

Abstract
The charge density in the cell wall microenvironment of Gram-positive bacteria is believed to influence the expression of heterologous proteins. To test this, the expression of a SpaP-S1 fusion protein, consisting of the surface protein SpaP of Streptococcus mutans and a pertussis toxin S1 fragment, was studied in the live vaccine candidate bacterium Streptococcus gordonii. Results showed that the parent strain PM14 expressed very low levels of SpaP-S1. By comparison, the dlt mutant strain, which has a mutation in the dlt operon preventing d-alanylation of the cell wall lipoteichoic acids, and another mutant strain, OB219(pPM14), which lacks the LPXTG major surface proteins SspA and SspB, expressed more SpaP-S1 than the parent. Both the dlt mutant and the OB219(pPM14) strain had a more negatively charged cell surface than PM14, suggesting that the negative charged cell wall played a role in the increase in SpaP-S1 production. Accordingly, the addition of Ca2+, Mg2+, and K+, presumably increasing the positive charge of the cell wall, led to a reduction in SpaP-S1 production, while the addition of bicarbonate resulted in an increase in SpaP-S1 production. The level of SpaP-S1 production could be correlated with the level of PrsA, a peptidyl-prolyl cis/trans isomerase, in the cells. PrsA expression appears to be regulated by the cell envelope stress two-component regulatory system LiaSR. The results collectively indicate that the charge density of the cell wall microenvironment can modulate heterologous SpaP-S1 protein expression in S. gordonii and that this modulation is mediated by the level of PrsA, whose expression is regulated by the LiaSR two-component regulatory system.
Streptococcus gordonii is a commensal bacterium of the human oral cavity. The bacterium has gained interest as a live oral vaccine vehicle (21). The expression of a number of vaccine antigens has been described previously; however, the levels of protein produced have been very low. The successful use of this bacterium as a vaccine vehicle depends on the ability to produce adequate amounts of heterologous proteins. Little is known about factors governing the yield of secreted proteins following transcription and translation. In our recent studies, we reported that the expression of a heterologous SpaP-S1 protein in S. gordonii could be increased using an inducible promoter (27). However, even under the optimal induction condition, the level of SpaP-S1 produced was very low. Similar problems have been reported in Bacillus subtilis, where yields of native proteins can be as much as grams per liter (11, 30) but the production of heterologous proteins is significantly lower (33).
It is believed that a number of posttranslational events that limit heterologous protein production exist, including the inefficient translocation and folding of proteins and degradation by proteases (38). Proper folding of secreted proteins is critical to protein production, because improperly folded proteins are highly susceptible to proteolytic degradation. In B. subtilis, the folding of secreted proteins is catalyzed by a membrane-associated lipoprotein named PrsA (17, 43). PrsA displays homology to peptidyl-prolyl cis/trans isomerases. PrsA homologues are found ubiquitously in all Gram-positive bacteria. In B. subtilis, the protein secretion rate was linearly proportional to levels of PrsA (42). In addition, overproduction of PrsA resulted in increased expression of the heterologous amylase Q from Bacillus amyloliquefaciens and the protective antigen from Bacillus anthracis in B. subtilis (17, 44). Similarly, increasing levels of PrtM, a PrsA-like protein in Lactococcus lactis, led to increased heterologous protein expression in L. lactis (28).
Misfolded proteins in the cell wall lead to cellular stress, and B. subtilis uses the CssRS two-component regulatory system to sense this stress and activate the CssRS regulon, which includes the htrA and htrB genes, coding for cell wall-associated serine proteases (8, 13). In Staphylococcus aureus, stress caused by cationic antimicrobial peptides led to PrsA upregulation mediated by the VraSR (LiaSR homologue) cell envelope stress two-component regulatory system (32). In Streptococcus mutans, LiaSR responds to stress from acidic pH (25).
In this study, we examined the role of the cell wall in modulating heterologous SpaP-S1 production in S. gordonii. The results suggest that a negatively charged cell wall can increase SpaP-S1 production. The increase in SpaP-S1 appears to be due to an increase in PrsA production, which is regulated by LiaSR.
MATERIALS AND METHODS
Bacteria and growth conditions.
The bacterial strains used in this study are listed in Table 1 . S. gordonii DL1 is the parent strain, and all mutants were derived from this strain. The dlt mutant lacks d-alanine residues on its lipoteichoic acids due to the inactivation of the dlt operon (5). OB219 lacks two LPXTG major surface proteins, SspA and SspB (26). Both the dlt mutant and OB219 were transformed with pPM14, which carries the SpaP-S1 fusion gene under the control of the tetracycline-inducible xyl-tetO promoter (27). spaP encodes the LPXTG major surface protein SpaP (also named P1, or antigen I/II) from S. mutans (24). The s1 gene encodes the 179-amino-acid N-terminal fragment of pertussis toxin subunit S1 and is fused to the middle part of spaP (23). S. gordonii RJM4 carries the SpaP-S1 gene on the chromosome (22) and was used to avoid the potential incompatibility of having two plasmids in the same cell.
TABLE 1.
Bacterial strains used in the study
| Bacterial strain | Description | Source/ reference |
|---|---|---|
| S. gordonii | ||
| DL1 | Parent strain, Challis | D. LeBlanc |
| PM14 | DL1 carrying pPM14 (Kanr) | 27 |
| OB219 | SspA- and SspB-negative mutant of DL1, sspAB::ermAM | 26 |
| dltA mutant | DltA-negative mutant of DL1, dltA::ermAM | 5 |
| RJM4 | DL1 carrying a single copy of the SpaP-S1 gene on the chromosome, Kanr | 22 |
| prsA mutant | PrsA-negative mutant of RJM4, prsA::ermAM | This study |
| SgPL2 | RJM4 carrying pSgPL2 (Spcr) | This study |
| liaR mutant | LiaR-negative mutant of RJM4, liaR::ermAM | This study |
| E. coli | ||
| XL1-Blue | Cloning strain | Stratagene |
| SgP1H | XL1-Blue carrying pSgP1H (Kanr) | This study |
| SgPL1 | XL1-Blue carrying pSgPL1 (Kanr) | This study |
| SgPL2 | XL1-Blue carrying pSgPL2 (Spcr) | This study |
S. gordonii strains were cultured on brain heart infusion (BHI) agar or in tryptone-vitamin glucose (TVG) broth (35 mg/ml tryptone, 1 μl/ml vitamin solution [0.04 μg/ml p-amino benzoic acid, 0.2 mg/ml thiamine, 1.0 μg/ml nicotinamide, and 0.2 μg/ml riboflavin], 5 mg/ml glucose, pH 7.8) buffered with 100 mM HEPES. When needed, 250 μg/ml of kanamycin, 250 μg/ml spectinomycin, or 10 μg/ml erythromycin was added to the media. The induction of SpaP-S1 expression was achieved by the addition of 10 ng/ml tetracycline to the growth medium (27). Escherichia coli was cultured in Luria-Bertani broth (1% tryptone, 0.5% yeast extract, and 1% NaCl, wt/vol) containing 50 μg/ml kanamycin, 50 μg/ml spectinomycin, or 10 μg/ml tetracycline.
For experiments to examine the effects of cations or bicarbonate on SpaP-S1 expression, filter-sterilized stock solutions of KCl, CaCl2·2H2O, MgCl2, or NaHCO3 were added to the growth media at the indicated concentrations (see Fig. 3) prior to inoculation. All cultures grew to the same cell density, indicating that the added ions did not impair growth.
SDS-PAGE and Western blotting.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed as described by Laemmli (19). Bacterial cultures were standardized to the same optical density at 600 nm (OD600) following growth. Cells were pelleted by centrifugation (10,000 × g, 5 min), resuspended in SDS-PAGE sample buffer, and boiled for 5 min. Alternatively, the entire bacterial cultures were precipitated with 5% trichloroacetic acid and then extracted with SDS-PAGE sample buffer as total cell extracts prior to electrophoresis. Following electrophoresis, the SDS-polyacrylamide gels were stained with Coomassie blue R-250 to ensure equal loading of proteins in the samples. For Western blotting, proteins were transferred to nitrocellulose membranes using methods described by Towbin et al. (39). The SpaP-S1 fusion protein was detected using human anti-pertussis toxin IgG antibody (1/10,000) (3) followed by goat anti-human IgG alkaline phosphatase-conjugated antibody (1/4,000; Sigma-Aldrich Chemical Co., Oakville, ON, Canada). The PrsA protein was detected using mouse anti-PrsA antibody (1/2,000) (see below) followed by goat anti-mouse IgG alkaline phosphatase conjugates (1/8,000; Sigma-Aldrich). The intensities of the full-length 187-kDa SpaP-S1 and 31-kDa PrsA immunoreactive bands were analyzed by digital imaging using the ImageJ program (NIH, http://rsb.info.nih.gov/ij).
Evaluation of bacterial surface charge.
Bacterial surface charge was evaluated by two methods. The first method measured the zeta potential values using a zeta potential analyzer (PanKem system 3000; PenKem Inc., Bedford Hills, NY) according to the method described by Speers et al. (35). Mid-exponential-phase (OD600 of approximately 0.5) cells were washed once in 20 mM morpholinepropanesulfonic acid (MOPS) buffer, pH 7, and resuspended in the same buffer to 3.5 × 107 CFU/ml. A minimum of 6 measurements was obtained for each triplicate culture of the three strains. The second method used the cationic dye alcian blue 8GX binding assay (4, 5). Mid-exponential-phase cells were washed once with 20 mM MOPS buffer and resuspended in the same buffer to a final OD600 of 0.5 (ca. 2.5 × 108 CFU/ml). Alcian blue was added to a final concentration of 65 μg/ml. Samples were rotated at room temperature for 10 min at 3 rpm. As a negative control, MOPS buffer with 65 μg/ml alcian blue was incubated under the same conditions without bacteria. After incubation, bacteria were removed by centrifugation (10,000 × g, 5 min). The unbound alcian blue in the supernatant fluids was measured at 530 nm using a spectrophotometer. The amount of alcian blue bound to the bacteria was calculated as (A530 of supernatant without bacteria − A530 of supernatant with bacteria)/A530 of supernatant without bacteria × 100.
Isolation of cellular fractions from S. gordonii.
Five hundred ml of late-exponential-phase bacterial cultures was centrifuged (10,000 × g, 15 min, 4°C). The supernatant was collected and stored at −80°C. The bacterial cells were washed once with phosphate-buffered saline (PBS), resuspended in 2 ml of PBS, and disrupted in a cell disintegrator (Mickle Laboratory Engineering Co. Ltd., Gonshall, Surrey, United Kingdom) for 1.5 h at 4°C to achieve 95% breakage. The supernatant was removed, and the glass beads were washed 4 times with 1 ml of PBS. The supernatant and wash fractions were combined and centrifuged (2,000 × g, 2 min) to pellet any remaining glass beads and whole cells. The supernatant was recovered and centrifuged (27,000 × g, 30 min, 4°C) to separate the cell wall from the cytoplasm and membranes. The cell wall pellet was resuspended in 5 ml of distilled H2O (dH2O) and centrifuged (27,000 × g, 30 min, 4°C). The resulting cell wall pellet was resuspended in 500 μl of PBS and stored at −80°C. The cell membrane/cytoplasm supernatant was centrifuged for 1 h at 50,000 × g to separate the cell membrane from the cytoplasm. The cell membrane pellet was resuspended in 500 μl of PBS and stored at −80°C. The supernatant, containing the cytoplasm, was also stored at −80°C. All cellular-fraction samples were thawed and standardized by volume prior to electrophoresis and Western immunoblotting.
Analysis of cell wall proteins.
Four hundred μl of cell wall was centrifuged (10,000 × g, 15 min), resuspended in 50 μl of dH2O, and freeze-dried. One mg of the dried cell wall was resuspended in 3× sample buffer (20 mM Tris [pH 6.8], 0.3% glucose [wt/vol], 6% SDS [wt/vol], and 0.15% β-mercaptoethanol) and boiled for 20 min to extract non-covalently-bound proteins. The samples were centrifuged (10,000 × g, 15 min). The cell wall pellet was washed 8 times with warmed (37°C) dH2O. The residual cell walls were incubated overnight at 37°C with 100 U of mutanolysin (Sigma-Aldrich) in 100 μl of 50 mM Tris buffer (pH 7.0) containing 10 mM MgCl2 and 2 mM phenylmethylsulfonyl fluoride. In parallel, 1 mg of unextracted cell wall was digested overnight with mutanolysin. Following digestion, the samples were boiled in sample buffer, centrifuged (10,000 × g, 5 min), and analyzed by SDS-PAGE and Western immunoblotting.
Reverse transcription-PCR.
Total RNA was extracted from cells using the hot acid phenol method of Peterson et al. (31) as described previously (40). Contaminating DNA was removed by digestion with amplification-grade DNase I (Invitrogen Life Technologies, Burlington, ON, Canada), and the resulting RNA was verified to be free of DNA by PCR for the 16S rRNA gene using primers SL525/SL697 (Table 2). cDNA was synthesized using random primers and SuperScript II reverse transcriptase (Invitrogen) following the manufacturer's instructions. The levels of 16S rRNA and SpaP-S1 gene transcripts were estimated by PCR with Taq DNA polymerase and primer pairs SL525/SL697 and SL642/SL643, respectively. The PCR products were incubated at 95°C for 3 min, followed by 30 cycles of 95°C for 30 s, 50°C for 30 s, and 72°C for 1 min and a final 5-min extension at 72°C. The PCR products were analyzed by gel electrophoresis, and the intensity of the 600-bp s1 gene fragment was normalized against that of the 16S rRNA gene fragment (500 bp) by using the ImageJ program, The 16S rRNA gene was chosen as the endogenous reference gene, as its expression is relatively stable irrespective of growth conditions (36).
TABLE 2.
Primers used in the study
| Primer name | Primer sequence (restriction enzyme)a | Primer function |
|---|---|---|
| SL525 | GAATTAAACCACATGCTCCACCGC | Reverse primer for 16S rRNA gene |
| SL550 | CTGGGCCCAGGCGGCCGGGCCCAAAATTTGTTTGAT (SfiI) | Forward primer for ermAM |
| SL551 | CCTGGCCGGCCTGGCCGGCAGCGACTCATAGAAT (SfiI) | Reverse primer for ermAM |
| SL642 | ATACATATGGATCCTCCCGCCACCG | Forward primer for s1 gene |
| SL643 | ATAGAATTCCGATGTGTAGGGGTTGGG | Reverse primer for s1 gene |
| SL651 | CTTGGCCCAGGCGGCCGACAAGGACATTATTACTA (SfiI) | Forward primer for prsA |
| SL667 | ATAGGTACCACAAAGGAGTTTTCATTAA (KpnI) | Forward primer for entire prsA gene, including ribosomal binding site and start codon |
| SL668 | ATAGAATTCTTATTTTGATGAAGAAGATG (EcoRI) | Reverse primer for prsA including stop codon |
| SL669 | CTTGGCCGGCCTGGCCTTTTGATGAAGAAGATGAAG (SfiI) | Reverse primer for prsA without stop codon |
| SL692 | ACTACATCTTCGTCCTACAG | Forward primer for liaR upstream fragment |
| SL693 | CCTGGCCGCCTGGGCCCGAACCATTTCGTGATCATC (SfiI) | Reverse primer for liaR upstream fragment |
| SL694 | CCTGGCCGGCCTGGCCTTCCTCAGGAAGACTTCTGA (SfiI) | Forward primer for liaR downstream fragment |
| SL695 | ACAGGTAGCTTCTGGTAAAC | Reverse primer for liaR downstream fragment |
| SL697 | ATTTATTGGGCGTAAAGCGAGCGC | Forward primer for 16S rRNA gene |
| SL700 | CCTGGCCGCCTGGGCCGCTTCATATGCAGCTTTATA (SfiI) | Reverse primer for prsA upstream fragment |
| SL701 | CCTGGCCGGCCTGGCCCACCAGAGGTAACTGCG (SfiI) | Forward primer for prsA downstream fragment |
Restriction enzyme sites are underlined.
Recombinant PrsA and generation of anti-PrsA antibodies.
To produce the recombinant PrsA, the 896-bp DNA fragment encoding the mature PrsA without the LXXC lipoprotein motif was amplified by PCR from the chromosome of DL1 using the primers SL651 and SL669 (Table 2). The PCR product was restricted with SfiI and cloned into the same sites on pSecPrn(−HA). pSecPrn(−HA) is a derivative of pSecCR1 (15); it carries a 0.6-kb DNA insert (Prn) instead of the anti-CR1 gene and the hexahistidine tag but lacks the hemagglutinin (HA) tag. The cloning put the prsA gene behind the tetracycline-inducible Pxyl-tetO promoter, the S. mutans spaP ribosomal binding site and signal sequence, and an in-frame His tag at the C terminus. The resulting plasmid was named pSgP1H and maintained in E. coli XL1-Blue.
Cells from 100-ml cultures of E. coli (pSgP1H) induced with tetracycline were harvested by centrifugation (10,000 × g, 10 min) and resuspended in 5 ml of NiCAM wash buffer (50 mM Na2HPO4, 300 mM NaCl, and 20 mM imidazole, pH 7). Cells were broken by sonication (Vibra Cell; Sonics & Materials Inc., Danbury, CT) and centrifuged (20,000 × g, 30 min). The clear supernatant was applied to a 4-ml NiCAM column (Sigma-Aldrich), which had been equilibrated with the NiCAM wash buffer. The column was washed with 20 ml of NiCAM wash buffer, and the bound PrsA was eluted with 10 ml of NiCAM wash buffer containing 250 mM imidazole. The eluted PrsA was >95% pure as judged by SDS-PAGE analysis (data not shown).
To generate a polyclonal antibody against PrsA, BALB/c mice (female, 5 weeks old, n = 5) were injected subcutaneously, on days 1, 14, and 21, with 10 μg PrsA absorbed to 2% (wt/vol) aluminum hydroxide gel (Sigma-Aldrich) (16). The animals were euthanized on day 33 and sera saved as the anti-PrsA antibody.
Construction of the prsA mutant and the liaR mutant.
The 5′ (446-bp) and 3′ (447-bp) regions of prsA were amplified from DL1 chromosomal DNA by PCR using the primer pairs SL667/SL700 and SL701/SL668, respectively (Table 2). An erythromycin resistance cassette (ermAM) was PCR amplified from a synthetic construct template (6) with the primer pair SL550/SL551. The prsA fragments were restricted with SfiI and ligated to the ermAM DNA separately. The ligated DNAs were PCR amplified to produce the 1.40-kb 5′ prsA-ermAM and 1.34-kb 3′ ermAM-prsA DNA using the SL667/SL551 and SL550/SL668 primer pairs, respectively. The PCR products were mixed in a 1:1 ratio and further amplified by overlapping PCR using the primer pair SL667/SL668 to obtain the final 1.88-kb prsA::ermAM construct, which was introduced into S. gordonii RJM4 via natural transformation (22). Transformants were selected on erythromycin-BHI agar and verified to carry an insertional inactivation of prsA by PCR and Western immunoblotting.
The liaR mutant was constructed similarly to the prsA mutant. The regions upstream (466 bp) and downstream (532 bp) of liaR were amplified from DL1 chromosomal DNA by PCR using the primer pairs SL692/SL693 and SL694/SL695, respectively (Table 2). The PCR products were ligated to ermAM, and the 1.6-kb ligated product was PCR amplified using SL692 and SL695. The resulting liaR::ermAM construct was transformed into RJM4 to obtain the liaR mutant. The mutant was verified by PCR using primer pair SL692/SL695 to carry the 1.6-kb liaR::ermAM, while the parent RJM4 had the intact liaR (data not shown).
Construction of the PrsA overexpression strain.
The full-length prsA gene, including its ribosomal binding site, signal sequence, and LXXC lipoprotein motif, was PCR amplified using primers SL667 and SL668. The PCR product was restricted with KpnI and EcoRI and ligated to the same sites on pSecPrn(−HA). The cloning created pSgPL1, with the expression of prsA under the control of the tetracycline-inducible Pxyl-tetO promoter. Because pSgPL1 confers kanamycin resistance, it is not suitable for use in RJM4. Therefore, the 1.95-kb SphI-EcoRI fragment carrying Pxyl-tetOprsA and the tetR repressor gene was subcloned into the E. coli-Streptococcus shuttle vector pDL278 (Spcr) (20) to obtain pSgPL2. pSgPL2 was transformed into RJM4 to obtain the PrsA overexpression strain.
Statistical analysis.
Statistical significance for the results was evaluated by repeated-measures analysis of variance (ANOVA). This was followed by a Tukey test. A P value that was equal to or less than 0.05 was considered to be significant.
RESULTS
S. gordonii dlt mutant and OB219(pPM14) express higher levels of SpaP-S1 than the parent strain.
To examine the effects of the cell wall microenvironment on heterologous protein expression, the levels of SpaP-S1 expression were compared between the parent PM14 and two mutant strains of S. gordonii. The mutant strains are a dlt mutant, which cannot d-alanylate lipoteichoic acids (5), and OB219, which lacks the major surface proteins SspA and SspB (26). All three strains carry plasmid pPM14. Results from zeta potential measurements and the cationic dye alcian blue 8GX binding assay showed that the mutants have an increase in negative surface charges. The zeta potential values for the dltA mutant and OB219(pPM14) were −28.01 ± 0.83 and −27.81 ± 0.29 mV, respectively, and were significantly more negative than that (−21.23 ± 0.49 mV) of PM14 (P < 0.001). The results of the alcian blue binding assay showed that the dlt mutant and OB219(pPM14) bound 72% ± 2% and 73% ± 1% of alcian blue, respectively, in comparison to 62% ± 2% binding by PM14 (P < 0.001). When the three strains were analyzed for SpaP-S1 expression, the dlt mutant and OB219(pPM14) produced 2.5- and 2.9-fold more SpaP-S1 than PM14, respectively (Fig. 1). Reverse transcription-PCR analysis showed that the three strains expressed the same level of SpaP-S1 gene transcript (data not shown).
FIG. 1.
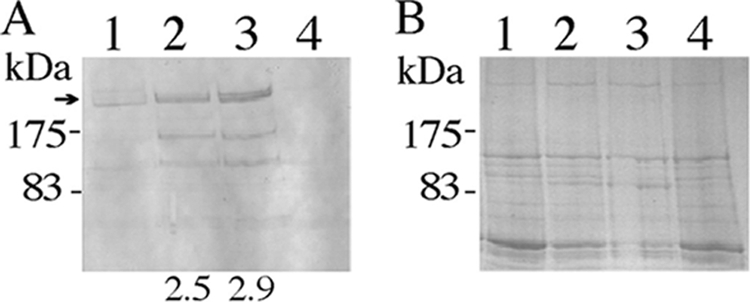
SpaP-S1 produced by S. gordonii PM14, the dlt mutant, and OB219(pPM14). (A) Western immunoblot of total cell extracts showing the full-length SpaP-S1 (arrow) and smaller-sized immunoreactive bands, presumably degradation products. (B) SDS-polyacrylamide gel (6.5%) showing equal loading of proteins in the samples. Lanes: 1, PM14; 2, dltA mutant; 3, OB219(pPM14); 4, DL1. DL1 does not carry pPM14 and thus does not produce the SpaP-S1 protein. Numbers below the blot indicate the fold increases in SpaP-S1 produced by the mutants compared to the level produced by PM14.
S. gordonii dlt mutant and OB219(pPM14) have equivalent amounts of SpaP-S1 anchored to the cell walls.
Since OB219(pPM14) lacks the LPXTG major surface proteins SspA and SspB, it is conceivable that more anchoring space is available in the cell wall, resulting in the increase in SpaP-S1 expression. To investigate this possibility, cellular fractions were isolated from the mutants and PM14. Western immunoblotting of the isolated fractions was performed to determine the location of SpaP-S1 within the bacterium. Results showed that >95% of the SpaP-S1 protein localized in the cell wall fraction, with the remaining located in the supernatant, the membrane and cytoplasm (data not shown). Consistent with earlier results, the cell walls of both the dlt mutant and OB219(pPM14) contained significantly more SpaP-S1 than that of PM14 (Fig. 2). Interestingly, the cell walls from the dlt mutant and OB219(pPM14) had similar amounts of covalently bound SpaP-S1. In addition, the cell walls of the dltA mutant and OB219(pPM14) contained more proteins than the PM14 cell wall.
FIG. 2.

SpaP-S1 anchored to the cell wall of PM14, the dlt mutant, and OB219(pPM14). (A) SDS-polyacrylamide gel (7.5%) of proteins from cell walls before hot-SDS extraction. Each sample is derived from the same quantity of cell wall treated with mutanolysin, as described in Materials and Methods. (B) Western blot of SpaP-S1 in cell wall samples shown in panel A. (C) Western blot of SpaP-S1 on residual cell wall after hot-SDS extraction. Lanes: M, prestained protein markers (New England BioLabs, Mississauga, ON, Canada); 1, PM14; 2, dlt mutant; 3, OB219(pPM14). The arrow indicates the full-length SpaP-S1 protein. The lower-mass immunoreactive bands are presumably degradation products.
Ca2+, Mg2+, and K+ decrease SpaP-S1 expression.
The finding that the dlt mutant and OB219(pPM14) have a more negatively charged cell surface and produced more SpaP-S1 than the parent suggests that the cell wall charge plays a role in SpaP-S1 expression. Thus, it is of interest to investigate whether SpaP-S1 expression can be modulated by the addition of cations to the cultures. As shown in Fig. 3A, SpaP-S1 production by OB219(pPM14) decreased with increasing concentrations of Ca2+. Mg2+ has a similar effect of lowering SpaP-S1 production (Fig. 3A). The addition of K+ also decreased SpaP-S1 expression, but the decrease was not as drastic as that achieved by Ca2+ or Mg2+ addition (Fig. 3B). Similar results exerted by Ca2+, Mg2+, and K+ were observed for the dlt mutant and PM14 (data not shown).
FIG. 3.

Effects of cations and bicarbonate on SpaP-S1 expression. (A) Immunoblot of SpaP-S1 produced by OB219(pPM14) following growth in the presence of Ca2+ or Mg2+ at the indicated concentrations. 0, no added Ca2+ or Mg2+. (B) Immunoblot of SpaP-S1 produced by OB219(pPM14) following growth in the presence of K+ at the indicated concentrations. 0, no addition. (C) Immunoblot of SpaP-S1 produced by S. gordonii in the presence (+) or absence (−) of 50 mM bicarbonate. Lanes: 1, PM14; 2, dlt mutant; 3, OB219(pPM14); 4, DL1. Numbers below the blots are percentages or fold changes in SpaP-S1 compared to the level for the no-addition control. Arrows indicate the full-length SpaP-S1 protein. The samples were also analyzed on 6.5% SDS-polyacrylamide gels to ensure equal loading of proteins (data not shown).
Bicarbonate ion increases SpaP-S1 expression.
Dorschner et al. (9) reported that the thickness of the bacterial cell wall is markedly decreased when cells are grown in the presence of a physiological concentration of bicarbonate. To determine whether bicarbonate has an effect on SpaP-S1 expression, S. gordonii strains were cultured in the presence of 50 mM NaHCO3. The results showed that an increase in SpaP-S1 expression was observed in all three strains (Fig. 3C). The parent strain produced 2.8-fold more SpaP-S1 when cultured in the presence of bicarbonate than when cultured without bicarbonate addition. Similarly, both the dltA mutant and OB219(pPM14) expressed more SpaP-S1 when grown in the presence of bicarbonate.
Role of PrsA in SpaP-S1 production.
Given that heterologous protein production could be increased by PrsA overproduction in B. subtilis (17, 44), it is conceivable that the observed increase in SpaP-S1 production in the dltA mutant and OB219(pPM14) of S. gordonii was due to an increase in PrsA expression. To examine this possibility, the levels of PrsA in PM14 and the two mutants were analyzed. The results showed that the dltA mutant and OB219(pPM14) produced 1.5- and 2-fold more PrsA, respectively, than PM14, suggesting that PrsA plays a role in SpaP-S1 production (Fig. 4A). To help confirm this, the prsA gene was insertionally inactivated in S. gordonii RJM4. The mutation was confirmed genetically by PCR, which showed the amplification of a 1.9-kb prsA::ermAM DNA from the chromosome by use of primer pair SL667/SL668 (data not shown). In addition, the mutant lacked the 31-kDa PrsA protein, as shown by Western blotting using anti-PrsA as the probe (Fig. 4B). The prsA mutant produced 35% less SpaP-S1 than the parent strain (Fig. 4B). To further provide support for the role of PrsA in SpaP-S1 production, the prsA gene was cloned into pDL278 and transformed into RJM4 to create a PrsA overproduction strain. The resulting strain produced 2.6-fold more PrsA and 3.5-fold more SpaP-S1 than the parent RJM4 (Fig. 4C). The above-described results collectively indicate that the observed increased SpaP-S1 production in the dltA mutant and OB219(pPM14) was due to PrsA overexpression.
FIG. 4.
PrsA and SpaP-S1 expression by S. gordonii. (A) Immunoblot of PrsA produced by PM14 (lane 1), OB219(pPM14) (lane 2), and the dltA mutant (lane 3). (B) Immunoblots of PrsA (left) and SpaP-S1 (right) produced by RJM4 (lane 1) and the prsA mutant (lane 2). (C) Immunoblots of PrsA (left) and SpaP-S1 (right) produced by RJM4 (lane 1) and RJM4 carrying pSgPL2 (lane 2). (D) Immunoblots of PrsA (left) and SpaP-S1 (right) produced by RJM4 (lane 1) and the liaR mutant (lane 2). Arrows indicate the full-length SpaP-S1 protein, and asterisks indicate PrsA. The samples were also analyzed on 6.5% or 12.5% SDS-polyacrylamide gels to ensure equal loading of proteins (data not shown).
To investigate whether the upregulation of PrsA was controlled by the cell envelope stress two-component regulatory system LiaSR, the response regulator LiaR in RJM4 was inactivated by the ermAM cassette. PrsA was upregulated 9.8-fold in the liaR mutant (Fig. 4D). In keeping with earlier results, SpaP-S1 production was increased 4.3-fold in the liaR mutant.
DISCUSSION
In this study, the role of the cell wall microenvironment in the expression of heterologous protein SpaP-S1 in S. gordonii was investigated. The results indicate that the overall charge density of the cell wall is important to SpaP-S1 production; when the cell wall becomes electronegative, more SpaP-S1 is produced. This statement is supported by results obtained using the dlt mutant and OB219(pPM14). Both strains have a more negatively charged cell wall than the parent PM14 and produced higher levels of SpaP-S1 than PM14. On the other hand, cation addition, which presumably makes the cell wall more electropositive, resulted in a decrease in SpaP-S1 production.
The observation that the dlt mutant has a more negative cell surface can easily be explained by the fact that lipoteichoic acids are not d-alanylated, due to the inactivation of the dlt operon. An increase in cell surface negative charge displayed by dlt mutants of other Gram-positive bacteria has been reported previously; thus, our results are consistent with the literature (5, 14, 18, 41). The observation that OB219(pPM14) also has a more negative cell surface is an unexpected result. However, given that SspA and SspB are calcium-binding proteins (10), it is conceivable that in the absence of these two proteins, calcium is not readily recruited to the cell wall, leading to a more negative cell surface. It has been proposed that the recruitment of calcium and other positively charged ions to the cell wall may promote bacterial aggregation by partially reducing the negative surface charge (34, 45).
Increased heterologous protein expression in several dlt mutants has been documented. The B. subtilis dlt mutant had increased expression of heterologous amylase Q from B. amyloliquefaciens and the protective antigen from B. anthracis (14, 37). Similar results for expression of other heterologous proteins were obtained in dlt mutants of Bacillus licheniformis and L. lactis (7, 29). Thus, our results obtained with the dlt mutant are consistent with the literature. However, the results obtained with OB219(pPM14) are novel. To the best of our knowledge, this is the first report that a mutant lacking two surface proteins can have an effect on heterologous protein expression. The results suggest that the increased negative charge in the cell wall is the reason for the increase in SpaP-S1 production and that the presumably increased availability of LPXTG anchoring sites vacated by SspA and SspB played no role. Our results further showed that the effect induced by dlt inactivation and sspA and sspB inactivation is not limited to SpaP-S1. The cell walls isolated from the two mutants contained more proteins than the parent PM14 cell wall, suggesting that the production of other cell wall-associated proteins is also increased.
The argument that the increase in cell wall negative charge is responsible for the increase in SpaP-S1 production is reinforced by the results of the cation experiments. Addition of Ca2+, Mg2+, and K+ resulted in a decrease in SpaP-S1 production. Although we are not able to provide results to show that the cell wall microenvironment became more positive from cation addition, it is reasonable to assume that this is the case. The cell wall is known to function as a cation reservoir (2, 12). With more cations present in the medium, more cations should be in the cell wall. The results of the cation addition experiment are in agreement with reports for Bacillus brevis showing that the yield of α-amylase was reduced in the presence of Ca2+, Mg2+, and K+ ions (1). Our results further showed that Ca2+ and Mg2+ ions have a greater effect on SpaP-S1 expression than K+, possibly due to the greater positive charge of the divalent cations than the monovalent ions.
The finding that SpaP-S1 production is increased by bicarbonate is interesting. The simple explanation for the effect of bicarbonate is that bicarbonate makes the cell wall more electronegative, thus leading to an increase in SpaP-S1 production. However, the effect of bicarbonate may be more complex. Bicarbonate can reduce the cell wall thickness via the modulation of cell wall biosynthesis gene expression (9). Thus, the observed increased SpaP-S1 production may be a combined effect of increased cell wall electronegativity and cell wall thickness.
The reason behind the increase in SpaP-S1 expression in the dltA mutant and OB219(pPM14) appears to be an increase in PrsA production. This statement is supported by the results that OB219(pPM14) and the dltA mutant produced more PrsA than the parent strain and that SpaP-S1 production was increased in the PrsA-overproducing strain and decreased in the PrsA-negative mutant. The role of PrsA in SpaP-S1 production is in keeping with observations made for B. subtilis and L. lactis that overproduction of PrsA led to larger amounts of foreign protein expression (17, 28, 44). Our results further showed that PrsA expression is upregulated in the liaR mutant, and coincident with the increase in PrsA expression, SpaP-S1 production was also increased. This result further supports the idea that PrsA plays a role in SpaP-S1 production. The result also indicates that LiaSR regulates PrsA expression, which is in agreement with findings in S. aureus that prsA expression is upregulated by LiaSR when cells are stressed with cationic antimicrobial peptides (32). We have attempted to obtain a viable liaSR clone in E. coli but failed; hence, complementation experiments cannot be performed. In S. mutans, LiaSR is needed for cell survival of acidic pH (25), indicating that LiaSR can sense and respond to cell envelope ion concentrations. Thus, it is reasonable to suggest that LiaSR senses and responds to cell wall charges and regulates the expression of PrsA in S. gordonii. PrsA modulates the level of SpaP-S1 production presumably by allowing more efficient protein folding.
In conclusion, heterologous SpaP-S1 protein production in the dltA mutant and OB219(pPM14) was higher than that in the parent strain. The increase can be correlated with an increase in the overall cell envelope negative charge and the PrsA level.
Acknowledgments
We thank Yi-Jing Li and BinYou Zheng for technical assistance, Alex Speers for assistance with zeta potential measurements, and Howard Jenkinson for providing the bacterial strain OB219.
E. Davis was a recipient of a Nova Scotia Health Research Foundation graduate studentship. D. Kennedy was a recipient of an IWK summer studentship. This study was supported by the Natural Sciences and Engineering Council of Canada and the Canadian Institutes of Health Research.
Footnotes
Published ahead of print on 30 December 2010.
REFERENCES
- 1.Adachi, T., H. Yamagata, N. Tsukagoshi, and S. Udaka. 1991. Repression of the cell wall protein gene operon in Bacillus brevis 47 by magnesium and calcium. J. Bacteriol. 173:4243-4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beveridge, T. J., and R. G. Murray. 1976. Uptake and retention of metals by cell walls of Bacillus subtilis. J. Bacteriol. 127:1502-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruss, J. B., and G. R. Siber. 1999. Protective effects of pertussis immunoglobulin (PIGIV) in the aerosol challenge model. Clin. Diagn. Lab. Immunol. 6:464-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Budak, Y., H. Demirci, M. Akdogan, and D. Yavuz. 2004. Erytrocyte membrane anionic charge in type 2 diabetic patients with retinopathy. BMC Ophthalmol. 4:14-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan, K. G., et al. 2007. The roles of d-alanylation of Streptococcus gordonii lipoteichoic acid in innate and adaptive immunity. Infect. Immun. 75:3033-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Claverys, J. P., A. Dintilhac, E. V. Pestova, B. Martin, and D. A. Morrison. 1995. Construction and evaluation of new drug-resistance cassettes for gene disruption mutagenesis in Streptococcus pneumoniae, using an ami test platform. Gene 164:123-128. [DOI] [PubMed] [Google Scholar]
- 7.Craynest, M., S. Jorgensen, M. Sarvas, and V. P. Kontinen. 2003. Enhanced secretion of heterologous cyclodextrin glycosyltransferase by a mutant of Bacillus licheniformis defective in the D-alanylation of teichoic acids. Lett. Appl. Microbiol. 37:75-80. [DOI] [PubMed] [Google Scholar]
- 8.Darmon, E., et al. 2002. A novel class of heat and secretion stress-responsive genes is controlled by the autoregulated CssRS two-component system of Bacillus subtilis. J. Bacteriol. 184:5661-5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorschner, R. A., et al. 2006. The mammalian ionic environment dictates microbial susceptibility to antimicrobial defense peptides. FASEB J. 20:35-42. [DOI] [PubMed] [Google Scholar]
- 10.Duan, Y., E. Fisher, D. Malamud, E. Golub, and D. R. Demuth. 1994. Calcium-binding properties of SSP-5, the Streptococcus gordonii M5 receptor for salivary agglutinin. Infect. Immun. 62:5220-5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fahnestock, S. R., and K. E. Fisher. 1986. Expression of the staphylococcal protein A gene in Bacillus subtilis by gene fusions utilizing the promoter from a Bacillus amyloliquefaciens alpha-amylase gene. J. Bacteriol. 165:796-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galdiero, F. 1966. The kinetics of potassium exchange in cells of Staphylococcus aureus. Biochim. Biophys. Acta 126:54-60. [DOI] [PubMed] [Google Scholar]
- 13.Hyyrylainen, H. K., M. Sarvas, and V. Kontinen. 2005. Transcriptome analysis of the secretion stress response of Bacillus subtilis. Appl. Microbiol. Biotechnol. 67:389-396. [DOI] [PubMed] [Google Scholar]
- 14.Hyyrylainen, H. L., et al. 2000. d-Alanine substitution of teichoic acids as a modulator of protein folding and stability at the cytoplasmic membrane/cell wall interface of Bacillus subtilis. J. Biol. Chem. 275:26696-26703. [DOI] [PubMed] [Google Scholar]
- 15.Knight, J. B., S. A. Halperin, K. A. West, and S. F. Lee. 2008. Expression of a functional single chain variable fragment antibody against the complement receptor 1 in Streptococcus gordonii. Clin. Vaccine Immunol. 15:925-931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knight, J. B., et al. 2006. Immunogenicity and protective efficacy of a recombinant filamentous hemagglutinin from Bordetella pertussis. Clin. Exp. Immunol. 144:5543-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kontinen, V. P., and M. Sarvas. 1993. The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol. Microbiol. 8:727-737. [DOI] [PubMed] [Google Scholar]
- 18.Koprivnjak, T., A. Peschel, M. H. Gelb, N. S. Liang, and J. P. Weiss. 2002. Role of charge properties of bacterial envelope in bactericidal action of human group IIA phospholipase A2 against Staphylococcus aureus. J. Biol. Chem. 277:47636-47644. [DOI] [PubMed] [Google Scholar]
- 19.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 20.LeBlanc, D. J., L. N. Lee, and A. Abu-Al-Jaibat. 1992. Molecular, genetic, and functional analysis of the basic replicon of pVA380-1, a plasmid of oral streptococcal origin. Plasmid 28:130-145. [DOI] [PubMed] [Google Scholar]
- 21.Lee, S. F. 2003. Oral colonization and immune responses to Streptococcus gordonii: potential use as a vector to induce antibodies against respiratory pathogens. Curr. Opin. Infect. Dis. 16:231-235. [DOI] [PubMed] [Google Scholar]
- 22.Lee, S. F., S. A. Halperin, H. Wang, and A. MacArthur. 2002. Oral colonization and immune responses to Streptococcus gordonii expressing a pertussis toxin S1 fragment in mice. FEMS Microbiol. Lett. 208:175-178. [DOI] [PubMed] [Google Scholar]
- 23.Lee, S. F., R. J. March, S. A. Halperin, G. Faulkner, and L. Gao. 1999. Surface expression of a protective recombinant pertussis toxin S1 subunit fragment in Streptococcus gordonii. Infect. Immun. 67:1511-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee, S. F., et al. 1989. Construction and characterization of isogenic mutants of Streptococcus mutans deficient in the major surface protein antigen P1. Infect. Immun. 57:3306-3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li, Y. H., et al. 2002. Novel two-component regulatory system involved in biofilm formation and acid resistance in Streptococcus mutans. J. Bacteriol. 184:6333-6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love, R. M., M. D. McMillan, and H. F. Jenkinson. 1997. Invasion of dentinal tubules by oral streptococci is associated with collagen recognition mediated by the antigen I/II family of polypeptides. Infect. Immun. 65:5157-5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mallaley, P. P., S. A. Halperin, A. Morris, A. MacMillan, and S. F. Lee. 2006. Expression of a pertussis toxin S1 fragment by inducible promoters in oral Streptococcus and the induction of immune responses during oral colonization in mice. Can. J. Microbiol. 52:436-444. [DOI] [PubMed] [Google Scholar]
- 28.Marugg, J. D., R. van Kranenburg, P. Laverman, G. A. Rutten, and W. M. de Vos. 1996. Identical transcriptional control of the divergently transcribed prtP and prtM genes that are required for proteinase production in Lactococcus lactis SK11. J. Bacteriol. 178:1525-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nouaille, S., et al. 2004. Influence of lipoteichoic acid d-alanylation on protein secretion in Lactococcus lactis as revealed by random mutagenesis. Appl. Environ. Microbiol. 70:1600-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palva, I. 1982. Molecular cloning of alpha-amylase gene from Bacillus amyloliquefaciens and its expression in B. subtilis. Gene 19:81-87. [DOI] [PubMed] [Google Scholar]
- 31.Peterson, S., R. T. Cline, H. Tettelin, V. Sharov, and D. A. Morrison. 2000. Gene expression analysis of the Streptococcus pneumoniae competence regulons by use of DNA microarrays. J. Bacteriol. 182:6192-6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pietiainen, M., et al. 2009. Transcriptome analysis of the responses of Staphylococcus aureus to antimicrobial peptides and characterization of the roles of vraDE and vraSR in antimicrobial resistance. BMC Genomics 10:429-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quax, W. J. 1997. Merits of secretion of heterologous proteins from industrial microorganisms. Folia Microbiol. 42:99-103. [DOI] [PubMed] [Google Scholar]
- 34.Rose, R. K. 2000. The role of calcium in oral streptococcal aggregation and the implications for biofilm formation and retention. Biochim. Biophys. Acta 1475:76-82. [DOI] [PubMed] [Google Scholar]
- 35.Speers, A., T. D. Durance, P. Odense, S. Owen, and M. A. Tung. 1993. Physical properties of commercial brewing yeast suspensions. J. Inst. Brew. 99:159-164. [Google Scholar]
- 36.Stipp, R. N., R. B. Goncalves, J. F. Hofling, D. J. Smith, and R. O. Mattos-Graner. 2008. Transcriptional analysis of gtfB, gtfC, and gbpB and their putative response regulators in several isolates of Streptococcus mutans. Oral Microbiol. Immunol. 23:466-471. [DOI] [PubMed] [Google Scholar]
- 37.Thwaite, J. E., et al. 2002. Optimization of the cell wall microenvironment allows increased production of recombinant Bacillus anthracis protective antigen from B. subtilis. Appl. Environ. Microbiol. 68:227-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tjalsma, H., et al. 2004. Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol. Mol. Biol. Rev. 68:207-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Towbin, H., T. Staehelin, and J. Gordon. 1992. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Biotechnology 24:145-149. [PubMed] [Google Scholar]
- 40.Tremblay, Y. N., H. Lo, Y. H. Li, S. A. Halperin, and S. F. Lee. 2009. Expression of the Streptococcus mutans essential two-component regulatory system VicRK is pH and growth-phase dependent and controlled by the LiaFSR three-component regulatory system. Microbiology 155:2856-2865. [DOI] [PubMed] [Google Scholar]
- 41.Vadyvaloo, V., et al. 2004. Cell-surface alterations in class IIa bacteriocin resistant Listeria monocytogenes strains. Microbiology 150:3025-3033. [DOI] [PubMed] [Google Scholar]
- 42.Vitikainen, M., et al. 2001. Quantitation of the capacity of the secretion apparatus and requirement for PrsA in growth and secretion of alpha-amylase in Bacillus subtilis. J. Bacteriol. 183:1881-1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wahlstrom, E., M. Vitikainen, V. P. Kontinen, and M. Sarvas. 2003. The extracytoplasmic folding factor PrsA is required for protein secretion only in the presence of the cell wall in Bacillus subtilis. Microbiology 149:569-577. [DOI] [PubMed] [Google Scholar]
- 44.Williams, R. C., et al. 2003. Production of Bacillus anthracis protective antigen is dependent on the extracellular chaperone, PrsA. J. Biol. Chem. 278:18056-18062. [DOI] [PubMed] [Google Scholar]
- 45.Yamashita, Y., A. Kunimori, and T. Takehara. 1991. Effect of calcium ions on cell surface electrostatics of Bacteroides gingivalis and other oral bacteria. Zentralbl. Bakteriol. 275:46-53. [DOI] [PubMed] [Google Scholar]