

Abstract
Pathogenic Yersinia species inject a panel of Yop virulence proteins by type III protein secretion into host cells to modulate cellular defense responses. This enables the survival and dissemination of the bacteria in the host lymphoid tissue. We have previously shown that YopE of the Y. enterocolitica serogroup O8 is degraded in the host cell through the ubiquitin-proteasome pathway. YopE normally manipulates rearrangements of the actin cytoskeleton and triggers phagocytosis resistance. To shed light into the physiological role of YopE inactivation, we mutagenized the lysine polyubiquitin acceptor sites of YopE in the Y. enterocolitica serogroup O8 virulence plasmid. The resulting mutant strain escaped polyubiquitination and degradation of YopE and displayed increased intracellular YopE levels, which was accompanied by a pronounced cytotoxic effect on infected cells. Despite its intensified activity on cultured cells, the Yersinia mutant with stabilized YopE showed reduced dissemination into liver and spleen following enteral infection of mice. Furthermore, the accumulation of degradation-resistant YopE was accompanied by the diminished delivery of YopP and YopH into cultured, Yersinia-infected cells. A role of YopE in the regulation of Yop translocation has already been described. Our results imply that the inactivation of YopE by the proteasome could be a tool to ensure intermediate intracellular YopE levels, which may effectuate optimized Yop injection into host cells. In this regard, Y. enterocolitica O8 appears to exploit the host ubiquitin proteasome system to destabilize YopE and to fine-tune the activities of the Yop virulence arsenal on the infected host organism.
The plasmid-encoded Ysc type III protein secretion system acts as a core determinant of Yersinia virulence (1, 33, 36). It is common to Y. pestis, the causative agent of bubonic plague, and to Y. enterocolitica and Y. pseudotuberculosis, which mediate gastrointestinal syndromes, lymphadenitis, and septicemia. The secretion system mediates the polarized translocation of Yersinia virulence proteins (Yersinia outer proteins [Yops]) inside eukaryotic host cells, where the Yops interfere with central signaling processes of host immunity (1, 33, 36). Four of the Yops, i.e., YopE, YopT, YopH, and YopO/YpkA, cooperatively inhibit rearrangements of the actin cytoskeleton and prevent the uptake and killing of Yersinia by phagocytic cells (1, 33, 36). Rho-GTPase family members, which critically regulate the actin cytoskeleton dynamics, are important Yop target molecules. YopE is a GTPase-activating protein (GAP) which switches Rho-GTPases into an inactive state by increasing their intrinsic Rho-GTPase activities. YopT is a cysteine protease that cleaves the C-terminal isoprenoid moieties of Rho-GTPases. The serine/threonine kinase YopO/YpkA mimics Rho guanidine nucleotide dissociation inhibitors (GDIs) to lock Rho-GTPases in an “off” state. YopH dismantles peripheral focal adhesion complexes by dephosphorylating host cell proteins, such as p130Cas and the focal adhesion kinase. Yersinia also represses the proinflammatory response and triggers apoptosis in macrophages (1, 33, 36). These effects are mediated by YopP (in Y. enterocolitica), or the homologous YopJ protein (in Y. pestis and Y. pseudotuberculosis). YopP/YopJ acetylates members of the mitogen-activated protein kinase (MAPK) kinase (MKK) superfamily and the NF-κB-activating IκB kinase β (IKK-β) to deactivate proinflammatory MAPK and NF-κB signaling (1, 33, 36). The last known effector, YopM, binds and activates ribosomal S6 kinase (RSK) 1 and protein kinase C-like (PRK) 2, with yet-unknown pathophysiological consequences for the host cell (1, 33, 36).
It is thought that the pathogenic Yersinia spp. emanated from a common ancestor strain that acquired a predecessor virulence plasmid which provided the bacteria with the ability to cause disease in mammals (17, 32, 39). The effector Yop arsenal is therefore remarkably conserved in Y. enterocolitica, Y. pseudotuberculosis, and Y. pestis. However, some substantial differences in the expression or the activities of individual Yops exist. YopT, for example, is not expressed by some serotypes of Y. pseudotuberculosis (37). YopP of the Y. enterocolitica serogroup O8 exhibits stronger proapoptotic activity than YopP from other Y. enterocolitica serogroups or YopJ from Y. pestis and Y. pseudotuberculosis (11, 27, 41). Size polymorphisms in YopM are found among different Yersinia species and serotypes (6). These observations fit into the concept that the diverse Yersinia species and serotypes have evolved separately after having acquired the predecessor virulence plasmid.
We have recently reported about another specificity in the regulation of the activity of an individual Yop: it was found that YopE from the Y. enterocolitica serotype O8, but not those from the serogroups O3 and O9, is modified by ubiquitination and degraded by the host cell proteasome (15). The proteasome, as the major nonlysosomal proteolytic system, mediates constitutive, controlled intracytoplasmic protein breakdown in eukaryotic cells (31). This protein-degradative function directed against YopE of Y. enterocolitica serogroup O8 contributes to reverse the antiphagocytic activities of Yersinia (15, 26). The inactivation of YopE by the proteasome could therefore be an immediate, innate immune reaction that fights bacterial infection. In line with this, other bacterial effector proteins have been demonstrated to be subjected to proteasomal degradation (5, 19, 22, 23, 30), which limits bacterium-related disease (5, 30). As a consequence, bacteria have developed mechanisms to evade or subvert processes of ubiquitination for their own benefit (4, 9, 16, 22, 29). We had identified K62 and K75 as the sites of polyubiquitination of YopE O8 (15). The modification with polyubiquitin allows the recognition and processing of proteins destined for destruction by the proteasome complex. K62 and K75 are unique to Y. enterocolitica serogroup O8 and are not found either in YopE protein species from the more distantly related serogroups O3 and O9 or in YopE from Y. pestis or Y. pseudotuberculosis (15). This tempts one to speculate that the two lysines may have specifically arisen in YopE O8 to fulfill a unique role in the pathogenesis of Y. enterocolitica serogroup O8 infection.
In this study, we investigated the physiological function of K62 and K75 during Y. enterocolitica serotype O8 infection. We mutagenized K62 and K75 in YopE encoded by the Yersinia virulence plasmid to R62 and Q75, respectively, in order to study the consequences of the ubiquitination and degradation of YopE in a physiological bacterial background, using a Yersinia strain harboring the complete virulence plasmid. The substitution mutation of K62 and K75 prevented the destabilization of virulence plasmid-encoded YopE O8 by the ubiquitin-proteasome pathway. Interestingly, the intracellular accumulation of degradation-resistant YopEK62R/K75Q was accompanied by the diminished delivery of other Yop effectors into the infected cells. This indicates that the reduction of translocated YopE by the host cell proteasome may specifically amplify the protein levels and anti-host cell activities of other effector Yops. The inactivation of YopE O8 by the proteasome pathway could therefore be a means of bacterial virulence that enables adequate Yop translocation. In line with this, the Yersinia strain producing degradation-resistant YopEK62R/K75Q was slightly attenuated in its ability to disseminate into liver and spleen in a mouse animal model of intragastric infection. This suggests that the susceptibility of YopE to polyubiquitination and proteasomal destabilization could fine-tune and optimize the activities of the Yersinia type III protein secretion system on the infected host.
MATERIALS AND METHODS
Bacterial strains and construction and characterization of mutants.
The Y. enterocolitica strains used in this study were the serogroup O8 wild-type strain WA (8, 14), its virulence plasmid-cured derivative WA-ΔpYV (14), and mutants of WA deficient for YopE (WA-ΔyopE [34]), YopP (WA-ΔyopP [25]), YopH (WA-ΔyopH [34]), or LcrD (WA-ΔlcrD [28]). For some experiments, WA-ΔyopE was complemented with plasmids encoding wild-type Y. enterocolitica serogroup O9 YopE (WA-ΔyopE/YopE-O9wt) or YopE O9 in which R62 and Q75 were replaced by lysines (WA-ΔyopE/YopE-O9R62K/Q75K). These plasmids have been described previously (15). We also generated a stable Y. enterocolitica YopE O8 mutant with mutagenized K62 and K75 (WA-YopEK62R/K75Q) and an isogenic control strain producing wild-type YopE (WA-YopEwt). The parent strain for this was WA-ΔyopE in which the yopE locus is replaced by a kanamycin resistance cassette (34). We restored the ability of this strain to produce YopE by reexchanging the kanamycin cassette with either wild-type or K62- and K75-mutated yopE. This was accomplished by an approach using the λ phage recombinases Redα and Redβ as described previously (10, 34). The recombinases are encoded together with Redγ, an inhibitor of bacterial exonucleases, on plasmid pKD46, which was transformed into WA-ΔyopE. Recombination functions were induced by the administration of 0.1% arabinose to the bacterial culture medium. The pKD46-bearing, arabinose-induced WA-ΔyopE strain was then made electrocompetent and transformed with the DNA recombination fragments for mutagenesis. The recombination fragments were amplified by PCR. They consisted of either the wild-type or mutagenized yopE gene, a chloramphenicol resistance marker integrated downstream from yopE, and homology arms flanking the 5′ and 3′ ends of the yopE-chloramphenicol resistance marker constructs. The homology arms carried 60 nucleotides upstream and 58 nucleotides downstream from the yopE coding region, respectively. Their sequences were derived from the published sequence of the Y. enterocolitica O8 virulence plasmid pYVa127/90 (Gen Bank accession number NC_004564) (12). A subcloned yopE gene, in which the coding regions for K62 (AAG) and K75 (AAA) were replaced by arginine (AGG) and glutamine (CAA) from YopE O9 (15), served as parent yopE DNA for WA-YopEK62R/K75Q. It was processed in the same manner with wild-type yopE O8, generating WA-YopEwt as control strain for WA-YopEK62R/K75Q. The primers used for amplification of the homology arms were 5′-GCCACCGGCTATTTTCCCACTAAG-3′ and 5′-GATGGTCAGGGAGTCAGTGGAAATCTACAACACGCGGCGACCGCATCTGTCGTTAAAA-3′, respectively. Clones that had undergone successful homologous recombination were selected by chloramphenicol resistance and kanamycin susceptibility. Two individual clones, producing either K62- and K75-mutagenized (WA-YopEK62R/K75Q) or wild-type (WA-YopEwt) YopE, were chosen, and the correct yopE insertion in these clones was verified by sequencing. Analysis of the Yop secretion profile of the strains into the bacterial culture medium was assessed as previously described (34). Yersiniae were grown overnight in Luria-Bertani broth at 27°C, diluted 1:20 in fresh medium, and grown for another 2 h at 37°C. Yop secretion was then induced by the addition of EGTA (5 mM) for Ca2+ chelation, MgCl2 (15 mM), and glucose (0.2%). After 3 h at 37°C, the bacteria were removed by centrifugation and proteins in the culture supernatant were precipitated with trichloroacetic acid, separated by SDS-PAGE, and stained with Coomassie blue.
Cell lines, infection conditions, and analysis of cell toxicity.
The human embryonic kidney cell line HEK293 was cultured in Dulbecco modified Eagle medium (DMEM) containing 10% heat-inactivated fetal calf serum (Invitrogen, Karlsruhe, Germany). Murine J774A.1 macrophages were grown in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum and 5 mM l-glutamine. Where indicated, the cells were treated with a 10 μM concentration of the proteasome inhibitor MG-132 (Z-Leu-Leu-Leu-CHO; Biomol, Plymouth Meeting, PA) 30 min prior to infection. The application of the proteasome inhibitor did not trigger apoptosis or cytotoxically alter the viability of the cells by another mechanism within the investigated time frames (26). For infection, overnight bacterial cultures grown at 27°C were diluted 1:20 in fresh Luria-Bertani broth and grown for another 2 h at 37°C. A shift of the growth temperature to 37°C initializes activation of the Yersinia type III secretion machinery for efficient Yop translocation upon cellular contact. To equalize and synchronize infection, bacteria were seeded on the cells by centrifugation at 400 × g for 5 min at a ratio of 50 bacteria per cell. For incubation times longer than 90 min, bacteria were killed by addition of gentamicin (100 μg/ml) after 90 min. Gentamicin preferentially kills extracellular bacteria; more than 90% of YopE-producing Yersinia strains were extracellular at that time point (data not shown), ensuring inactivation of the majority of the bacteria. Yersinia-conferred cell toxicity resulting from actin cytoskeleton disruption was monitored by quantifying the numbers of cells with a completely rounded phenotype. For every condition, three separate experiments were performed, and at least 200 cells from each experiment were scored in a blinded manner. Mean percentages of rounded versus total numbers of cells ± standard deviations (SD) were determined, and P values were calculated by using a two-tailed, unpaired Student t test.
Immunoprecipitation and immunoblotting.
For assessment of YopE ubiquitination, HEK293 cells were infected in six-well cell culture plates with Yersinia strains in the presence of 10 μM MG-132. MG-132 inhibits the proteasome activity to prevent the degradation of ubiquitinated proteins (31). Cells were processed for immunoprecipitation 75 min after onset of infection by lysing the cells in a buffer containing 50 mM Tris (pH 7.5), 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 μM MG-132, a cocktail of protease inhibitors (Roche, Basel, Switzerland), and a 10 μM concentration of the deubiquitinase inhibitor N-ethylmaleimide (26). The cleared lysates were preabsorbed to protein A/G-agarose (Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at 4°C and then incubated with rabbit polyclonal antibodies against YopE for 16 h at 4°C to precipitate YopE from the infected cells (15, 26). The immune complexes were collected with protein A/G-agarose (Santa Cruz Biotechnology, Santa Cruz, CA), washed five times with lysis buffer, subjected to SDS-PAGE, and transferred to a polyvinylidene difluoride (PVDF) membrane. Ubiquitin-YopE conjugates were detected by immunoblotting with the monoclonal mouse antiubiquitin antibody FK2 (Biomol International, Plymouth Meeting, PA). Immunoreactive bands were visualized using appropriate secondary antibodies and enhanced chemiluminescence detection reagents (Amersham Pharmacia Biotech, Inc., Piscataway, NJ). The membrane was then stripped in 62.5 mM Tris (pH 6.7)-0.1 mM 2-mercaptoethanol-2% SDS for 30 min at 50°C and reprobed with the anti-YopE polyclonal antibody to verify the successful precipitation of YopE.
To determine the overall levels of cellular YopE, YopP, or YopH, infected cells were solubilized with a buffer containing 10 mM HEPES (pH 7.8), 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 0.1% NP-40, 1 mM dithiothreitol, and phosphatase and protease inhibitors (Roche, Mannheim, Germany). In another set of experiments, the infected cells were treated with 1% digitonin (Sigma-Aldrich, Munich, Germany), a nonionic detergent that preferentially disrupts cholesterol-containing membranes. This ensures preferable solubilization of the eukaryotic plasma membrane but not of bacteria (20), which helps to reduce the levels of nontranslocated Yops that may originate from cell-associated yersiniae. The lysates were cleared from cell detritus and bacteria by centrifugation, separated by SDS-PAGE, and subjected to immunoblotting using polyclonal anti-YopE-, YopP-, or YopH-antibodies (26). Where indicated, the membrane fraction of the digitonin-permeabilized cell lysates was additionally separated from the cytoplasmic fraction by ultracentrifugation at 100,000 × g. The stripping method described above was applied to recycle the membranes for successive detection of actin (mouse monoclonal antibody; Millipore, Billerica, MA) in order to control equal protein loading with cellular lysates. Phospho-specific immunoblotting against p38 and p130Cas was performed as described above using a monoclonal antibody directed against phosphorylated threonine-180 and tyrosine-182 of p38 or phosphorylated tyrosine-249 of p130Cas (Cell Signaling Technology, Danvers, MA). The total cellular pool of p38 and p130Cas was labeled by stripping the membrane and successive immunoblotting with global anti-p38 (Cell Signaling Technology) or p130Cas antibody (Enzo Life Sciences, Plymouth Meeting, PA), respectively. The data shown are from one experiment that is representative of at least three performed. The phosphorylation of p130Cas in relation to total p130Cas was quantified by densitometry using Multi Gauge V3 Fujifilm software.
In vitro GAP assay.
The in vitro GAP assay was performed using the RhoGAP assay kit (Cytoskeleton, Denver, CO). RhoG, YopEwt, and YopEK62R/K75Q were expressed and purified as glutathione S-transferase (GST)-fused proteins as described previously (24). RhoG (1.5 μg) and the YopE proteins (1.5 μg) were incubated with 200 mM GTP for 20 min at 37°C. The reactions were then stopped, and the kit detection reagent was added to determine free phosphate generated by the hydrolysis of GTP. Absorbance was measured at 650 nm, and specific GAP activity was determined in relation to a phosphate standard. P values were calculated by using a two-tailed, unpaired Student t test.
Mouse infections.
Six- to 8-week-old female BALB/c mice (Charles River Laboratories, Sulzfeld, Germany) were infected with 2 × 108 bacteria orogastrically or 104 bacteria intravenously from frozen bacterial stock suspensions (34). This procedure facilitates the administration of identical bacterial counts in separate, consecutive experiments. The stock suspensions were prepared by growing the bacteria to the stationary phase in Luria-Bertani medium at 27°C, followed by freezing in 15% glycerol. Bacteria were washed twice with phosphate-buffered saline (PBS) and resuspended at an appropriate dilution in PBS before infection. A 250-μl portion of the suspension was orogastrically administered using a stomach sonde, or 100 μl was intravenously injected into the lateral tail vein. Mice were subjected to fasting 2 h prior to enteral infection. The dose actually administered was determined by plating serial dilutions on Mueller-Hinton agar for 36 h at 27°C and counting CFU. Mice were sacrificed by CO2 asphyxiation. Liver, spleen, and Peyer's patches were aseptically removed and homogenized in 5 ml (liver), 2 ml (spleen), or 0.5 ml (Peyer's patches) PBS-Tween 20 (0.05%). Loosely attached bacteria were removed beforehand from the Peyer's patches by rinsing them with PBS. To determine the numbers of CFU per organ, serial dilutions of homogenates were plated on Yersinia selective CIN agar (BD Biosciences, Heidelberg, Germany). The limits of detection were 50 CFU in the liver, 20 CFU in the spleen, and 5 CFU in the Peyer's patches. P values were determined by using a two-tailed, unpaired Student t test. P values of <0.05 were considered significant. To analyze the Yop secretion patterns of Yersinia strains before and after the passage through mice, the bacteria were orogastrically inoculated and recovered from the mouse spleen 3 days after onset of infection as described above.
RESULTS
Mutagenesis of YopE K62 and K75 in the Y. enterocolitica serogroup O8 virulence plasmid.
Our previous studies have shown that YopE of the Y. enterocolitica serogroup O8 is destabilized by the ubiquitin-proteasome pathway of the host (15). In this context, we had identified K62 and K75 as the polyubiquitin acceptor sites of YopE O8 that mediate the targeting of YopE to the host cell proteasome (Fig. 1). These previous studies were conducted with Yersinia strains that overproduced YopE in the absence of any other effector Yop (15, 26). To gain more insights into the physiological consequences of the ubiquitination and degradation of YopE, we generated a Yersinia YopE mutant strain that was mutagenized at K62 and K75 in the yopE coding region of the pYV virulence plasmid. This strain should be fully competent in Yop production. The parent strain for this construct was a Yersinia mutant that was initially negative for YopE due to the replacement of the yopE locus by a kanamycin resistance cassette (strain WA-ΔyopE). We restored the ability of this strain to produce YopE by reexchanging the kanamycin cassette with either wild-type or K62- and K75-mutated yopE. The selection of clones that have undergone successful homologous recombination was enabled by a chloramphenicol resistance marker integrated downstream from the yopE coding region in the recombining DNA fragment. Two individual clones that produced either wild-type YopE (WA-YopEwt) or YopE mutagenized at K62 and K75 (WA-YopEK62R/K75Q) were selected. The correct insertion of the yopE genes in these clones was verified by sequencing. In the YopE-mutagenized strain, K62 and K75 were replaced by arginine and glutamine, which are found at these positions in Y. enterocolitica serogroup O9. The selected clones did not differ in their growth behaviors and in their in vitro Yop secretion profiles from the parent YopE-negative mutant strain WA-ΔyopE, except that the complemented strains WA-YopEwt and WA-YopEK62R/K75Q have regained the ability to produce YopE (Fig. 2). The Yop spectrum shown in Fig. 2 was monitored in the bacterial culture medium upon Ca2+ removal, which artificially triggers Yop release by yersiniae. The two Yersinia strains investigated secreted comparable amounts of YopE and other effector Yops. The Yop secretion patterns of the individual strains did not significantly differ before and after the passage through mice (Fig. 2). For mouse passage, the bacteria were recovered from the spleens of orogastrically infected mice at day 3 postinfection.
FIG. 1.

Comparison of the ubiquitination sites of YopE from Y. enterocolitica O8 with corresponding sequences form other Yersinia clades. The deduced YopE sequences between amino acids 55 and 80 from Y. enterocolitica serogroup O8 (GenBank accession no. NP_783702), Y. enterocolitica serogroup O9 (NP_052427), Y. pseudotuberculosis IP32953 (YP_068436), and Y. pestis KIM (NP_857762) are aligned. The ubiquitination sites of YopE from Y. enterocolitica O8 are underlined (K62 and K75) and compared to the corresponding amino acids of the other Yersinia clades (boldface).
FIG. 2.

Yop secretion profiles of the investigated Yersinia strains. The Coomassie blue-stained SDS-polyacrylamide gel shows proteins released by Yersinia upon Ca2+ removal into the bacterial growth medium. The spectra of Yop release were analyzed for the YopE-negative mutant WA-ΔyopE and the mutant strain recomplemented with either wild-type YopE (WA-YopEwt) or K62- and K75-mutagenized YopE (WA-YopEK62R/K75Q) before and after the passage through mice. For mouse passage, the bacteria were recovered from the spleens of orogastrically infected mice at day 3 postinfection. Molecular size marker proteins are shown in the first lane.
Mutagenesis of K62 and K75 prevents the degradation and inactivation of virulence plasmid-encoded YopE by the host proteasome.
The YopE ubiquitination patterns of the Yersinia strains were characterized following host cell infection. Accordingly, HEK293 cells were infected with WA-YopEwt and WA-YopEK62R/K75Q in the presence of the proteasome inhibitor MG-132, a procedure that causes the accumulation of proteins destined for proteasomal degradation (31). The YopE proteins were immunoprecipitated from cellular lysates with anti-YopE antibodies and immunoblotted with antiubiquitin to detect YopE protein species modified with ubiquitin. The Y. enterocolitica serogroup O8 wild-type strain WA and its virulence plasmid-cured counterpart WA-ΔpYV served as positive and negative controls, respectively. Figure 3 A shows that a typical pattern of antiubiquitin immunoreactive bands with increasing slower electrophoretic mobilities precipitated with YopE after infection with WA and WA-YopEwt. The appearance of such higher-molecular-weight proteins is consistent with a modification of YopE by polyubiquitination (26). No such bands were detected in the YopE precipitates from WA-YopEK62R/K75Q-infected cells (Fig. 3A). Thus, YopEwt produced by WA-YopEwt is subjected to polyubiquitination similarly to YopE originating from wild-type WA. Mutagenized YopEK62R/K75Q, in contrast, evades polyubiquitin modification after its delivery into host cells.
FIG. 3.

Mutagenesis of K62 and K75 prevents ubiquitination and proteasomal degradation of YopE from Y. enterocolitica serogroup O8. (A) Ubiquitination of YopE O8 protein species. HEK293 cells were left noninfected (φ) or infected with the wild-type Y. enterocolitica serogroup O8 strain WA, its virulence plasmid-cured derivative WA-ΔpYV, or the mutant strain recomplemented with either wild-type YopE (WA-YopEwt) or K62- and K75-mutagenized YopE (WA-YopEK62R/K75Q). Infections were performed in the presence of the proteasome inhibitor MG-132. Cellular extracts were prepared 75 min after onset of infection and immunoprecipitated using anti-YopE antibody. The immunoprecipitates were first immunoblotted with antiubiquitin for the detection of polyubiquitin-modified proteins (Ubpoly, upper panel). Subsequently, the membrane was stripped and reprobed with anti-YopE to control successful YopE precipitation (YopE, lower panel). The asterisk denotes the position of the H chain of the precipitating antibody. Molecular masses of standard marker proteins are indicated. (B) Time-dependent destabilization of wild-type YopE O8 in Y. enterocolitica-infected cells. HEK293 cells were infected with WA-YopEwt or WA-YopEK62R/K75Q. Ninety minutes later, yersiniae were killed by addition of gentamicin. At the denoted times after onset of infection, cellular lysates were prepared, cleared by centrifugation, and subjected to immunoblotting using anti-YopE antibody. (C) Stabilization of wild-type YopE O8 by proteasome inhibition. HEK293 cells were left uninfected (φ) or infected with the indicated Yersinia strains in the absence or presence of the proteasome inhibitor MG-132 as for panel B. At 3.5 h after onset of infection, cellular lysates were prepared, cleared by centrifugation, and analyzed on the cellular YopE level by immunoblotting. (D) Localization of YopE protein species to the cytoplasmic fraction of infected cells. HEK293 cells were infected with WA-YopEwt, WA-YopEK62R/K75Q, or the Yop secretion-defective mutant WA-ΔlcrD in the presence of MG-132. Ninety minutes later, cellular membranes were solubilized with 1% digitonin, and cleared from nonlysed cells, and bacteria by centrifugation. The cleared lysates were then subjected to ultracentrifugation. The YopE levels in the resulting cytoplasmic and membrane fractions were analyzed by immunoblotting (left panel). As a control for selective lysis of eukaryotic cells, bacteria were treated in the same manner with 1% digitonin in the absence of HEK293 cells or with 1% SDS to generate total bacterial cell lysates (right panel). (E) YopE protein species prepared from the cytoplasmic host cell fraction, as described for panel D, appear in higher resolution as a double band. (F) Differential stability of YopEwt and YopEK62R/K75Q in J774A.1 macrophages. J774A.1 cells were infected with WA-YopEwt or WA-YopEK62R/K75Q. At 3.5 h after onset of infection, cellular lysates were prepared and analyzed on the cellular YopE level by immunoblotting as for panel B. Equal loading of the gels with cellular, cytoplasmic lysates was controlled by stripping of the membranes and subsequent immunoblotting against actin.
We subsequently investigated whether the differences in the ubiquitination patterns of YopEwt and YopEK62R/K75Q may correlate with different stabilities of translocated YopE. HEK293 cells were infected with either Yersinia strain. Gentamicin was added 90 min after the onset of infection to prevent bacterial overgrowth. The protein levels of YopE were monitored in cell lysates prepared at different time points. Figure 3B shows that the amount of translocated YopEK62R/K75Q was increased compared to that of YopEwt after 2.5 h. Comparable results were obtained when reduced bacterial counts were used for infection and gentamicin was omitted (data not shown). Importantly, the levels of YopEK62R/K75Q in the following incubation period remained nearly constant, whereas wild-type YopEwt was substantially reduced within 7 h (Fig. 3B). The destabilization of YopE under these conditions has been shown to occur by proteasomal degradation (15, 26). In accordance, addition of the proteasome inhibitor MG-132 to the infected cells significantly increased the protein levels of YopEwt, while the amount of YopEK62R/K75Q was less affected (Fig. 3C). The escape of YopEK62R/K75Q from ubiquitination and proteasomal degradation may result from impaired translocation inside the host cell. However, elevated YopEK62R/K75Q levels were also detected in cytoplasmic fractions from cell lysates prepared with digitonin and cleared from membrane-associated YopE by ultracentrifugation (Fig. 3D, left panel). Digitonin preferentially solubilizes the eukaryotic plasma membrane but not the bacteria (20). Furthermore, YopEK62R/K75Q and YopEwt appeared as subtle double bands in the cytoplasmic fraction of digitonin-lysed cells (Fig. 3E), reflecting a not-yet-characterized posttranslational modification observed previously (15). This modification was not detected in total bacterial cell lysates (Fig. 3D, right panel) or in lysates prepared from cells infected with a secretion-defective ΔlcrD Yersinia mutant (WA-ΔlcrD) (Fig. 3D, left panel) or with a translocation-impaired YopD-negative strain (data not shown). YopE consequently appears to be translocated and intracellularly subjected to a second form of posttranslational modification irrespective of the exchange of K62R and K75Q. Thus, the ubiquitin-proteasome pathway governs the stability of YopE encoded by the Y. enterocolitica serogroup O8 virulence plasmid. The loss of K62 and K75 renders YopE insensitive to proteasomal degradation and enables the intracellular persistence of YopE. YopE also seems to undergo comparable destabilization in other cell types, because YopEK62R/K75Q was detected in a much larger amount than YopEwt also in the macrophage cell line J774A.1 (Fig. 3F).
The persistence of YopE is associated with sustained cytotoxicity of Yersinia on infected HEK293 cells (15, 26). YopE triggers disruption of the actin cytoskeleton structure and thereby induces a typical contracted-to-rounded morphology of the infected cells. The time-dependent degradation of wild-type YopEwt (Fig. 3B) correlated with a reduction of the cytotoxic cell alterations mediated by Yersinia: The rounding of cells was less severe following 4.5 h of infection with WA-YopEwt than after infection with WA-YopEK62R/K75Q (Fig. 4A). When the degradation of YopE was prevented by the application of the proteasome-inhibitory compound MG-132, the cytotoxic effects of YopEwt were enhanced and approached those conferred by WA-YopEK62R/K75Q (Fig. 4A). The prominent cell-rounding activity of WA-YopEK62R/K75Q, in contrast, was not affected by MG-132, which correlates with the resistance of YopEK62R/K75Q to proteasomal degradation (Fig. 3). Together, these data indicate that the subjection of virulence plasmid-encoded, wild-type YopE to degradation by the host ubiquitin-proteasome pathway physiologically reduces its cytotoxic activity on the host cell. To rule out that the increased cytotoxicity of WA-YopEK62R/K75Q may result simply from stronger biochemical GAP activity of YopE, we performed an in vitro GTP hydrolysis assay to compare the activities of YopEwt and YopEK62R/K75Q on recombinant RhoG (24). YopEwt and YopEK62R/K75Q both stimulated the intrinsic GTPase activity of RhoG in comparable manners, indicating that they do not differ in their biochemical activity (Fig. 4B). This reinforces the idea that the stronger cytotoxic cell alteration triggered by YopEK62R/K75Q is related to its increased intracellular protein stability.
FIG. 4.

Wild-type YopE O8 triggers diminished, proteasome inhibitor-sensitive cytotoxic alterations of infected cells. (A) Differential sensitivity of YopE-conferred cytotoxicity to proteasome inhibition. HEK293 cells were left untreated or treated with the proteasome inhibitor MG-132 prior to infection with virulence plasmid-cured yersiniae (WA-ΔpYV) or yersiniae producing either wild-type (WA-YopEwt) or K62- and K75-mutagenized (WA-YopEK62R/K75Q) YopE O8. Ninety minutes after onset of infection, the yersiniae were killed by addition of gentamicin. The cells were fixed, and cellular morphologies were microscopically analyzed after a total incubation period of 4.5 h. The numbers of cells with a completely rounded phenotype were quantified from three separate experiments, and mean percentages of rounded versus total numbers of cells ± SD are indicated. Differences in cell rounding were statistically significant for WA-YopEwt versus WA-YopEK62R/K75Q in the absence of MG-132 and for WA-YopEwt with versus without MG-132 (P < 0.005). (B) Comparable in vitro GAP activities of YopEwt and YopEK62R/K75Q. Recombinant, purified GST-fused YopEwt and YopEK62R/K75Q were subjected to in vitro GAP assay using recombinant RhoG. GTP hydrolysis and release of free phosphate were measured at 650 nm after coincubation of RhoG and the YopE protein species for 20 min at 37°C. GST protein was used as negative control. φ, background phosphate levels in the absence of GST or GST-YopE. Specific GAP activity from two independent, representative experiments was determined with a phosphate standard and is indicated as mean nmol ± SD. The increase in GAP activity triggered by GST-YopEwt and GST-YopEK62R/K75Q compared to background levels (φ) was statistically significant (P < 0.03).
Loss of K62 and K75 in YopE entails diminished dissemination of yersiniae into lymphatic organs after enteral infection.
We wondered whether the destabilization of YopE may affect the pathogenicity of Yersinia and influence the infection process in vivo. We therefore compared the abilities of the strains WA-YopEwt and WA-YopEK62R/K75Q to colonize the lymphatic organs of mice following Yersinia infection. In the first set of experiments, groups of BALB/c mice were intragastrically fed with 2 × 108 CFU of WA-YopEwt or WA-YopEK62R/K75Q. The orogastric application route reflects the physiologic mode of enteropathogenic Yersinia infection. Tissue homogenates of Peyer's patches, liver, and spleen were prepared and plated in serial dilutions to determine CFU and the number of vital bacteria. Figure 5 shows that both strains had efficiently colonized the Peyer's patches of the animals at day 3 postinfection. However, the Yersinia strain producing YopEwt was more efficient in colonizing liver and spleen at this time point than YopEK62R/K75Q-producing yersiniae. Accordingly, significantly more bacteria were recovered from livers and spleens of WA-YopEwt-infected mice than from those of mice infected with WA-YopEK62R/K75Q (Fig. 5). This indicates that mutagenesis of K62 and K75 and concomitant persistence of translocated YopE lead to a disadvantage in the dissemination of the bacteria into peripheral lymphoid organs following enteral Yersinia infection. This effect of YopE, related to K62 and K75, was less evident after intravenous Yersinia infection. After intravenous infection, a tendency for higher bacterial loads in liver and spleen with strain WA-YopEwt was noticed at day 1, but the difference from infection with WA-YopEK62R/K75Q was not significant (Fig. 6). This suggests that the destabilization of YopE by the ubiquitin-proteasome pathway may preferentially contribute to facilitate spread of the bacteria from the Peyer's patches to other lymphoid tissues. Once the bacteria had colonized liver and spleen, the ubiquitination of YopE seemed not to decisively influence the course of infection anymore because the colonization rates in liver and spleen were similar for both Yersinia strains at day 5 after intragastric and day 3 after intravenous application (data not shown). These data show that the degradation of YopE has a subtle but significant effect on the early colonization of liver and spleen in the orogastrically infected host.
FIG. 5.
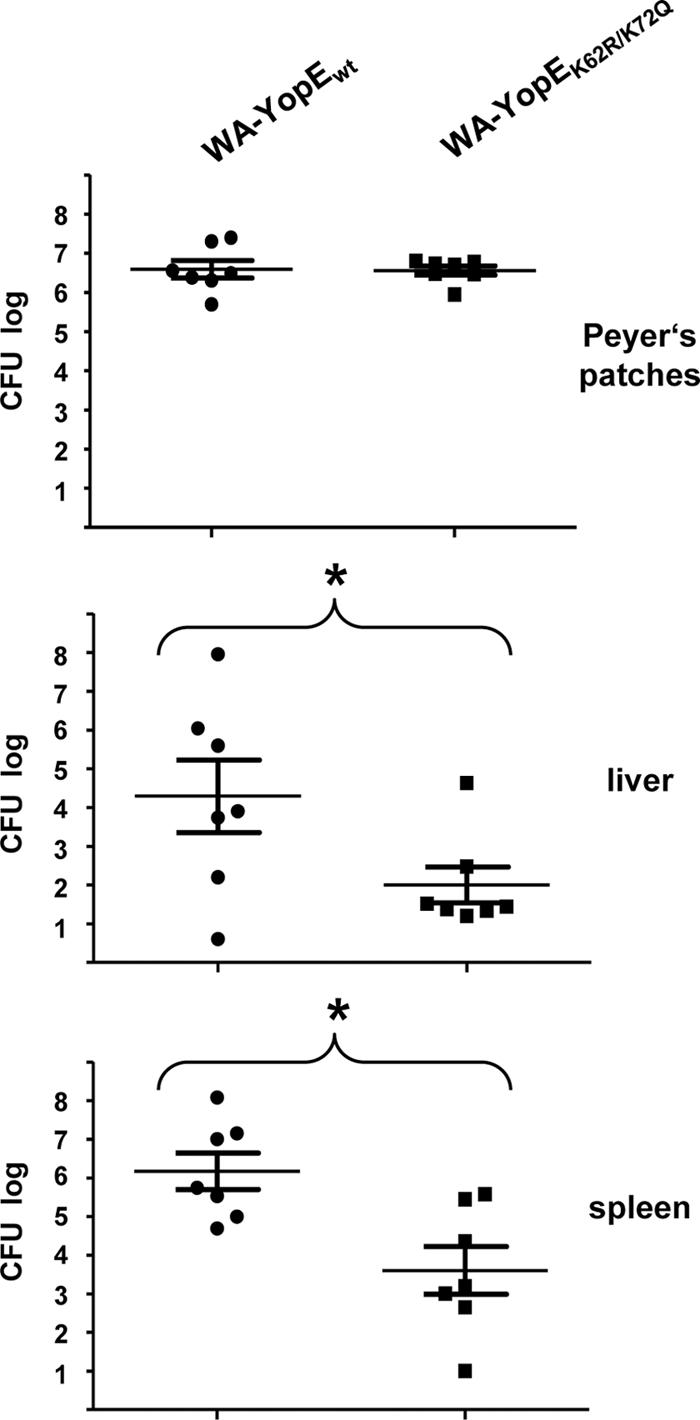
Differential colonization of mouse lymphatic organs with YopEwt- and YopEK62R/K75Q-producing yersiniae following orogastric mouse infection. Equal groups of BALB/c mice were orogastrically infected with 2 × 108 yersiniae producing either wild-type (WA-YopEwt) or K62- and K75-mutagenized (WA-YopEK62R/K75Q) YopE O8. Peyer's patches, livers, and spleens were removed at day 3 postinfection, homogenized, and plated to determine bacterial CFU. Values represent the average log CFU per organ for seven mice, with standard errors of the means indicated by error bars. Asterisks denote statistical significances (P < 0.05) in the colonization values between the two strains (liver, P = 0.0487; spleen, P = 0.0065).
FIG. 6.
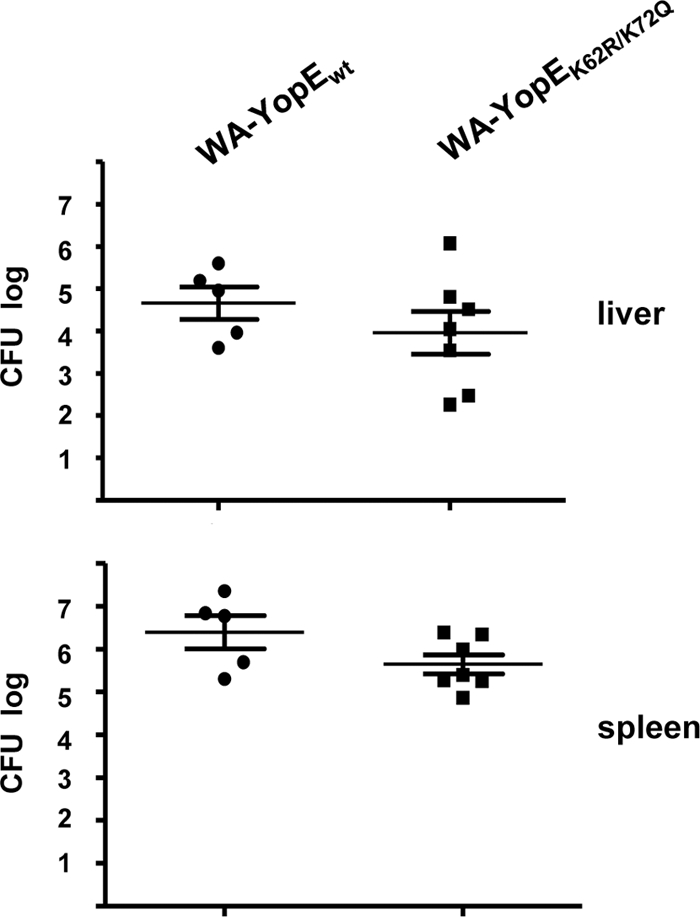
Colonization of liver and spleen with YopEwt- and YopEK62R/K75Q-producing yersiniae after intravenous mouse infection. Equal groups of BALB/c mice were intravenously infected with 104 yersiniae producing either wild-type (WA-YopEwt) or K62- and K75-mutagenized (WA-YopEK62R/K75Q) YopE O8. Livers and spleens were removed at 1 day postinfection, homogenized, and plated to determine bacterial CFU. Values represent the average log CFU per organ for five (WA-YopEwt) and seven (WA-YopEK62R/K75Q) mice, with standard errors of the means indicated by error bars. Two mice were deceased after infection with WA-YopEwt.
The sensitivity of YopE to ubiquitination fine-tunes Yop delivery into host cells.
The results obtained in the mouse infection model indicate that the ubiquitination and degradation of YopE fulfill a particular role in the pathogenesis of yersiniosis. We wondered how the destabilization of YopE might contribute to regulation of Yersinia virulence. Previous studies have shown that YopE, besides directly acting as an effector Yop on the host cells, balances the secretion and translocation of other Yop effectors (3, 18, 21, 35). The effector Yops are injected inside the host cells through a translocation pore that is formed by specific components of the Yersinia type III secretion apparatus (1, 33, 36). The action of YopE on Rho-GTPases counteracts the formation of pores and reduces the injection of Yops inside the cell (3, 18, 21, 35). As such, the ubiquitination of YopE may exert a regulatory role in Yersinia type III effector translocation. We consequently assessed whether differences in the translocation patterns of other Yops might exist between strains WA-YopEwt and WA-YopEK62R/K75Q. HEK293 cells were infected with WA-YopEwt, WA-YopEK62R/K75Q, and the wild-type strain WA as control. Cell lysates were prepared, and the protein levels of YopH, YopP, and YopE were compared by immunoblotting. Interestingly, the amounts of YopP and YopH were reduced in cells infected with WA-YopEK62R/K75Q compared to WA-YopEwt- and WA-infected cells (Fig. 7A). The YopE levels, as expected, behaved contrarily: YopE accumulated upon infection with WA-YopEK62R/K75Q, whereas it was diminished after infection with WA and WA-YopEwt (Fig. 7A). The differences in the YopE levels likely result from the destruction of wild-type YopE by the proteasome pathway, while K62- and K75-mutagenized YopEK62R/K75Q resists proteasomal degradation and accumulates in the cells (Fig. 3). In fact, administration of the proteasome inhibitor MG-132 to WA-YopEwt-infected cells reduced YopP translocation, while the intracellular YopP levels were unaffected by proteasome inhibitor treatment in case of infection with WA-YopEK62R/K75Q (Fig. 7B). These results demonstrate that the levels of translocated YopE and of YopP and YopH are reciprocally regulated. The ubiquitination and degradation of wild-type YopE apparently increase the translocation of other Yop effectors. In line with this conclusion, the total absence of YopE in case of infection with YopE-negative mutant WA-ΔyopE led to hypertranslocation of YopP (Fig. 7C). An increase in the translocation of YopH by a YopE-negative strain has been previously reported (2). These results confirm that YopE is implicated in the control of Yop translocation. To test whether YopE from the Y. enterocolitica serogroup O9, which lacks K62 and K75, may comparably affect the translocation of YopP in the Y. enterocolitica O8 background, the YopE-deficient mutant WA-ΔyopE was complemented either with wild-type YopE O9 (strain WA-ΔyopE/YopE-O9wt) or with YopE O9 in which R62 and Q75 were replaced by lysines (giving strain WA-ΔyopE/YopE-O9R62K/Q75K). Again, the production of degradation-resistant YopE-O9wt was accompanied by reduced cellular levels of YopP, whereas more intracellular YopP was detected when degradation-sensitive YopE-O9R62K/Q75K was expressed (Fig. 7D). These data show that the destabilization of YopE through the ubiquitin-proteasome pathway affects the regulatory circuit of Yop translocation, apparently helping to increase the efficiency of Yop delivery into host cells. The degradation and inactivation of YopE by the host cell might therefore be a bacterial mechanism that adjusts the translocation of Yop effectors and fine-tunes their activities on the host cell.
FIG. 7.

The stability of YopE controls the translocation levels of other Yop effectors. Reverse regulation of intracellular levels of YopH and YopP by YopE is shown. (A) HEK293 cells were left untreated (φ) or infected with yersiniae producing either wild-type YopE (WA and WA-YopEwt) or K62- and K75-mutagenized YopE O8 (WA-YopEK62R/K75Q). (B) The cells were additionally treated with the proteasome inhibitor MG-132 where indicated. (C) Yersinia mutants defective for YopP (WA-ΔyopP) or YopE (WA-ΔyopE) were additionally used. (D) YopE-deficient WA-ΔyopE was complemented either with wild-type YopE O9 (WA-ΔyopE/YopE-O9wt) or with YopE O9 harboring lysines instead of R62 and Q75 (WA-ΔyopE/YopE-O9R62K/Q75K). Cellular lysates for all panels were prepared 3.5 to 4 h after onset of infection, and the amounts of Yops in the lysates were determined by immunoblotting with the respective anti-Yop antibodies. Equal loading of the gels with cell lysates was controlled by reprobing the membranes with antiactin antibody. A possibly unspecific band appears below YopP in panel A.
To assess whether the destabilization of YopE helps to control the anti-host cell activities of YopP, we analyzed the phosphorylation levels of the p38 kinase in infected HEK293 cells. p38 is a member of the MAPK family that is deactivated by YopP during Yersinia infection. YopP acetylates and inhibits the MKKs, which act as upstream activators of MAPKs, including p38 (1, 33, 36). The phosphorylation and activation status of p38 was monitored by immunoblotting with an antibody that recognizes the active form of p38 phosphorylated at T180/Y182. It was found that p38 was phosphorylated after infection with the YopP-negative mutant WA-ΔyopP (Fig. 8A). The p38 phosphorylation levels, in contrast, were diminished in cells infected with wild-type WA or WA-YopEwt. This confirms that YopP-negative yersiniae induce the p38 pathway, whereas the presence of YopP counteracts the phosphorylation and activation of p38 (1, 33, 36). WA-YopEK62R/K75Q, on the other hand, was substantially impaired in its ability to suppress p38 phosphorylation. Accordingly, the p38 phosphorylation levels were enhanced and resembled those of WA-ΔyopP-infected cells (Fig. 8A). These results indicate that the adequate translocation of YopP, which is ensured by the proteasomal degradation of YopE in case of infection with WA and WA-YopEwt, regulates the action of YopP on p38 phosphorylation. When the amount of translocated YopP is reduced due to intracellular persistence of YopEK62R/K75Q, p38 is largely preserved in a phosphorylated and activated state. Similarly, YopH, which impairs the phosphorylation of p130Cas triggered by YopH-negative strains (1, 36), was not as efficient in silencing p130Cas phosphorylation in case of infection with WA-YopEK62R/K75Q as it was in WA-YopEwt infection (Fig. 8B). The YopH-deficient mutant WA-ΔyopH, as expected, triggered remarkable p130Cas tyrosine phosphorylation. This indicates that the sensitivity of YopE to polyubiquitination and proteasomal degradation governs the activity of other Yop effectors on the host cell.
FIG. 8.

The stability of YopE influences the activities of YopP and YopH. (A) Effect of YopE on YopP-dependent inactivation of p38. HEK293 cells were left untreated or infected with WA, WA-ΔyopP, WA-YopEwt, or WA-YopEK62R/K75Q. Cellular lysates were prepared 4 h after onset of infection, and the phosphorylation and activation status of the MAPK p38 was assessed by immunoblotting with an antibody that recognizes active p38 phosphorylated at T180/Y182. (B) Effect of YopE on YopH-dependent inhibition of p130Cas phosphorylation. HEK293 cells were left untreated or infected the YopH-deficient mutant WA-ΔyopH, WA-YopEwt, or WA-YopEK62R/K75Q. Cellular lysates were prepared after 3.5 h of infection, and phosphorylation of p130Cas was assessed by immunoblotting with an antibody recognizing Y249-phosphorylated p130Cas. The total pools of p38 and p130Cas in the cell lysates for panels A and B were controlled by reprobing the membranes with general anti-p38 and anti-p130Cas antibody, respectively. The phosphorylation of p130Cas in relation to total p130Cas was quantified by densitometry. Values are expressed as percentages of p130Cas phosphorylation relative to infection with WA-ΔyopH (100%) and the background immunoblot signal (0%).
DISCUSSION
The species Y. enterocolitica comprises a biochemically, serologically, and genetically heterogenous group of organisms. In Europe, the strains most frequently isolated belong to the serogroups O3 and O9, whereas other serogroups, such as O8, O20, and O21, are encountered predominantly in North America. The heterogenous distribution of the serotypes reflects the independent evolution of the different Y. enterocolitica lineages (17, 32, 38, 39). At the level of the central common Yersinia type III secretion system Ysc, we had identified a surprising uniqueness for the North American Y. enterocolitica serogroup O8: it was found that YopE O8 becomes ubiquitinated at K62 and K75, which mediates its degradation by the host cell proteasome (15). The susceptibility of YopE to degradation is accompanied by a reduced cytotoxic activity of YopE O8 on the host cell cytoskeleton. The YopE proteins of the Old World Y. enterocolitica lineages O3 and O9 lack the two lysines and resist ubiquitination and proteasomal degradation. YopE O3 and O9 consequently exert a pronounced cytotoxic effect on infected host cells (15). The lysines K62 and K75 are furthermore not found in YopE from Y. pestis and Y. pseudotuberculosis, which are closely related. These observations imply that K62 and K75 may serve a specific function in the activity of YopE O8 that could influence the pathogenicity of Y. enterocolitica serogroup O8.
To test this hypothesis, we specifically mutagenized K62 and K75 in YopE encoded by the Y. enterocolitica O8 virulence plasmid. K62 and K75 were replaced by arginine and glutamine according to the sequence of YopE O9. As anticipated, the substitution mutation of K62 and K75 prevented the ubiquitination of YopE O8 and led to the accumulation of translocated YopE, which was followed by an increased cytotoxicity of Y. enterocolitica O8 on infected host cells. Interestingly, the pronounced cytotoxic alterations triggered by YopEK62R/K75Q-mutagenized Y. enterocolitica O8 in cultured cells were not accompanied by an increase in virulence in a mouse model of infection. When BALB/c mice were orogastrically infected with Y. enterocolitica O8 producing either wild-type or K62- and K75-mutagenized YopE, it was revealed that the YopE mutant WA-YopEK62R/K75Q was modestly but significantly impaired in its ability to localize to liver and spleen at day 3 postinfection. This suggests that the resistance of YopE to ubiquitination and inactivation through the proteasome pathway is disadvantageous for the dissemination of the bacteria in the early infection process, or, conversely, the degradation of YopE by the host cell proteasome may facilitate the colonization of deeper lymphoid organs. This advantage was evident only for enteral Yersinia infection, because no significant differences in the colonization of liver and spleen were observed after intravenous infection with the two strains. These results indicate that the ubiquitination and inactivation of wild-type YopE may play a role in the early stage of bacterial spread and tissue colonization following invasion of the intestinal mucosa. Although YopE is degraded, a certain amount of YopE appears to be required for optimized Yersinia virulence. The complete deficiency of YopE in case of infection with a YopE-negative Y. enterocolitica O8 mutant seemed to diminish bacterial virulence more severely than infection with strains producing either wild-type or degradation-resistant, K62- and K75-mutagenized YopE (reference 34 and data not shown). The translocated YopE levels may therefore not fall below a threshold limit to preserve effective pathogenicity of Y. enterocolitica O8.
The mechanisms by which the inactivation of YopE through the ubiquitin-proteasome pathway could contribute to support bacterial dissemination are less clear. Importantly, a functional role of YopE in regulating Yop translocation inside the host cells has been demonstrated (3, 18, 21, 35). The effector Yops are injected through the host cell membrane via a translocation pore that is formed by specific components of the Yersinia type III secretion apparatus. The pore is stabilized by the activation of Rho-GTPases and resultant actin cytoskeleton rearrangements. YopE, and likely YopT, counteracts this process by impairing Rho-GTPase members (21, 35). This may delimit pore formation and terminate the delivery of Yops into the cells. From that it was speculated that the importance of YopE for Yersinia virulence may be related predominantly to its ability to regulate Yop translocation, rather than acting directly as an immediate virulence factor on host cell defense mechanisms (3, 18). Our data support the view that YopE has a major regulatory role in controlling the translocation of Yops. We found that the deficiency of K62 and K75 in YopE O8 was accompanied by the reduced detection of YopP and YopH in infected cells. The translocated levels of these Yop effectors thus behaved reciprocally to the cellular amount of YopE. Furthermore, the inhibitory actions of YopP and YopH on p38 MAPK and p130Cas phosphorylation, respectively, were attenuated by the stabilization of YopE. This indicates that the ubiquitination and degradation of YopE could be a means to adjust the intracellular levels of translocated Yops along with their activities on the host cell. This coherence could be relevant for the observed in vivo effect that the mutation of K62 and K75 in YopE exerted on bacterial dissemination after orogastric ingestion. Our results could be seen as another example of the tight and balanced regulation of the activity of the Yersinia type III protein secretion system on the mammalian host (13). For instance, it was previously shown that YopP/YopJ require a delicate, balanced activity for an optimized effect on the outcome of Yersinia virulence (7, 41). The replacement of less active YopJ/YopP by another isotype with stronger activity does not automatically lead to enhanced virulence. On the contrary, when YopJ in Y. pseudotuberculosis is replaced by more cytotoxically active YopP from Y. enterocolitica, the pathogenicity of the resulting Yersinia strain is attenuated. These studies indicated that intermediate levels of YopJ-dependent cytotoxicity are necessary for maximal systemic virulence of Y. pseudotuberculosis (7, 41). The destruction of YopE O8 through the ubiquitin-proteasome pathway could in the same manner help to keep the amount of translocated YopE at a level that optimizes Yop translocation and Yersinia virulence. This regulatory phenomenon seems to be specific for Y. enterocolitica serogroup O8, because K62 and K75 have been hitherto verified only in YopE from Y. enterocolitica O8. This suggests that the degradation and inactivation of YopE are dispensable for effective virulence of other Yersinia species and serotypes. Y. enterocolitica O9 and Y. pseudotuberculosis have been shown to translocate minor amounts of YopP/YopJ into host cells compared to Y. enterocolitica O8 (11, 40). This feature could be related to the specific role that the inactivation of YopE O8 plays in Yop translocation. However, the reduced Yop translocation levels are obviously also compatible with Yersinia virulence. The Yop arsenal of Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica O9 seems to effectuate full bacterial virulence. In these circumstances, the stronger activity of YopE, resulting from YopE accumulation, may outmatch other Yop activities in pathogen-mediated immunomodulation. In fact, in vivo studies have indicated that loss of YopE could affect bacterial virulence more severely in Y. pestis and Y. pseudotuberculosis infection (1, 36) than in infection with Y. enterocolitica O8 (34). From that it may be speculated that YopE has a more direct anti-host cell activity in the pathogenicity of Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica O9, whereas YopE from Y. enterocolitica O8 may have a predominant regulatory role in Yop translocation, at least in the early phase of systemic infection. Y. enterocolitica serogroup O8 therefore appears to engage a unique virulence strategy. It exploits the proteasome, a given host cell pathway normally required to maintain cellular homeostasis, in order to manipulate the host immune response most efficiently for colonization of the host organism.
Acknowledgments
This work was supported by grants (DFG Ru788/2 and DFG Ru788/3) from the Deutsche Forschungsgemeinschaft.
We thank Jürgen Heesemann for fruitful scientific input and discussions.
Editor: J. B. Bliska
Footnotes
Published ahead of print on 13 December 2010.
REFERENCES
- 1.Aepfelbacher, M., C. Trasak, and K. Ruckdeschel. 2007. Effector functions of pathogenic Yersinia species. Thromb. Haemost. 98:521-529. [PubMed] [Google Scholar]
- 2.Aili, M., et al. 2008. Regulation of Yersinia Yop-effector delivery by translocated YopE. Int. J. Med. Microbiol. 298:183-192. [DOI] [PubMed] [Google Scholar]
- 3.Aili, M., E. L. Isaksson, B. Hallberg, H. Wolf-Watz, and R. Rosqvist. 2006. Functional analysis of the YopE GTPase-activating protein (GAP) activity of Yersinia pseudotuberculosis. Cell. Microbiol. 8:1020-1033. [DOI] [PubMed] [Google Scholar]
- 4.Angot, A., A. Vergunst, S. Genin, and N. Peeters. 2007. Exploitation of eukaryotic ubiquitin signaling pathways by effectors translocated by bacterial type III and type IV secretion systems. PLoS Pathog. 3:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balachandran, P., et al. 2007. The ubiquitin ligase Cbl-b limits Pseudomonas aeruginosa exotoxin T-mediated virulence. J. Clin. Invest. 117:419-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boland, A., S. Havaux, and G. R. Cornelis. 1998. Heterogeneity of the Yersinia YopM protein. Microb. Pathog. 25:343-348. [DOI] [PubMed] [Google Scholar]
- 7.Brodsky, I. E., and R. Medzhitov. 2008. Reduced secretion of YopJ by Yersinia limits in vivo cell death but enhances bacterial virulence. PLoS Pathog. 4:e1000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter, P. B., C. F. Varga, and E. E. Keet. 1973. New strain of Yersinia enterocolitica pathogenic for rodents. Appl. Microbiol. 26:1016-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins, C. A., and E. J. Brown. Cytosol as battleground: ubiquitin as a weapon for both host and pathogen. Trends Cell Biol. 20:205-213. [DOI] [PubMed]
- 10.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denecker, G., et al. 2002. Effect of low- and high-virulence Yersinia enterocolitica strains on the inflammatory response of human umbilical vein endothelial cells. Infect. Immun. 70:3510-3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foultier, B., and G. R. Cornelis. 2003. DNA sequence and analysis of the pYVa127/90 virulence plasmid of Yersinia enterocolitica strain A127/90. Res. Microbiol. 154:553-557. [DOI] [PubMed] [Google Scholar]
- 13.Galan, J. E. 2009. Common themes in the design and function of bacterial effectors. Cell Host Microbe 5:571-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heesemann, J., and R. Laufs. 1983. Construction of a mobilizable Yersinia enterocolitica virulence plasmid. J. Bacteriol. 155:761-767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hentschke, M., K. Trulzsch, J. Heesemann, M. Aepfelbacher, and K. Ruckdeschel. 2007. Serogroup-related escape of Yersinia enterocolitica YopE from degradation by the ubiquitin-proteasome pathway. Infect. Immun. 75:4423-4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hicks, S. W., and J. E. Galan. Hijacking the host ubiquitin pathway: structural strategies of bacterial E3 ubiquitin ligases. Curr. Opin. Microbiol. 13:41-46. [DOI] [PMC free article] [PubMed]
- 17.Howard, S. L., et al. 2006. Application of comparative phylogenomics to study the evolution of Yersinia enterocolitica and to identify genetic differences relating to pathogenicity. J. Bacteriol. 188:3645-3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isaksson, E. L., et al. 2009. The membrane localization domain is required for intracellular localization and autoregulation of YopE in Yersinia pseudotuberculosis. Infect. Immun. 77:4740-4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kubori, T., and J. E. Galan. 2003. Temporal regulation of Salmonella virulence effector function by proteasome-dependent protein degradation. Cell 115:333-342. [DOI] [PubMed] [Google Scholar]
- 20.Lee, V. T., D. M. Anderson, and O. Schneewind. 1998. Targeting of Yersinia Yop proteins into the cytosol of HeLa cells: one-step translocation of YopE across bacterial and eukaryotic membranes is dependent on SycE chaperone. Mol. Microbiol. 28:593-601. [DOI] [PubMed] [Google Scholar]
- 21.Mejia, E., J. B. Bliska, and G. I. Viboud. 2008. Yersinia controls type III effector delivery into host cells by modulating Rho activity. PLoS Pathog. 4:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munro, P., G. Flatau, and E. Lemichez. 2007. Bacteria and the ubiquitin pathway. Curr. Opin. Microbiol. 10:39-46. [DOI] [PubMed] [Google Scholar]
- 23.Patel, J. C., K. Hueffer, T. T. Lam, and J. E. Galan. 2009. Diversification of a Salmonella virulence protein function by ubiquitin-dependent differential localization. Cell 137:283-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roppenser, B., A. Roder, M. Hentschke, K. Ruckdeschel, and M. Aepfelbacher. 2009. Yersinia enterocolitica differentially modulates RhoG activity in host cells. J. Cell Sci. 122:696-705. [DOI] [PubMed] [Google Scholar]
- 25.Ruckdeschel, K., et al. 2001. Yersinia outer protein P of Yersinia enterocolitica simultaneously blocks the nuclear factor-kappa B pathway and exploits lipopolysaccharide signaling to trigger apoptosis in macrophages. J. Immunol. 166:1823-1831. [DOI] [PubMed] [Google Scholar]
- 26.Ruckdeschel, K., et al. 2006. The proteasome pathway destabilizes Yersinia outer protein E and represses its antihost cell activities. J. Immunol. 176:6093-6102. [DOI] [PubMed] [Google Scholar]
- 27.Ruckdeschel, K., K. Richter, O. Mannel, and J. Heesemann. 2001. Arginine-143 of Yersinia enterocolitica YopP crucially determines isotype-related NF-κB suppression and apoptosis induction in macrophages. Infect. Immun. 69:7652-7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruckdeschel, K., A. Roggenkamp, S. Schubert, and J. Heesemann. 1996. Differential contribution of Yersinia enterocolitica virulence factors to evasion of microbicidal action of neutrophils. Infect. Immun. 64:724-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rytkonen, A., and D. W. Holden. 2007. Bacterial interference of ubiquitination and deubiquitination. Cell Host Microbe 1:13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schnupf, P., D. A. Portnoy, and A. L. Decatur. 2006. Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: role of the PEST-like sequence. Cell Microbiol. 8:353-364. [DOI] [PubMed] [Google Scholar]
- 31.Shabek, N., and A. Ciechanover. Degradation of ubiquitin: the fate of the cellular reaper. Cell Cycle 9:523-530. [DOI] [PubMed]
- 32.Snellings, N. J., M. Popek, and L. E. Lindler. 2001. Complete DNA sequence of Yersinia enterocolitica serotype 0:8 low-calcium-response plasmid reveals a new virulence plasmid-associated replicon. Infect. Immun. 69:4627-4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trosky, J. E., A. D. Liverman, and K. Orth. 2008. Yersinia outer proteins: Yops. Cell. Microbiol. 10:557-565. [DOI] [PubMed] [Google Scholar]
- 34.Trulzsch, K., T. Sporleder, E. I. Igwe, H. Russmann, and J. Heesemann. 2004. Contribution of the major secreted yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infect. Immun. 72:5227-5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Viboud, G. I., and J. B. Bliska. 2001. A bacterial type III secretion system inhibits actin polymerization to prevent pore formation in host cell membranes. EMBO J. 20:5373-5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viboud, G. I., and J. B. Bliska. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59:69-89. [DOI] [PubMed] [Google Scholar]
- 37.Viboud, G. I., E. Mejia, and J. B. Bliska. 2006. Comparison of YopE and YopT activities in counteracting host signalling responses to Yersinia pseudotuberculosis infection. Cell. Microbiol. 8:1504-1515. [DOI] [PubMed] [Google Scholar]
- 38.Virdi, J. S., and P. Sachdeva. 2005. Molecular heterogeneity in Yersinia enterocolitica and ‘Y. enterocolitica-like’ speciesi-implications for epidemiology, typing and taxonomy. FEMS Immunol. Med. Microbiol. 45:1-10. [DOI] [PubMed] [Google Scholar]
- 39.Wren, B. W. 2003. The yersiniae—a model genus to study the rapid evolution of bacterial pathogens. Nat. Rev. Microbiol. 1:55-64. [DOI] [PubMed] [Google Scholar]
- 40.Zauberman, A., et al. 2006. Interaction of Yersinia pestis with macrophages: limitations in YopJ-dependent apoptosis. Infect. Immun. 74:3239-3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zauberman, A., et al. 2009. Yersinia pestis endowed with increased cytotoxicity is avirulent in a bubonic plague model and induces rapid protection against pneumonic plague. PLoS One 4:e5938. [DOI] [PMC free article] [PubMed] [Google Scholar]