

Abstract
The protozoan pathogen responsible for the most severe form of human malaria, Plasmodium falciparum, replicates asexually in erythrocytes within a membrane-bound parasitophorous vacuole (PV). Following each round of intracellular growth, the PV membrane (PVM) and host cell membrane rupture to release infectious merozoites in a protease-dependent process called egress. Previous work has shown that, just prior to egress, an essential, subtilisin-like parasite protease called PfSUB1 is discharged into the PV lumen, where it directly cleaves a number of important merozoite surface and PV proteins. These include the essential merozoite surface protein complex MSP1/6/7 and members of a family of papain-like putative proteases called SERA (serine-rich antigen) that are implicated in egress. To determine whether PfSUB1 has additional, previously unrecognized substrates, we have performed a bioinformatic and proteomic analysis of the entire late asexual blood stage proteome of the parasite. Our results demonstrate that PfSUB1 is responsible for the proteolytic processing of a range of merozoite, PV, and PVM proteins, including the rhoptry protein RAP1 (rhoptry-associated protein 1) and the merozoite surface protein MSRP2 (MSP7-related protein-2). Our findings imply multiple roles for PfSUB1 in the parasite life cycle, further supporting the case for considering the protease as a potential new antimalarial drug target.
Malaria is a huge global health threat, causing immense suffering and up to 2.7 million fatalities per annum worldwide, largely in children below the age of five. There is no licensed malaria vaccine, and while some effective drugs are available at present, the parasite often rapidly acquires resistance in the face of drug pressure. Recent reports of resistance to some of the latest artemisinin-based antimalarial drugs are particularly alarming (22, 76), stressing the need to expand our understanding of the basic biology of the malaria parasite and to seek parasite-specific processes that can be exploited as new drug targets. Malaria is caused by obligate intracellular protozoan parasites of the genus Plasmodium, transmitted by female Anopheline mosquitoes. Of the five Plasmodium species that cause human malaria, Plasmodium falciparum is responsible for the most acute disease.
Like other apicomplexan pathogens, P. falciparum is an obligate intracellular parasite, and all the clinical manifestations of malaria result from replication of the parasite in circulating erythrocytes. Following erythrocyte invasion, the parasite occupies a membrane-bound parasitophorous vacuole (PV) where it divides asexually to form a mature schizont containing 16 or more daughter merozoites. In a poorly understood process called egress, the enclosing PV and residual host erythrocyte membranes eventually rupture, releasing the merozoites, which at once invade fresh erythrocytes to perpetuate the cycle. Successive cycles of replication lead to increasing parasitemia and pathology. Both egress and subsequent invasion can be blocked by broad-spectrum inhibitors of serine and cysteine proteases, indicating a key role for proteases (reviewed in reference 7).
A number of serine proteases belonging to the subtilisin-like family (clan SB) (62) have been identified in apicomplexan parasites. These include enzymes of unknown function in Neospora (47) and Babesia (54), one in Cryptosporidium that may be involved in host cell invasion (23, 74), and two enzymes in Toxoplasma (51, 52), at least one of which also plays a role in invasion (43). The Plasmodium genome contains genes for just three subtilisin-like proteases, of which two—SUB1 and SUB2—appear to be indispensable in asexual blood stages (73, 78). Recent work in P. falciparum (78) has begun to reveal details of a proteolytic pathway in which egress is triggered by the discharge of P. falciparum SUB1 (PfSUB1) into the PV lumen (10, 77). There, PfSUB1 directly mediates the proteolytic maturation of a family of abundant, soluble, papain-like putative proteases called the serine-rich antigen (SERA) family, previously implicated in egress (1, 58, 78). Pharmacological inhibition of PfSUB1 activity very effectively blocks egress, indicating a direct role for PfSUB1 in regulating egress, possibly through activation of the SERA enzymes (3, 78). Work from this laboratory (42) has subsequently shown that PfSUB1 is also required for merozoite maturation. Upon its release into the PV just before egress, PfSUB1 additionally carries out the well described primary proteolytic processing of the most abundant merozoite surface component, an essential, glycolipid-anchored, heteromultimeric protein complex called MSP1/6/7 (merozoite surface proteins 1, 6, and 7) (40). MSP1, the largest protein in the complex, is cleaved into 4 fragments which remain noncovalently associated (30, 49), while both MSP6 and MSP7 (which are products of distinct genes) are also cleaved at one or more positions (56, 57, 70, 72). Correct regulation of MSP1 processing is essential for parasite viability (16), and merozoites released under conditions where this surface protein processing is even partially blocked are not invasive (42), implying that processing by PfSUB1 of the MSP1/6/7 merozoite surface complex in some way prepares or primes the parasite surface for invasion. The involvement of PfSUB1 in both maturation of merozoite surface proteins and merozoite release provides the first known mechanistic link between egress per se and the development of invasive parasites. However, our understanding of the molecular events that regulate and mediate egress is poor. Nothing is known of the mechanisms leading to destabilization of the PV and host cell membranes or how PfSUB1-mediated modifications to merozoite surface proteins modulate parasite invasiveness.
Besides MSP1/6/7 and some SERA family proteins, numerous parasite proteins have been localized to the merozoite surface and the PV lumen and membrane (PVM). Our work implicating PfSUB1 in MSP1/6/7 processing identified it as a multifunctional enzyme and raised the possibility that PfSUB1 might have additional substrates which would become available to it following its release into the PV. Here, we have explored this possibility. In a systematic, three-pronged approach, we first exploited existing knowledge of the enzyme's substrate specificity, based on known macromolecular and small-peptide substrates of PfSUB1, to perform an in silico bioinformatic search of the entire predicted P. falciparum proteome. This resulted in the identification of a set of candidate substrates containing one or more putative PfSUB1 cleavage sites. In a second, experimental proteomic step, we made use of the stringent specificity of the enzyme to identify blood stage parasite proteins that are susceptible to cleavage by PfSUB1 under native conditions ex vivo. Finally, we used antibodies specific to selected candidate proteins of particular interest to confirm that these are indeed subject to endogenous PfSUB1-mediated processing in vivo during the parasite life cycle. Our results demonstrate that PfSUB1 has multiple substrates that include proteins of the merozoite, PV, and PVM, suggesting hitherto unsuspected roles for PfSUB1 in the asexual blood stage life cycle of the parasite.
MATERIALS AND METHODS
Parasite culture.
Maintenance in vitro of asexual blood stages of P. falciparum clone 3D7 and purification of mature schizonts were as described previously (8).
Protease and prodomain production.
Enzymatically active recombinant PfSUB1 (rPfSUB1) and the PfSUB1 prodomain (PfSUB1PD; a selective, nanomolar inhibitor of PfSUB1 activity) were expressed in insect cells or Escherichia coli, respectively, and purified and quantified as previously described (29, 36, 77). One unit (U) of rPfSUB1 is defined as the amount of protease that hydrolyzes 1 pmol of the fluorogenic peptide substrate SERA5st1F-6R (78) in 1 min at a substrate concentration of 0.2 μM in digestion buffer {25 mM Tris-HCl, pH 8.2, 12 mM CaCl2, 25 mM CHAPS [(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} at 21°C.
Antibodies and Western blot analysis.
Western blots were performed as described previously (36), probing with monoclonal antibody (MAb) X509 to detect MSP1 and its primary, 42-kDa processing product (12), MAb 24C6.1F1 (21) to detect cleavage of SERA5, MAb 2.29 (17) (a kind gift of Jana McBride, University of Edinburgh, United Kingdom) to detect processing of rhoptry-associated protein 1 (RAP1), and a polyclonal antibody to MSP7-related protein-2 (MSRP2) (38) (kindly provided by Madhu Kadekoppala, NIMR).
PoPS search parameters and in silico protein analysis.
A PfSUB1 cleavage site specificity model was created in the online application PoPS (Prediction of Protease Specificity; http://pops.csse.monash.edu.au/pops-cgi/) (13, 14) by combining information from previous analysis of PfSUB1 specificity using peptide substrates (42, 77) and established PfSUB1 cleavage sites in validated substrates (42, 78). The entire predicted P. falciparum proteome was downloaded from PlasmoDB (www.plasmoDB.org) in FASTA format, uploaded into PoPS, and then analyzed using the PfSUB1 cleavage site model. SignalP (http://www.cbs.dtu.dk/services/SignalP/) was used for prediction of classical signal peptides. TMPred (http://www.ch.embnet.org/software/TMPRED_form.html) was used to predict transmembrane helices, NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to identify similarity to other proteins, Interpro (http://www.ebi.ac.uk/interpro/) was used to search for known domains within the proteins (35), and JPred (http://www.compbio.dundee.ac.uk/∼www-jpred/) was used to predict secondary structure features and accessibility of cleavage site residues (18). The physical and chemical parameters of proteins were examined using the ProtParam online tool (http://www.expasy.ch/tools/protparam.html).
PfSUB1 digestion of parasite proteins.
For identification of membrane-associated putative PfSUB1 substrates, the purification of schizonts, treatment with a cocktail of protease inhibitors, saponin lysis, and washing of the permeabilized cells were as described previously (42). Treated schizonts were stored frozen in aliquots at −70°C until required. Approximately 2 × 109 (∼150 μl) saponin-treated schizonts were thawed into 1.2 ml ice-cold 25 mM HEPES, pH 7.4, 12 mM CaCl2 (digestion buffer), supplemented with protease inhibitors E64 (10 μM), pepstatin A (1 μM), and leupeptin and antipain (10 μg ml−1 each). The schizonts were pelleted and then washed once more before being finally resuspended into 400 μl of the same buffer and divided into two equal aliquots. One of these was supplemented with 30 μl (45 U) purified rPfSUB1 (sample PT+), while the other was supplemented with 20 μl (168 pmol) purified PfSUB1PD (sample PT−). Both samples were then incubated at 37°C for 2 h, with occasional mixing, before being solubilized by the addition of 1.6 ml 8 M urea, 25 mM CHAPS, 20 mM dithiothreitol (DTT) in 10 mM Tris-HCl, pH 8.2. Samples were mixed at room temperature for 45 min and then clarified by centrifugation and filtered (0.45-μm Nanosep MF GHP; Pall Life Sciences). Just prior to analysis by reversed-phase high-pressure liquid chromatography (RP-HPLC), samples were acidified by the addition of 3.2 μl neat trifluoroacetic acid (TFA; 0.2% [vol/vol] final concentration). For analysis of nonmembrane-associated putative PfSUB1 substrates, ∼4 × 109 purified schizonts were snap-frozen without prior treatment with protease inhibitors, then thawed into 1.6 ml ice-cold digestion buffer containing protease inhibitors as described above and clarified by centrifugation at 13,000 × g for 20 min at 4°C, and the supernatant recovered and divided into two equal aliquots. One of these was supplemented with 120 U purified rPfSUB1 as described above (sample ST+), while the other was supplemented with 60 μl (504 pmol) purified PfSUB1PD (sample ST−). Both samples were then incubated at 37°C for 1 h with occasional mixing, before being acidified by the addition of neat TFA to 0.2% (vol/vol) final concentration.
RP-HPLC protein fractionation and MS.
Samples were chromatographed on a Vydac 4.6-mm by 150-mm 214TP C4 RP-HPLC column at a flow rate of 1 ml min−1, using a gradient of 0 to 18% (vol/vol) acetonitrile over 20 min and then 18 to 63% (vol/vol) acetonitrile over the ensuing 40 min, all in 0.1% (vol/vol) TFA. Eluate fractions (65 fractions per run, 1 ml each) were collected, dried in a SpeedVac on low heat, and then each taken up in 40 μl of reducing SDS-PAGE sample buffer and subjected to SDS-PAGE on 1-mm-thick 8-to-16% linear-gradient gels (Invitrogen). Gel-separated proteins were visualized by staining with InstantBlue (Generon). Excision, alkylation, and in-gel tryptic digestion of selected polypeptides and analysis by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (MS) were performed as described previously (77). For LC-tandem mass spectrometry (MS-MS), polyacrylamide gel slices (1 to 2 mm) containing the purified proteins were prepared for analysis by using the Janus liquid handling system (PerkinElmer, United Kingdom). Briefly, the excised protein gel pieces were placed in wells of a 96-well microtiter plate, destained with 50% (vol/vol) acetonitrile and 50 mM ammonium bicarbonate, reduced with 10 mM DTT, and alkylated with 55 mM iodoacetamide. Alkylated proteins were digested with 6 ng μl−1 trypsin (Promega, United Kingdom) overnight at 37°C. The resulting peptides were extracted in 1% (vol/vol) formic acid, 2% (vol/vol) acetonitrile and analyzed by nanoscale capillary LC-MS-MS using a nanoAcquity UPLC (Waters, United Kingdom) to deliver a flow of 300 nl min−1. A C18 Symmetry 5-μm, 180-μm by 20-mm μ-precolumn (Waters, United Kingdom) trapped the peptides prior to separation on a C18 BEH130 1.7-μm, 75-μm by 250-mm analytical UPLC column (Waters, United Kingdom). The peptides were eluted with a gradient of acetonitrile. The analytical column outlet was interfaced with a Triversa NanoMate microfluidic chip for mass spectrometric analysis (Advion, United Kingdom). Mass spectrometric information was obtained using an orthogonal-acceleration quadrupole-time of flight mass spectrometer (Synapt HDMS; Waters, United Kingdom). Data-dependent analysis was carried out, for which automatic MS-MS data on the 8 most intense, multiply charged precursor ions in the m/z range of 400 to 1,500 were acquired. MS-MS data were acquired over the m/z range of 50 to 1,995. LC-MS-MS data were then searched against the UniProt KB (release 15.5) protein database using the Mascot search engine program (Matrix Science, United Kingdom) (60).
Peptide substrate assays.
For determination of cleavage specificity by rPfSUB1, N-acetylated (Ac) decapeptides based on RAP1 and MSRP2 processing sites were synthesized using standard Fmoc (9-fluorenylmethoxy carbonyl) chemistry, and stock solutions (80 mM in dimethyl sulfoxide) stored at −20°C. Incubation with rPfSUB1, fractionation of partially digested peptides by RP-HPLC, and identification of digestion products by electrospray mass spectrometry were as described previously (9, 77).
Purification and N-terminal sequence analysis of MSRP225.
Approximately 2.5 liters of culture supernatant was collected from P. falciparum cultures in which schizont rupture and reinvasion had been allowed to take place into protein-free medium, as described previously (33). Clarified supernatant was first supplemented with 1 M Tris-HCl, pH 8.2, to 50 mM and applied to a HiPrep Q XL 16/10 anion exchange column (GE Healthcare) to remove tightly binding anionic components. The flowthrough was then diluted 5-fold into 10 mM Tris-HCl, pH 8.9, and applied to a second HiPrep Q XL 16/10 column, and bound proteins eluted with a 400-ml gradient of 0 to 400 mM NaCl in 10 mM Tris-HCl, pH 8.9. The fractions containing the 25-kDa species of MSRP2 (MSRP225) (identified by Western blot analysis) were chromatographed on a Superdex 75 prep grade 26/60 column equilibrated in 20 mM Tris-HCl, 150 mM NaCl before being acidified by the addition of TFA to 0.2% (vol/vol) and fractionated on a Vydac 4.6-mm by 150-mm 214TP C4 RP-HPLC column as described above. Fractions containing purified MSRP225 were lyophilized and subjected to SDS PAGE, and proteins transferred electrophoretically to polyvinylidene difluoride membrane before being analyzed by Edman degradation by Mike Weldon at the Protein and Nucleic Acid Chemistry Facility, Cambridge University, United Kingdom, as previously described (34).
RESULTS
Bioinformatic identification of new putative PfSUB1 substrates.
Maturation of SERA5 by PfSUB1 involves cleavage at two nonpolymorphic positions, designated site 1 and site 2, which flank the central papain-like domain of SERA5, as well as at a third, polymorphic site called site 3 (78). Multiple alignment of all 8 members of the P. falciparum SERA family that are expressed in asexual blood stages (2, 50, 53) has shown the presence of homologous sequences at the positions corresponding to sites 1 and 2 in all gene products. This suggested that all of these other SERA proteins may also be substrates for PfSUB1, although experimental evidence for this has only been obtained for SERA4 and SERA6 (78). Processing of MSP1, MSP6, and MSP7 by PfSUB1 takes place at sequence motifs that in all cases share similarity with the known and putative SERA cleavage sites (42). Together with other data from analysis of PfSUB1 autoproteolytic maturation (66, 77), as well as from studies of a range of cleavable and noncleavable synthetic peptide substrates (42, 77), this has allowed us to propose a consensus PfSUB1 recognition motif of Ile/Leu/Val/Thr-Xaa-Gly/Ala-Paa(not Leu)↓Xaa (where Xaa is any amino acid residue and Paa tends to be a polar residue). We also observed a tendency for acidic residues, as well as Ser or Thr, at one or more of the proximal 5 positions on the prime side of the scissile bond (Fig. 1A). In the present study, we set out to make use of this information to perform an in silico search of the predicted P. falciparum proteome for additional parasite proteins containing putative PfSUB1 cleavage sites, anticipating that this would provide clues as to new candidate physiological PfSUB1 substrates.
FIG. 1.

Predicted substrate preference of PfSUB1 and its application to an in silico search for new substrates. (A) Graphical representation in single-letter code of a multiple-sequence alignment of amino acid residues flanking known and predicted PfSUB1 cleavage sites within SERA5 (sites 1, 2, and 3), SERA4 and SERA6 (predicted sites 1 and 2), and MSP1, MSP6, and MSP7 (a total of 17 different sequences), plus the internal PfSUB1 autocatalytic processing site at which cleavage occurs during protease maturation (66). The overall height of each stack of residues indicates the degree of sequence conservation at that position, while the height of each residue within the stack indicates the relative frequency of each amino acid residue at that position. Residues are color coded according to the chemical nature of their side chains (red, acidic [D and E]; blue, basic [K, R, and H]; orange, aliphatic [L, V, and I]; black, small [G and A]; green, uncharged polar [S, T, Y, N, and Q]; and purple, nonpolar, nonaliphatic [F and P]). The scissile bond is indicated by an arrow. Residue numbering is according to the system of Schechter and Berger (68). The figure was produced using the WebLogo facility at http://weblogo.berkeley.edu/logo.cgi and annotated with Adobe Photoshop. (B) Overview of PoPS-based in silico screen for candidate new PfSUB1 substrates. TMD, transmembrane domain.
Proteases usually recognize their substrates in an extended β-strand conformation, but in addition to simple primary sequence, surrounding secondary or tertiary structural elements in macromolecular substrates can be important in preventing or facilitating access to scissile bonds. We considered it likely, for example, that many potential PfSUB1 cleavage sites might be inaccessible in folded polypeptides, but also that characteristics other than simple primary sequence may be important determinants of susceptibility to cleavage. Relatively few of the structures of Plasmodium proteins have been determined at atomic resolution, but secondary structural features can be predicted in silico. In preliminary work to assess whether secondary structure characteristics could be incorporated into our predictions of new PfSUB1 substrates, the secondary structure of the 40 amino acid residues flanking the scissile bond(s) in each of the 18 known and putative PfSUB1 protein substrates referred to above was analyzed using the secondary structure prediction algorithm Jpred. Several sites, including those in MSP7, SERA4 site 2, and SERA6 site 1, were found to lie within what are predicted to be completely disordered regions (not shown). In other cases, while the scissile bond itself lies in a predicted disordered region, other residues forming part of the putative recognition sequence form part of an α-helix or β-strand; examples of these are the autocatalytic cleavage sites within PfSUB1, SERA5 site 1, SERA5 site 2, SERA5 site 3, SERA6 site 2, and SERA4 site 2 (not shown). Since these results indicated substantial diversity in the secondary structural features flanking established PfSUB1 cleavage sites, it was concluded that secondary structure predictions could not be used as criteria in subsequent screens for new PfSUB1 substrates.
PoPS (Prediction of Protease Specificity) is a computational application that enables modeling of protease specificity and the in silico prediction of potential protease substrates (13, 14). PoPS has been widely used for the prediction and identification of novel protease substrates (e.g., see references 25, 26, and 69). A PfSUB1 specificity model (see Table S1 in the supplemental material) was assembled for use in PoPS using the compiled substrate data shown in Fig. 1A and used to computationally scan the predicted proteome of P. falciparum (Fig. 1B). Application of the model to the entire set of predicted proteins annotated in the P. falciparum PlasmoDB database at the time of this study (n = 5,679) resulted in the identification of 2,086 primary hits (36.7%) containing one or more predicted PfSUB1 cleavage sites. To further constrain the list of possible PfSUB1 substrates, each primary hit was assessed according to a set of characteristics common to all previously validated PfSUB1 substrates. Most well-established merozoite surface, PV, and PVM proteins possess a predicted N-terminal secretory signal peptide, so primary hits lacking this feature were excluded, as were proteins with more than 1 predicted transmembrane domain. Proteins with a predicted molecular mass of ≥200 kDa were also excluded, as it was considered that these would be difficult to validate experimentally. Following these initial filters, the remaining primary hits were then included or excluded on the basis of evidence in the literature for expression in asexual blood stage schizonts. This included data from previous large-scale microarray or proteomic studies for transcription or protein expression in blood stage parasites (24, 27, 44, 45). All primary hits that were among a set of soluble proteins previously identified as components of the PV (55) were also included. The application of these criteria resulted in a final short list of most-likely candidate PfSUB1 substrates (see Table S2 in the supplemental material) comprising just 76 proteins (1.4% of predicted P. falciparum proteins). As expected, this final list contained the SERA family members SERA1 to -3, SERA7, and SERA9. It also contained a number of interesting new candidates, including the merozoite surface proteins MSP3, Pf12, Pf92, and MTRAP; members of the families of MSP7-like (MSRP) and MSP3-like merozoite/PV proteins; the rhoptry proteins RhopH3, RAMA, rhoptry-associated protein 1 (RAP1), and RAP2; the PV proteins GBP130 and S-antigen; and the PVM proteins EXP1 and PTEX150. Several of these have previously been shown to undergo proteolytic processing (see Table S2 in the supplemental material). Importantly, in two cases, those of MSP3 (59) and RAP1 (see below), the PfSUB1 processing sites predicted by our PoPS-based screen are identical to those experimentally mapped by N-terminal sequence analysis of purified cleavage products. The list of putative substrates also included several proteins, including hypothetical proteins, about which little or nothing is known. These results supported our starting hypothesis, indicating the presence in the parasite proteome of additional proteins that possess putative PfSUB1 cleavage sites and that also exhibit a temporal and spatial expression profile consistent with them being potential substrates for the protease.
Proteomic identification of new PfSUB1 substrates.
In previous work (42), we developed a simple cell-based assay suitable for the identification of membrane-associated PfSUB1 substrates, such as MSP1/6/7. Intact P. falciparum schizonts were first treated with a cocktail of broad-spectrum protease inhibitors (many of which are membrane permeable) designed to inhibit the activity of endogenous proteases, including PfSUB1. The schizonts were then exposed to the detergent saponin, which permeabilizes the host cell membrane and PVM, releasing most of the soluble erythrocyte cytosol and PV components and resulting in a preparation in which constituents of the intraerythrocytic merozoite surface, PV lumen, and PVM are accessible to exogenous solvent. The addition of rPfSUB1 to these preparations and incubation at 37°C resulted in efficient conversion of MSP1/6/7 precursors to smaller fragments indistinguishable from those on the surface of naturally released merozoites, thus mimicking natural PfSUB1-mediated processing (42). In view of the earlier success of this approach, we considered that it could form the basis in this work for a more extensive, proteome-wide search for additional potential PfSUB1 substrates, providing data that could be used to complement and validate the predictions of the in silico strategy described above. In an important modification of the original method, control schizont extract samples that were not incubated with rPfSUB1 were instead incubated with recombinant PfSUB1 prodomain (PfSUB1PD), a potent and selective PfSUB1 inhibitor (36), to suppress any residual endogenous PfSUB1 activity not inactivated by the initial treatment with broad-spectrum protease inhibitors. The workflow used for sample preparation of schizont membrane extracts is depicted in Fig. 2A. In preliminary experiments evaluating this protocol, careful examination of Coomassie blue-stained, SDS-PAGE fractionated, rPfSUB1-treated (PT+) and control (PT−) schizont membrane extracts (see Fig. S1A in the supplemental material) revealed that only a very small subset of the total protein population was detectably modified by incubation with rPfSUB1, attesting to the high specificity of rPfSUB1. Western blot analysis of the same samples (see Fig. S1B in the supplemental material) showed that, as expected, this subset of modified proteins included the previously identified PfSUB1 substrate MSP1. In order to examine the set of soluble parasite proteins depleted from the membrane fraction described above, a parallel workflow (Fig. 2B) was also implemented in which soluble proteins released from schizonts by simple freeze-thaw were also examined. In this case, schizonts were not pretreated with protease inhibitors, since it would not have been possible to remove residual inhibitors from the protein preparations by washing. The resulting rPfSUB1-treated and control soluble schizont extracts are referred to as ST+ and ST−, respectively (Fig. 2B). Western blot analysis of these extracts showed, as expected, the presence of the SERA5 P126 precursor (which is a predominantly soluble component of the PV); this was present in full-length form in the ST− preparations and in processed form in the ST+ samples as a result of processing by rPfSUB1 (see Fig. S1C in the supplemental material). Encouraged by all these observations, we decided to use this approach to perform a proteomic search for additional potential PfSUB1 substrates.
FIG. 2.

Sample workflow for proteomic identification of schizont proteins susceptible to digestion ex vivo by rPfSUB1. (A) Preparation, rPfSUB1 digestion, and evaluation of schizont membrane extracts. (B) Preparation, rPfSUB1 digestion, and evaluation of schizont soluble protein extracts. (C) Solubilization, RP-HPLC fractionation, comparative SDS-PAGE, and mass spectrometric analysis of control (PT− and ST−) and PfSUB1-digested (PT+ and ST+) samples. +/−, with or without.
Attempts to identify PfSUB1-modified proteins by direct one-dimensional SDS-PAGE fractionation of total treated schizont extracts (such as those depicted in Fig. S1A in the supplemental material) followed by in-gel tryptic digestion and mass spectrometry proved unsuccessful, probably due to comigration of multiple proteins in the very complex protein profiles obtained. Accordingly, to reduce the complexity of the protein profiles, as well as to provide an additional means of visualizing differences between rPfSUB1-treated and untreated samples, schizont extracts were fractionated by RP-HPLC prior to SDS-PAGE (Fig. 2C). The major advantages of this approach compared to other two-dimensional (2D) protein separation methodologies, such as 2D-gel electrophoresis, include the high binding capacity of silica-based C18 reversed-phase matrices, their insensitivity to the high urea and detergent concentrations used for membrane protein solubilization, the high resolution of gradient-elution RP-HPLC, and the use of entirely volatile solvents for column elution. The results in Fig. 3A show that, consistent with the SDS-PAGE profiles of the total extracts, small but reproducible differences were evident in the RP-HPLC elution profiles of the rPfSUB1-treated and control samples. This was confirmed by comparative SDS-PAGE analysis of corresponding RP-HPLC fractions (Fig. 3B), where clear differences were observed between equivalent RP-HPLC fractions from the rPfSUB1-treated and control untreated schizont extracts. Bands that visibly differed between equivalent pairs of fractions, representing proteins that were likely either substrates or products of PfSUB1 digestion, were excised and tryptic digests analyzed by MALDI-TOF MS or LC-MS-MS.
FIG. 3.

Fractionation of rPfSUB1-digested schizont extracts reveals protein substrates of PfSUB1. (A) Comparative C4 RP-HPLC elution profiles of rPfSUB1-digested (PT+) and control (PT-) schizont membrane extracts. The profiles were largely very similar, consistent with cleavage of only a small fraction of the total protein population by rPfSUB1 (see Fig. S1A in the supplemental material). However, some differences were evident (arrows). Similar results were obtained upon RP-HPLC fractionation of ST+ and ST− samples (data not shown). (B) Representative InstantBlue-stained SDS-PAGE gel of RP-HPLC-fractionated PT+ and PT− samples, indicating species modified by incubation with rPfSUB1 (arrows) as determined by visual comparison of equivalent RP-HPLC eluate fractions. The complete set of SDS-PAGE-analyzed RP-HPLC fractions, annotated with protein identities as determined by LC-MS-MS of excised bands, is shown in Fig. S2 in the supplemental material. MW, molecular weight.
A full set of representative SDS-PAGE gels from analysis of the RP-HPLC-fractionated PT+, PT−, ST+, and ST− samples are shown in Fig. S2 in the supplemental material. Details of the 25 different proteins identified are summarized in Table 1. The initially most striking feature of the primary data set was that many of the bands observed to shift in response to rPfSUB1 treatment (43 out of a total of 102 bands examined; 42%) were identified as precursors or fragments of the previously known PfSUB1 substrates SERA4, SERA5, MSP1, and MSP7. This observation alone strongly supports the validity of our approach. The fact that these particular proteins were so readily detectable is consistent with previous proteomic data indicating that they are all relatively abundant schizont proteins (44, 45). Several of the additional, new parasite proteins identified as being susceptible to cleavage by PfSUB1 are unlikely to represent authentic substrates for the enzyme, as they probably exist in predominantly nuclear or cytosolic locations in the parasite. Notable examples of these include the putative DNA/RNA binding protein Alba (PF08_0074), a putative histone binding protein (PFL0280c), a putative 60S ribosomal protein (PFE0845c), and elongation factor 1-α (PF13_0304). Merozoite capping protein 1 (PF10_0268) may be another example of this class of protein, as it has been reported to localize to the merozoite cytoskeleton or inner membrane complex, flattened cisternae that lie beneath the merozoite plasma membrane (41). All five of these proteins were found to contain predicted PfSUB1 sites in our initial PoPS-based screen but were excluded as possible substrates on the basis of their predicted or established location and so are not included in the “most likely” list shown in Table S2 in the supplemental material. Similarly, MESA/PfEMP2 is thought to be expressed primarily in association with the cytoskeleton of the parasitized host erythrocyte (6). Although it is certainly not possible to rule out any of these proteins as potential PfSUB1 substrates, false positives arising from nonspecific cleavage in the in vitro system used here were not unexpected, given that it very likely allowed exposure to the rPfSUB1 of a range of parasite proteins that would not under normal physiological conditions come into contact with endogenous PfSUB1. In contrast, of the other new proteins identified by the proteomic analysis, several are known to localize primarily to the PV (e.g., GBP130, MSP9/ABRA, and S-antigen) or the PVM (EXP1 and PTEX150). Intriguingly, three of the identified proteins (RAP1, RAMA, and RhopH3) localize to the rhoptries, paired, flask-shaped secretory organelles that reside at the apical end of the merozoite; all three of these proteins have been previously reported to be subjected to proteolytic processing in asexual blood stages (Table 1 and below; also see Table S2 in the supplemental material). Of further interest, the newly identified proteins included human α- and β-spectrin, abundant components of the erythrocyte cytoskeleton. These were the only erythrocyte components detected in the screen. Overall, of the additional new putative PfSUB1 substrates identified by the proteomic analysis, 12 out of the 23 parasite-derived proteins (52%) were also predicted as substrates by the PoPS-based in silico screen described above. Collectively, our results provide evidence for the existence of a set of parasite and host erythrocyte proteins that are susceptible to PfSUB1-mediated proteolysis in their native state. These could form a new set of physiological PfSUB1 substrates, in addition to the previously recognized MSP1/6/7 and SERA family proteins.
TABLE 1.
Putative PfSUB1 substrates identified by proteomic analysis
| ID no.a | PlasmoDB (or NCBIb) accession no. | Gene product | Present in indicated fraction |
Contains predicted cleavage site (PoPS) | Predicted PfSUB1 substratec | Known or probable subcellular locationd | Published evidence of processing | |
|---|---|---|---|---|---|---|---|---|
| ST (soluble) | PT (membrane) | |||||||
| 1 | PF10_0159 | GBP130 | ✓ | ✓ | ✓ | ✓ | PV | 61 |
| 2 | PF10_0361 | Unknown protein | ✗ | ✓ | ✓ | ✗ | ND | |
| 3 | PFB0340c | SERA5 | ✓ | ✓ | ✓ | ✓ | PV | 19, 20, 46, 78 |
| 4 | PF08_0074 | DNA/RNA binding protein Alba | ✗ | ✓ | ✓ | ✗ | ND | |
| 5 | PF11_0111 | Asparagine-rich antigen | ✗ | ✓ | ✓ | ✗ | ND | |
| 6 | PFE0845c | 60S ribosomal protein L8 | ✗ | ✓ | ✓ | ✗ | ND | |
| 7 | PFE0040c | MESA/PfEMP2 | ✓ | ✗ | ✗ | ✗ | MS | |
| 8 | PFL0105w | Unknown protein | ✗ | ✓ | ✗ | ✗ | ND | |
| 9 | PF14_0344 | PTEX150 (translocon protein) | ✗ | ✓ | ✓ | ✓ | ND | |
| 10 | PF13_0304 | Elongation initiation factor 1α | ✓ | ✓ | ✓ | ✗ | ND | |
| 11 | PFI1475w | MSP1 | ✓ | ✓ | ✓ | ✓ | MS | 11, 12, 31, 48 |
| 12 | PFB0345c | SERA4 | ✓ | ✓ | ✓ | ✓ | PV | 78 |
| 13 | PF13_0197 | MSP7 | ✓ | ✓ | ✓ | ✓ | MS | 56, 57 |
| 14 | PF10_0268 | MCP1 | ✗ | ✓ | ✓ | ✓ | Cytosol | |
| 15 | PF14_0700 | DNAJ protein | ✗ | ✓ | ✓ | ✗ | ND | |
| 16 | PFL0280c | Histone binding protein | ✗ | ✓ | ✗ | ✗ | ND | |
| 17 | PF08_0054 | Heat shock protein 70 | ✓ | ✓ | ✓ | ✗ | PV | |
| 18 | Q5VYL1 | Human α-spectrin | ✗ | ✓ | Erythrocyte | |||
| 18 | Q59FP5 | Human β-spectrin | ✗ | ✓ | Erythrocyte | |||
| 19 | PF11_0224 | EXP1 | ✗ | ✓ | ✓ | ✓ | PVM | |
| 20 | PFL1385c | MSP9/ABRA | ✓ | ✓ | ✗ | ✗ | PV | |
| 21 | PF14_0102 | RAP1 | ✓ | ✓ | ✓ | ✓ | Rhoptry | 32, 65 |
| 22 | PF10_0343 | S antigen | ✓ | ✗ | ✓ | ✓ | PV | |
| 23 | PFI0265c | RhopH3 | ✗ | ✓ | ✓ | ✓ | Rhoptry | 67 |
| 24 | P04264 | Human keratin (type II cytoskeletal) | ✓ | ✗ | ||||
| 25 | MAL7P1.208 | RAMA | ✓ | ✗ | ✓ | ✓ | Rhoptry | 63, 71 |
ID, identification; See Fig. S2 in the supplemental material.
NCBI accession number provided for nonparasite proteins only.
See Table S2 in the supplemental material.
PV, parasitophorous vacuole; PVM, parasitophorous vacuole membrane; MS, merozoite surface; ND, not determined.
Confirmation of MSRP2 as an authentic PfSUB1 substrate.
To test the above hypothesis, we examined in detail two of the new predicted PfSUB1 substrates. MSP7, one of the previously known PfSUB1 substrates, forms a protein complex together with MSP1 and MSP6 which uniformly coats the merozoite surface. MSP7 is encoded by one of a cluster of six related genes on chromosome 13 of the P. falciparum genome. The other 5 members of this gene family are referred to as MSP7-related proteins, or MSRPs (see reference 37 for a recent review). Recent work from Kadekoppala et al. (38) has shown that the protein product of only one MSRP, MSRP2 (MAL13P1.174), is detectable in asexual blood stage parasites and that this localizes to the PV, where it exists predominantly in two forms, of 35 kDa (thought to be the full-length protein) and 28 kDa. Upon schizont rupture, MSRP2 is released into culture medium as a fully soluble 25-kDa species (MSRP225), presumably resulting from proteolytic truncation of the PV-located forms. The timing of this conversion to the terminal 25-kDa form is consistent with a role for PfSUB1 in this step. Our proteomic analysis did not detect MSRP2 in schizont extracts, but in view of the fact that MSRP2 was identified by our PoPS bioinformatic analysis as a putative PfSUB1 substrate, we decided to experimentally test this possibility. As shown in Fig. 4A, Western blot analysis of the soluble schizont extracts with antibodies raised against the C-terminal 146 residues of MSRP2 identified two proteins corresponding to the 28- and 35-kDa forms of the protein. Incubation with rPfSUB1 resulted in the conversion of these to a 25-kDa product of similar mobility to that naturally released into P. falciparum culture supernatants. This suggests that MSRP225 is indeed a product of PfSUB1 activity at or around the point of egress. The PoPS bioinformatic analysis (see Table S2 in the supplemental material) predicted two putative PfSUB1 cleavage sites in MSRP2. Previous work has shown that short synthetic peptides based on authentic PfSUB1 cleavage sites can act as good substrates for the protease, so to test our predictions, peptides based on the two predicted MSRP2 sites were assayed for sensitivity to cleavage. Peptide Ac-SLKGESEDNT was efficiently cleaved by rPfSUB1 at the predicted Glu-Ser bond (Fig. 4B). In contrast, a peptide based on the other predicted site, Ac-DIIGQGIFSL, which is less hydrophilic overall, was not cleaved (not shown). To attempt to unambiguously define the site of cleavage to produce MSRP225, we developed a protocol to purify the protein from parasite culture supernatants, using a combination of ion-exchange, gel filtration, and RP-HPLC chromatography (no monoclonal antibodies are available to enable affinity purification). N-terminal sequence analysis of partially purified MSRP225 isolated from ∼250 ml of culture supernatant identified three major signals at each cycle of Edman degradation (Fig. 4C), indicating heterogeneity likely derived from comigrating contaminating proteins. Of the possible sequences these signals correspond to, only SEDNTK (Ser95 to Lys100) is a match to the MSRP2 sequence. Collectively, these results strongly support our prediction of MSRP2 as an authentic PfSUB1 substrate (Fig. 4D) and suggest that cleavage to produce MSRP25 takes place at the 90SLKGE↓SEDNTK100 motif (the arrow indicates the scissile bond). The calculated molecular mass of the sequence extending from Ser95 to Ile281 of MSRP2, representing the predicted C-terminal product of cleavage, is 21,846 Da, smaller than the apparent mass of the MSRP225 product. However, like many malarial proteins, MSRP2 probably migrates anomalously on SDS-PAGE gels, since the predicted mass of the full-length protein (32,800 Da) is considerably less than that of the putative full-length MSRP235 form detected on Western blots of schizont extracts.
FIG. 4.

MSRP2 is an authentic PfSUB1 substrate. (A) Western blot analysis of MSRP2 processing. Soluble schizont extracts without incubation (START) or after incubation for 1 h at 37°C with purified PfSUB1PD (−) or rPfSUB1 (+) were fractionated by SDS-PAGE alongside total SDS extracts of merozoites, mature schizonts, or parasite culture supernatants. The blot was probed with a polyclonal antibody specific for the C-terminal region of MSRP2. The positions of migration of the various processed forms of MSRP2 described previously (38) are indicated; note that, as found in that same study, none of the MSRP2 species are found associated with merozoites. Furthermore, MSRP225 is not detected in schizonts but only in culture supernatants (s/n) following egress, consistent with it resulting from PfSUB1 activity. Asterisks indicate nonspecific bands detected by the anti-MSRP2 antibody, as described previously (38). (B) Analytical RP-HPLC fractionation of N-acetylated synthetic peptide Ac-SLKGESEDNT, incubated without (top) or with (bottom) rPfSUB1 to allow partial cleavage. Identities of major peaks are indicated. The N-terminal product of cleavage at the Glu-Ser bond was identified by electrospray mass spectrometry. The predicted C-terminal product of cleavage (NH2-SEDNT) was not observed in the RP-HPLC elution profile; it was assumed not to bind to the column due to its hydrophilic nature, as previously found for several PfSUB1 peptide substrates (42). (C) Purification and N-terminal sequencing of MSRP225 (arrow) isolated from parasite culture supernatants as described in Materials and Methods. The identity of the purified protein, shown after SDS-PAGE and Coomassie blue staining (right, run alongside approximately 1 μg each of molecular mass marker proteins), was confirmed by Western blot (left-hand side), probing with the same antibodies used in the experiment whose results are shown in panel A. The identities of the first 6 residues identified by Edman degradation are listed; three major phenylhydantoin signals (∼1 pmol each) were obtained at each cycle. Only the sequence SEDNTK matches that of MSRP2, and BLAST searches showed that it is not found elsewhere in the predicted P. falciparum proteome (data not shown). (D) Model of MSRP2 processing. Both MSRP235 and MSRP228 are present in mature schizonts, the latter arising from the former as a result of processing by an unknown enzyme (?). Conversion to MSRP225 takes place just prior to egress as a result of cleavage by PfSUB1 at 90SLKGE↓SEDNT99.
Confirmation of RAP1 as an authentic PfSUB1 substrate.
RAP1, which has been extensively studied by several groups for over 2 decades, was initially identified as an abundant component of the rhoptries, secretory organelles involved in host erythrocyte invasion and PV formation. RAP1 is synthesized as a short-lived, 84-kDa precursor which subsequently undergoes sequential proteolytic N-terminal truncation to form products of 82 kDa (p82) and 67 kDa (p67) (sometimes via a rare ∼70-kDa intermediate). The p82-to-p67 processing step appears to take place late in schizont maturation (15, 32), and as a result, p67 is abundant only in very mature schizonts and free merozoites. RAP1 forms heterodimeric interactions with one of two lower-molecular-mass rhoptry proteins, RAP2 and RAP3, and gene disruption studies have shown that truncation of RAP1 abolishes its interaction with these proteins, leading to a loss of RAP2 trafficking to rhoptries (4). The RAP1/2 or RAP1/3 complex is in turn trafficked through interactions between the N-terminal region of RAP1 (amino acid residues 22 to 55) and an essential glycosyl phosphatidylinositol-anchored rhoptry protein called RAMA, and recent evidence suggests that cleavage of RAP1 is required to release it from RAMA once in the rhoptry bulb (63). The protease that mediates this cleavage is unknown. Direct N-terminal amino acid sequence analysis by Ridley et al. (64) has shown that the p67 RAP1 product is a result of cleavage at residue Ala190 within the sequence 186GIVGA↓DEEAP195 (Fig. 5A). This RAP1 cleavage site is precisely the same as that predicted by our PoPS analysis to be susceptible to PfSUB1 cleavage (see Table S2 in the supplemental material). To address whether RAP1 is an authentic substrate for PfSUB1, PT+ and PT− samples similar to those used for the proteomic analysis were examined by Western blot in parallel with extracts of mature schizonts and merozoites taken directly from culture, probing with a monoclonal antibody against RAP1. As shown by the results in Fig. 5B, incubation of the PT extracts in vitro with rPfSUB1 resulted in conversion of the RAP1 p82 to a product indistinguishable from the authentic, endogenous, parasite-derived p67 present in merozoites and mature schizonts. This supports the prediction that RAP1 is a PfSUB1 substrate. To obtain further evidence for this, a synthetic decapeptide based on the sequence encompassing the RAP1 p67 cleavage site was examined for susceptibility to cleavage by rPfSUB1. As shown by the results in Fig. 4C, the peptide was efficiently and specifically cleaved at the Ala-Asp bond. These results strongly support our contention that PfSUB1 is the endogenous parasite protease responsible for conversion of RAP1 to its p67 form.
FIG. 5.
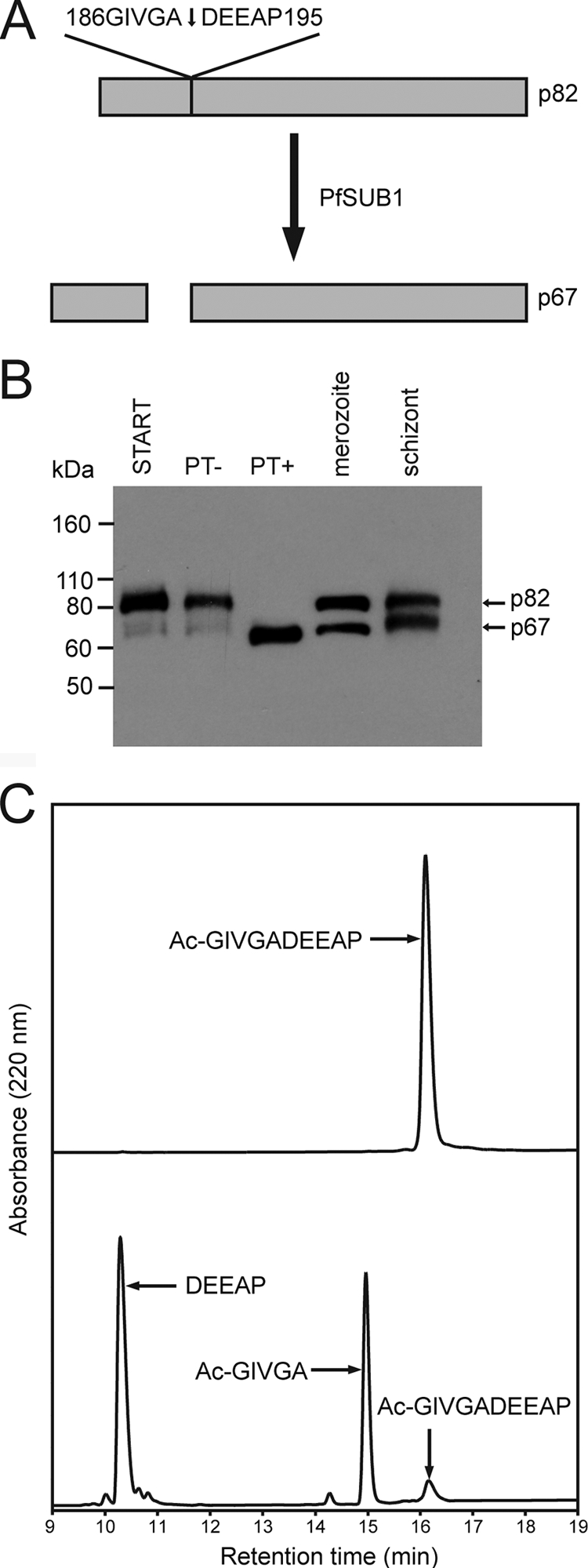
RAP1 is an authentic PfSUB1 substrate. (A) Schematic depicting conversion of RAP1 p82 to p67, based on published literature referenced in the text. The known processing site, 186GIVGA↓DEEAP195, is a putative PfSUB1 cleavage site as predicted by the PoPS analysis (see Table S1 in the supplemental material). (B) Western blot analysis of RAP1 processing. Schizont membrane extracts without incubation (START) or after incubation for 1 h at 37°C with purified PfSUB1PD (PT−) or rPfSUB1 (PT+) were fractionated by SDS-PAGE alongside total SDS extracts of merozoites or mature schizonts. The blot was probed with the RAP1-specific MAb 2.29. The positions of migration of the processed forms of RAP1 are indicated. (C) Analytical RP-HPLC fractionation of N-acetylated synthetic peptide Ac-GIVGADEEAP, incubated without (top) or with (bottom) rPfSUB1 to allow partial cleavage. Identities of major substrate and product peaks (established by electrospray mass spectrometry) are indicated.
DISCUSSION
In earlier work, we demonstrated that the release of PfSUB1 into the PV in the moments prior to egress enables it to access and modify a number of parasite surface and PV proteins implicated in either egress or host cell invasion. The present study has now extended those observations, presenting evidence that PfSUB1 has additional substrates. These findings expand the potential role(s) of PfSUB1 to modification of proteins of the rhoptries and, possibly, even the PVM and parasitized host cell cytoskeleton.
Our study relied on two conceptually simple but complementary approaches: first, to use existing knowledge of PfSUB1 substrate specificity to search the predicted P. falciparum proteome for proteins bearing potential PfSUB1 cleavage sites, and second, to exploit the stringent specificity of rPfSUB1 to experimentally identify parasite proteins susceptible to cleavage under native conditions in vitro. The significant potential for false positives using both approaches was apparent to us from the start. Our current understanding of PfSUB1 substrate specificity is likely incomplete, based as it is on only a rather limited number (18 at the inception of this work) of known substrate sequences. We were therefore not surprised to obtain a large number of hits in our initial PoPS-based screen, representing nearly 38% of all predicted P. falciparum proteins. However, by subjecting these candidates to a series of filters, we arrived at a short list of 76 most-likely candidate substrates, each of which contain 1 or more predicted cleavage sites. In the case of two of these proteins, MSP3 and RAP1, the PoPS-based screen predicted cleavage by PfSUB1 at precisely the same bonds as those previously experimentally determined by N-terminal sequencing studies (59, 64). As with the PoPS-based search, we also fully expected that our in vitro digestion approach used to experimentally identify parasite proteins susceptible to cleavage by rPfSUB1 could produce false positives, due to the expected PfSUB1-mediated cleavage of proteins that, in vivo, would not be accessible to PfSUB1. These could include parasite cytosolic, nuclear, and host cell proteins. Remarkably, however, the results in fact strongly suggested that this was not a common occurrence; the very fact that a large proportion of the species that shifted in mobility following digestion turned out to correspond to previously known or suspected substrates (e.g., SERA5, MSP1, and MSP7) attests to the specificity of PfSUB1 and supports the validity of the approach. Of the other PfSUB1-sensitive proteins identified by the proteomic analysis, while some were considered possible false positives, others included known PV and merozoite proteins, several of which have been reported to be subject to proteolytic processing at some point(s) in the asexual blood stage parasite life cycle. Importantly, several of these were also present in our list of PoPS-derived predictions.
Of the new putative PfSUB1 substrates identified, we chose two to examine in more detail. MSRP2 was identified as a putative substrate by the PoPS prediction but was not detected in the proteomic analysis, perhaps due to relatively low abundance. MSRP2 is of interest because it is a member of a family of proteins that are structurally related to MSP7, a component of the merozoite surface MSP1/6/7 complex. Unlike MSP7, however, MSRP2 exists predominantly as a soluble component of the PV lumen. We showed that the two forms of MSRP2 present in schizonts could be converted by rPfSUB1 to a smaller form indistinguishable from that shed into culture supernatants following egress, consistent with a role for PfSUB1 in its endogenous processing. In a further demonstration of the utility of the PoPS model, a peptide based on one of the two predicted cleavage sites within MSRP2, 90SLKGE↓SEDNT99, was specifically cleaved by rPfSUB1 at the predicted bond, suggesting that this corresponds to the site at which cleavage occurs to produce the terminal MSRP225 species. Confirmation of this was finally obtained by N-terminal sequencing of MSRP225 isolated from parasite culture supernatants.
The second predicted substrate that we examined in detail was the rhoptry protein RAP1. In this case, the site at which cleavage occurs to convert the p82 form to p67 had already been established by Ridley and colleagues (64). We showed that rPfSUB1 could mediate this conversion in vitro and that a peptide based on the known site was correctly cleaved by the protease. We conclude that RAP1 is an authentic PfSUB1 substrate. The function of RAP1 cleavage is unknown, although there is evidence from others that erythrocyte invasion-inhibitory MAbs (including MAb 2.29 used in this study), which recognize epitopes on RAP1 adjacent to the cleavage site, act by directly interfering with this cleavage (28), in turn suggesting a role for RAP1 in invasion. Arguing against this is evidence from gene disruption studies that the expression of full-length RAP1 is not essential for maintenance of the asexual parasite blood stage life cycle (4). It is noteworthy that two other rhoptry proteins, RAMA and RhopH3, were also identified by both the PoPS prediction and the proteomic analysis. There is evidence that RAMA interacts with both the RAP1/2/3 complex and a high-molecular-weight rhoptry protein complex that includes RhopH3 (71). It is conceivable that RAP1, RAMA, and RhopH3 are processed simultaneously upon interaction with PfSUB1. More work will be required to elucidate the role of PfSUB1-mediated processing in the fate and function of these proteins.
An obvious question that arises from our identification of rhoptry proteins as PfSUB1 substrates is that of how they could be accessible to PfSUB1 in the parasite. Three possibilities occur to us. First, since the rhoptry membrane is effectively continuous with the merozoite plasma membrane once fusion has occurred at the apical duct (5), it is conceivable that soluble PfSUB1, once released from exonemes into the lumen of the PV, could have direct access from there to rhoptry-resident proteins. Alternatively, it is possible that some release of rhoptry components may take place prior to egress, as is the case with certain microneme proteins (39, 75); this would also render the rhoptry proteins accessible to PV-located PfSUB1. A third provocative possibility is that exoneme discharge involves their fusion not just with the parasite plasma membrane but also directly with the cytoplasmic face of the rhoptry membrane (which would topologically be equivalent to fusion with the plasma membrane). This would provide a reliable mechanism for timely and tightly regulated processing of rhoptry proteins just prior to their subsequent discharge during invasion. Further work will be required to address these possibilities.
Egress involves the destabilization and rupture of at least two membranes—the PVM and the host erythrocyte membrane. It is therefore of particular interest that two established PVM proteins, EXP1 and PTEX150, were identified as putative PfSUB1 substrates here, by both the PoPS prediction and proteomic approaches. Proteolytic processing of either protein has not been reported, so the significance of this finding is unclear, but it is conceivable that PfSUB1 could be directly involved in modification of the PVM at egress. Similarly interesting is the evidence that human α- and β-spectrin, abundant components of the erythrocyte cytoskeleton, are susceptible to PfSUB1-mediated cleavage. Bearing in mind the caveats expressed above regarding the possibility of false positives, this observation is of interest since degradation of the erythrocyte cytoskeleton has been suggested to play a role in rupture of the host cell during egress (for a recent review of this, see reference 7). Future work will address the significance of these findings. For the present, our findings are intriguing, but we believe that extreme caution is required in considering spectrin, EXP1, or PTEX150 as an authentic PfSUB1 substrate.
In conclusion, this study has shown that PfSUB1 may have several distinct roles in the modification of parasite and, possibly, even host proteins. A focus of ongoing work is to elucidate which of these many PfSUB1-mediated processing steps are critical for parasite survival. PfSUB1 is a “druggable” enzyme (78), so the evidence that PfSUB1 has multiple functions strengthens the case for considering it as a target for antimalarial drug development, since the capacity for coevolution of the protease and its many substrates in response to protease inhibitor-based drugs is minimal. This would be expected to limit the rate of emergence of parasites resistant to anti-PfSUB1 drugs.
Supplementary Material
Acknowledgments
This work was supported by the UK Medical Research Council (grant U117532063) and Cancer Research UK, as well as the European Union Framework Programme 7 grant MALSIG (grant 223044). N.C.S.D.M. is in receipt of a Medical Research Council Ph.D. studentship. M.G.C. is supported by a fellowship from the Fundação para a Ciência ea Tecnologia (FCT) (SFRH/BPD/36651/2007).
We are grateful to Steve Howell for performing the MALDI-TOF analysis, to Mike Weldon for the N-terminal sequencing, and to Jana McBride for the gift of MAb 2.29.
Editor: J. H. Adams
Footnotes
Published ahead of print on 10 January 2011.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Aly, A. S., and K. Matuschewski. 2005. A malarial cysteine protease is necessary for Plasmodium sporozoite egress from oocysts. J. Exp. Med. 202:225-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoki, S., et al. 2002. Serine repeat antigen (SERA5) is predominantly expressed among the SERA multigene family of Plasmodium falciparum, and the acquired antibody titers correlate with serum inhibition of the parasite growth. J. Biol. Chem. 277:47533-47540. [DOI] [PubMed] [Google Scholar]
- 3.Arastu-Kapur, S., et al. 2008. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat. Chem. Biol. 4:203-213. [DOI] [PubMed] [Google Scholar]
- 4.Baldi, D. L., et al. 2000. RAP1 controls rhoptry targeting of RAP2 in the malaria parasite Plasmodium falciparum. EMBO J. 19:2435-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bannister, L. H., J. M. Hopkins, R. E. Fowler, S. Krishna, and G. H. Mitchell. 2000. Ultrastructure of rhoptry development in Plasmodium falciparum erythrocytic schizonts. Parasitology 121(Pt. 3):273-287. [DOI] [PubMed] [Google Scholar]
- 6.Black, C. G., et al. 2008. In vivo studies support the role of trafficking and cytoskeletal-binding motifs in the interaction of MESA with the membrane skeleton of Plasmodium falciparum-infected red blood cells. Mol. Biochem. Parasitol. 160:143-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blackman, M. J. 2008. Malarial proteases and host cell egress: an “emerging” cascade. Cell. Microbiol. 10:1925-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackman, M. J. 1994. Purification of Plasmodium falciparum merozoites for analysis of the processing of merozoite surface protein-1. Methods Cell Biol. 45:213-220. [DOI] [PubMed] [Google Scholar]
- 9.Blackman, M. J., et al. 2002. Structural and biochemical characterization of a fluorogenic rhodamine-labeled malarial protease substrate. Biochemistry 41:12244-12252. [DOI] [PubMed] [Google Scholar]
- 10.Blackman, M. J., et al. 1998. A subtilisin-like protein in secretory organelles of Plasmodium falciparum merozoites. J. Biol. Chem. 273:23398-23409. [DOI] [PubMed] [Google Scholar]
- 11.Blackman, M. J., H. G. Heidrich, S. Donachie, J. S. McBride, and A. A. Holder. 1990. A single fragment of a malaria merozoite surface protein remains on the parasite during red cell invasion and is the target of invasion-inhibiting antibodies. J. Exp. Med. 172:379-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blackman, M. J., H. Whittle, and A. A. Holder. 1991. Processing of the Plasmodium falciparum major merozoite surface protein-1: identification of a 33-kilodalton secondary processing product which is shed prior to erythrocyte invasion. Mol. Biochem. Parasitol. 49:35-44. [DOI] [PubMed] [Google Scholar]
- 13.Boyd, S. E., M. Garcia de la Banda, R. N. Pike, J. C. Whisstock, and G. B. Rudy. 2004. PoPS: a computational tool for modeling and predicting protease specificity, p. 372-381. Proceedings of the 2l004 IEEE Computational Systems Bioinformatics Conference, International IEEE Computer Society, Los Alamitos, CA. [DOI] [PubMed]
- 14.Boyd, S. E., R. N. Pike, G. B. Rudy, J. C. Whisstock, and M. Garcia de la Banda. 2005. PoPS: a computational tool for modeling and predicting protease specificity. J. Bioinform. Comput. Biol. 3:551-585. [DOI] [PubMed] [Google Scholar]
- 15.Bushell, G. R., L. T. Ingram, C. A. Fardoulys, and J. A. Cooper. 1988. An antigenic complex in the rhoptries of Plasmodium falciparum. Mol. Biochem. Parasitol. 28:105-112. [DOI] [PubMed] [Google Scholar]
- 16.Child, M. A., C. Epp, H. Bujard, and M. J. Blackman. 2010. Regulated maturation of malaria merozoite surface protein-1 is essential for parasite growth. Mol. Microbiol. 78:187-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark, J. T., R. Anand, T. Akoglu, and J. S. McBride. 1987. Identification and characterisation of proteins associated with the rhoptry organelles of Plasmodium falciparum merozoites. Parasitol. Res. 73:425-434. [DOI] [PubMed] [Google Scholar]
- 18.Cole, C., J. D. Barber, and G. J. Barton. 2008. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36:W197-W201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debrabant, A., et al. 1992. Intramolecular mapping of Plasmodium falciparum P126 proteolytic fragments by N-terminal amino acid sequencing. Mol. Biochem. Parasitol. 53:89-95. [DOI] [PubMed] [Google Scholar]
- 20.Delplace, P., et al. 1988. Protein p126: a parasitophorous vacuole antigen associated with the release of Plasmodium falciparum merozoites. Biol. Cell 64:215-221. [DOI] [PubMed] [Google Scholar]
- 21.Delplace, P., J. F. Dubremetz, B. Fortier, and A. Vernes. 1985. A 50 kilodalton exoantigen specific to the merozoite release-reinvasion stage of Plasmodium falciparum. Mol. Biochem. Parasitol. 17:239-251. [DOI] [PubMed] [Google Scholar]
- 22.Dondorp, A. M., et al. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361:455-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng, X., D. E. Akiyoshi, G. Widmer, and S. Tzipori. 2007. Characterization of subtilase protease in Cryptosporidium parvum and C. hominis. J. Parasitol. 93:619-626. [DOI] [PubMed] [Google Scholar]
- 24.Florens, L., et al. 2002. A proteomic view of the Plasmodium falciparum life cycle. Nature 419:520-526. [DOI] [PubMed] [Google Scholar]
- 25.Furmonaviciene, R., et al. 2007. The protease allergen Der p 1 cleaves cell surface DC-SIGN and DC-SIGNR: experimental analysis of in silico substrate identification and implications in allergic responses. Clin. Exp. Allergy 37:231-242. [DOI] [PubMed] [Google Scholar]
- 26.Golubkov, V. S., et al. 2005. Membrane type-1 matrix metalloproteinase (MT1-MMP) exhibits an important intracellular cleavage function and causes chromosome instability. J. Biol. Chem. 280:25079-25086. [DOI] [PubMed] [Google Scholar]
- 27.Hall, N., et al. 2005. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science 307:82-86. [DOI] [PubMed] [Google Scholar]
- 28.Harnyuttanakorn, P., J. S. McBride, S. Donachie, H. G. Heidrich, and R. G. Ridley. 1992. Inhibitory monoclonal antibodies recognise epitopes adjacent to a proteolytic cleavage site on the RAP-1 protein of Plasmodium falciparum. Mol. Biochem. Parasitol. 55:177-186. [DOI] [PubMed] [Google Scholar]
- 29.Harris, P. K., et al. 2005. Molecular identification of a malaria merozoite surface sheddase. PLoS Pathog. 1:241-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holder, A. A., et al. 1992. A malaria merozoite surface protein (MSP1)—structure, processing and function. Mem. Inst. Oswaldo Cruz 87(Suppl. 3):37-42. [DOI] [PubMed] [Google Scholar]
- 31.Holder, A. A., et al. 1987. Processing of the precursor to the major merozoite surface antigens of Plasmodium falciparum. Parasitology 94(Pt. 2):199-208. [DOI] [PubMed] [Google Scholar]
- 32.Howard, R. F., D. L. Narum, M. Blackman, and J. Thurman. 1998. Analysis of the processing of Plasmodium falciparum rhoptry-associated protein 1 and localization of Pr86 to schizont rhoptries and p67 to free merozoites. Mol. Biochem. Parasitol. 92:111-122. [DOI] [PubMed] [Google Scholar]
- 33.Howell, S. A., et al. 2003. A single malaria merozoite serine protease mediates shedding of multiple surface proteins by juxtamembrane cleavage. J. Biol. Chem. 278:23890-23898. [DOI] [PubMed] [Google Scholar]
- 34.Howell, S. A., C. Withers-Martinez, C. H. Kocken, A. W. Thomas, and M. J. Blackman. 2001. Proteolytic processing and primary structure of Plasmodium falciparum apical membrane antigen-1. J. Biol. Chem. 276:31311-31320. [DOI] [PubMed] [Google Scholar]
- 35.Hunter, S., et al. 2009. InterPro: the integrative protein signature database. Nucleic Acids Res. 37:D211-D215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jean, L., F. Hackett, S. R. Martin, and M. J. Blackman. 2003. Functional characterisation of the propeptide of Plasmodium falciparum subtilisin-like protease-1. J. Biol. Chem. 278:28572-28579. [DOI] [PubMed] [Google Scholar]
- 37.Kadekoppala, M., and A. A. Holder. 2010. Merozoite surface proteins of the malaria parasite: the MSP1 complex and the MSP7 family. Int. J. Parasitol. 40:1155-1161. [DOI] [PubMed] [Google Scholar]
- 38.Kadekoppala, M., S. A. Ogun, S. Howell, R. S. Gunaratne, and A. A. Holder. 2010. Systematic genetic analysis of the Plasmodium falciparum MSP7-like family reveals differences in protein expression, location, and importance in asexual growth of the blood stage parasite. Eukaryot. Cell 9:1064-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kafsack, B. F., et al. 2009. Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science 323:530-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kauth, C. W., et al. 2006. Interactions between merozoite surface proteins 1, 6, and 7 of the malaria parasite Plasmodium falciparum. J. Biol. Chem. 281:31517-31527. [DOI] [PubMed] [Google Scholar]
- 41.Klotz, F. W., et al. 1989. A 60-kDa Plasmodium falciparum protein at the moving junction formed between merozoite and erythrocyte during invasion. Mol. Biochem. Parasitol. 36:177-185. [DOI] [PubMed] [Google Scholar]
- 42.Koussis, K., et al. 2009. A multifunctional serine protease primes the malaria parasite for red blood cell invasion. EMBO J. 28:725-735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lagal, V., E. M. Binder, M. H. Huynh, B. F. Kafsack, P. K. Harris, R. Diez, D. Chen, R. N. Cole, V. B. Carruthers, and K. Kim. 2010. Toxoplasma gondii protease TgSUB1 is required for cell surface processing of micronemal adhesive complexes and efficient adhesion of tachyzoites. Cell. Microbiol. 12:1792-1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lasonder, E., et al. 2002. Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature 419:537-542. [DOI] [PubMed] [Google Scholar]
- 45.Le Roch, K. G., et al. 2003. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301:1503-1508. [DOI] [PubMed] [Google Scholar]
- 46.Li, J., H. Matsuoka, T. Mitamura, and T. Horii. 2002. Characterization of proteases involved in the processing of Plasmodium falciparum serine repeat antigen (SERA). Mol. Biochem. Parasitol. 120:177-186. [DOI] [PubMed] [Google Scholar]
- 47.Louie, K., R. Nordhausen, T. W. Robinson, B. C. Barr, and P. A. Conrad. 2002. Characterization of Neospora caninum protease, NcSUB1 (NC-P65), with rabbit anti-N54. J. Parasitol. 88:1113-1119. [DOI] [PubMed] [Google Scholar]
- 48.Lyon, J. A., R. H. Geller, J. D. Haynes, J. D. Chulay, and J. L. Weber. 1986. Epitope map and processing scheme for the 195,000-dalton surface glycoprotein of Plasmodium falciparum merozoites deduced from cloned overlapping segments of the gene. Proc. Natl. Acad. Sci. U. S. A. 83:2989-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McBride, J. S., and H. G. Heidrich. 1987. Fragments of the polymorphic Mr 185,000 glycoprotein from the surface of isolated Plasmodium falciparum merozoites form an antigenic complex. Mol. Biochem. Parasitol. 23:71-84. [DOI] [PubMed] [Google Scholar]
- 50.McCoubrie, J. E., et al. 2007. Evidence for a common role for the serine-type Plasmodium falciparum serine repeat antigen proteases: implications for vaccine and drug design. Infect. Immun. 17:5565-5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller, S. A., E. M. Binder, M. J. Blackman, V. B. Carruthers, and K. Kim. 2001. A conserved subtilisin-like protein TgSUB1 in microneme organelles of Toxoplasma gondii. J. Biol. Chem. 276:45341-45348. [DOI] [PubMed] [Google Scholar]
- 52.Miller, S. A., V. Thathy, J. W. Ajioka, M. J. Blackman, and K. Kim. 2003. TgSUB2 is a Toxoplasma gondii rhoptry organelle processing proteinase. Mol. Microbiol. 49:883-894. [DOI] [PubMed] [Google Scholar]
- 53.Miller, S. K., et al. 2002. A subset of Plasmodium falciparum SERA genes are expressed and appear to play an important role in the erythrocytic cycle. J. Biol. Chem. 277:47524-47532. [DOI] [PubMed] [Google Scholar]
- 54.Montero, E., et al. 2006. A conserved subtilisin protease identified in Babesia divergens merozoites. J. Biol. Chem. 281:35717-35726. [DOI] [PubMed] [Google Scholar]
- 55.Nyalwidhe, J., and K. Lingelbach. 2006. Proteases and chaperones are the most abundant proteins in the parasitophorous vacuole of Plasmodium falciparum-infected erythrocytes. Proteomics 6:1563-1573. [DOI] [PubMed] [Google Scholar]
- 56.Pachebat, J. A., et al. 2007. Extensive proteolytic processing of the malaria parasite merozoite surface protein 7 during biosynthesis and parasite release from erythrocytes. Mol. Biochem. Parasitol. 151:59-69. [DOI] [PubMed] [Google Scholar]
- 57.Pachebat, J. A., et al. 2001. The 22 kDa component of the protein complex on the surface of Plasmodium falciparum merozoites is derived from a larger precursor, merozoite surface protein 7. Mol. Biochem. Parasitol. 117:83-89. [DOI] [PubMed] [Google Scholar]
- 58.Pang, X. L., T. Mitamura, and T. Horii. 1999. Antibodies reactive with the N-terminal domain of Plasmodium falciparum serine repeat antigen inhibit cell proliferation by agglutinating merozoites and schizonts. Infect. Immun. 67:1821-1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pearce, J. A., A. N. Hodder, and R. F. Anders. 2004. The alanine-rich heptad repeats are intact in the processed form of Plasmodium falciparum MSP3. Exp. Parasitol. 108:186-189. [DOI] [PubMed] [Google Scholar]
- 60.Perkins, D. N., D. J. Pappin, D. M. Creasy, and J. S. Cottrell. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551-3567. [DOI] [PubMed] [Google Scholar]
- 61.Ravetch, J. V., J. Kochan, and M. Perkins. 1985. Isolation of the gene for a glycophorin-binding protein implicated in erythrocyte invasion by a malaria parasite. Science 227:1593-1597. [DOI] [PubMed] [Google Scholar]
- 62.Rawlings, N. D., A. J. Barrett, and A. Bateman. MEROPS: the peptidase database. Nucleic Acids Res. 38:D227-D233. [DOI] [PMC free article] [PubMed]
- 63.Richard, D., et al. 2009. Identification of rhoptry trafficking determinants and evidence for a novel sorting mechanism in the malaria parasite Plasmodium falciparum. PLoS Pathog. 5:e1000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ridley, R. G., H. W. Lahm, B. Takacs, and J. G. Scaife. 1991. Genetic and structural relationships between components of a protective rhoptry antigen complex from Plasmodium falciparum. Mol. Biochem. Parasitol. 47:245-246. [DOI] [PubMed] [Google Scholar]
- 65.Ridley, R. G., et al. 1990. Characterisation and sequence of a protective rhoptry antigen from Plasmodium falciparum. Mol. Biochem. Parasitol. 41:125-134. [DOI] [PubMed] [Google Scholar]
- 66.Sajid, M., C. Withers-Martinez, and M. J. Blackman. 2000. Maturation and specificity of Plasmodium falciparum subtilisin-like protease-1, a malaria merozoite subtilisin-like serine protease. J. Biol. Chem. 275:631-641. [DOI] [PubMed] [Google Scholar]
- 67.Sam-Yellowe, T. Y., H. Shio, and M. E. Perkins. 1988. Secretion of Plasmodium falciparum rhoptry protein into the plasma membrane of host erythrocytes. J. Cell Biol. 106:1507-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schechter, I., and A. Berger. 1967. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27:157-162. [DOI] [PubMed] [Google Scholar]
- 69.Scott, F. L., et al. 2008. Caspase-8 cleaves histone deacetylase 7 and abolishes its transcription repressor function. J. Biol. Chem. 283:19499-19510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stafford, W. H., et al. 1996. A 22 kDa protein associated with the Plasmodium falciparum merozoite surface protein-1 complex. Mol. Biochem. Parasitol. 80:159-169. [DOI] [PubMed] [Google Scholar]
- 71.Topolska, A. E., A. Lidgett, D. Truman, H. Fujioka, and R. L. Coppel. 2004. Characterization of a membrane-associated rhoptry protein of Plasmodium falciparum. J. Biol. Chem. 279:4648-4656. [DOI] [PubMed] [Google Scholar]
- 72.Trucco, C., et al. 2001. The merozoite surface protein 6 gene codes for a 36 kDa protein associated with the Plasmodium falciparum merozoite surface protein-1 complex. Mol. Biochem. Parasitol. 112:91-101. [DOI] [PubMed] [Google Scholar]
- 73.Uzureau, P., J. C. Barale, C. J. Janse, A. P. Waters, and C. B. Breton. 2004. Gene targeting demonstrates that the Plasmodium berghei subtilisin PbSUB2 is essential for red cell invasion and reveals spontaneous genetic recombination events. Cell Microbiol. 6:65-78. [DOI] [PubMed] [Google Scholar]
- 74.Wanyiri, J. W., et al. 2009. Role of CpSUB1, a subtilisin-like protease, in Cryptosporidium parvum infection in vitro. Eukaryot. Cell 8:470-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Waters, A. P., et al. 1990. A merozoite receptor protein from Plasmodium knowlesi is highly conserved and distributed throughout Plasmodium. J. Biol. Chem. 265:17974-17979. [PubMed] [Google Scholar]
- 76.White, N. J. 2008. Qinghaosu (artemisinin): the price of success. Science 320:330-334. [DOI] [PubMed] [Google Scholar]
- 77.Withers-Martinez, C., et al. 2002. Expression of recombinant Plasmodium falciparum subtilisin-like protease-1 in insect cells: characterization, comparison with the parasite protease, and homology modelling. J. Biol. Chem. 277:29698-29709. [DOI] [PubMed] [Google Scholar]
- 78.Yeoh, S., et al. 2007. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell 131:1072-1083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.