ATP (27). This region is highly conserved and characteristic of the p-loop nucleoside triphosphatase (NTPase) superfamily (16).
A putative DNA polymerase I is encoded by orf63. The region corresponding to the N-terminal protein region encodes a 3′-5′ exonuclease motif (Pfam01612), and that corresponding to the C-terminal region encodes a DNA polymerase A motif (Pfam00476), as well as a DNA-binding motif. This gene appears to encode the DNA polymerase responsible for phage DNA replication. The gene cluster from orf66 to orf76 is transcribed in the opposite direction to orf56 to orf65. The majority of gene products in this cluster display no significant identity to known proteins (Orf66, Orf67, Orf68, Orf69, Orf70, Orf71, Orf73, Orf74, and Orf76). Two of the genes (orf72 and orf76) share similarity to genes of unknown function in Mycobacterium phages, while Orf75 appears to be an endonuclease of unknown function.
The final gene cluster (orf77 to orf8) is transcribed in the same direction and appears to form a large operon. A putative rho-independent transcriptional terminator is located between orf77 and orf78 (Table 4), suggesting that orf78 may be separately regulated. The product of orf78 is a putative metallophoesterase based on its transcribed amino acid sequence and the presence of the COG4186 motif. The remainder of putative genes in this cluster encode products of unknown function (Table 2).
Evolutionary events that contributed to the TPA2 genome.
The transcribed products of the TPA2 genome share amino acid similarities with proteins found in bacterial members of the Corynebacterineae and to Mycobacterium and Corynebacterium phages. Interestingly, some proteins share high amino acid identity but display only low BLAST E values, since their genes appear to be chimeric. An excellent example of this apparent chimeric gene structure is seen in the tape measure protein Orf33. The first 1,280 amino acids share a 36% identity with a tape measure protein from Mycobacterium abscessus ATCC 19977 (accession no. YP_001702534). The next 270 amino acids are most similar to the tape measure protein in Rhodococcus phage ReqiPine5 (accession no. ADD81132), followed by 150 amino acids with no identity to any known protein. The subsequent 100 amino acids share 39% identity with the G5 domain from Bifidobacterium gallicum (accession no. ZP_05966447), and finally, the last 580 amino acids are 39% identical to gp23 of the Mycobacterium phage Nigel (accession no. YP_002003862). A further example is the lysA gene, encoding the putative phage endolysin. Its gene product is split into two sections, with the first 154 amino acid sequence sharing 46% identity with that of a hypothetical protein from Mycobacterium phage TM4 (accession no. AAD17596), followed by a sequence of 30 amino acids of unknown function. The final 374 amino acids share 59% identity with a hypothetical protein from Nocardia farcinica (accession no. YP_120121). These mosaic gene structures were also noted for other genes in the TPA2 genome (data not shown), suggesting that this arrangement is a common feature of TPA2 genome evolution.
Other complex recombination events appear to have occurred within TPA2. Evidence for this is apparent when its genome sequence is compared with that of the Mycobacterium phage Rosebush. This phage encodes 90 putative proteins, gp1 to gp90 (35). In TPA2, terL encodes a terminase most closely related to gp7 in phage Rosebush. Immediately adjacent to terL is orf10 of no known function, although its closest relative is gp71 from the Mycobacterium phage Rosebush. This region of sequence similarity occurs over the center portion of the protein only, which suggests that the genes within the TPA2 genome, despite their conserved modular arrangement, can recombine and form new modular arrangements.
Comparative phage genomics reveals that many phages appear to be mosaics of each other (36). The genome sequence of TPA2 reported here also reveals such a chimeric structure, with both individual genes and operonic modules displaying a striking mosaic architecture. Interestingly, TPA2 has a gene organization similar to that of Mycobacterium phage Rosebush and can be grouped into the cluster B division constructed by Hatfull et al. (20). Although this cluster B is subdivided into four subclusters, it is difficult to allocate TPA2 to any of these because of the lack of nucleotide sequence identity with those currently located there. Any such identity is shared with all the mycobacteriophages in cluster B.
Conclusions.
This report describes the first complete genome sequence of a Tsukamurella phage. Bioinformatic analysis reveals that TPA2 is novel lytic phage, with only 30% of its genome being related to other phages at the DNA level, and that it has many previously unreported features. This phage targets a wide range of Tsukamurella species. It may be possible to use the TPA2 phage for the biocontrol of Tsukamurella stabilized foams in activated sludge plants or other environments where Tsukamurella spp. are problematic.
This study expands our understanding of the ecology of phages in activated sludge. While it is known that bacteriophages play an important role in controlling the population densities of their bacterial hosts (46a), their impact on the community structure of activated sludge systems is largely unknown. The repeated isolations of this phage more than 9 years apart suggest that it is probably widespread and not ephemeral but is an important member of the activated sludge phage metapopulation. Such a persistence demonstrates the need to better understand phage ecology within such systems and understand how populations may change with plant operational conditions. The genome sequence described here will enable specific targeted quantitative PCR methods to be developed for this purpose. Equally important is understanding the role that TPA2 and other phages might have in promoting gene transfer within activated sludge communities. The evidence presented here suggests that this is likely to be substantial, but the genomes of more activated sludge phages clearly need to be analyzed before this can be elucidated. This work is currently in progress.
Supplementary Material
Acknowledgments
We thank Robert Glashier, Jason McKenzie, and Glenys Shirley for their assistance with the transmission electron microscope and the anonymous reviewers for their insightful comments and suggestions.
The research was supported by an Australian Research Council (ARC) Linkage grant (LP0774913) together with Melbourne Water (David Gregory) and South East Water (Graham Short), whom we thank for their financial support. S. Petrovski was funded by ARC Linkage and La Trobe University grants.
Footnotes
Published ahead of print on 23 December 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Ackermann, H. W. 2003. Bacteriophage observations and evolution. Res. Microbiol. 154:245-251. [DOI] [PubMed] [Google Scholar]
- 2.Adams, M. H. 1959. Bacteriophages. Intersciences Publishers, Inc., New York, NY.
- 3.Adindla, S., K. K. Inampudi, K. Guruprasd, and L. Guruprasad. 2004. Identification and analysis of novel tandem repeats in the cell surface proteins of archaeal and bacterial genomes using computational tools. Comp. Funct. Genomics 5:2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker, B., et al. 1997. Head morphogenesis genes of the Bacillus subtilis bacteriophage SPP1. J. Mol. Biol. 268:822-839. [DOI] [PubMed] [Google Scholar]
- 5.Bidnenko, E., C. Mercier, J. Tremblay, P. Tailliez, and S. Kulakauskas. 1998. Estimation of the state of the bacterial cell wall by fluorescent in situ hybridization. Appl. Environ. Microbiol. 64:3059-3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brüssow, H. 2005. Phage therapy: the Escherichia coli experience. Microbiology 151:2133-2140. [DOI] [PubMed] [Google Scholar]
- 7.Brüssow, H., and F. Desiere. 2001. Comparative phage genomics and the evolution of Siphovirdae: insights from dairy phages. Mol. Microbiol. 39:213-222. [DOI] [PubMed] [Google Scholar]
- 8.Catalano, C. E. 2000. The terminase enzyme from bacteriophage lambda: a DNA-packaging machine. Cell. Mol. Life Sci. 57:128-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, C. L., et al. 2008. Genome sequence of the lytic bacteriophage P1201 from Corynebacterium glutamicum NCHU 87078: evolutionary relationships to phages from Corynebacterineae. Virology 378:226-232. [DOI] [PubMed] [Google Scholar]
- 10.Cheng, X., X. Zhang, J. W. Pflugrath, and F. W. Studier. 1994. The structure of bacteriophage T7 lysozyme, a zinc amidase and an inhibitor of T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 91:4034-4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daniel, A., P. E. Bonnen, and V. A. Fischetti. 2007. First complete genome sequence of two Staphylococcus epidermidis bacteriophages. J. Bacteriol. 189:2086-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de los Reyes, F. L. 2010. Foaming, p. 215-258. In R. Seviour and P. H. Nielsen (ed.), Microbial ecology of activated sludge. IWA Publishing, Norfolk, United Kingdom.
- 13.Deng, Z. X., T. Kieser, and D. A. Hopwood. 1987. Activity of a Streptomyces transcriptional terminator in Escherichia coli. Nucleic Acids Res. 15:2665-2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drummond, A. J., et al. 2010. Geneious v5.1. Biomatters Ltd., Auckland, New Zealand.
- 15.Finn, R. D., et al. 2010. The Pfam protein families database. Nucleic Acids Res. 38:D211-D222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frickey, T., and A. N. Lupas. 2004. Phylogenetic analysis of AAA proteins. J. Struct. Biol. 146:2-10. [DOI] [PubMed] [Google Scholar]
- 17.Fujisawa, H., and M. Morita. 1997. Phage DNA packaging. Genes Cells 2:537-545. [DOI] [PubMed] [Google Scholar]
- 18.Goodfellow, M., and L. A. Maldonado. 2006. The families Dietziaceae, Gordoniaceae, Nocardiaceae and Tsukamurellaceae, p. 843-888. In M. Dworkin, S. Flakow, E. Rosenberg, K. H. Schleifer, and E. Stackebrandt, (ed.), The prokaryotes, 3rd ed., vol. 3. Archaea. Bacteria: firmicutes, actinomycetes. Springer, New York, NY. [Google Scholar]
- 19.Hatfull, G. F., S. G. Cresawn, and R. W. Hendrix. 2008. Comparative genomics of the mycobacteriophages: insights into bacteriophage evolution. Res. Microbiol. 159:332-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatfull, G. F., et al. 2010. Comparative genomic analysis of 60 mycobacteriophage geneomes: genome clustering, gene acquisition and gene size. J. Mol. Biol. 397:119-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatfull, G. F., et al. 2006. Expoloring the mycobacteriophage metaproteome: phage genomics as an educational platform. PLoS Genet. 2:e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inouye, M., N. Arnheim, and R. Sternglanz. 1973. Bacteriophage T7 lysozyme is a N-acetylmuramyl-l-alanine amidase. J. Biol. Chem. 248:7247-7252. [PubMed] [Google Scholar]
- 23.Kattar, M. M., et al. 2001. Tsukamurella strandjordae sp. nov., a proposed new species causing sepsis. J. Clin. Microbiol. 39:1467-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kragelund, C., et al. 2007. Ecophysiology of mycolic acid-containing Actinobacteria (Mycolata) in activated sludge foams. FEMS Micriobiol. Ecol. 61:174-184. [DOI] [PubMed] [Google Scholar]
- 25.Laslett, D., and B. Canback. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lesnik, E. A., et al. 2001. Prediction of rho-independent transcriptional terminators in Escherichia coli. Nucleic Acids Res. 29:3583-3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lohman, T. M., and K. P. Bjornson. 1996. Mechanisms of helicase-catalysed DNA unwinding. Annu. Rev. Biochem. 65:169-214. [DOI] [PubMed] [Google Scholar]
- 28.Lowther, W. T., and B. W. Matthews. 2000. Structure and function of the methionine aminopeptidases. Biochim. Biophys. Acta 1477:157-167. [DOI] [PubMed] [Google Scholar]
- 29.Lu, Z., E. Altermann, F. Breidt, and S. Kozyavkin. 2010. Sequence analysis of Leuconostoc mesenteroides bacteriophage 1-A4 isolated from an industrial vegetable fermentation. Appl. Environ. Microbiol. 76:1955-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mott, M. L., and J. M. Berger. 2007. DNA replication initiation: mechanisms and regulation in bacteria. Nat. Rev. Microbiol. 5:343-354. [DOI] [PubMed] [Google Scholar]
- 31.Nam, S. W., W. Kim, J. Chun, and M. Goodfellow. 2004. Tsukamurella pseudospumae sp. nov., a novel actinomycete isolated from activated sludge foam. Int. J. Syst. Evol. Microbiol. 54:1209-1212. [DOI] [PubMed] [Google Scholar]
- 32.Neal, R. J., and K. F. Chater. 1991. Bidirectional promoter and terminator regions bracket mmr, a resistance gene embedded in the Streptomyces coelicolor A3(2) gene cluster encoding methylenomycin production. Gene 100:75-83. [DOI] [PubMed] [Google Scholar]
- 33.Otawa, K., et al. 2007. Abundance, diversity, and dynamics of viruses on microorganisms in activated sludge processes. Microb. Ecol. 53:143-152. [DOI] [PubMed] [Google Scholar]
- 34.Payne, K., Q. Sun, J. Sacchettini, and G. F. Hatfull. 2009. Mycobacteriophage lysine B is a novel mycolylarabinogalactan esterase. Mol. Microbiol. 73:367-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pedulla, M. L., et al. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171-182. [DOI] [PubMed] [Google Scholar]
- 36.Pham, T. T., D. Jacobs-Sera, M. L. Pedulla, R. W. Hendrix, and G. F. Hatfull. 2007. Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology 153:2711-2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piuri, M., and G. F. A. Hatfull. 2006. Peptidoglycan hydrolase motif within the mycobacteriophage TM4 tape measure protein promotes efficient infection of stationary phase cells. Mol. Microbiol. 62:1569-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rao, V. B., and M. Feiss. 2008. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 42:647-681. [DOI] [PubMed] [Google Scholar]
- 39.Seviour, R. J., et al. 2008. Ecophysiology of the Actinobacteria in activated sludge systems. Antonie Van Leeuwenhoek 94:21-33. [DOI] [PubMed] [Google Scholar]
- 40.Sharples, G. J., L. M. Corbett, and P. McGlynn. 1999. DNA structure specificity of Rap endonuclease. Nucleic Acids Res. 27:4121-4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soddell, J. A. 1999. Foaming, p. 161-202. In R. J. Seviour and L. L. Blackall (ed.), Microbiology of activated sludge. Kluwer, Dordrecht, Netherlands.
- 42.Soddell, J. A., and R. J. Seviour. 1990. Microbiology of foaming in activated sludge plants—a review. J. Appl. Bacteriol. 69:145-176. [Google Scholar]
- 43.Stanley, T., et al. 2006. The potential misidentification of Tsukamurella pulmonis as an atypical Mycobacterium species: a cautionary tale. J. Med. Microbiol. 55:475-478. [DOI] [PubMed] [Google Scholar]
- 44.Steinhaus, E. A. 1941. A study of bacteria associated with thirty species of insects. J. Bacteriol. 42:757-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas, J. A. 2005. Actinophages in activated sludge. Ph.D. thesis. La Trobe University, Bendigo, Victoria, Australia.
- 46.Thomas, J. A., J. A. Soddell, and D. I. Kurtböke. 2002. Fighting foam with phages. Water Sci. Technol. 46:511-553. [PubMed] [Google Scholar]
- 46a.Weinbauer, M. G. 2004. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 28:127-181. [DOI] [PubMed] [Google Scholar]
- 47.White, M. F., M. Giraud-Panis, J. R. G. Pöhler, and D. M. J. Lilley. 1997. Recognition and manipulation of branched DNA structure by junction-resolving enzymes. J. Mol. Biol. 269:647-664. [DOI] [PubMed] [Google Scholar]
- 48.Withey, S. E., E. Cartmell, L. M. Avery, and T. Stephenson. 2005. Bacteriophages potential for application in wastewater treatment processes. Sci. Total Environ. 339:1-18. [DOI] [PubMed] [Google Scholar]
- 49.Xu, J., R. W. Hendrix, and R. L. Duda. 2004. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol. Cell 16:11-21. [DOI] [PubMed] [Google Scholar]
- 50.Zuber, S., et al. 2008. Decreasing Enterobacter sakazakii (Cronobacter spp.) food contamination level with bacteriophages: prospects and problems. Microb. Biotechnol. 1:532-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.