

Abstract
Wine Saccharomyces cerevisiae strains producing a new killer toxin (Klus) were isolated. They killed all the previously known S. cerevisiae killer strains, in addition to other yeast species, including Kluyveromyces lactis and Candida albicans. The Klus phenotype is conferred by a medium-size double-stranded RNA (dsRNA) virus, Saccharomyces cerevisiae virus Mlus (ScV-Mlus), whose genome size ranged from 2.1 to 2.3 kb. ScV-Mlus depends on ScV-L-A for stable maintenance and replication. We cloned and sequenced Mlus. Its genome structure is similar to that of M1, M2, or M28 dsRNA, with a 5′-terminal coding region followed by two internal A-rich sequences and a 3′-terminal region without coding capacity. Mlus positive strands carry cis-acting signals at their 5′ and 3′ termini for transcription and replication similar to those of killer viruses. The open reading frame (ORF) at the 5′ portion codes for a putative preprotoxin with an N-terminal secretion signal, potential Kex2p/Kexlp processing sites, and N-glycosylation sites. No sequence homology was found either between the Mlus dsRNA and M1, M2, or M28 dsRNA or between Klus and the K1, K2, or K28 toxin. The Klus amino acid sequence, however, showed a significant degree of conservation with that of the product of the host chromosomally encoded ORF YFR020W of unknown function, thus suggesting an evolutionary relationship.
Saccharomyces cerevisiae killer strains produce and secrete protein toxins that are lethal to sensitive strains of the same or related yeast species. These toxins have been grouped into three types, K1, K2, or K28, based on their killing profiles and lack of cross-immunity. Members of each group can kill nonkiller yeasts as well as killer yeasts belonging to the other types. They are immune, however, to their own toxin or to toxins produced by strains of the same killer type (for reviews, see references 21, 32, 33, and 47).
K1, K2, and K28 killer toxins are genetically encoded by medium-size double-stranded RNA (dsRNA) viruses grouped into three types, M1, M2, and M28, of 1.6, 1.5, and 1.8 kb, respectively. Only one strand (the positive strand) has coding capacity. In each case, the 5′-end region contains an open reading frame (ORF) that codes for the toxin precursor, or preprotoxin (pptox), which also provides immunity. The three toxin-coding M dsRNAs show no sequence homology to each other (35). M viruses depend on a second large (4.6-kb) dsRNA helper virus, L-A, for maintenance and replication. L-A provides the capsids in which both L-A and M dsRNAs are separately encapsidated (reviewed by Schmitt and Breinig [33]). L-BC virus is an L-A-related virus, with a similar 4.6-kb genome size, which coexists with L-A in most killer and nonkiller S. cerevisiae strains (1, 37). L-BC shows no sequence homology with L-A, and it has no known helper activity. L-A and L-BC, however, share the same genomic organization. They code for two proteins, the major coat protein Gag and a minor Gag-Pol fusion protein translated by a −1 ribosomal frameshifting mechanism (7, 10, 17, 26). These viruses, called Saccharomyces cerevisiae viruses (ScVs), belong to the Totiviridae family and are cytoplasmically inherited, spreading horizontally by cell-cell mating or by heterokaryon formation (47). In addition to the M dsRNA-encoded killer toxins, other S. cerevisiae killer toxins, named KHR and KHS, showing weak killer activity, are encoded on chromosomal DNA (13, 14).
The positive strands of both L-A and M viruses contain cis signals in their 3′-terminal regions essential for packaging and replication (46). The signal for transcription initiation has been proposed to be present in the first 25 nucleotides (nt) of L-A, probably in the 5′-terminal sequence itself (5′-GAAAAA). This sequence, designated the 5′-terminal recognition element (11), is also present in M1, M2, and M28 5′ ends.
The M1, M2, or M28 positive-strand-encoded ORF is translated into a pptox that subsequently enters the secretory pathway for further processing, leading to the secretion of the mature toxin. The unprocessed toxin precursor consists of an N-terminal signal sequence necessary for its import into the lumen of the endoplasmic reticulum (ER), followed by the α and β subunits of the mature toxin separated from each other, in the case of K1 and K28, by a potentially N-glycosylated γ sequence. The signal peptide is removed in the ER, and N-glycosylation and disulfide bond formation occur. Then, in a late Golgi compartment, protease processing takes place involving Kex1 and Kex2 proteases. Finally, the toxin is secreted as an active α/β heterodimer, with both subunits being covalently linked by one or more disulfide bonds (33, 46).
S. cerevisiae viral toxins kill sensitive yeast cells in a receptor-mediated process by interacting with receptors located in the yeast cell wall and cytoplasmic membrane. After reaching the plasma membrane, ionophoric virus toxins, such as K1 and most likely K2, disrupt cytoplasmic membrane function by forming cation-selective ion channels. K28 toxin, after interacting with a sensitive cell, is taken up by endocytosis and travels the secretion pathway in reverse until it reaches the cytosol. This toxin blocks DNA synthesis and arrests cells in the early S phase of the cell cycle (33). Among other dsRNA-encoded killer toxins, zygocin from the osmotolerant yeast Zygosaccharomyces bailii has been the most studied. It is a monomeric protein with a broad antifungal killing spectrum that kills sensitive cells by disrupting the cytoplasmic membrane (34, 44, 45).
The aim of the present work is the characterization of a new killer toxin encoded by an M dsRNA virus, ScV-Mlus, found in wine yeasts from the Ribera del Guadiana region in Spain. Toward this end, we have (i) examined the antifungal spectrum of the Klus toxin, (ii) cloned and sequenced Mlus, (iii) analyzed Mlus genome organization in comparison with other M dsRNAs, (iv) performed functional analysis of the Mlus preprotoxin ORF expressed from a vector and demonstrated killer activity, and (v) shown homology between the Klus toxin amino acid sequence and that of the host chromosomally encoded ORF YFR020W protein of unknown function. The possible evolutionary relationship between the two ORFs is discussed.
MATERIALS AND METHODS
Yeast strains and media.
All Klus phenotype wine yeasts isolated from spontaneous fermentations of grapes from vineyards of the Ribera del Guadiana region in Spain were Saccharomyces cerevisiae prototrophic and homothallic strains. The yeast strains used in this work are summarized in Table 1. To study Mlus dependency on L-A during the meiotic segregation of L-A dsRNA in the MAK10/mak10 hybrid MM375 or MM392, parental strain F200 was first transformed with plasmid pI2L2 or with the empty vector pI2, respectively, and then mated with the Mlus-carrying strain MMR359. Plasmid pI2L2 contains the entire L-A cDNA sequence from pTIL05 cloned downstream of the constitutive PGK1 promoter in the 2μm derivative multicopy plasmid pI2 (17, 48).
TABLE 1.
S. cerevisiae and other yeast strains used in this study
| Strain | Genotype/relevant phenotype | Source and/or origin |
|---|---|---|
| EX33 | MATa/α HO/HO [K10 K20 K280 Klus0] | J. A. Regodóna (from wine) |
| EX73 | MATa/α HO/HO L-A M2 [K2+] | J. A. Regodóna (from wine) |
| F166 | MATα leu1 kar1 L-A-HNB M1 [K1+] | J. C. Ribasb (from R. B. Wickner) |
| F182 | MATα his2 ade1 leu2-2 ura3-52 ski2-2 L-A M28 [K28+] | J. C. Ribasb (from M. Schmitt) |
| EX436 | MATa/α HO/HO L-A Mlus [Klus+] | This study (from wine) |
| EX122 | MATa/α HO/HO L-A Mlus [Klus+] | This study (from wine) |
| EX198 | MATa/α HO/HO L-A Mlus [Klus+] | This study (from wine) |
| EX229 | MATa/α HO/HO L-A Mlus [Klus+] | This study (from wine) |
| E304 | MATaura3 leu2 can1 cyh2 M1 [K1+] | M. Ramírezc |
| F200 | MATahis3 ade1 trp1 ρ0mak10 L-BC [K0] | J. C. Ribasb |
| MMR209 | MATa/α URA3/ura3 LEU2/leu2 cyh2 L-A Mlus [Klus+] | Cross EX229 × E304 |
| MMR209-10B | MATα ura3 cyh2 L-A Mlus [Klus+] | Spore clone from MMR209 |
| MMR247 | MATa/α URA3/ura3 LEU2/leu2 HIS3/his3 ADE1/ade1 TRP1/trp1 cyh2 MAK10/mak10 L-A L-BC Mlus [Klus+] | Cross F200 × MMR209-10B |
| MMR340 | MATahis3 ade1 trp1 ρ0mak10 L-BC [K0] pI2 [L-A-o TRP+] | F200 transformed with pI2 |
| MMR383 | MATahis3 ade1 trp1 ρ0mak10 L-BC [K0] pI2L2 [L-A- HNB PGK TRP+] | F200 transformed with pI2L2 |
| MMR359 | MATα ura3 his3 trp1 Mlus [Klus+] | Spore clone from MMR247 |
| MMR375 | MATa/α URA3/ura3 his3/his3 ADE1/ade1 trp1/trp1MAK10/mak10 Mlus [Klus+] pI2 [L-A-o TRP+] | Cross MMR340 × MMR359 |
| MMR392 | MATa/α URA3/ura3 his3/his3 ADE1/ade1 trp1/trp1 MAK10/mak10 Mlus [Klus+] pI2L2 [L-A PGK TRP+] | Cross MMR383 × MMR359 |
| 5x47 | MATa/α his1/+ trp1/+ ura3/+ | R. Esteband (from R. B. Wickner) |
| RE108 | MATα lys2 mkt1 ski7-1 can1 cyh2 L-A M2 [K2+] | R. Esteband |
| 1101 | MATα his 4 kar1-1 L-A L-BC M1 [K1+] | R. Esteband (from R. B. Wickner) |
| 2928 | MATaura3 trp1 his3 L-A-o, L-BC | R. Esteband (from R. B. Wickner) |
| 2927 | MATahis3 ski2-2 L-o | R. Esteband (from R. B. Wickner) |
| Candida albicans 10231 | Pathogen; 87% of membrane hidrofobicity at 37°C and 4% at 22°C | C. Lópeze |
| C. kefir | Pathogen | C. Lópeze |
| C. glabrata | Pathogen | C. Lópeze |
| C. dubliniensis | Pathogen | C. Lópeze |
| C. krusei | Pathogen | C. Lópeze |
| C. parapsilosis | Pathogen | C. Lópeze |
| C. tropicalis | Pathogen | C. Lópeze |
| C. albicans wt 5314C | Pathogen | J. Correaf |
| C. albicans CAF | (wt URA3+/−); pathogen | J. Correaf |
| Yarrowia lipolytica wt a | L. M. Hernándezg | |
| Y. lipolytica mnn9 a | Mutation mnn9 truncates carbohydrate outer chain of the cell wall mannoproteins of S. cerevisiae | L. M. Hernándezg |
| Y. lipolytica SA1-5 wt | A. Domínguezh | |
| Kluyveromyces lactis | Killer phenotype encoded by a gene on a dsDNA plasmid (pGKL1) | A. Domínguezh |
| Hansenula mrakii 22 wt | Killer, toxin HM1 encoded in a chromosomal gene | J. C. Ribasb |
| Schizosaccharomyces pombe 33 wt 972h− | J. C. Ribasb | |
| Hanseniaspora 5 | Killer against S. cerevisiae | M. Ramírezc |
J. A. Regodón, Departamento de Química Analítica, Universidad de Extremadura, Badajoz, Spain. Isolated from D.O. Ribera del Guadiana, Spain.
J. C. Ribas, Departamento de Microbiología y Genética, Universidad de Salamanca, Spain.
M. Ramírez, Departamento de Ciencias Biomédicas, Área de Microbiología, Universidad de Extremadura, Badajoz, Spain.
R. Esteban, Instituto de Microbiología-Bioquímica CSIC/Universidad de Salamanca, Salamanca, Spain.
C. López, Grupo Microbiología de Medicina, Departamento de Ciencias Biomédicas, Área de Microbiología, Universidad de Extremadura, Badajoz, Spain.
J. Correa, Departamento de Ciencias Biomédicas, Área de Microbiología, Universidad de Extremadura, Badajoz, Spain.
L. M. Hernández, Departamento de Ciencias Biomédicas, Área de Microbiología, Universidad de Extremadura, Badajoz, Spain.
A. Domínguez, Departamento de Microbiología y Genética, Universidad de Salamanca, Salamanca, Spain.
Standard culture media were used for yeast growth (16). Yeast extract-peptone-dextrose (YEPD) agar contained 1% yeast extract, 2% peptone, 2% glucose, and 2% agar. YEPD plus cyh is YEPD agar supplemented with cycloheximide (cyh) to a final concentration of 2 μg/ml. Synthetic minimal medium (SD) contained a 0.67% yeast nitrogen base (without amino acids, with ammonium sulfate; Difco, Detroit, MI), 2% glucose, and 2% agar. Uracil (20 mg/liter), l-leucine (30 mg/liter), l-histidine-HCl (20 mg/liter), and l-methionine (20 mg/liter) were added when necessary. For selection of transformants, tryptophan- or uracil-omitted synthetic medium (H-trp or H-ura) was used.
Standard yeast genetic procedures were used for sporulation of cultures and dissection of asci (18). Cells grown on YEPD plates for 2 days at 30°C were transferred to sporulation plates (1% potassium acetate, 0.1% yeast extract, 0.05% glucose, 2% agar) and incubated for 7 to 20 days at 25°C until more than 80% of the cells had sporulated. Twenty-four asci from each yeast strain were dissected on YEPD plates and incubated for 5 days at 30°C to obtain the spore clones.
Killer activity was tested on low-pH (pH 4 or 4.7) methylene blue (4 MB or 4.7 MB) plates (18) seeded with 100 μl of a 48-h culture of the sensitive strain (29). Depending on the experiments, strains being tested for killer activity were loaded as 4-μl aliquots of stationary-phase cultures, patched from solid cultures, or replica plated onto the seeded low-pH MB plates. Then the plates were incubated for 4 days at 20°C, 28°C, or 30°C.
Total nucleic acid preparation.
For routine dsRNA minipreps, the cells were suspended in 10 mM Tris-HCl (pH 7.5) buffer containing 0.1 M NaCl, 10 mM EDTA, and 0.2% SDS; thereafter, an equal volume of phenol (pH 8.0) was added. This mixture was incubated at room temperature for 30 min, with shaking. After centrifugation, the nucleic acids recovered in the aqueous phase were precipitated with isopropanol, washed with 70% ethanol, dried, and dissolved in Tris-EDTA (TE) buffer, pH 8.0 (22). Digestion of DNA was done with DNase I (RNase-free) from Fermentas Life Sciences according to the manufacturer's specifications. Digestion of RNA was performed with RNase A (Sigma-Aldrich) following the manufacturer's indications. For selective degradation of single-stranded RNA, samples were incubated with RNase A (10 μg/ml) at 37°C for 30 min in the presence of 0.5 M NaCl. Samples were then processed through phenol-chloroform-isoamyl alcohol extraction to inactivate the enzyme before analysis through an agarose gel.
For Northern experiments, total nucleic acids were prepared by breaking the cells with glass beads followed by phenol extraction and ethanol precipitation.
Northern blot hybridization and probes.
For Northern blot analysis, total nucleic acids were separated on 1.2% agarose gels, blotted onto neutral nylon membranes (Hybond-N; Amersham Biosciences), and hybridized with 32P-labeled specific probes (11a). The L-A probe was made by T3 runoff transcription from plasmid pRE691 (which contains the L-A sequence from nucleotide 1783 to nucleotide 2647) and the L-BC probe by T7 transcription from plasmid pRE442, predigested with appropriate restriction enzymes, as described previously (8). Mlus probes were made by T7 transcription from plasmid pMlus-8 (which contains the Mlus sequence from nucleotide 28 to nucleotide 576) or pMlus-11 (which contains the Mlus sequence from nucleotide 119 to nucleotide 935, numbering from the 3′ terminus), predigested in both cases with EcoRV (see Results). In the case of the Southern experiment, the probes were obtained by random priming using the Ready-To-Go DNA labeling beads kit (GE Healthcare) and [α-32P]dCTP. Plasmid pRE1213 (see below) or plasmid pRE1122 (pBluescript KS+ vector containing the entire SKI4 gene) was used to obtain the DNA fragments for random priming.
cDNA synthesis, cloning, and sequencing of Mlus dsRNA.
Mlus cDNA synthesis was carried out using the Universal Riboclone cDNA synthesis system kit from Promega based on the method of Gubler and Hoffman (15). Mlus dsRNA was obtained from strain EX229 by CF-11 cellulose chromatography as described elsewhere (42) and further separated from other dsRNAs in the same strain by agarose gel electrophoresis and electroelution. For cDNA synthesis, approximately 0.5 μg of Mlus dsRNA dissolved in water was denatured in the presence of 1 μg random hexameric primers by boiling the sample for 3 min and was followed by quick cooling in an ice-water bath. The conditions for first- and second-strand synthesis were as recommended by Promega. Afterwards, the sample was treated with T4 DNA polymerase, extracted with phenol-chloroform-isoamyl alcohol, and ethanol precipitated. cDNA fragments corresponding to Mlus dsRNA without further purification were blunt end ligated into the unique SmaI site of a pBluescript KS+ vector (Stratagene, San Diego, CA) and introduced into competent Escherichia coli DH5α cells. Transformants containing inserts were sequenced.
To clone the 3′ ends of positive and negative strands of Mlus, we used 3′ rapid amplification of cDNA ends (3′ RACE). First the 3′ ends of the molecule were A-tailed using poly(A) polymerase as described elsewhere (30). Then the sample was mixed with 1 μl (100 pmol) of an oligo(dT) primer (5′-GACTCGAGTCGAGCGGCCGCTTTTTTTTTTTTTTTTT-3′) and H2O up to a 12.5-μl final volume, boiled for 2 min, and then chilled on ice. Then cDNA synthesis was carried out at 42°C for 1 h using Superscript reverse transcriptase (Invitrogen) in a 20-μl reaction volume as recommended by the supplier. After a reverse transcription (RT) reaction, the sample was heated to 70°C for 15 min to inactivate the reverse transcriptase and used directly for PCR (2 μl in a 50-μl total PCR volume). For PCR amplification, we used primer NR68 (5′-GACTCGAGTCGAGCGGCCGC-3′) and either primer NR73 (5′-AATTAGCGGCCGCCCAGTGATAAGACGGTAG-3′) or NR66 (5′-AATTAGCGGCCGCCAGCAAGGTGGCCTACAT-3′) that annealed near the 5′ or 3′ end of the molecule, respectively. The PCR products were digested with NotI, ligated into the unique NotI site of the pBluescript KS+ vector, and sequenced.
Expression of the preprotoxin ORF.
A DNA fragment of 726 nucleotides containing the putative preprotoxin-encoding ORF was obtained by RT-PCR of Mlus dsRNA using oligonucleotides NR74 (5′-AATTAGGATCCATGCATTTAAAAAGTTCT-3′) and NR75 (5′-AATTAGGTACCCTAACTAGAGCATGTGTA-3′) and subcloned into plasmid pEMBLyex4 downstream of the inducible GAL1-CYC hybrid promoter (6), to obtain plasmid pRE1213. The preprotoxin initiation and stop codons, where the oligonucleotides annealed, are in bold type and underlined. This plasmid was used to transform strain 2928 or 2927, and transformants were tested for toxin production using 4.7 MB plates containing galactose instead of glucose and seeded with the sensitive EX33 or 5x47 strain.
Miscellaneous.
DNA manipulations (enzyme digestions, cloning procedures, and Southern analysis) were done according to standard methods described in reference 31. Most of the enzymes were purchased from Promega. Synthetic oligonucleotides were purchased from Thermo. Yeast cells were transformed using lithium acetate to permeabilize the cells (12), and transformants were selected in H-trp or H-ura medium.
Nucleotide sequence accession number.
The Mlus cDNA sequence and the sequence of the encoded Klus preprotoxin, or toxin precursor, gene appear in NCBI/GenBank under accession no. GU723494.
RESULTS
Phenotypic characterization of Klus killer yeasts.
We analyzed the killer phenotype in 1,114 prototrophic and homothallic Saccharomyces cerevisiae wine yeasts isolated from 110 spontaneous fermentations of grapes collected in vineyards of the Ribera del Guadiana region in Spain. A total of 38% of these yeasts were killer, and the rest nonkiller (neutral or sensitive). Most of the killer yeasts were K2, while 7% showed a new killer phenotype that we called Klusitaneae (Klus). The Klus strains killed the three known K1, K2, and K28 strains, but they did not kill other Klus yeasts (Fig. 1A). They were also lethal to yeast species other than S. cerevisiae, such as Hanseniaspora sp., Kluyveromyces lactis, Candida albicans, Candida dubliniensis, Candida kefir, and Candida tropicalis (Fig. 1B). To confirm that the sometimes faint inhibition zone was indeed due to killer factor and not to some other metabolic activity, a control assay using the cured Klus strain was done, and no halos at all were observed (as shown in Fig. 2C). The Klus strains were, in turn, sensitive to killer toxins produced by Hansenula mrakii and Hanseniaspora sp. (not shown). In spite of the wide Klus spectrum of killing activity, we observed that, in general, the killer activity of Klus was weaker than those of the K1, K2, and K28 strains. Klus's strongest activity was at pH 4 to 4.7 and 28 to 30°C against C. tropicalis and S. cerevisiae K2 strains, although we observed slight variations depending on the Klus killer strain tested (not shown).
FIG. 1.

Killer phenotype of Klus yeasts. (A) Killer phenotype assay of Klus wine yeasts against S. cerevisiae strains. The assay was done on methylene blue agar plates seeded with standard killer-sensitive (EX33) yeast strain or killer K2 (EX73), K1 (F166), K28 (F182), and Klus (EX198) yeast strains. The assay conditions (pH and temperature) are given on the right. (B) Killer phenotype assay of Klus wine yeasts against non-Saccharomyces yeasts. The assay was done at pH 4.7 and 28°C. The digital images of the killer assay plates were taken with a color digital camera, Nikon Coolpix 900. The digital images were changed to black and white images, properly composed, and contrast manipulated to highlight the weak zones, by using Microsoft PowerPoint software. Some panels are composed of images coming from different plates. wt, wild type.
FIG. 2.

Genetic determinants of Klus toxin. (A) Presence of L and M molecules in Klus strains. Nucleic acids were obtained from killer K1 (F166), K2 (EX73), K28 (F182), and Klus strains of different isotypes (Mlus-1 to Mlus-4) and separated by agarose gel electrophoresis. The ethidium bromide staining of the gel is shown. (B) Nuclease treatments. Nucleic acids from strains K2 and Klus-3 untreated, after DNase I digestion, or after RNase A treatment under high- or low-salt conditions were separated in an agarose gel. (C) Cycloheximide curing of killer viruses. Agarose gel electrophoresis of nucleic acids from killer K1 (F166), K2 (EX73), K28 (F182), and Klus strain EX229 before and after virus curing with cycloheximide (top); killer phenotype assay of the same strains (bottom). EX229-1 and EX229-2 are two cured clones from EX229. The assay was done on methylene blue agar plates (pH 4.7, 28°C) seeded with killer K2 (EX73) strain.
Genotypic characterization of Klus killer yeasts.
All Klus killer strains carried two nucleic acid molecules that showed mobility in agarose gels similar to those of dsRNAs from other killer yeasts, including those of (i) a slower-moving band, similar in size to the dsRNA genome of the L-A virus (4.6 kb), and (ii) a faster-moving band, similar in size to the genomes of M viruses (1.5 to 2.3 kb) (Fig. 2A). On the basis of the mobility of the faster moving bands, we distinguished four isotypes, Mlus-1, Mlus-2, Mlus-3, and Mlus-4, whose sizes ranged from 2.1 to 2.3 kb (Fig. 2A). The size of each dsRNA isotype did not vary after 20 serial transfers on YEPD plates at 30°C, roughly 100 cell doublings (29). The dsRNA nature of the two nucleic acid molecules was confirmed by DNase I and RNase A treatments. On the one hand, mitochondrial DNA (mtDNA) disappeared after DNase I treatment, while L-A dsRNA and M2 dsRNA used as controls and the bands of similar sizes present in the Klus strain remained (Fig. 2B). The intensity of the L-A and M bands observed with the gel decreased after DNase I treatment, since part of the sample was probably lost during the sample processing to eliminate the enzyme before the agarose electrophoresis. However, the most intense mitochondrial DNA bands fully disappeared, while the less intense M bands remained. On the other hand, L-A dsRNA, M2 dsRNA, and the Klus strain bands disappeared after RNase A treatment, while mtDNA remained unaffected. The RNA molecules were fairly resistant to RNase A digestion in the presence of 0.5 M NaCl, as expected for dsRNA but not for single-stranded RNA (ssRNA) (2, 4, 43) (Fig. 2B). Mlus dsRNAs were lost during the growth of Klus strains in the presence of cycloheximide, and we observed a concomitant loss of their killer activity (Fig. 2C). This suggests that the Klus killer phenotype is encoded by the Mlus dsRNAs. A similar situation has been described for the killer toxins encoded by M1, M2, and M28 dsRNAs (32, 47).
A Northern hybridization with an Mlus-4 probe from plasmid pMlus-11, corresponding to the 3′ region of the molecule (see Materials and Methods), showed that the four Klus yeast isotypes carry the same ScV-Mlus virus, independent of their size, while there was no hybridization to M1, M2, or M28 dsRNA (Fig. 3A). We observed the same result when a 5′-end-specific probe was used (not shown). Hybridization with L-A- or L-BC-specific probes showed that all Klus yeasts carry L-A, while only half of the strains analyzed carry L-BC (Fig. 3B), thus indicating that Mlus does not depend on L-BC for maintenance in the yeast cell. The L-A probe gives a much weaker signal in strains carrying Mlus than in laboratory strain 1101. This is due to a significant difference between the nucleotide sequences of L-A in Klus strains and those of L-A of laboratory strains, which share 75% identity (N. Rodriguez-Cousiño and R. Esteban, unpublished results).
FIG. 3.
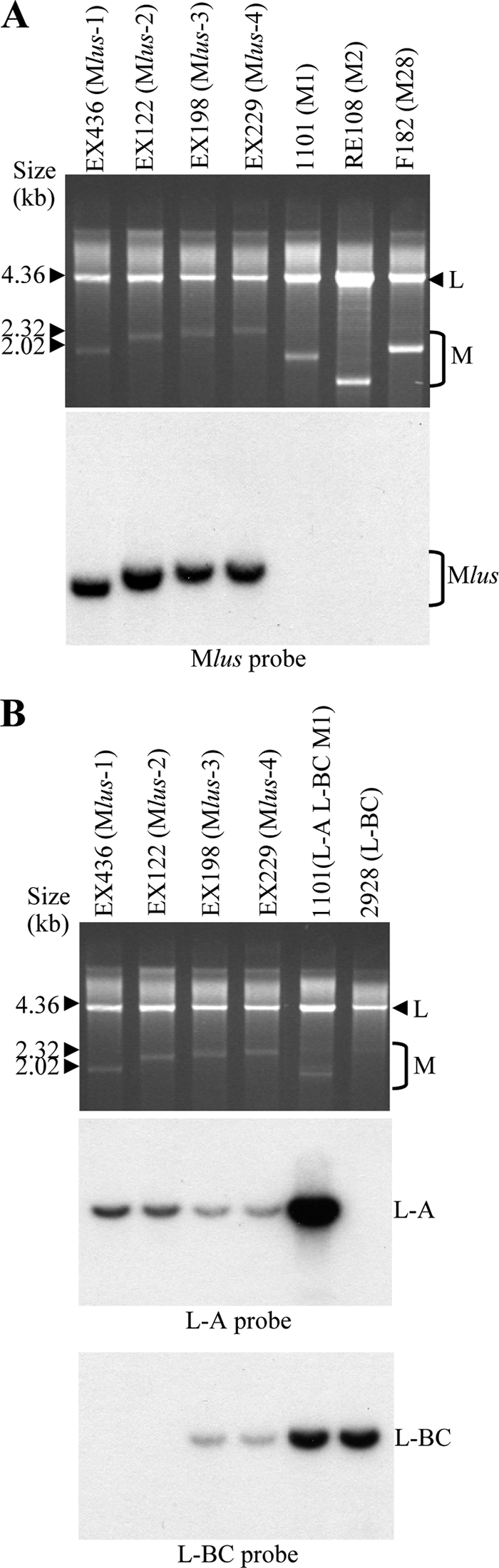
Characterization of the Klus yeast dsRNAs. (A) Mlus is different from M1, M2, or M28 dsRNAs. Nucleic acids from killer yeasts K1 (1101), K2 (RE108), and K28 (F182) or from the four Klus isotypes (EX436, EX122, EX198, and EX229) were separated on an agarose gel. (Top) Ethidium bromide staining of the gel; (bottom) Northern hybridization with a Mlus probe. (B) Presence of L-A and L-BC dsRNAs in Klus strains. Total nucleic acids from the same Klus strains shown in panel A and from standard laboratory strains 1101 (L-A, L-BC, and M1) and 2928 (L-BC) were analyzed by agarose gel electrophoresis (top; ethidium bromide staining of the gel). The RNA molecules were blotted onto a nylon membrane and hybridized with specific probes for L-A (middle) or for L-BC (bottom). Τhe size markers correspond to lambda DNA digested with HindIII.
Analysis of the Mlus dependency on L-A virus.
To test whether Mlus is a satellite RNA of L-A virus, we analyzed Mlus and L-A dsRNA segregation after sporulation and tetrad dissection of heterozygous MAK10/mak10 Klus hybrids. The MAK10 gene is required for ScV-L-A maintenance in the yeast cell (20). The mak10 mutation results in a decrease (or even the disappearance) of ScV-L-A (38), further resulting in the disappearance of its satellite M dsRNA and the killer phenotype. Half of the spore clones of each tetrad from the MMR247 hybrid (MAK10/mak10 L-A L-BC Mlus [Klus+]) lost the Klus killer phenotype and the Mlus dsRNA (Fig. 4A). Also the amount of L dsRNA decreased substantially because of the decrease or disappearance of L-A dsRNA. The low-intensity L dsRNA band that was detected most likely belongs to the L-BC dsRNA present in the MMR247 hybrid and its parent yeast strains (MMR20910B and F200). The defect of Mlus maintenance in mak10 mutants was circumvented by the expression of L-A viral particles (Gag and Gag-Pol) in trans from the expression vector pI2L2, as described previously (48), while this did not occur in the control cells carrying the empty vector (Fig. 4B and C). This again indicates that Mlus depends on L-A for its maintenance.
FIG. 4.

Mlus dsRNA depends on the coexistence of L-A dsRNA. (A) Killer phenotype and Mlus and L-A dsRNA segregation of heterozygous MAK10/mak10 Klus hybrids. Agarose gel electrophoresis of nucleic acids (top) and killer assay (bottom) of MMR247 hybrid (MAK10/mak10 L-A L-BC Mlus [Klus+]), its parent strains (F200 and MMR209-10B), and the spore clones from one tetrad. (B) Killer assay of MMR375 hybrid (MAK10/mak10 L-A L-BC Mlus [Klus+] pI2 [L-A0 TRP+]) and the spore clones from one tetrad. (C) Killer assay of MMR392 hybrid (MAK10/mak10 L-A L-BC Mlus [Klus+] pI2L2 [L-A PGK TRP+]) and the spore clones from one tetrad. The killer assays were done on the sensitive EX33 strain.
Analysis of Mlus dsRNA sequence.
We cloned Mlus dsRNA from strain EX229, Mlus-4. cDNA clones were obtained by random priming using purified Mlus as a template for first-strand cDNA synthesis. We cloned and sequenced 22 independent clones, which covered different parts of the Mlus genome. Most of the viral genome was obtained by alignment of the resulting sequences. In this way we determined a 5′ fragment of 906 nucleotides and a 3′ fragment of 1,054 nucleotides. The 5′-3′ orientation refers to the strand with coding capacity (see below). The sequence between the two fragments was determined by RT-PCR using oligonucleotides that annealed at the two sides of the gap. The sequences corresponding to the very 5′ and 3′ ends were obtained by 3′ RACE and were identical in five independent clones in each case. The full-length Mlus cDNA sequence thus determined was 2,033 nucleotides, which is smaller than the 2.3-kb size estimated for Mlus-4 dsRNA, as visualized on agarose gel. There is one open reading frame in the most 5′-end region (from nucleotides 112 to 840) encoding a protein of 242 amino acids, the putative preprotoxin. The central part of the molecule is characterized by the presence of two A-rich regions, which differ in the exact number of adenine residues. Clones varied from 17 to 48 adenine residues in the first stretch and from 24 to 43 adenine residues in the second stretch. The rest of the molecule lacks coding capacity. The 3′ region is presumed to provide structural elements required for RNA replication. In particular, we found the 3′ terminal recognition element (TRE) from nucleotide 1 to 18 (numbering from the 3′ end), with a free energy (ΔG value) of −4.2 kcal/mol. This stem-loop structure is quite similar to the one present in L-A, with the last 4 nucleotides identical to L-A's. However, we found no putative viral binding site (VBS) with a typical stem-loop structure interrupted by an unpaired protruding A residue, although VBSs with this characteristic have been reported for L-A, M1, and M28 dsRNAs. Overall, the genome organization of Mlus dsRNA resembles that of other toxins encoding satellite viruses, like ScV-M1, ScV-M2, ScV-M28, or Zygosaccharomyces bailii Mzb dsRNA ZbV-M (Fig. 5) (32, 44).
FIG. 5.

Genomic organization and coding capacity of the positive strands of ScV-Mlus, ZbV-M, ScV-M1, and ScV-M28. Preprotoxin ORFs are located at the 5′ ends (rectangles). The conserved 5′ GAAAA sequence is shown in bold type. The intramolecular poly(A)-rich stretches are indicated by “(A),” and the potential cis-acting 3′ sequences required for in vivo RNA packaging and replication (VBS, viral binding site; IRE, internal replication enhancer; TRE, 3′ terminal recognition element) are also shown. Numbers indicate the size (in nucleotides) of the virus transcripts and their coding and noncoding regions.
Preprotoxin ORF expression.
As mentioned before, the 5′ region of Mlus cDNA contains an ORF (from nucleotides 112 to 840) encoding a protein of 242 amino acids. This ORF product contains a stretch of hydrophobic amino acids at the amino terminus according to the Kyte-Doolittle hydrophilicity plot (19) (a possible N-terminal secretion signal), as well as potential Kex2p/Kexlp processing sites and potential sites for protein N-glycosylation, with all these features suggestive of it being a preprotoxin (see Fig. 7A). However, there is an out-of-frame upstream AUG codon, at nucleotides 21 to 23. A secondary structure prediction of the 5′-terminal 57 nucleotides of Mlus, including the first AUG, using the MFOLD program (50) revealed a highly structured region with a ΔG value of −13.1 kcal/mol (Fig. 6A). This first AUG might not be accessible to ribosomes, and thus, translation could initiate at the second internal AUG (nucleotides 112 to 114). To prove that the ORF encodes the 242-amino acid preprotoxin, we amplified a 0.72-kb fragment encompassing this region by RT-PCR, with oligonucleotides NR74 and NR75 and Mlus-4 dsRNA as a template, and cloned it under the control of the galactose-inducible GAL1 promoter to obtain plasmid pRE1213. Two yeast strains, strain 2928 and strain 2927, were transformed either with plasmid pRE1213 or with the vector alone. Transformants were streaked for single-colony isolation on H-ura plates and then replica plated on 4.7 MB plates seeded with sensitive strain 5x47 or EX33, with galactose as a carbon source to induce plasmid expression. A clear halo of growth inhibition in the lawn was observed around the clones carrying plasmid pRE1213 compared to those with the vector alone (Fig. 6B).
FIG. 6.
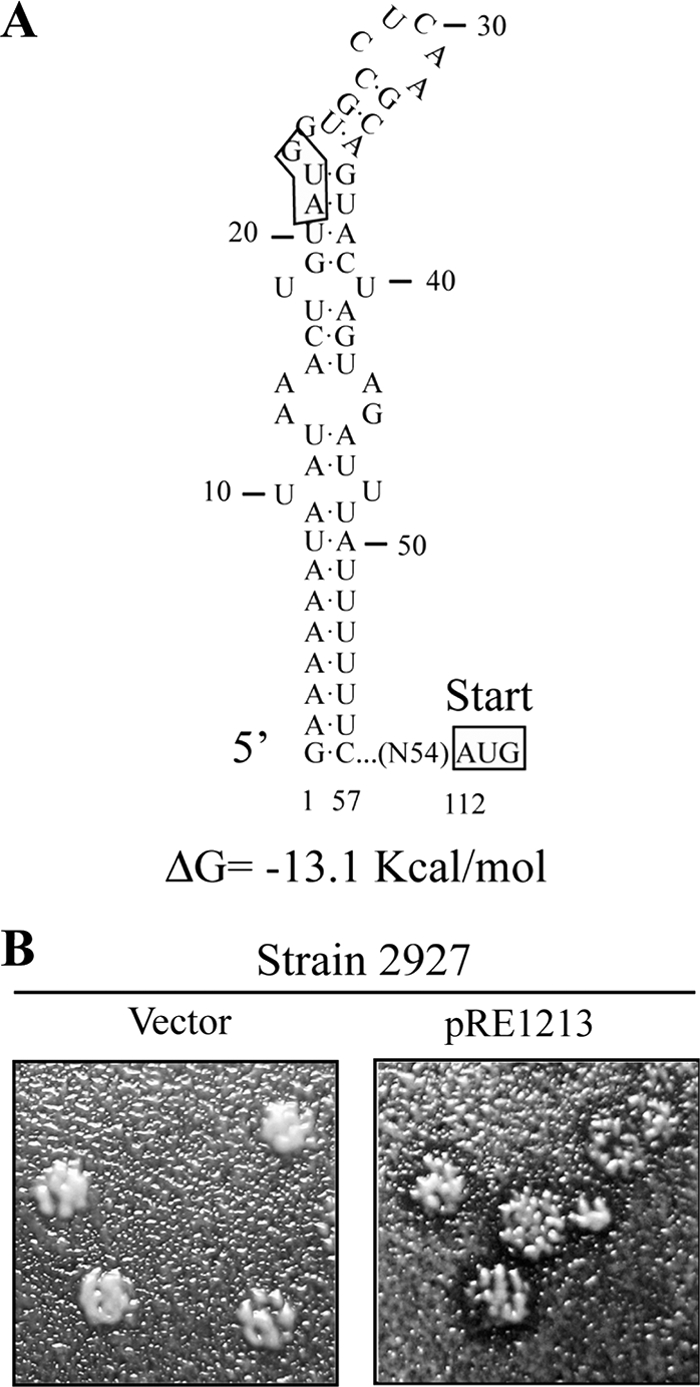
ORF expression from the Mlus 2nd AUG confers killer activity. (A) Secondary structure prediction of the 5′-terminal 57 nucleotides of Mlus positive strand using MFOLD (50). (B) Killer assay of strain 2927 transformed either with plasmid pRE1213 (containing the ORF, encoding the 242-amino acid protein, without the 5′-most-upstream AUG codon) or with the vector alone using 4.7 MB plates seeded with sensitive strain 5x47, with galactose as a carbon source to induce the expression of Klus toxin from the plasmid.
Mlus preprotoxin similarity to YRF020W ORF protein.
A BLAST search for similarities between the Mlus ORF-encoded protein and proteins in data banks showed a 32% identity and 51% similarity with an S. cerevisiae chromosomally encoded ORF protein of 232 amino acids (YFR020W) of unknown function. The identity is greater (44.4%) in the C-terminal half of the protein (Fig. 7A). This prompted us to test whether the ORF of the preprotoxin encoded by Mlus dsRNA had a counterpart in the genome of Klus strain EX229. To this end, we performed a Southern analysis. We could not find any hybridization signal with a probe of 0.43 kb corresponding to the internal DraI-EcoRI fragment of the Mlus ORF (nt 120 to 555), whereas a control probe of 1.0 kb from the single-copy gene CSL4/SKI4 detected in the same Southern blot a band of the expected size (not shown). This indicates that there is no copy of the Mlus ORF in the genome of strain EX229. Neither did the Mlus probe hybridize with the fragment of the yeast genome that contains the YRF020W gene (not shown). We confirmed that the YFR020W ORF was present in all our Klus strains by PCR amplification of a 0.7-kb fragment with oligonucleotides flanking the coding sequence. In addition, we confirmed that the YFR020W ORF was also present in standard laboratory strains 2928 and 1101 (Table 1). The lack of cross-hybridization between Mlus cDNA and the chromosomal yeast gene YFR020W can be explained by the low degree of conservation between the two sequences, with no more than 9 identical nucleotides in a single row (Fig. 7B). Additionally, we used quite stringent conditions in our Southern analysis.
FIG. 7.

Relationship between Mlus and YFR020W protein. (A) Comparison of the amino acid sequences of the Klus putative preprotoxin and the YFR020W ORF protein. The comparison was done using the ClustalW multiple sequence alignment program (41). Asterisks (*) indicate identical amino acids; double dots (:) and single dots (.) indicate conserved and semiconserved substitutions of amino acids, respectively. Identical amino acids are also marked in bold and shaded. The C-terminal 117-amino-acid region that displays a 44.4% identity between both proteins is boxed. The short arrow indicates the putative processing site, after the signal peptide. Long arrows indicate the location of putative Kex2 endopeptidase sites, after KR amino acids. Potential N-glycosylation sites are marked by overlining of the respective sequences. (B) Comparison of the nucleotide sequences of the Klus ORF and the YFR020W ORF. The comparison was done using ClustalW as described in the legend to panel A. The Klus ORF protein amino acid sequence is displayed above the nucleotide sequence. In bold and shaded are the amino acids identical to those of the YFR020W protein. Note that in the boxed area, the unchanged amino acids are coded for either by identical triplets or by triplets in which the first and second nucleotides are identical and the third (wobble position) is modified.
A direct comparison of the nucleotide sequences encoding the most conserved amino acid region between the two proteins showed that, of 52 identical amino acids, 29 (55.7%) are encoded by triplets in which the 1st and 2nd nucleotides were unchanged and the nucleotide at the wobble position (3rd nucleotide) in each triplet was modified (Fig. 7B). These conservative changes at the nucleotide level clearly indicate that the two sequences are evolutionarily related.
With respect to the similarity to other killer toxins encoded by dsRNA viruses, no sequence homology was found between the Klus and the K1, K2, or K28 toxins. In the case of the zygocin from Z. bailii, there are few amino acids conserved in the C-terminal regions of the two toxins (not shown).
DISCUSSION
Characterization of the new Klus killer yeasts.
The Klus killer strains are fairly well represented among the wine yeasts of spontaneous must fermentations. Because they can kill other S. cerevisiae strains as well as many other yeast species, they may have a beneficial effect during grape must fermentation, as has been shown for K2 killer strains (27). Moreover, given the broad antifungal spectrum of the Klus toxin (Fig. 1B), these yeasts may be used in food fermentation processes to avoid undesirable spoilage yeasts, in contrast to K1, K2, or K28 yeasts that kill sensitive cells only of the same or some congeneric species (21). The Klus optimum killer activity varied depending on the pH, temperature, and the pair of killer and sensitive strains tested. Similar to other killer toxins (28, 49), the Klus toxin is active under conditions typical of the environment in which these yeasts grow—acid (pH 3.5 to 5.5)-fermenting sugar-rich substrates at mild temperatures (18 to 28°C).
Genetic characterization of Mlus virus.
The Klus killer yeasts contained two dsRNA molecules, corresponding to a new Mlus dsRNA and the genome of its helper virus L-A. Mlus sizes were constant for isolates of a given strain but varied among different isolates (Fig. 2B). The fact that all Klus yeasts contained L-A but only half contained L-BC indicates that Mlus does not depend on L-BC, but rather on L-A. This putative dependency of Mlus on L-A was confirmed by a 2+:2− meiotic segregation of the Mlus dsRNA and the Klus killer phenotype from heterozygous MAK10/mak10 Klus hybrids. Also in this mak10 background, Mlus was maintained in trans when L-A coat proteins were expressed from plasmid pI2L2 as described previously (48), thus confirming that Mlus depends on L-A. The nucleotide sequence of L-A from laboratory strains and the L-A present in the Klus wine strains showed about 75% identity; however, at the amino acid level this identity rose to 91% (N. Rodriguez-Cousiño and R. Esteban, unpublished results). This may explain why the viral proteins expressed from pI2L2, although slightly different from those of the endogenous EX229 strain L-A, can support Mlus dsRNA replication.
Mlus dsRNA organization and the encoded Klus preprotoxin.
We determined the complete cDNA sequence of Mlus-4 dsRNA, including the 5′ and 3′ ends. The length of the sequence obtained (2,033 bp) is somewhat smaller than the 2.3 kb visualized for Mlus-4 dsRNA on agarose gel. In our experiments, we found that Mlus cDNA fragments with stretches of more than 50 adenine residues in a row were difficult to clone and sequence, probably because these repetitive sequences may facilitate sliding or jumping of either the reverse transcriptase or the Taq polymerase used in RT-PCR. Thus, we believe that variable numbers of adenine residues in the central A-rich regions account for the different sizes observed with the four Mlus isotypes. In comparison with the other M killer viruses so far described, in Mlus there are two A-rich regions of variable size, instead of a single continuous one (Fig. 5). In any case, this region is not important for killer toxin production, as the only ORF found in the 5′ half of the Mlus dsRNA is sufficient for killer activity when expressed from a vector (see below).
In Mlus the AUG initiation codon for the putative preprotoxin ORF is not near the 5′ end of the molecule (nt 112 to 114), unlike in the M1, M28, and MZb preprotoxin AUG initiation codons (Fig. 5) (23, 35, 36, 44). It resembles the M dsRNA encoding the KP4 toxin in Ustilago maydis (25). However, in KP4, translation occurs from the first AUG, whereas in Mlus there is an out-of-frame upstream AUG codon at nt 21 to 23. Since this first AUG in Mlus is buried in a highly structured region (Fig. 6A), it is conceivable that translation initiation occurs at the second internal AUG, by a so-far-unknown mechanism. We confirmed that, indeed, the Klus activity of Mlus does not require the most 5′ upstream sequence, by expressing the ORF encoding a 242-amino-acid protein (starting from the 2nd AUG) from a plasmid. Two different nonkiller strains transformed with this plasmid produced a killing zone when tested with a sensitive strain (Fig. 6B). A strong secondary structure has been proposed for the 5′ terminus of M1 (39, 40), although no effect on translation has been ascribed to it.
The organization of the Mlus ORF resembles that of other killer preprotoxins, such as those of M1, M2, and M28 viruses. It contains a stretch of hydrophobic amino acids at the amino terminus, potential Kex2p/Kexlp processing sites, and potential sites for protein N glycosylation (Fig. 7A). Thus, theoretically, proteolytic cleavage of the Klus preprotoxin by signal peptidase and Kex2 protease could produce three putative peptides. According to the disulfide bond formation prediction (5), there are two potential disulfide bonds inside the larger C-terminal domain (putative β subunit, 144 amino acids). Experiments with the purified Klus toxin will be needed, however, to determine whether the toxin is an α/β heterodimer like K1, K2, and K28 toxins or a monomer like zygocin.
We found a relevant homology of the Klus ORF protein amino acid sequence with that of an S. cerevisiae chromosomally encoded 232-amino-acid ORF protein of unknown function, YFR020W (Fig. 7). At present, we do not know whether the YFR020W gene product has killer toxin activity. If so, Klus killer assays should have been done on a strain deleted of this gene because of possible self-immunity. Preliminary data, however, suggest that there is no significant difference between a strain deleted of the YFR020W ORF from the EUROFAN deletion collection and the wild-type BY4741 strain, with regard to their sensitivity to the Klus toxin (not shown).
The YFR020W ORF is flanked by two long-terminal-repeat (LTR) elements. It has recently been reported that the KHR1 gene, which encodes a heat-resistant killer toxin, is present in the genome of wine strain EC1118 and is absent in other yeast strains, such as the reference genome strain S288c (3, 24). Also the KHR1 gene is present in a 6.1-kb fragment flanked by two LTR elements. Although Mlus is an RNA replicon and has no DNA counterpart, as shown by Southern blot analysis, the similarity between the Mlus-encoded preprotoxin and the YFR020W ORF product suggests an evolutionary relationship between them and gives a glimpse of the events that could produce a dsRNA-encoded toxin from a chromosomally encoded ORF protein. It is possible that during a transposition event the YFR020W ORF could acquire extra sequences needed for encapsidation and replication on L-A virions by recombination. Once independent of the DNA replication, the Mlus ORF could have evolved at a much higher rate than its genome counterpart, because its replication became dependent on the L-A-encoded RNA-dependent RNA polymerase with a much higher mutation rate. Alternatively, a cDNA copy of the preprotoxin coding sequence from a viral RNA fragment could have been integrated into the DNA chromosome. There is a recent report about the evolutionary capture of extracellular genetic elements, including dsRNA viruses by yeast nuclear chromosomes (9). In any case, the conservation of the nucleotide sequences between the Klus preprotoxin gene and the YFR020W ORF (Fig. 7B) is suggestive of divergent evolution.
Acknowledgments
This work was funded by grants 2PR01B002 and 2PR04B003 from the Extremadura Regional Government and in part by grant BFU2007-60057 from the Spanish Ministry of Education and Science. Matilde Maqueda was the recipient of a studentship from the Spanish Ministry of Education and Science.
Footnotes
Published ahead of print on 14 January 2011.
REFERENCES
- 1.Ball, S. G., C. Tirtiaux, and R. B. Wickner. 1984. Genetic control of L-A and L-(BC) dsRNA copy number in killer systems of Saccharomyces cerevisiae. Genetics 107:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berrye, A., and A. Bevane. 1972. A new species of double-stranded RNA from yeast. Nature 239:279-280. [DOI] [PubMed] [Google Scholar]
- 3.Borneman, A. R., A. H. Forgan, I. S. Pretorius, and P. J. Chambers. 2008. Comparative genome analysis of a Saccharomyces cerevisiae wine strain. FEMS Yeast Res. 8:1185-1195. [DOI] [PubMed] [Google Scholar]
- 4.Cansado, J., J. Barros Velázquez, C. Sieiro, M. Gacto, and T. G. Villa. 1999. Presence of non-suppressive, M2-related dsRNAs molecules in Saccharomyces cerevisiae strains isolated from spontaneous fermentations. FEMS Microbiol. Lett. 181:211-215. [DOI] [PubMed] [Google Scholar]
- 5.Ceroni, A., A. Passerini, A. Vullo, and P. Frasconi. 2006. DISULFIND: a disulfide bonding state and cysteine connectivity prediction server. Nucleic Acids Res. 34:W177-W181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cesareni, G., and J. A. H. Murray. 1987. Plasmid vectors carrying the replication origin of filamentous single-stranded phages, p. 135-154. In J. Setlow and A. Hollaender (ed.), Genetic engineering: principles and methods, vol. 9. Plenum Press, New York, NY. [Google Scholar]
- 7.Dinman, J. D., T. Icho, and R. B. Wickner. 1991. A-1 ribosomal frameshift in a double-stranded RNA virus of yeast forms a gag-pol fusion protein. Proc. Natl. Acad. Sci. U. S. A. 88:174-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esteban, R., L. Vega, and T. Fujimura. 2005. Launching of the yeast 20S RNA narnavirus by expressing the genomic or antigenomic viral RNA in vivo. J. Biol. Chem. 280:33725-33734. [DOI] [PubMed] [Google Scholar]
- 9.Frank, A. C., and K. H. Wolfe. 2009. Evolutionary capture of viral and plasmid DNA by yeast nuclear chromosomes. Eukaryot. Cell 8:1521-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujimura, T., J. C. Ribas, A. M. Makhov, and R. B. Wickner. 1992. Pol of gag-pol fusion protein required for encapsidation of viral RNA of yeast L-A virus. Nature 359:746-749. [DOI] [PubMed] [Google Scholar]
- 11.Fujimura, T., and R. B. Wickner. 1989. Reconstitution of template-dependent in vitro transcriptase activity of a yeast double-stranded RNA virus. J. Biol. Chem. 264:10872-10877. [PubMed] [Google Scholar]
- 11a.Fujimura, T., R. Esteban, L. M. Esteban, and R. B. Wickner. 1990. Portable encapsidation signal of the L-A double-stranded-RNA virus of Saccharomyces cerevisiae. Cell 62:819-828. [DOI] [PubMed] [Google Scholar]
- 12.Gietz, R. D., R. H. Schiestl, A. R. Willems, and R. A. Woods. 1995. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11:355-360. [DOI] [PubMed] [Google Scholar]
- 13.Goto, K., H. Fukuda, K. Kichise, K. Kitano, and S. Hara. 1991. Cloning and nucleotide sequence of the KHS killer gene of Saccharomyces cerevisiae. Agric. Biol. Chem. 55:1953-1958. [PubMed] [Google Scholar]
- 14.Goto, K., et al. 1990. Isolation and properties of a chromosome-dependent KHR killer toxin in Saccharomyces cerevisiae. Agric. Biol. Chem. 54:505-509. [PubMed] [Google Scholar]
- 15.Gubler, U., and B. J. Hoffman. 1983. A simple and very efficient method for generating cDNA libraries. Gene 25:263-269. [DOI] [PubMed] [Google Scholar]
- 16.Guthrie, C., and G. R. Fink. 1991. Guide to yeast genetics and molecular biology. Methods Enzymol. 194:3-57. [PubMed] [Google Scholar]
- 17.Icho, T., and R. B. Wickner. 1989. The double-stranded RNA genome of yeast virus L-A encodes its own putative RNA polymerase by fusing two open reading frames. J. Biol. Chem. 264:6716-6723. [PubMed] [Google Scholar]
- 18.Kaiser, C., S. Michaelis, and A. Mitchell. 1994. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 19.Kyte, J., and R. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105-132. [DOI] [PubMed] [Google Scholar]
- 20.Lee, Y. J., and R. B. Wickner. 1992. MAK10, a glucose-repressible gene necessary for replication of a dsRNA virus of Saccharomyces cerevisiae, has T cell receptor alpha-subunit motifs. Genetics 132:87-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magliani, W., S. Conti, M. Gerloni, D. Bertolotti, and L. Polonelli. 1997. Yeast killer systems. Clin. Microbiol. Rev. 10:369-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maqueda, M., E. Zamora, N. Rodríguez-Cousiño, and M. Ramírez. 2010. Wine yeast molecular typing using a simplified method for simultaneously extracting mtDNA, nuclear DNA and virus dsRNA. Food Microbiol. 27:205-209. [DOI] [PubMed] [Google Scholar]
- 23.Meskauskas, A. 1990. Nucleotide sequence of cDNA to yeast M2-1 dsRNA segment. Nucleic Acids Res. 18:6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Novoa, M., et al. 2009. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proc. Natl. Acad. Sci. U. S. A. 106:16333-16338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park, C. M., et al. 1994. Structure and heterologous expression of the Ustilago maydis viral toxin KP4. Mol. Microbiol. 11:155-164. [DOI] [PubMed] [Google Scholar]
- 26.Park, C. M., J. D. Lopinski, J. Masuda, T. H. Tzeng, and J. A. Bruen. 1996. A second double-stranded RNA virus from yeast. Virology 216:451-454. [DOI] [PubMed] [Google Scholar]
- 27.Pérez, F., M. Ramírez, and J. A. Regodón. 2001. Influence of killer strains of Saccharomyces cerevisiae on wine fermentation. Antonie Van Leeuwenhoek 79:393-399. [DOI] [PubMed] [Google Scholar]
- 28.Pfeiffer, P., and F. Radler. 1984. Comparison of the killer toxin of several yeasts and the purification of a toxin of type K2. Arch. Microbiol. 137:357-361. [DOI] [PubMed] [Google Scholar]
- 29.Ramírez, M., et al. 2004. Genetic instability of heterozygous hybrid populations of natural wine yeasts. Appl. Environ. Microbiol. 70:4686-4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodríguez-Cousiño, N., A. Solórzano, T. Fujimura, and R. Esteban. 1998. Yeast positive-stranded virus-like RNA replicons. J. Biol. Chem. 273:20363-20371. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 32.Schmitt, M. J., and F. Breinig. 2002. The viral killer system in yeast: from molecular biology to application. FEMS Microbiol. Rev. 748:1-20. [DOI] [PubMed] [Google Scholar]
- 33.Schmitt, M. J., and F. Breinig. 2006. Yeast viral killer toxins: lethality and self-protection. Nat. Rev. Microbiol. 4:212-221. [DOI] [PubMed] [Google Scholar]
- 34.Schmitt, M. J., and F. Neuhausen. 1994. Killer toxin-secreting double-stranded RNA mycoviruses in the yeasts Hanseniaspora uvarum and Zygosaccharomyces bailii. J. Virol. 68:1765-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmitt, M. J., and D. J. Tipper. 1995. Sequence of the M28 dsRNA: preprotoxin is processed to an α/β heterodimeric protein. Virology 213:341-351. [DOI] [PubMed] [Google Scholar]
- 36.Skipper, N., D. Y. Thomas, and P. C. K. Lau. 1984. Cloning and sequencing of the preprotoxin-coding region of the yeast M1 double-stranded RNA. EMBO J. 3:107-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sommer, S. S., and R. B. Wickner. 1982. Yeast L dsRNA consists of at least three distinct RNAs; evidence that the non-Mendelian genes [HOK], [NEX] and [EXL] are on one of these dsRNAs. Cell 31:429-441. [DOI] [PubMed] [Google Scholar]
- 38.Sommer, S. S., and R. B. Wickner. 1982. Co-curing of plasmids affecting killer double-stranded RNAs of Saccharomyces cerevisiae: [HOK], [NEX], and the abundance of L are related and further evidence that Ml requires L. J. Bacteriol. 150:545-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thiele, D. J., and M. J. Leibowitz. 1982. Structural and functional analysis of separated strands of killer double-stranded RNA of yeast. Nucleic Acids Res. 10:6903-6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thiele, D. J., R. W. Wang, and M. J. Leibowitz. 1982. Separation and sequence of the 3′ termini of M double-stranded RNA from killer yeast. Nucleic Acids Res. 10:1661-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson, J. D., T. J. Gibson, and D. G. Higgins. 2003. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics 2003:2.3.1-2.3.22. [DOI] [PubMed] [Google Scholar]
- 42.Toh-E, A., P. Guerry, and R. B. Wickner. 1978. Chromosomal superkiller mutants of Saccharomyces cerevisiae. J. Bacteriol. 136:1002-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vodkin, M. H., and G. R. Fink. 1973. A nucleic acid associated with a killer strain of yeast. Proc. Natl. Acad. Sci. U. S. A. 70:1069-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weiler, F., K. Rehfeldt, F. Bautz, and M. J. Schmitt. 2002. The Zygosaccharomyces bailii antifungal virus toxin zygocin: cloning and expression in a heterologous fungal host. Mol. Microbiol. 46:1095-1105. [DOI] [PubMed] [Google Scholar]
- 45.Weiler, F., and J. M. Schmitt. 2003. Zygocin, a secreted antifungal toxin of the yeast Zygosaccharomyces bailii, and its effect on sensitive fungal cells. FEMS Yeast Res. 3:69-76. [DOI] [PubMed] [Google Scholar]
- 46.Wickner, R. B., H. Bussey, T. Fujimura, and R. Esteban. 1995. Viral RNA and the killer phenomenon of Saccharomyces, p. 221-226. In U. Kück (ed.), The Mycota, vol. II. Genetics and biotechnology. Springer Verlag, Berlin, Germany. [Google Scholar]
- 47.Wickner, R. B. 1991. Yeast RNA virology: the killer systems, p. 263-296. In The molecular and cellular biology of the yeast Saccharomyces: genome dynamics, protein synthesis, and energetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 48.Wickner, R. B., T. Icho, T. Fujimura, and W. R. Winder. 1991. Expression of yeast L-A double-stranded RNA virus proteins produces derepressed replication: a ski− phenocopy. J. Virol. 65:155-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Young, T. W. 1987. Killer yeasts, p. 131-164. In A. H. Rose and J. S. Harrison (ed.), The yeasts, vol. 2. Academic Press, London, United Kingdom. [Google Scholar]
- 50.Zuker, M., D. H. Mathews, and D. H. Turner. 1999. Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide., In J. Barciszewski and B. F. C. Clark (ed.), RNA biochemistry and biotechnology. Kluwer Academic Publishers, Dordrecht, Netherlands.