

Abstract
The nifJ gene codes for pyruvate:ferredoxin oxidoreductase (PFOR), which reduces ferredoxin during fermentative catabolism of pyruvate to acetyl-coenzyme A (acetyl-CoA). A nifJ knockout mutant was constructed that lacks one of two pathways for the oxidation of pyruvate in the cyanobacterium Synechococcus sp. strain PCC 7002. Remarkably, the photoautotrophic growth rate of this mutant increased by 20% relative to the wild-type (WT) rate under conditions of light-dark cycling. This result is attributed to an increase in the quantum yield of photosystem II (PSII) charge separation as measured by photosynthetic electron turnover efficiency determined using fast-repetition-rate fluorometry (Fv/Fm). During autofermentation, the excretion of acetate and lactate products by nifJ mutant cells decreased 2-fold and 1.2-fold, respectively. Although nifJ cells displayed higher in vitro hydrogenase activity than WT cells, H2 production in vivo was 1.3-fold lower than the WT level. Inhibition of acetate-CoA ligase and pyruvate dehydrogenase complex by glycerol eliminated acetate production, with a resulting loss of reductant and a 3-fold decrease in H2 production by nifJ cells compared to WT cells. Continuous electrochemical detection of dissolved H2 revealed two temporally resolved phases of H2 production during autofermentation, a minor first phase and a major second phase. The first phase was attributed to reduction of ferredoxin, because its level decreased 2-fold in nifJ cells. The second phase was attributed to glycolytic NADH production and decreased 20% in nifJ cells. Measurement of the intracellular NADH/NAD+ ratio revealed that the reductant generated by PFOR contributing to the first phase of H2 production was not in equilibrium with bulk NADH/NAD+ and that the second phase corresponded to the equilibrium NADH-mediated process.
Although cyanobacteria are oxygenic photoautotrophs, many are capable of performing limited anaerobic metabolism in the dark (autofermentation). As with many other bacteria, autofermentation results in catabolism of stored energy reserves (photosynthate) coupled to excretion of H2 and organic acids and/or alcohols. The pathways by which these products are formed in oxygenic photoautotrophs are not well understood and are the subject of this report, with a particular focus on the redirection of the reductants into H2 production by genetic engineering.
Pyruvate:ferredoxin (flavodoxin) oxidoreductase (PFOR), encoded by the nifJ gene, catalyzes the oxidation of pyruvate to acetyl-coenzyme A (acetyl-CoA) and carbon dioxide and is an oxygen-sensitive enzyme that is ubiquitous in anaerobic bacteria. The electron acceptor for PFOR is usually either ferredoxin (Fd) or flavodoxin (Flvd) (35), but rubredoxin, the iron-sulfur protein, has also been shown to be an alternative electron acceptor (47). PFOR is also present in certain strains of facultatively anaerobic bacteria (39), and a similar oxidoreductase exists in some aerobic bacteria (33). PFOR catalyzes the reverse reaction, the reductive carboxylation of acetyl-CoA to pyruvate, in certain strains of bacteria that grow on acetate (e.g., the methanogen Methanococcus maripaludis) (21).
Because cyanobacteria are capable of facultative fermentative metabolism under anoxic conditions, it is not surprising that about 40% of fully sequenced cyanobacterial genomes contain nifJ genes (29). PFOR in cyanobacteria has been shown to be more closely related to the enzymes found in anaerobes than to those in aerobes (39). Although expression of PFOR under oxic conditions in cyanobacteria has been previously reported (39), the enzyme is inactivated by O2 (31). Ferredoxin has been reported to be the electron acceptor for PFOR in cyanobacteria (19). Although the full physiological function, including the fermentative function, of this enzyme in cyanobacteria has not been extensively explored, PFOR has been shown to catalyze reduction of ferredoxin for nitrogen fixation in diazotrophic, heterocystous cyanobacteria (31).
The oxidation of pyruvate to acetate in cyanobacteria can alternatively be catalyzed by the pyruvate dehydrogenase complex (PDC), which consists of the pyruvate dehydrogenase enzyme (PdhA), dihydrolipoyl transacetylase, and dihydrolipoyl dehydrogenase (9, 29). PDC ultimately reduces NAD+, and it cannot catalyze the reverse set of reactions to produce pyruvate from acetyl-CoA and CO2. PDC is typically considered to function under oxic conditions but has been shown to contribute to fermentative metabolism in Enterococcus (Streptococcus) faecalis, which contains both PDC and PFOR (8). These reports suggest that only PDC is expected to be available to oxidize pyruvate to acetyl-CoA during photoautotrophic growth in cyanobacteria, because the oxygen sensitivity of PFOR suggests that it should not function during oxic growth. Conversely, the involvement of PDC during autofermentation in cyanobacteria has not been extensively explored, although pyruvate dehydrogenase activity is reportedly absent during autofermentation in the cyanobacterium Microcystis sp. strain PCC 7806 (28). Figure 1 summarizes the reactions potentially catalyzed by PFOR and PDC during pyruvate metabolism in Synechococcus sp. strain PCC 7002.
FIG. 1.
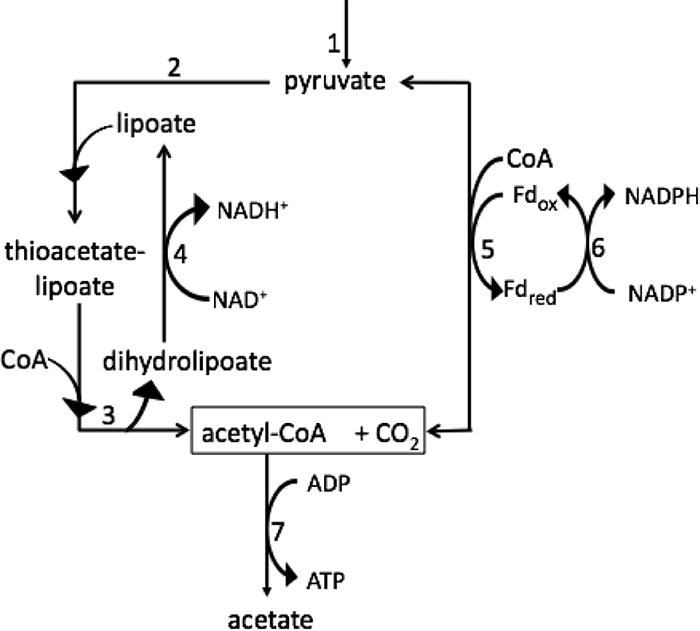
Schematic of metabolism of pyruvate to acetate during autofermentation in Synechococcus sp. strain PCC 7002. Thiamine pyrophosphate (TPP; also known as thiamine diphosphate) is a cofactor of both the pyruvate dehydrogenase enzyme and PFOR; lipoate is an intermediate in the reactions of PDC. 1, enzymes of glycolysis; 2, pyruvate dehydrogenase; 3, dihydrolipoyl transacetylase; 4, dihydrolipoyl dehydrogenase; 5, PFOR (reversible); 6, Fd:NADP+ oxidoreductase (FNR); 7, acetate-CoA ligase.
In strains of bacteria that synthesize both PFOR and either nitrogenase or bidirectional hydrogenase, the reduction of ferredoxin by PFOR during fermentation is a potential source of reductant for H2 production. Diazotrophic bacteria can utilize the reduced Fd generated by PFOR for nitrogen fixation and H2 production via nitrogenase. PFOR was shown to be necessary for in vivo nitrogen fixation in the proteobacterium Klebsiella pneumoniae (14, 36). Additionally, PFOR is required when iron is a limiting factor in the diazotrophic cyanobacterium Anabaena sp. strain PCC 7120 (3). The bidirectional [NiFe] hydrogenase found in some cyanobacteria contains 5 subunits: hoxHY comprises the catalytic hydrogenase domain, whereas hoxEFU comprises the diaphorase domain that exchanges hydride with NAD+ (11). NADH (E0 = −315 mV), rather than Fd (−350 ≤ E0 ≤ −455 mV) or Fld (−415 ≤ EM10 ≤ −440 mV; −180 ≤ EM20 ≤ −210 mV), is the only established electron donor for cyanobacterial NiFe hydrogenases (44). Interestingly, all fully sequenced cyanobacteria that contain a bidirectional hydrogenase also contain PFOR, although the involvement of PFOR in H2 production has never been directly demonstrated. Because [NiFe] hydrogenases are not known to utilize ferredoxin as an electron donor, both the ferredoxin:NADP+ oxidoreductase (FNR) and NAD(P)+:NAD(P)H transhydrogenase together are believed to be required to equilibrate the pool of Fdred/Fdox and NADH/NAD+. Surprisingly, the reduced iron-sulfur center(s) in PFOR is able to evolve H2 at very low rates in vitro in the absence of an alternative electron acceptor(s) other than protons (27). However, this low activity is not thought to occur in vivo.
Synechococcus sp. strain PCC 7002 is a fully sequenced, unicellular, euryhaline cyanobacterium that contains PFOR, PDC, and a bidirectional hydrogenase but lacks nitrogenase (GenBank accession number NC_010475). In the experiments described here, an nifJ null mutant of Synechococcus sp. strain PCC 7002 was constructed and effects on photoautotrophic growth, photosynthetic efficiency, and fermentative metabolism were studied using metabolite analysis by nuclear magnetic resonance (NMR), liquid chromatography-mass spectrometry (LC-MS), and fluorescence methods. Based on continuous electrochemical monitoring, the first observations concerning the temporal dynamics of H2 metabolism in this organism are reported.
MATERIALS AND METHODS
Growth conditions.
Cultures of Synechococcus sp. strain PCC 7002 were grown photoautotrophically in medium A+ (42) supplemented with 2 μM NiCl2 and sparged with 2% (vol/vol) CO2 in air. Cells were grown for 3 days at 35°C in 500-ml flasks to a late-exponential-phase optical density at 550 nm (OD550) of ∼2.5 by the application of fluorescent light from one side of the culture vessel at 200 μmol of photons m−2 s−1. Glycerol-adapted cultures (designated “+G”) were gradually adapted to grow mixotrophically as increasing amounts of glycerol were added to the medium. Fully adapted cultures were grown on 10 mM glycerol to high density (OD550 > 10).
Construction of the nifJ mutant.
A 1,600-bp fragment of nifJ gene coding sequence was amplified by PCR using primers NifJF (5′ CTCCCAAGGACAGCCAAATCTC 3′) and NifJR (5′ CACCAGAGAGGGCAAAGAAACAG 3′). This fragment contains one HindIII site. The antibiotic resistance cartridge containing the aphAII gene, which confers kanamycin resistance, was introduced into this HindIII site. The HindII site was digested and subsequently ligated to aphAII to generate the mutagenesis construct (see Fig. S1a in the supplemental material). Transformants were selected on plates containing medium A+ (42) supplemented with 50 μg of kanamycin ml−1. Single transformant colonies were repeatedly streaked on plates containing 300 μg of kanamycin ml−1. The segregation status of transformants was assessed by analytical PCR with primers NifJchkF (5′ GGATTTGCCCTGTTGTCGTCTG 3′) and NifJchkR (5′ GCCTTTCTTGCCGTAAGTCTTCTG 3′); full segregation of the nifJ and nifJ::aphA2 alleles was detected by failure to amplify a DNA fragment corresponding to the size of the nifJ amplicon produced from a wild-type (WT) DNA template (see Fig. S1b in the supplemental material).
Quantitative RT-PCR.
Cells were grown to an OD550 of 1 and harvested and disrupted by three freeze-thaw cycles. Total RNA was isolated by using a High Pure RNA isolation kit (Roche). The purity and concentration of the resulting total RNA were determined with a NanoDrop (ND-1000) spectrophotometer. Primers and probes for the nifJ gene were designed by the Genomics Core Facility of The Pennsylvania State University using real-time PCR Primer Express probe-primer design software (version 2.0; Applied Biosystems). DNase-treated RNA was subjected to reverse transcription (RT) using a High Capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCR was performed using 10 or 20 ng of cDNA in a reaction with TaqMan Universal PCR Master Mix (Applied Biosystems) in a volume of 20 μl. Primers were added at a concentration of 400 nM and a probe at a concentration of 200 nM; the probe was labeled at the 5′ end with 6-carboxyfluorescein (FAM) and at the 3′ end with Black Hole Quencher (Biosearch Tech). The amplification protocol consisted of 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C, using a 7300 real-time PCR system. Threshold cycle (CT) values of the targeted gene and the reference gene were used together with the ΔCT method to determine relative transcript levels of the gene. The data collected were analyzed and interpreted by the use of REST (Relative Expression Software Tool) software.
Fermentative conditions.
Fermentative conditions were induced as previously described (25). Briefly, cells were concentrated by centrifugation and resuspended in nitrate-free medium A at similar cell densities. Final cell densities were measured by determination of optical density (OD) at 550 nm (38). The final OD550 values were 2.05 for WT cells and 1.91 for nifJ mutant cells. Glycerol-adapted cultures were grown mixotrophically to the stationary phase in the presence of 10 mM glycerol. The final OD550 values were 14 for WT+G and 16 for nifJ+G cultures. Data for the WT and nifJ mutant strains were normalized on the basis of cell numbers, with 1.0 ± 0.2 × 108 cells per ml = OD550 of 1.0 (38). Dark, anoxic conditions were created to induce autofermentation. At each time point, the headspace gas, cells, and media were assayed as described below.
Analytical assays.
The headspaces of vials containing fermenting cultures were sampled with a gas-tight syringe, and H2 levels were measured by gas chromatography (GC) using a Gow Mac series 300 instrument with a thermal conductivity detector and argon as the carrier gas. The metabolite concentrations in the extracellular medium of fermenting cells were quantified using proton nuclear magnetic resonance (1H NMR) spectroscopy as described previously and 30 μg ml−1 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt (TSP) as the internal standard (6). Hydrogenase activity was measured via the methyl viologen assay as described previously (5, 25). Activity was determined immediately following the onset of fermentative conditions. Dissolved H2 concentrations were measured using a membrane-covered rate (Clark-type) electrode as described previously (1), except that cultures were allowed to ferment directly on the membrane at 35°C. Total sugars were assayed using anthrone reagent (13) as described previously (25). Fast-repetition-rate fluorometry (FRRF) experiments were performed using single-turnover pulses as described previously (4). A laser diode excitation source (λ = 655 nm) at a light intensity of 32 mmol of photons m−2 s−1 was used as the excitation source. The sample was flashed with 25 pulses (flashlettes), each 1 μs in duration, followed by a 1-μs dark interval prior to the next flashlette. These pulse “trains” were repeated at a frequency of 100 Hz for 50 trains. Sampling was repeated after dark adaptation (>10 s), and the signal was averaged over 24 h or in 12-h increments as indicated.
LC-MS.
Internal metabolite extracts for LC-MS analysis were prepared as described previously (25). Briefly, aliquots (4 ml) of cultures from fermentation vials were removed via syringe in the presence of argon and were subjected to rapid vacuum filtration. The filter was immediately submerged in 1.8 ml of 80:20 MeOH:H2O quench-extraction solvent at −20°C for 15 min. The cell debris was extracted twice and stored at −80°C until analysis. LC-MS metabolite quantification was conducted using an Agilent 6410 QQQ mass spectrometer coupled to a 1200-series chromatography system. A Phenomenex Synergy C-18 Hydro-RP column was employed at 40°C, with 10 mM tributylamine and 10 mM acetic acid in H2O as mobile phase A and methanol as mobile phase B.
RESULTS
Growth and transcription of nifJ.
The nifJ mutant had a growth rate similar to that of the WT during continuous illumination for 7 days under the conditions specified (Fig. 2). During diurnal growth (12 h of light and 12 h of dark), the mutant culture grew an average of 1.2-fold faster than the WT over a period of 115 h (Fig. 2). Transcription of the nifJ gene in WT cells was investigated by quantitative reverse transcription-PCR (RT-PCR) amplification. RNA was prepared from cells of three replicate cultures grown to the late exponential phase under standard growth conditions (OD550 = 1.0) and from those same three cultures incubated in the dark for 30 min after sparging with 1% (vol/vol) CO2 and 99% N2 to initiate autofermentation. The control RNA samples from photoautotrophically grown cells produced a relative nifJ transcript level of 1.00 ± 0.06. The samples incubated in the dark under anoxic conditions for 30 min had much (130- ± 23-fold) higher nifJ transcript levels (Fig. 3). Although nifJ transcript levels increased dramatically under fermentative conditions, these transcript levels were only about 24% of the level seen with the most abundant cellular protein, i.e., phycocyanin (encoded by cpcB), under the same conditions.
FIG. 2.

Growth of nifJ (□) and WT (⋄) cultures under conditions of constant illumination (left panel) or a diurnal cycle of 12 h of light and 12 h of dark (right panel) as measured by optical density at 550 nm. Dark periods of the diurnal cycle are indicated by shading. The nifJ cultures grew 1.2-fold faster than the WT cultures.
FIG. 3.
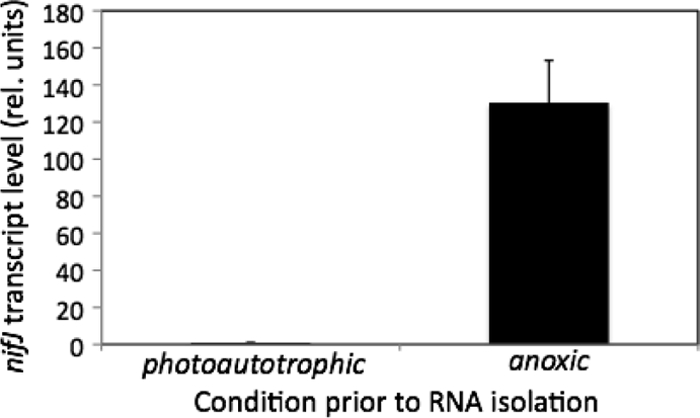
Relative transcript levels of nifJ quantified via RT-PCR after photoautotrophic growth (OD550 = 1.0) or 30 min of growth under anoxic conditions. Threshold count (CT) values of the targeted gene were used to determine relative transcript levels of the gene. Error bars represent ± one standard deviation for the results of three biological replicate experiments.
Variable fluorescence.
The steady-state chlorophyll variable fluorescence yield, Fv/Fm, averaged over 24 h was 5% higher in the nifJ cells (0.409 ± 0.002) than in WT cells (0.389 ± 0.002) (Fig. 4). The transient FRRF response to single turnover pulses at a rate of 100 Hz revealed oscillations in Fv/Fm arising from modulation of the Chl yield by S-state cycling in the photosystem II (PSII) water oxidation complex (2). The period of the transient oscillations (P) was obtained by Fourier transformation (2, 15), and the analysis revealed that the cycling period of nifJ cells (P = 4.25) was shorter than that of the WT cells (P = 4.31). The oscillation amplitude was fit to the Kok model to extract the miss (α) and double-hit (β) parameters (41); nifJ displayed 13% lower α and 24% lower β values. All three of the transient Kok parameters and the steady-state Fv/Fm value were internally self-consistent and indicated that PSII turns over more efficiently in nifJ cells than in WT cells. This corresponded to a 5% higher quantum yield when sampled at a turnover rate of 100 Hz. To determine the stability of this phenotype in the dark, a comparison of transient Fv/Fm data averaged in successive 12-h increments up to 48 h revealed that, whereas the Fv/Fm of WT cells remained constant, the Fv/Fm of nifJ cells decreased progressively to reach the same level as that of the WT cells (0.389 ± 0.002) by 48 h (see Fig. S2 in the supplemental material). It should be noted that the net level of photon flux from the measuring fluorometer over the dark period is a tiny fraction of that of the integrated light received during an equal period of continuous illumination for growth.
FIG. 4.

Fast-repetition-rate fluorometry of WT (Δ) and nifJ (□) cells grown photoautotrophically in air. Each excitation flash sequence (λ = 655 nm) was 50 μs long and was composed of 25 identical flashlettes of 1 μs each and 1 μs dark interval equally spaced prior to the next flash. The flash sequence was repeated at a frequency of 100 Hz for 50 flashes, followed by a dark adaptation period of 120 s prior to the next sequence of 50 flashes. Each flash train was averaged over 24 h. Inset: Period, turnover period; α, Kok miss parameter; β, Kok double-hit parameter.
Autofermentation.
As measured in vitro by the methyl viologen assay at 25°C, hydrogenase activity was slightly higher for nifJ cells at 5.5 ± 0.3 mol H2 evolved h−1 per 1017 cells than for WT cells at 4.7 ± 0.8 mol H2 evolved h−1 per 1017 cells. Photoautotrophically grown cultures were concentrated and allowed to ferment in the absence of added nitrate on a membrane-covered H2 rate electrode at 35°C that measures the current due to oxidation of H2. The electrode consumes all H2 in the thin sample within a few minutes and so produces results that differ from the GC measurements of H2 in the headspace. WT cells displayed two kinetic phases of H2 production during fermentation as determined using the electrode (Fig. 5 A). The first phase increased slowly within 5 h of the onset of anoxic conditions and lasted 18 ± 5 h, with the highest rate typically occurring at 15 ± 1 h and then quickly declining to the baseline level. The second phase appeared abruptly, immediately after the first, after ∼20 h, reaching a maximum within about 5 h and then continuing at a constant rate. The nifJ cells, however, lacked the characteristic peak in the first phase and instead increased monotonically to a lower level under the first 23 h of anoxic conditions (Fig. 5B). The integrated area of phase 1, corresponding to the amount of H2 produced, was (2 ± 0.3)-fold lower in nifJ cells. The second phase in nifJ cells began sharply at 23 ± 2 h after the onset of anoxic conditions and was also lower; the integrated area was 18% ± 6% smaller in nifJ cells. These kinetic measurements were replicated in triplicate with independent cultures.
FIG. 5.

Rate of extracellular H2 concentration change in solution following exposure to dark, anoxic conditions at 35°C. The rate of change of dissolved H2 concentration was measured using a membrane-covered electrode that irreversibly oxidizes all H2. (A) WT cells display two distinct phases of H2 production. (B) nifJ cells display a decreased phase 1 value, with the integrated area (2 ± 0.3)-fold lower. The integrated area represented by phase 2 is 18% ± 6% lower in nifJ cells. Data represent the results of one of three replicate experiments that exhibited comparable changes. (C) Model for H2 production in Synechococcus sp. strain PCC 7002. The redox poise at the diaphorase site may be increased by the presence of ferredoxin, which exchanges with NADH via transhydrogenase and FNR.
Cells were grown in continuous light and then allowed to autoferment in sealed vials in order to measure cumulative H2 production and concentrations of excreted metabolites over 4 days of fermentation (Fig. 6). At the onset of anoxic conditions and as measured by the anthrone assay, WT cells contained 110 ± 10 mol reduced carbohydrate per 1017 cells and nifJ cells contained 94 ± 4 mol reduced carbohydrate per 1017 cells. After 4 days of fermentation, WT cells catabolized 31 ± 14 mol carbohydrate per 1017 cells and nifJ cells catabolized 26 ± 5 mol carbohydrate per 1017 cells (see Fig. S3 in the supplemental material). The amount of carbohydrate catabolized can be converted into equivalents of carbon and reductant generated from glucose equivalents during glycolytic catabolism. Assuming that the pentose phosphate pathway is not contributing to catabolism, one catabolized glucose equivalent is equal to 6 carbon equivalents and 2 NADH (reductant) equivalents. After 4 days of fermentation, WT cells catabolized carbohydrate to a stoichiometry of 186 carbon equivalents and 62 reductant equivalents and nifJ cells catabolized carbohydrate to 156 carbon and 52 reductant equivalents. These fluxes into glycolysis are annotated as negative values in Table 1. After 4 days of fermentation, WT cultures accumulated 2.9 ± 0.3 mol H2 per 1017 cells, 6.5 ± 0.4 mol acetate per 1017 cells, 60 ± 7 mol lactate per 1017 cells, and 1.8 ± 0.8 mol alanine per 1017 cells (Fig. 6). After 4 days of fermentation, nifJ cultures accumulated 1.3-fold less H2 (2.3 ± 0.2 mol H2 per 1017 cells), 2.0-fold less acetate (3.0 ± 0.4 mol acetate per 1017 cells), 1.2-fold less lactate (51 ± 7 mol lactate per 1017 cells), and 1.7-fold more alanine (3.0 ± 1.0 mol alanine per 1017 cells) than WT cultures. The excreted products can be converted into the equivalents of the carbon and reductant consumed in order to generate them, with H2, lactate, and alanine consuming one reductant (NADH) equivalent and acetate + CO2, lactate, and alanine consuming 3 carbon equivalents. A summary of excreted metabolites appears in Table 1. Equivalents were determined by multiplying observed concentrations by the associated equivalent; e.g., row 5 = (row 1) * (row 3). WT cells excreted 200 equivalents of carbon and 59 equivalents of reductant, whereas nifJ cells excreted 170 equivalents of carbon and 53 equivalents of reductant, with both corresponding to the amount of carbohydrate catabolized.
FIG. 6.

Autofermentative metabolite excretion from wild-type WT (⋄) and nifJ (□) cultures. Lactate, alanine, and acetate concentrations were determined by 1H NMR. H2 concentrations in the headspace were measured by gas chromatography. The ordinate gives the concentrations per 1017 cells. Error bars represent ± one standard deviation calculated from the results of three biological replicate experiments.
TABLE 1.
Summary of catabolized carbohydrate and excreted products after 4 days of autofermentation
| Category | Value (mol × 1017 cells−1) for: |
||||||
|---|---|---|---|---|---|---|---|
| Intracellular R.S.C.a | Hydrogen excreted | Acetate excreted | Lactate excreted | Alanine excreted | CO2 excretedc | Excess equivalents excretedd | |
| WT cells (measured) | 31 ± 14 | 2.9 ± 0.3 | 6.5 ± 0.4 | 60 ± 7 | 1.8 ± 0.8 | 6.5 | |
| nifJ cells (measured) | 26 ± 5 | 2.3 ± 0.2 | 3.0 ± 0.4 | 51 ± 7 | 3.0 ± 1.0 | 3 | |
| Carbon equivalents per moleculeb | −6 | 0 | 2 | 3 | 3 | 1 | |
| NADH equivalents per moleculeb | −2 | 1 | −1 | 1 | 1 | 0 | |
| WT carbon equivalents | −186 | 0 | 13 | 180 | 5.4 | 6.5 | 18.9 |
| nifJ carbon equivalents | −156 | 0 | 6 | 153 | 9 | 3 | 15 |
| WT NADH equivalents | −62 | 2.9 | −6.5 | 60 | 1.8 | 0 | −3.8 |
| nifJ NADH equivalents | −52 | 2.3 | −3 | 51 | 3 | 0 | 1.3 |
R.S.C., reduced sugars catabolized.
Values represent theoretical equivalents of carbon (row 3) and reductant (row 4 [as NADH]) per catabolized glucose equivalent during fermentation.
CO2 values were not directly measured and are set equal to the acetate concentration, since CO2 forms stoichiometrically and concomitantly with acetyl-CoA via the PFOR or PDC reaction (Fig. 1). Additionally, the reductant formed in this reaction is accounted for in the acetate column only to avoid double counting.
Excreted equivalents that were excreted in excess of the carbohydrate catabolized. Based on the systematic error determined for each detection method, the propagated systematic error for this column is ±15 for carbon equivalents (rows 5 and 6) and ±5 for NADH equivalents (rows 7 and 8) based on the systematic error for each detection method.
Fermentation with glycerol.
WT and nifJ cultures were adapted to grow mixotrophically on 10 mM glycerol under conditions of constant illumination and then were allowed to autoferment under dark, anoxic conditions for 7 days in the presence of 10 mM glycerol (Fig. 7). Cells grown mixotrophically on glycerol displayed lower in vitro hydrogenase activity than cells grown photoautotrophically. Under these conditions, WT cells displayed in vitro hydrogenase activity, as measured by the methyl viologen assay, of 1.2 ± 0.1 mol H2 evolved per 1017 cells per h, whereas nifJ cells displayed in vitro activity of 2.2 ± 0.8 mol H2 evolved per 1017 cells per h (Table 2). Additionally, the mixotrophically grown cultures produced less H2 in vivo than the photoautotrophically grown cultures. Mixotrophically grown WT cells produced 0.5 ± 0.01 mol H2 per 1017 cells in the headspace after 7 days, whereas nifJ cells produced 0.18 ± 0.05 mol H2 per 1017 cells in the headspace after 3 days, at which point the culture stopped producing H2. The presence of glycerol during the fermentation also inhibited the production of acetate during fermentation by the nifJ cells, which excreted only trace amounts of acetate that were below the limit of accurate quantification (10 μM), while WT cells excreted acetate at the same rate as photoautotrophically grown WT cells, as indicated in Table 2. WT+G cells excreted d-lactate at the same rate as photoautotrophic WT cells, whereas nifJ+G cells excreted d-lactate at a 1.6-fold-higher rate. Photoautotrophically grown cells excreted no succinate, whereas WT+G and nifJ+G cells excreted small amounts of succinate. WT+G and nifJ+G cells excreted alanine at similar rates.
FIG. 7.

Autofermentative metabolite excretion from glycerol-adapted WT+G (⋄) and nifJ+G (□) cultures. Lactate, alanine, and acetate concentrations were determined by 1H NMR. H2 in the headspace was measured by gas chromatography. The nifJ+G cultures excreted 1.6-fold more lactate and 1.7-fold more succinate than the WT cultures, demonstrating the redistribution of carbon from acetate to these alternative pathways. Alanine excretion results were not significantly different between WT+G and nifJ+G cultures. The H2 production by nifJ+G cultures was decreased 3-fold compared to WT+G culture results. The ordinate gives the concentrations per 1017 cells. Error bars represent ± one standard deviation as calculated from the results of three biological replicate experiments.
TABLE 2.
Hydrogenase activities and average rates of excretion
| Culture | Value (mol × 1017 cells−1 day−1) for: |
||||
|---|---|---|---|---|---|
| Hydrogenase activitya | Hydrogen rateb | Acetate rate | Lactate rate | Alanine rate | |
| WT | 113 ± 19 | 0.73 ± 0.08 | 1.6 ± 0.2 | 15 ± 2 | 0.4 ± 0.2 |
| nifJ | 132 ± 7 | 0.57 ± 0.05 | 0.8 ± 0.1 | 13 ± 2 | 0.7 ± 0.3 |
| WT+G | 30 ± 3 | 0.08 ± 0.01 | 1.8 ± 0.1 | 14 ± 1 | 1.2 ± 0.1 |
| nifJ+G | 53 ± 19 | 0.06 ± 0.02 | 0 | 22 ± 1 | 1.3 ± 0.1 |
The methyl viologen in vitro hydrogenase values are reported in the text as mol per 1017 cells per h, while the average rates of excreted products for WT, nifJ, WT+G, and nifJ+G cultures are reported in units of mol per 1017 cells per day.
The indicated hydrogen rate for nifJ+G cells is for the first 3 days of autofermentation. The rate after 3 days was zero.
Intracellular pyridine nucleotides.
Intracellular NAD(P)(H) levels were quantified during the first 3 days of fermentation. At the onset of anoxic conditions (t = 0), NADH levels were below the detection limit (10 nM) for both WT and nifJ cultures. The NADPH levels for WT and nifJ cultures at the onset of fermentation were 0.04 ± 0.01 and 0.03 ± 0.01 mol per 1017 cells, respectively. The NADP+ levels for WT and nifJ cultures at the onset of fermentation were 0.09 ± 0.01 and 0.07 ± 0.03 mol per 1017 cells, respectively. NADPH and NADP+ levels remained constant throughout the fermentation. Over 3 days of fermentation, the intracellular concentration of NADH increased monotonically and concomitantly with a parallel decrease in the NAD+ level for both WT and nifJ cultures (Fig. 8). These changes resulted in a substantial increase in reductant poise for both cultures over the course of autofermentation (Fig. 8).
FIG. 8.
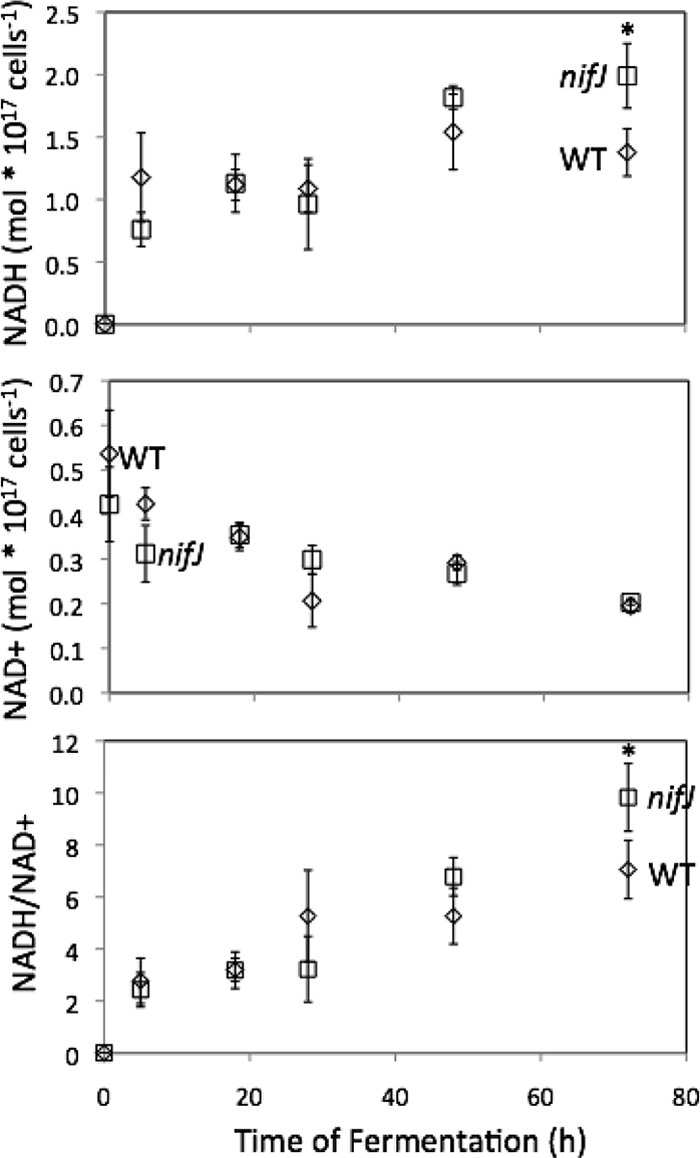
Intracellular concentrations of NADH and NAD+ and ratio of NADH/NAD+ for WT and nifJ cells of Synechococcus sp. strain PCC 7002 determined by LC/MS. Time zero was measured at the onset of anoxic conditions. Concentrations of NAD+ and NADH were measured with LC/MS. Error bars represent ± one standard deviation calculated from the results of three biological replicate experiments. Asterisks indicate data that are not significantly different (P = 0.1; 2-tailed t test).
DISCUSSION
The absence of a significant growth defect for nifJ mutant cells under continuous photoautotrophic growth conditions indicated that PFOR activity is not essential under these growth conditions. Either PFOR is inactive under such conditions or other pathways can perhaps compensate for its absence. Surprisingly, the nifJ mutant cells grew 20% faster than WT cells when both were grown under conditions of a diurnal cycle of 12 h of light and 12 h of dark. This phenotype may arise from a higher photosynthetic efficiency in the nifJ cells that is exaggerated when cells are grown on a diurnal cycle. Metabolism of cyanobacteria cells, during both growth and fermentation, is extremely flexible and can be controlled metabolically or genetically. This metabolic flexibility can lead to unexpected phenotypes in mutants.
The steady-state and transient FRRF data of cells grown under conditions of continuous illumination revealed that Fv/Fm, the relative quantum yield for photosynthetic charge separation at a 100-Hz sampling rate (turnover frequency), is 5% higher in nifJ cells (Fig. 4). Because the cells were adapted to dark (though exposed to air and therefore not anoxic) in this experiment and were exposed to only short pulses of light, PFOR may have been transcribed in WT cells, thereby producing an increase in the ratio of reduced to oxidized Fd. Indeed, transcriptional studies have revealed that nifJ expression increases significantly when cells are shifted from photosynthetic (i.e., light oxic) to respiratory (dark oxic) conditions, which is similar to the conditions used in this experiment (23). Because there is a strong thermodynamic driving force for reduced Fd (E0 = −0.42 V) to deliver electrons to the plastoquinone (PQ) pool (E0 = 0 V) through FNR (classical cyclic electron transfer), this transfer reduces the PQ pool, which is the endogenous electron acceptor pool for PSII (22). Correspondingly, nifJ cells that lack this flux of reductant from carbohydrate catabolism via Fd to the PQ pool are expected to exhibit a higher PSII quantum yield (an elevated Fv/Fm ratio), in agreement with our observations. While the Fv/Fm ratio for WT cells remained stable for a full 48 h for dark-adapted samples, the Fv/Fm ratio for nifJ cells was stable over 12 h but then decreased to the level seen with the WT after 48 h in the dark (see Fig. S2 in the supplemental material). The origin of this transition is unknown but may reflect changes in the regulation of cyclic electron transfer during this extended dark period. Since the diurnally grown cells are “reset” into the light after 12 h of darkness, this effect did not occur during the growth experiment. The increased photosynthetic efficiency and growth rate under diurnal cycling conditions have important implications for increasing biomass yields in phototrophs used for biofuel production. The nifJ mutant is currently being investigated further in an effort to understand the basis for the phenomena described here.
The increased hydrogenase activity associated with the PFOR mutant is not expected to increase the growth rate and quantum yield of photosynthetic charge separation efficiency, because the hydrogenase is inactive aerobically. In a mutant of Synechocystis sp. strain PCC 6803 which overexpresses the hydrogenase enzyme, no increase in growth was demonstrated (10).
Quantitative RT-PCR experiments showed that the nifJ mRNA levels increased rapidly and to very high (130-fold) levels within 30 min after WT cells were transferred from standard photoautotrophic (light oxic) conditions to dark anoxic (fermentative) conditions (Fig. 3). Assuming that this transcriptional difference also reflects a difference in enzyme activity, this result strongly suggests that Synechococcus sp. strain PCC 7002 catabolizes pyruvate under respiratory and fermentative conditions by using different enzymes. A recent study corroborated these results, showing a more than 100-fold increase in nifJ transcription 1 h after the cells were shifted from light oxic (photosynthetic) to dark anoxic (autofermentative) incubation (23). Additionally, the cells showed a decrease in transcription of the PDC components after 1 h of dark, anoxic incubation, which supports the idea of the partitioning of these two pathways under different conditions. Inactivation of the pdcA gene resulted in a mutant auxotrophic for acetate (46). This result indicates that, under aerobic conditions, nifJ expression or PFOR activity (or both) is unable to provide sufficient acetyl-CoA to support the autotrophic growth of this cyanobacterium. While the expression of ferredoxins and flavodoxins in cyanobacteria has been studied under various photosynthetic conditions (17, 20, 24, 34), the anoxic expression profile has not yet been reported. Future studies are planned to attempt to demonstrate whether some of these electron transport proteins are specifically associated with catabolic enzymes such as PFOR under fermentative growth conditions.
The observed 2-fold decrease in fermentative acetate production by the nifJ mutant after 4 days under conditions of dark anoxia indicated that PFOR does contribute to the fermentative metabolism of pyruvate. However, the nonzero level of acetate indicates that PDC must also contribute to this anoxic catabolism, at least in the absence of PFOR. Although the nifJ mutant cells produced 1.3-fold less H2 than the WT cells, PFOR was not the sole source of reductant for H2 production by the bidirectional hydrogenase. The presence of glycerol altered the fermentative metabolism of both the WT and the nifJ mutant cells. Glycerol completely inhibited acetate formation in nifJ mutant cells and further altered the distribution of both carbon products and H2 (Fig. 7). Notably, there was an increase in catabolism in the nifJ+G culture compared with the WT+G culture, and the redistribution of reductant flux toward lactate in the nifJ+G culture more than accounts for the decrease in reductant flux to H2. Glycerol decreases the fermentative formation of acetate from pyruvate at two possible points: acetate-CoA ligase (acetate-CoA synthase) and PDC have been shown to be inhibited by glycerol in chloroplasts and yeast, respectively (18, 37). It has additionally been demonstrated that PDC is inhibited by an elevated NADH:NAD+ ratio in Gram-negative bacteria (9) and that the redox poise is expected to be elevated in mixotrophic cyanobacteria, as evidenced by the reduction state of the PQ pool during dark incubation of cells supplemented with exogenous sugar (16). We observed no decrease in acetate production in WT cells during autofermentation in the presence of glycerol, suggesting that inhibition is specific to PDC. Conversely, the addition of glycerol under conditions of dark anoxia in nifJ cells eliminated all acetate excretion, forcing carbon flux into lactate and the reductive branch of the TCA pathway and indicating that PDC is responsible for acetyl-CoA production in the absence of glycerol.
H2 production from a constitutively expressed bidirectional [NiFe] hydrogenase occurs under dark, anoxic conditions in the cyanobacterium Arthrospira maxima. This has previously been shown to occur in two temporally resolved phases in fermenting cells derived from exponentially growing cultures (1). Because a respiratory incubation period prior to fermentation eliminates the first phase of H2 production in A. maxima, this transient phase had been attributed to accumulated photosynthetic reductant (Fd, NADPH) that is exhausted during respiration. The second phase of H2 production in A. maxima is fueled by NADH generated from glycolysis and occurs only after an accumulation of excess NADH:NAD+ sufficient for the thermodynamic production of H2 (1). The level of intracellular reductant pools correlates with H2 production in certain strains of bacteria, including cyanobacteria (7, 25, 30). NADH has been shown to be the preferred exogenous electron donor for the cyanobacterial NiFe hydrogenase (44). Further studies are required to establish the mechanisms and regulation of electron exchange among the major reductant sources (Fdred, NADPH, and NADH) under dark, fermentative conditions.
Synechococcus sp. strain PCC 7002 cells also display two temporal phases of H2 production. However, only phase 2 H2 is generated from glycolytically generated NADH, in analogy to A. maxima results. In contrast, phase 1 H2 differs qualitatively from H2 from the first phase in A. maxima, in which it rises to a substantial level within seconds after anoxic conditions are presented. Phase 1 H2 in A. maxima was shown to originate from Fd or NADPH previously generated by photosynthesis (1). In contrast, in Synechococcus sp. strain PCC 7002, phase 1 H2 levels increased over several hours after the onset of dark anoxic incubation and peaked at about 18 h before the sharp onset of phase 2 H2 production at 22 h (Fig. 8). This indicates either a residual photosynthetic reductant source different from that present in A. maxima or a new anoxic reductant source distinct from glycolytic NADH. A possible explanation that we favor is that the reductant for phase 1 H2 in Synechococcus sp. strain PCC 7002 does not originate primarily from residual photosynthetically derived Fd or NADPH but instead originates primarily from reduced ferredoxin formed during anoxic catabolism of pyruvate by PFOR. The several hours required to achieve the maximum H2 production rate would in this case be attributed to the time scale for catabolism of biosynthetic precursors to form pyruvate. While glycolysis reduces NADH during fermentation, the NADH:NAD+ ratio does not attain the level necessary for H2 production until later (phase 2 H2). The bidirectional hydrogenase, coded by the hox operon, is expected to be constitutively expressed, as it is in A. maxima and some other cyanobacteria (45). Additionally, the activity may be altered by maturation or activation of the enzyme following oxygen inactivation, as occurs in the [NiFe] hydrogenases of other bacteria (26, 32, 43).
In order to investigate further the influence of the PFOR enzyme on the reductive poise of cells, we measured the intracellular concentrations of NADH and NAD+ by LC-MS during fermentation in Synechococcus sp. strain PCC 7002. There was no significant difference between WT and nifJ mutant reductant levels over 3 days of autofermentation. At the onset of dark anoxia, the absence of detectable NADH in both WT and nifJ mutant cells (Fig. 8) suggested that the reductant for phase 1 H2 is not NADH. Instead, pyruvate oxidation by the PFOR enzyme appears to result in formation of a substantial portion of the reductant responsible for phase 1 H2, from the Fdred generated via the PFOR reaction, and PDC may be contributing the remainder. The low-level intrinsic hydrogenase activity reported for an isolated PFOR enzyme in vitro is probably not relevant under in vivo conditions (27). Reduced ferredoxins are stronger reducing agents (−350 ≤ E0 ≤ −455 mV) than NADH (E0 = −315 mV) and could, in principle, reduce hydrogenase. Although there is no precedent in the literature that documents direct electron transfer from reduced Fd to a cyanobacterial bidirectional [NiFe] hydrogenase, we cannot completely exclude this possibility. The lack of a difference between the WT and nifJ NADH/NAD+ levels supports the interpretation that there is not full redox equilibrium between NADH and the Fdred pool and that the ferredoxin reduced by PFOR may potentially serve as a direct electron donor to the [NiFe] hydrogenase. Interestingly, there is experimental evidence for the existence of a class of FeFe hydrogenases that require both NADH and Fdred (40), demonstrating that the mechanisms of some hydrogenases are not fully understood and are still emerging. Alternatively, Fdred and NAD(P)H are known to equilibrate via the combined activities of FNR and transhydrogenases (12, 48), resulting in reduction of NADH, which is the commonly accepted hydride donor to the cyanobacterial hydrogenase. In Fig. 5C, we show this known route, with Fdred equilibrating with NAD+. However, formation of phase 1 H2 cannot involve a single pool of NADH/NAD+ at equilibrium, as the LC-MS data (Fig. 8) show that this ratio had not yet increased sufficiently to allow H2 production to occur. Thus, we conclude that phase 1 H2 must arise from formation of a reduced electron donor that is channeled to the hydrogenase, possibly via the diaphorase subunit. Examination of the genome of Synechococcus sp. strain PCC 7002 reveals evidence for only one copy of the petH gene encoding FNR, which is specific for NADPH rather than NADH (GenBank accession number NC_010475). Additionally, there are only single genes for pntA, pntB, and pntC, which together encode the pyridine nucleotide transhydrogenase.
Phase 2 H2 exhibits a sharp rise at onset that indicates that it may be triggered by an increase of the NADH:NAD+ ratio to a high enough level for the H2 formation reaction to become thermodynamically favorable. The formation of phase 2 H2 in cyanobacteria is fueled primarily by NADH from carbohydrate (glycogen) catabolism, as proposed previously (1) and as corroborated in these studies by the increased redox poise required for its formation (caused by an increase in NADH levels and a decrease in NAD+, a possible inhibitor) for both WT and nifJ cells during fermentation (Fig. 8). Additional NADH is generated via metabolism of pyruvate, either directly via PDC or via PFOR (from Fdred, as mediated by FNR and transhydrogenase).
The data presented herein show that a role for the bidirectional hydrogenase in Synechococcus sp. strain PCC 7002 is to serve as a valve to reoxidize excess NADH formed during glycolysis. However, this is clearly one of at least two roles, as here the hydrogenase is also shown to oxidize reductant generated from Fdred produced by PFOR.
Supplementary Material
Acknowledgments
We thank Marcus Ludwig for sharing his transcription profiling results prior to publication. We thank David Vinyard for his help with FRRF and Agilent Technologies for instrumentation support.
This work was supported by the Air Force Office of Scientific Research (MURI grant FA9550-05-1-0365), which is gratefully acknowledged.
Footnotes
Published ahead of print on 11 February 2011.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Ananyev, G., D. Carrieri, and G. C. Dismukes. 2008. Optimization of metabolic capacity and flux through environmental cues to maximize hydrogen production by the cyanobacterium Arthrospira (Spirulina) maxima. Appl. Environ. Microbiol. 74:6102-6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ananyev, G., and G. C. Dismukes. 2005. How fast can photosystem II split water? Kinetic performance at high and low frequencies. Photosynth. Res. 84:355-365. [DOI] [PubMed] [Google Scholar]
- 3.Bauer, C. C., L. Scappino, and R. Haselkorn. 1993. Growth of the cyanobacterium Anabaena on molecular nitrogen: nifJ is required when iron is limited. Proc. Natl. Acad. Sci. U. S. A. 90:8812-8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carrieri, D., G. Ananyev, T. Brown, and G. C. Dismukes. 2007. In vivo bicarbonate requirement for water oxidation by photosystem II in the hypercarbonate-requiring cyanobacterium Arthrospira maxima. J. Inorg. Biochem. 101:1865-1874. [DOI] [PubMed] [Google Scholar]
- 5.Carrieri, D., G. Ananyev, A. M. Garcia Costas, D. A. Bryant, and G. C. Dismukes. 2008. Renewable hydrogen production by cyanobacteria: nickel requirements for optimal hydrogenase activity. Int. J. Hydrogen Energy 33:2014-2022. [Google Scholar]
- 6.Carrieri, D., et al. 2009. Identification and quantification of water-soluble metabolites by cryoprobe-assisted nuclear magnetic resonance spectroscopy applied to microbial fermentation. Magn. Reson. Chem. 47:S138-S146. [DOI] [PubMed] [Google Scholar]
- 7.Datta, R., and J. G. Zeikus. 1985. Modulation of acetone-butanol-ethanol fermentation by carbon monoxide and organic acids. Appl. Environ. Microbiol. 49:522-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deibel, R. H., and C. F. Niven, Jr. 1964. Pyruvate fermentation by Streptococcus faecalis. J. Bacteriol. 88:4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Kok, A., A. F. Hengeveld, A. Martin, and A. H. Westphal. 1998. The pyruvate dehydrogenase multi-enzyme complex from Gram-negative bacteria. Biochim. Biophys. Acta 1385:353-366. [DOI] [PubMed] [Google Scholar]
- 10.Germer, F., et al. 2009. Overexpression, isolation, and spectroscopic characterization of the bidirectional [NiFe] hydrogenase from Synechocystis sp. PCC 6803. J. Biol. Chem. 284:36462-36472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghirardi, M. L., et al. 2007. Hydrogenases and hydrogen photoproduction in oxygenic photosynthetic organisms. Annu. Rev. Plant Biol. 58:71-91. [DOI] [PubMed] [Google Scholar]
- 12.Hald, S., B. Nandha, P. Gallois, and G. N. Johnson. 2008. Feedback regulation of photosynthetic electron transport by NADP(H) redox poise. Biochim. Biophys. Acta 1777:433-440. [DOI] [PubMed] [Google Scholar]
- 13.Hassid, W. Z., and S. Abraham. 1957. Chemical procedures for analysis of polysaccharides. Methods Enzymol. 3:34-50. [Google Scholar]
- 14.Hill, S., and E. P. Kavanagh. 1980. Roles of nifF and nifJ gene products in electron transport to nitrogenase in Klebsiella pneumoniae. J. Bacteriol. 141:470-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolling, D. R. J., T. S. Brown, G. Ananyev, and G. C. Dismukes. 2009. Photosynthetic oxygen evolution is not reversed at high oxygen pressures: mechanistic consequences for the water-oxidizing complex. Biochemistry 48:1381-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kujat, S. L., and G. W. Owttrim. 2000. Redox-regulated RNA helicase expression. Plant Physiol. 124:703-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laudenbach, D. E., M. E. Reith, and N. A. Straus. 1988. Isolation, sequence analysis, and transcriptional studies of the flavodoxin gene from Anacystis nidulans R2. J. Bacteriol. 170:258-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence, C. L., C. H. Botting, R. Antrobus, and P. J. Coote. 2004. Evidence of a new role for the high-osmolarity glycerol mitogen-activated protein kinase pathway in yeast: regulating adaptation to citric acid stress. Mol. Cell. Biol. 24:3307-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leach, C. K., and N. G. Carr. 1971. Pyruvate:ferredoxin oxidoreductase and its activation by ATP in the blue-green alga Anabaena variabilis. Biochim. Biophys. Acta 245:165-174. [DOI] [PubMed] [Google Scholar]
- 20.Leonhardt, K., and N. A. Straus. 1992. An iron stress operon involved in photosynthetic electron transport in the marine cyanobacterium Synechococcus sp. PCC 7002. J. Gen. Microbiol. 138:1613-1621. [DOI] [PubMed] [Google Scholar]
- 21.Lin, W. C., Y.-L. Yang, and W. B. Whitman. 2003. The anabolic pyruvate oxidoreductase from Methanococcus maripaludis. Arch. Microbiol. 179:444-456. [DOI] [PubMed] [Google Scholar]
- 22.Livingston, A. K., J. A. Cruz, K. Kohzuma, A. Dhingra, and D. M. Kramer. 2010. An Arabidopsis mutant with high cyclic electron flow around photosystem I (hcef) involving the NADPH dehydrogenase complex. Plant Cell 22:221-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ludwig, M., and D. A. Bryant. Transcription profiling of the model cyanobacterium Synechococcus sp. strain PCC 7002 by Next-Gen (SOLiD) sequencing of cDNA. Front. Microbiol., in press. [DOI] [PMC free article] [PubMed]
- 24.Mazouni, K., F. Domain, F. Chauvat, and C. Cassier-Chauvat. 2003. Expression and regulation of the crucial plant-like ferredoxin of cyanobacteria. Mol. Microbiol. 49:1019-1029. [DOI] [PubMed] [Google Scholar]
- 25.McNeely, K., Y. Xu, N. Bennette, D. A. Bryant, and G. C. Dismukes. 2010. Redirecting reductant flux into hydrogen production via metabolic engineering of fermentative carbon metabolism in a cyanobacterium. Appl. Environ. Microbiol. 76:5032-5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menon, A. L., and R. L. Robson. 1994. In vivo and in vitro nickel-dependent processing of the [NiFe] hydrogenase in Azotobacter vinelandii. J. Bacteriol. 176:291-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menon, S., and S. W. Ragsdale. 1996. Unleashing hydrogenase activity in carbon monoxide dehydrogenase/acetyl-CoA synthase and pyruvate:ferredoxin oxidoreductase. Biochemistry 35:15814-15821. [DOI] [PubMed] [Google Scholar]
- 28.Moezelaar, R., and L. J. Stal. 1994. Fermentation in the unicellular cyanobacterium Microcystis PCC7806. Arch. Microbiol. 162:63-69. [Google Scholar]
- 29.Nakao, M., et al. 2010. CyanoBase: the cyanobacteria genome database update 2010. Nucleic Acids Res. 38:D379-D381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakashimada, Y., M. A. Rachman, T. Kakizono, and N. Nishio. 2002. Hydrogen production of Enterobacter aerogenes altered by extracellular and intracellular redox states. Int. J. Hydrogen Energy 27:1399-1405. [Google Scholar]
- 31.Neuer, G., and H. Bothe. 1982. The pyruvate:ferredoxin oxidoreductase in heterocysts of the cyanobacteriuim Anabaena cylindrica. Biochim. Biophys. Acta 716:358-365. [DOI] [PubMed] [Google Scholar]
- 32.Ogata, H., et al. 2005. Activation process of [NiFe] hydrogenase elucidated by high-resolution X-ray analyses: conversion of the ready to the unready state. Structure 13:1635-1642. [DOI] [PubMed] [Google Scholar]
- 33.Plaga, W., F. Lottspeich, and D. Oesterhelt. 1992. Improved purification, crystallization and primary structure of pyruvate:ferredoxin oxidoreductase from Halobacterium halobium. Eur. J. Biochem. 205:391-397. [DOI] [PubMed] [Google Scholar]
- 34.Plas, J., et al. 1988. Genes encoding ferredoxins from Anabaena sp. PCC 7937 and Synechococcus sp. PCC 7942: structure and regulation. Photosynth. Res. 18:179-204. [DOI] [PubMed] [Google Scholar]
- 35.Ragsdale, S. W. 2003. Pyruvate ferredoxin oxidoreductase and its radical intermediate. Chem. Rev. 103:2333-2346. [DOI] [PubMed] [Google Scholar]
- 36.Roberts, G. P., T. MacNeil, D. MacNeil, and W. J. Brill. 1978. Regulation and characterization of protein products coded by the nif (nitrogen fixation) genes of Klebsiella pneumoniae. J. Bacteriol. 136:267-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roughan, P. G., and J. B. Ohlrogge. 1994. On the assay of acetyl-CoA synthetase activity in chloroplasts and leaf extracts. Anal. Biochem. 216:77-82. [DOI] [PubMed] [Google Scholar]
- 38.Sakamoto, T., and D. A. Bryant. 1997. Growth at low temperature causes nitrogen limitation in the cyanobacterium Synechococcus sp. PCC 7002. Arch. Microbiol. 169:10-19. [DOI] [PubMed] [Google Scholar]
- 39.Schmitz, O., J. Gurke, and H. Bothe. 2001. Molecular evidence for the aerobic expression of nifJ, encoding pyruvate:ferredoxin oxidoreductase, in cyanobacteria. FEMS Microbiol. Lett. 195:97-102. [DOI] [PubMed] [Google Scholar]
- 40.Schut, G. J., and M. W. W. Adams. 2009. The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J. Bacteriol. 191:4451-4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shinkarev, V. P. 2003. Oxygen evolution in photosynthesis: simple analytical solution for the Kok model. Biophys. J. 85:435-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stevens, S. E., C. O. P. Patterson, and J. Myers. 1973. The production of hydrogen peroxide by blue-green algae: a survey. J. Phycol. 9:427-430. [Google Scholar]
- 43.Theodoratou, E., A. Paschos, S. Mintz-Weber, and A. Böck. 2000. Analysis of the cleavage site specificity of the endopeptidase involved in the maturation of the large subunit of hydrogenase 3 from Escherichia coli. Arch. Microbiol. 173:110-116. [DOI] [PubMed] [Google Scholar]
- 44.Troshina, O., L. Serebryakova, M. Sheremetieva, and P. Lindblad. 2002. Production of H2 by the unicellular cyanobacterium Gloeocapsa alpicola CALU 743 during fermentation. Int. J. Hydrogen Energy 27:1283-1289. [Google Scholar]
- 45.Wünschiers, R., M. Batur, and P. Lindblad. 2003. Presence and expression of hydrogenase specific C-terminal endopeptidases in cyanobacteria. BMC Microbiol. 3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu, Y. 2010. Ph.D. thesis. The Pennsylvania State University, University Park, PA.
- 47.Yoon, K.-S., R. Hille, C. Hemann, and F. R. Tabita. 1999. Rubredoxin from the green sulfur bacterium Chlorobium tepidum functions as an electron acceptor for pyruvate ferredoxin oxidoreductase. J. Biol. Chem. 274:29772-29778. [DOI] [PubMed] [Google Scholar]
- 48.Zhao, H., P. Wang, E. Huang, Y. Ge, and G. Zhu. 2008. Physiologic roles of soluble pyridine nucleotide transhydrogenase in Escherichia coli as determined by homologous recombination. Ann. Microbiol. 58:275-280. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.