

Abstract
Lyme disease, caused by the bacterium Borrelia burgdorferi, is the most widespread tick-borne infection in the northern hemisphere that results in a multistage disorder with concomitant pathology, including arthritis. During late-stage experimental infection in mice, B. burgdorferi evades the adaptive immune response despite the presence of borrelia-specific bactericidal antibodies. In this study we asked whether B. burgdorferi could invade fibroblasts or endothelial cells as a mechanism to model the avoidance from humorally based clearance. A variation of the gentamicin protection assay, coupled with the detection of borrelial transcripts following gentamicin treatment, indicated that a portion of B. burgdorferi cells were protected in the short term from antibiotic killing due to their ability to invade cultured mammalian cells. Long-term coculture of B. burgdorferi with primary human fibroblasts provided additional support for intracellular protection. Furthermore, decreased invasion of B. burgdorferi in murine fibroblasts that do not synthesize the β1 integrin subunit was observed, indicating that β1-containing integrins are required for optimal borrelial invasion. However, β1-dependent invasion did not require either the α5β1 integrin or the borrelial fibronectin-binding protein BBK32. The internalization of B. burgdorferi was inhibited by cytochalasin D and PP2, suggesting that B. burgdorferi invasion required the reorganization of actin filaments and Src family kinases (SFK), respectively. Taken together, these results suggest that B. burgdorferi can invade and retain viability in nonphagocytic cells in a process that may, in part, help to explain the phenotype observed in untreated experimental infection.
Borrelia burgdorferi sensu lato is the causative agent of Lyme disease and the most widespread arthropod infection in North America, Europe, and Asia (3, 60, 80, 81). The disease is transmitted via Ixodes ticks and presents as a multistage disorder that is initially characterized by a flu-like illness and a painless rash known as erythema migrans (60, 79-81). Subsequently the disease can involve cardiac, neurologic, and joint abnormalities if therapeutic approaches are not sought early in the infectious process (60, 80, 81). If treated early, patients generally respond well to antibiotic therapy (80, 81).
During experimental infection in mice, persistence is observed in the absence of antibiotic therapy. These experimentally infected animals have high-titered antibodies that kill B. burgdorferi in vitro; however, we know strikingly little as to how these spirochetes evade humoral clearance under these conditions. One contributing factor may be that B. burgdorferi invades nonphagocytic mammalian cells in vivo, thereby providing an immunoprotected niche. Along these lines, several groups have reported that B. burgdorferi is able to internalize into different eukaryotic cells, including endothelial cells, fibroblasts, neuronal, and neuroglial cells (24, 46, 52, 53). However, the fate of internalized spirochetes and the mechanisms employed by B. burgdorferi to trigger internalization, as well as the intracellular signaling pathways involved in this process, remain largely unknown.
In the recent past it has become apparent that both Staphylococcus aureus and Streptococcus pyogenes, although more commonly known as extracellular pathogens, have an intracellular niche that protects them from host and antibiotic-based clearance (32, 33, 54, 76, 77, 84). The internalization observed with the Gram-positive pathogens utilizes host fibronectin, whereby the bacterium engages soluble fibronectin, via fibronectin-binding proteins (FnBPs) in S. aureus and SfbI in S. pyogenes (32, 33, 35, 54, 76, 77, 84), binds α5β1 integrins on eukaryotic cells, and is internalized in a process that involves focal adhesion and Src family kinases (SFK) (1, 2, 32, 63). Since B. burgdorferi binds to fibronectin using the tandem β-zipper model in a manner similar to that seen for Staphylococcus and Streptococcus (17, 37, 44, 65, 68, 71, 72), we were interested in testing whether B. burgdorferi-fibronectin binding was also associated with borrelial invasion.
In this report we demonstrate that B. burgdorferi is internalized both within immortalized fibroblasts and within primary fibroblasts and endothelial cells, all at a low multiplicity of infection, in a process that is not entirely dependent on BBK32 and the fibronectin-binding integrin α5β1. Despite the muted involvement of α5β1 and fibronectin binding in borrelial internalization, herein we show that other as-yet-unknown β1 integrins are involved in the invasion of B. burgdorferi into host cells. Furthermore, these studies indicate that the internalization of B. burgdorferi is dependent on both actin reorganization and SFK activity. The results presented provide the foundation to further delineate the role of borrelia-based invasion within the context of B. burgdorferi infection.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All B. burgdorferi strains and various transformants are listed in Table 1. All cultures were grown in complete Barbour-Stoenner-Kelly II (BSK-II) liquid medium supplemented with 6% normal rabbit serum, at 32°C, 1% CO2. For B. burgdorferi, the following antibiotic concentrations were used in the BSK-II medium: 50 μg/ml streptomycin, 50 μg/ml gentamicin, and 300 μg/ml kanamycin.
TABLE 1.
Bacterial strains and plasmid constructs used in this study
| Strain or plasmid | Commentsa | Reference(s) or source |
|---|---|---|
| B. burgdorferi strains | ||
| MSK5 | B. burgdorferi strain B31; contains all known plasmids | 47 |
| ML23/pBSVφ(flaBp-gfp) | B. burgdorferi strain B31 lacking only lp25; expresses the cycle 3 gfp allele | 14, 47 |
| ML23/pEW159 | B. burgdorferi strain B31 lacking only lp25; expresses the cycle 3 gfp allele and contains bbe22 | This study |
| JS315/pEW159 | ML23 derivative carrying bbk32::Strr; expresses the cycle 3 gfp allele and contains bbe22 | This study |
| ML23/pJW201 | B. burgdorferi strain B31 lacking only lp25; expresses the gfp mut1 allele and contains bbe22 | This study |
| JS315/pJW201 | ML23 derivative carrying bbk32::Strr; expresses the gfp mut1 allele and contains bbe22 | This study |
| JS315/pJW202 | ML23 derivative carrying bbk32::Strr; expresses the gfp mut1 allele and contains bbe22 and bbk32 | This study |
| E. coli strains | ||
| TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| DH5α | F− φ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17 (rK− mK+) supE44 thi-1 gyrA96 relA1 λ− | Invitrogen |
| Plasmids | ||
| pBSVφ(flaBp-gfp) | Borrelial shuttle vector containing constitutively expressed cycle 3 gfp allele; Kanr | 14 |
| pBBE22 | Borrelial shuttle vector carrying bbe22; Kanr | 67 |
| pCR8/GW/TOPO | Gateway PCR cloning/entry vector; Spcr | Invitrogen |
| pJW101 | pCR8/GW/TOPO containing constitutively expressed cycle 3 gfp allele from pBSVΦ(flaBp-gfp); Spcr | This study |
| pBBE22gate | pBBE22 modified into a Gateway destination vector containing attR sites; Camr Kanr | 87 |
| pEW159 | Constitutively expressed cycle 3 gfp allele recombined via Gateway system into pBBE22gate; Kanr | This study |
| pTM61 | Modified borrelial shuttle vector pTM49 that contains the gfp mut1 allele; Genr | 59 |
| pJW201 | bbe22 region cloned into pTM61; Genr | This study |
| pCR2.1/BbrecA | B. burgdorferi recA cloned into pCR2.1-TOPO; Kanr Ampr | This study; D. Shaw |
Camr, chloramphenicol resistance; Genr, gentamicin resistance; Strr, streptomycin resistance; Ampr, ampicillin resistance; Kanr, kanamycin resistance; Spcr, spectinomycin resistance.
Escherichia coli strains TOP10 and DH5α (Invitrogen, Carlsbad, CA) were used for plasmid propagation under the following antibiotic selection: 5 μg/ml gentamicin, 50 μg/ml kanamycin, 100 μg/ml spectinomycin, and 15 μg/ml chloramphenicol. All E. coli strains were grown at 37°C with aeration.
Plasmid construction.
All plasmids used and constructed in this study are listed in Table 1. A constitutively expressed gfp gene (the cycle 3 gfp allele from plasmid pcDNA3.1/CT-GFP-TOPO; Invitrogen, Carlsbad, CA) (14), under the control of the B. burgdorferi flaB promoter, was PCR amplified from plasmid pBSVΦ(flaBp-gfp) (kindly provided by James Carroll) using the following oligonucleotide primers (5′ to 3′): proFlaB F, TGT CTG TCG CCT GTG GCT TCC; and GFP reverse, CTG CTA TTG TCT TCC CAA TCC TCC. The resulting 1,360-bp band was cloned into the entry vector pCR8/GW/TOPO (Invitrogen) and was designated pJW101. The pCR8/GW/TOPO vector contains attL1 and attL2 sites for recombination-based transfer of the constitutively expressed cycle 3 gfp allele into the destination vector pBBE22gate (87). To facilitate the directed recombination event, an LR clonase-dependent reaction was performed with pJW101 and pBBE22gate as previously described (87). The resulting plasmid containing the constitutively expressed cycle 3 gfp allele in pBBE22gate was designated pEW159.
To generate a shuttle vector containing the gfp mut1 allele, pBBE22 (generously provided by Steve Norris) (67) was digested with KpnI, and a 2.0-kb fragment containing bbe22 and bbe23 was ligated into KpnI-digested pTM61 (generously provided by Tara Moriarty and George Chaconas) (59). The resulting plasmid was designated pJW201.
To track B. burgdorferi genomic copies and transcripts, borrelial recA was PCR amplified with the following oligonucleotide primers: recA-F, 5′-ACG CAA ATT TTC CAT ATT ACT CAG ATT-3′; and recA-R, 5′-ACG CAA TTT AAG AAT GTC AAA GTT AAA-3′. The resulting product was cloned into pCR2.1-TOPO (Invitrogen) to obtain pCR2.1/BbrecA (kindly provided by Dana Shaw). Mouse fibroblast genomic copies and transcripts were determined as described previously using murine β-actin as a standard (87).
The nucleotide sequence of the aforementioned plasmid constructs isolated was determined by the Gene Technologies Laboratory, Institute of Developmental and Molecular Biology, Texas A&M University.
PCR.
A PCR was performed to verify plasmid construction and to track B. burgdorferi plasmid profiles following transformation. All PCRs were performed as previously described using Taq polymerase (Supermix or Supermix HiFi; Invitrogen, Carlsbad, CA) (39-41, 73, 74). The oligonucleotide primers used to profile borrelial plasmids have been described previously (47).
Eukaryotic cell culture.
The mouse fibroblast cell line L929 and the mouse fibroblast cell lines GD25 and GD25β1 (27, 32), as well as the primary normal human dermal fibroblast (NHDF) cells, were cultivated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine at 37°C and 5% CO2. GD25β1 cells were also supplemented with 10 μg/ml puromycin. Human umbilical vein endothelial cells (HUVECs) were cultured in M199 medium supplemented with 15% FBS, human epidermal growth factor, hydrocortisone, and bovine brain extract (Clonetics, Cambrex Bio Science Walkersville, Inc.) at 37°C in 5% CO2 (86). Prior to the addition of HUVECs to the flask, 1% gelatin (in phosphate-buffered saline [PBS]) was added to promote adherence.
Bacterial adhesion and internalization assays.
Prior to infection, 1 × 106 L929 murine fibroblasts, HUVECs, and NHDF cells were seeded on glass coverslips and grown at 37°C in 5% CO2 for 18 h. Prior to B. burgdorferi infection, cells were washed gently with PBS. Subsequently, 500 μl of fresh DMEM or M199 medium (depending on the eukaryotic cells used) supplemented with 10% FBS was added to the wells. The eukaryotic cells were then infected with gfp-expressing B. burgdorferi strains at a multiplicity of infection (MOI) of 1 to 5 for 2 to 18 h. Following infection, cells were washed three times with prewarmed PBS and then fixed with 3.7% paraformaldehyde for 30 min at room temperature. In experiments to address the involvement of actin in invasion, 10 nM cytochalasin D (Sigma/Aldrich, St. Louis, MO) was added to the cell culture medium for 2 h prior to the addition of B. burgdorferi. To evaluate the requirement of Src family kinases, 10 μM PP2 or 10 nM SU6656 (both from Gibco Corp., Camarillo, CA) was added to the cell culture medium for 2 h prior to the addition of B. burgdorferi. In order to test the role of the α5β1 integrin in borrelial internalization, 10 μg/ml of the rat monoclonal antibody MAB2575 against mouse α5β1 integrin (Chemicon, Temecula, CA) was added to the cell culture medium 1 h prior to infection.
Fluorescence and confocal microscopy.
To distinguish intracellular from extracellular B. burgdorferi cells, fixed cells were washed 5 times with cold PBS and 1% BSA, and the monoclonal antibody BOR-018-48316, specific for B. burgdorferi OspA (Capricorn Products, Inc., Portland, ME), was added to the samples. After a 1-h incubation, cells were washed again with cold PBS and 1% BSA and incubated with goat anti-mouse secondary IgG antibody conjugated with Alexa 594 (Invitrogen) at room temperature for 1 h. The samples were then washed 5 times with cold PBS containing 1% BSA, and the coverslips mounted on glass slides containing DAPI (4′,6-diamidino-2-phenylindole) and visualized by epifluorescence using a Nikon Eclipse E800 microscope. Extracellular bacteria fluoresced both red and green (merged subsequently to yellow) when viewed at the appropriate wavelength (488 nm versus 594 nm for green fluorescent protein [GFP] and Alexa 594, respectively), whereas intracellular bacteria fluoresced only green. For confocal microscopy, L929 cells infected with strain ML23/pEW159 were washed, fixed, and stained as described above. Samples were visualized using a Zeiss Stallion dual detector imaging system (Carl Zeiss MicroImaging, Thornwood, NY) with SlideBook software (Intelligent Imaging Innovations, Inc., Denver, CO). A series of z stacks of images were acquired at 0.5-μm intervals with the 63× lens objective and were followed by fluorescence deconvolution.
Viability assay.
To test the viability of B. burgdorferi cultured in cell culture medium, B. burgdorferi strain B31 derivative MSK5, at 5 × 107 cells/ml, were pelleted at 4,000 × g for 20 min, washed with PBS, and then cultured in 10 ml DMEM supplemented with 10% FBS at 37°C in 5% CO2. A 1-ml sample was removed each day up to 1 week, and the samples were split into 0.5-ml volumes. One sample was treated at 65°C for 30 min to kill B. burgdorferi to serve as a positive control for cell death. The parallel sample was assayed using the LIVE/DEAD BacLight bacterial viability kit (Invitrogen Detection Technologies, Eugene, OR) according to the manufacturer's protocol. The LIVE/DEAD stain assay was also used to determine the viability of B. burgdorferi in supplemented DMEM and treated with either 100 or 200 μg/ml gentamicin for 5 h.
Gentamicin protection assay.
For invasion studies, 1 × 106 L929 fibroblasts were seeded in a 12-well plate overnight. Prior to B. burgdorferi infection, L929 cells were washed gently with PBS, after which 1 ml fresh DMEM with 10% FBS was added to the wells. The cells were then infected with B. burgdorferi MSK5 at an MOI of 5 for 18 h. Following this time frame, the supernatant was removed and placed in new wells, and the remaining L929 cells were washed and resuspended in fresh DMEM with 10% FBS. Next, either 100 μg/ml or 200 μg/ml gentamicin was added to the supernatant or L929 cells for 5 h, respectively. A total of 10 μl of the supernatant fraction was then diluted in 18 ml BSK-II medium and plated in a 96-well plate. The infected cells were washed with warm PBS three times, processed with a sterile pestle to release B. burgdorferi, and then resuspended in 1 ml of BSK-II medium. A total of 10 μl of processed cells was diluted in 18 ml BSK-II medium and plated in a 96-well plate. Plates were incubated at 32°C, 1% CO2, and after 2 to 3 weeks a final growth determination was made.
To test if the attached B. burgdorferi could be killed by gentamicin, cells were treated with 10 μM PP2 to inhibit invasion for 2 h prior to coculture with MSK5 incubated with 5-(and 6-)carboxyfluorescein diacetate succinimidyl ester (CFDA SE; Invitrogen) according to the manufacturer's suggestions, to fluorescently label live B. burgdorferi. After MSK5 infection and treatment with 200 μg/ml gentamicin for 5 h, the cells were treated with Invitrogen's LIVE/DEAD stain and scored for viability.
Long-term recovery assay.
A total of 1 × 105 NHDF cells were seeded in 6-well plates overnight and then washed in fresh supplemented DMEM. Subsequently, these cells were infected with B. burgdorferi strain MSK5 at an MOI of 10. In some experiments, cultured cells were propagated with 10 μM PP2 for 18 h prior to the addition of B. burgdorferi strain B31 derivative MSK5.
In experiments designed to determine whether B. burgdorferi could be sustained by soluble components released from host cells, NHDF cells were also seeded in a 0.4-μm-pore-size Transwell-COL chamber (Corning Costar Corp., Cambridge, MA) and placed in a 6-well plate, and DMEM supplemented with 10% FBS and 2 mM l-glutamine was added to both the upper and lower compartments. A total of 1 × 106 MSK5 cells were added to the lower compartment and separated from the NHDF cells by a transwell insert. As a negative control, B. burgdorferi MSK5 cells alone were also cultured in DMEM in a 6-well plate. For designated samples, 100 μg/ml gentamicin was added for 1 h following the 18-h coincubation. Fresh DMEM was added every 2 days, with the cells that initially received PP2 added again to the media at the same concentration. After 4 weeks, 100 μl from the samples with B. burgdorferi alone in DMEM, as well as from the lower compartment of the transwell chamber, was diluted in 18 ml BSK-II medium and plated in 96-well plates. B. burgdorferi-infected NHDF cells treated with gentamicin alone, PP2 alone, or gentamicin and PP2 together were washed with warm PBS three times and processed with a sterile pestle to release B. burgdorferi. The samples were then resuspended in 1 ml of BSK-II medium. A 100-μl aliquot of the processed cell samples was diluted in 18 ml of complete BSK-II medium and plated in 96-well plates. Plates were incubated at 32°C in 1% CO2 and, following a 2-week incubation, were scored for growth.
Real-time reverse transcriptase PCR (RT-PCR).
To detect borrelial transcripts within infected fibroblasts, 1 × 106 L929 fibroblasts were grown and treated as described above and infected with B. burgdorferi MSK5 at an MOI of 1 for 18 h. Following this time frame, the supernatant was removed and placed in new wells, and the remaining L929 cells were washed three times with PBS and fed in freshly supplemented DMEM. To half of the samples, 200 μg/ml gentamicin was added to the supernatant and L929 cells for 5 h. The other samples were not treated with antibiotic. B. burgdorferi cells in the supernatant were pelleted at 4,000 × g for 20 min and washed twice with diethyl pyrocarbonate (DEPC)-treated PBS. The infected fibroblasts were also washed twice with DEPC-treated PBS. Both RNA and DNA were extracted using Tri Reagent (Molecular Research Center, Inc., Cincinnati, OH) according to the manufacturer's instructions. The infection of fibroblasts with MSK5 and the subsequent isolation of both RNA and DNA were done three separate times independently.
To isolate RNA, samples were treated with a Turbo DNA-free kit (Ambion, Austin, TX) to eliminate DNA contamination and were subsequently reverse transcribed into cDNA using the TaqMan reverse transcription reagents kit (Roche, Branchburg, NJ). For all RNA samples obtained, a PCR without reverse transcriptase was carried out to test for DNA contamination. The quantification of live B. burgdorferi spirochetes from both the supernatant and pelleted cells was assayed using SYBR green PCR master mix (Applied Biosystems Corp., Foster City, CA) in an Applied Biosystems ABI 7500 fast real-time PCR system.
Both real-time RT-PCR and PCR were conducted, using either cDNA or total DNA, respectively, in concert with the oligonucleotide primers specific for B. burgdorferi recA. The primer sequences (5′ to 3′) were as follows: nTM17FrecA, GTG GAT CTA TTG TAT TAG ATG AGG CTC TCG; and nTM17RrecA, GCC AAA GTT CTG CAA CAT TAA CAC CTA AAG. A B. burgdorferi recA construct, designated pCR2.1/BbrecA (kindly provided by Dana Shaw), was serially diluted starting at 105 copies, subjected to PCR with the recA-specific primers listed above, and used to construct a standard curve to convert the threshold cycle (CT) value of the samples tested into the number of borrelial RNA (cDNA) and DNA copies as previously described (39). A corresponding standard curve was also generated for β-actin as described previously (87).
Statistical analyses.
All comparisons of the data presented were performed using a two-tailed Student t test. For all experiments conducted, the means of three individual experiments were analyzed and standard deviations were calculated. Differences were considered statistically significant when the P values were <0.05.
RESULTS
Nonprofessional phagocytic cells differentially internalize B. burgdorferi.
Previous studies have shown that macrophage cells degrade phagocytized B. burgdorferi (9, 20, 29, 55, 56, 58, 69, 70, 81) and that the infection of monocytic cells induces apoptosis (20). However, despite these potent killing mechanisms by innate immune cells, B. burgdorferi is still able to colonize and maintain an infectious focus even following a potent adaptive immune response (80, 81). One possible mechanism employed by B. burgdorferi to avoid immune clearance might be to invade nonprofessional phagocytic cells near the site of deposition in the host, such as endothelial cells and/or fibroblasts. To test this hypothesis, GFP-expressing B. burgdorferi cells were added at an MOI of 1 to 5 to either L929 fibroblasts, normal human dermal fibroblast (NHDF) cells, or human umbilical vein endothelial cells (HUVECs). To evaluate the internalization of gfp-expressing B. burgdorferi following the infection of fibroblasts or endothelial cells, we incubated the infected host cells with a monoclonal antibody specific for OspA, coupled with an Alexa 594-tagged secondary antibody, to distinguish extracellular from intracellular B. burgdorferi cells (Fig. 1). Under these experimental conditions, intracellular B. burgdorferi bacteria fluoresce green due to GFP production, whereas extracellular B. burgdorferi bacteria fluoresce both red and green due to their concomitant production of GFP and their ability to bind antibodies to OspA (Fig. 1). Using the aforementioned criteria, nearly 4,000 host cells from each sample were scored in triplicate for both attachment and invasion as shown in Fig. 1 and documented in Fig. 2.
FIG. 1.
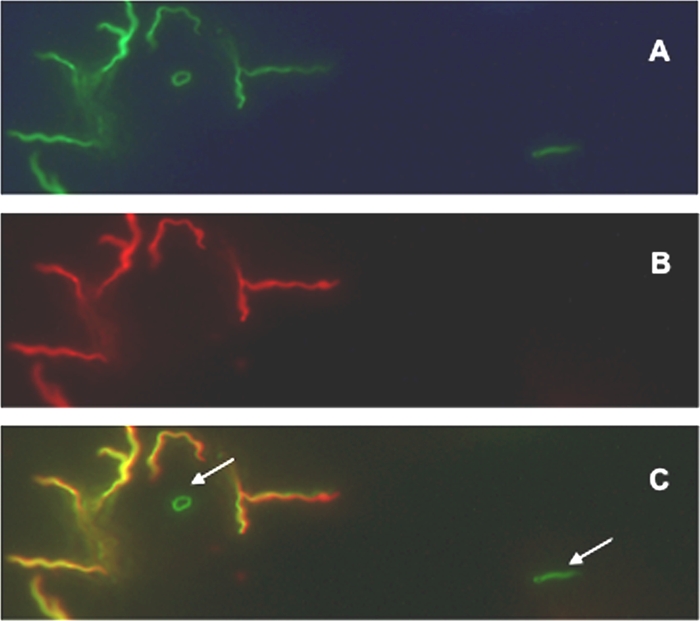
Invasion of Borrelia burgdorferi into fibroblast cells. A total of 1 × 106 L929 fibroblasts were cultured and infected with B. burgdorferi strain ML23/pBSVφ at an MOI of 5 for 2 h. After the cells were washed and fixed, the samples were incubated with mouse anti-OspA antibody and an Alexa 594-tagged secondary antibody. Total GFP fluorescence (A), detection of only the Alexa 594 signal (B), and merge of images (C) shown in panels A and B. Arrows in panel C denote intracellular B. burgdorferi.
FIG. 2.
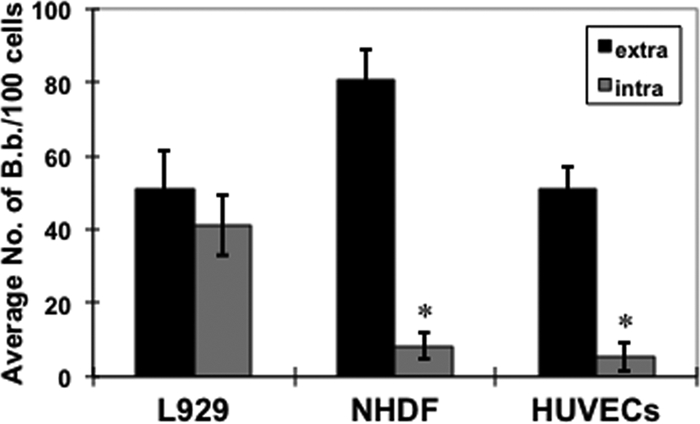
Borrelia burgdorferi invades immortalized and primary fibroblasts, as well as primary endothelial cells. Fibroblasts (L929 or NHDF) or endothelial cells (HUVECs) were infected with ML23/pEW159 at an MOI of 5 for 18 h. Extracellular B. burgdorferi bacteria (B.b.) were detected with anti-OspA coupled with an Alexa Fluor 594 secondary antibody. Data shown for extracellular and intracellular B. burgdorferi are the averages from 3 independent assays from >4,000 host cells analyzed per group per assay (total of >12,000 analyzed). Error bars depict standard deviation. Asterisks indicate a statistically significant difference of internalization in NHDF and HUVECs relative to L929 cells (P < 0.001).
Each cell type tested exhibited comparable attachment levels of B. burgdorferi (Fig. 2); however, the L929 fibroblasts exhibited a 5- or 8-fold increase in spirochetal internalization relative to the NHDF or HUVEC cells, respectively (Fig. 2). Whereas the normal morphology of the spirochetes was retained for extracellular bacteria, the morphology of some of the spirochetes changed dramatically when they were internalized (Fig. 1). These anomalous configurations may be due to the volume constraints of the infected cells or may be reminiscent of nutritionally restricted quiescent forms of borrelial cells that have a cyst-like morphology (4). To confirm the subcellular localization of the internalized B. burgdorferi, z-series confocal microscopy was employed (see Fig. S1 in the supplemental material). This analysis indicated that B. burgdorferi was within cells and readily distinguishable from cells that were attached to the cell surface (i.e., those that were bound by antibody to OspA). It is important to note that the morphology of B. burgdorferi within fibroblasts is distinct from that observed in neuronal and neuroglial cells (in which the spirochetes appear more intact [52]), suggesting that different cell types may provide more hospitable domains for B. burgdorferi due to alternative processing and/or trafficking within these different eukaryotic cells. It is also important to note that in all cases the internalization of B. burgdorferi did not change the viability of the mammalian cells, based on trypan blue exclusion (data not shown).
Actin reorganization and Src family kinases are involved in B. burgdorferi invasion.
To determine if the invasion of B. burgdorferi into L929 fibroblasts involves actin rearrangement, the effect of cytochalasin D, an inhibitor of actin microfilament reorganization, was used. When cytochalasin D was added to the cell culture medium for 2 h prior to the addition of B. burgdorferi, spirochetal invasion was significantly reduced (Fig. 3); however, attachment was not adversely affected.
FIG. 3.
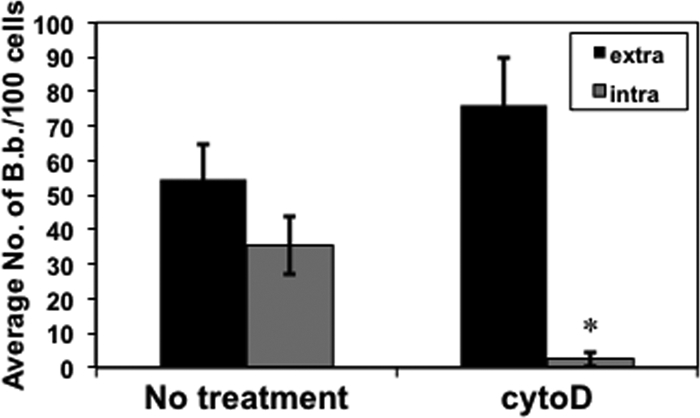
Inhibition of actin polymerization significantly reduces borrelial invasion of fibroblasts. Cytochalasin D was added to a final concentration of 10 nM for 2 h prior to infection of fibroblasts with ML23/pEW159. The infected cells were processed, and B. burgdorferi was visualized as indicated in the legend to Fig. 2. Addition of cytochalasin D did not affect fibroblast viability for the duration of the experiment (not shown). The data shown for extracellular and intracellular B. burgdorferi are the averages from 3 independent assays from >4,000 host cells analyzed per group per assay (total of >12,000 analyzed). Error bars depict standard deviation. Asterisks indicate a statistically significant difference in the internalization of B. burgdorferi in fibroblasts with or without cytochalasin D (P < 0.001).
Given that Src kinases are involved in the internalization of the Gram-positive pathogens Staphylococcus aureus and Streptococcus pyogenes following integrin binding (1, 2, 32, 33, 35, 54, 61, 64), we reasoned that the uptake of B. burgdorferi may also use a similar mechanism. To test this hypothesis, L929 cells were treated with PP2, a broad-spectrum Src kinase inhibitor, for 2 h prior to B. burgdorferi infection. Interestingly, internalization of B. burgdorferi was completely inhibited in cells given PP2 relative to those with none added (Fig. 4 A). Another Src family kinase inhibitor, SU6656, which inhibits a subset of Src kinases (10, 75, 89), significantly inhibited the internalization of B. burgdorferi but, in contrast to the PP2 treatment, did not abolish it (Fig. 4B). Taken together, these findings show that both actin reorganization and Src family kinases are required for the internalization of B. burgdorferi in vitro.
FIG. 4.
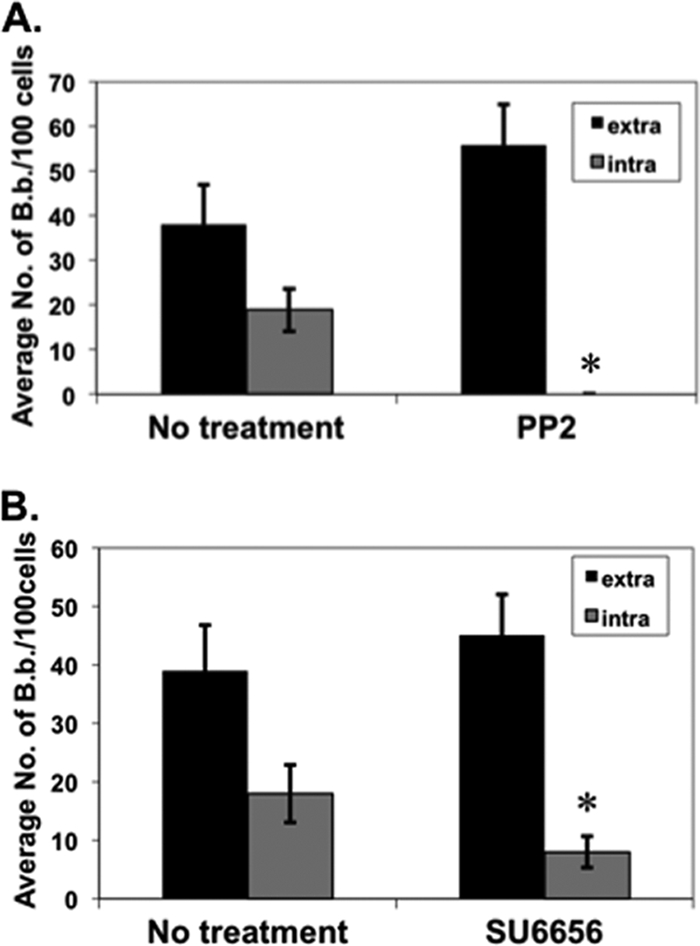
Inhibition of Src kinases significantly reduces borrelial invasion of fibroblasts. Fibroblasts were infected with gfp-expressing B. burgdorferi strain ML23/pEW159 as indicated in the legends to Fig. 2 and 3. The inhibitors PP2 (A) and SU6656 (B) were added to final concentrations of 10 μM and 10 nM for 2 h prior to infection, respectively. The infected cells were processed, and B. burgdorferi was visualized as indicated in the legends to Fig. 2 and Fig. 3. Addition of the inhibitors did not affect fibroblast viability for the duration of the experiment (not shown). The extracellular and intracellular B. burgdorferi data shown are the averages from 3 independent assays from >4,000 host cells analyzed per group per assay (total of >12,000 analyzed). Error bars depict standard deviation. Asterisks indicate a statistically significant difference in the internalization of B. burgdorferi in fibroblasts with and without PP2 (P < 0.00001, in panel A) or with or without SU6656 (P < 0.001, in panel B).
Internalization of B. burgdorferi in eukaryotic cells is not dependent on BBK32.
The invasion of host cells by Staphylococcus aureus requires fibronectin binding via the α5β1 integrin (1, 2, 33, 35, 54, 62, 77); therefore, by analogy, we hypothesized that the borrelial fibronectin binding protein BBK32 (30, 44, 50, 66, 68, 74) was needed for the internalization of B. burgdorferi. To test this, strain JS315, an ML23 derivative that contains a streptomycin resistance cassette inserted into bbk32 (74), was utilized. Accordingly, JS315 does not make BBK32 and exhibits reduced binding to host cells (74). To allow for easy visualization via epifluorescence, the parent strain ML23 and the bbk32::Strr strain JS315 were transformed with the gfp-containing plasmid pEW159, and we used an approach similar to that described above to distinguish between attached versus internalized B. burgdorferi. ML23/pEW159 and JS315/pEW159 showed a significant difference in attachment to L929 fibroblasts, consistent with the importance of the BBK32-dependent attachment to cultured eukaryotic cells (74) (Fig. 5 A). There was also a significant decrease in internalization into fibroblasts of both B. burgdorferi strains; however, the ratios of extracellular to intracellular B. burgdorferi bacteria of ML23/pEW159 and JS315/pEW159 were nearly identical, indicating that the internalization of B. burgdorferi is directly proportional to its degree of attachment.
FIG. 5.
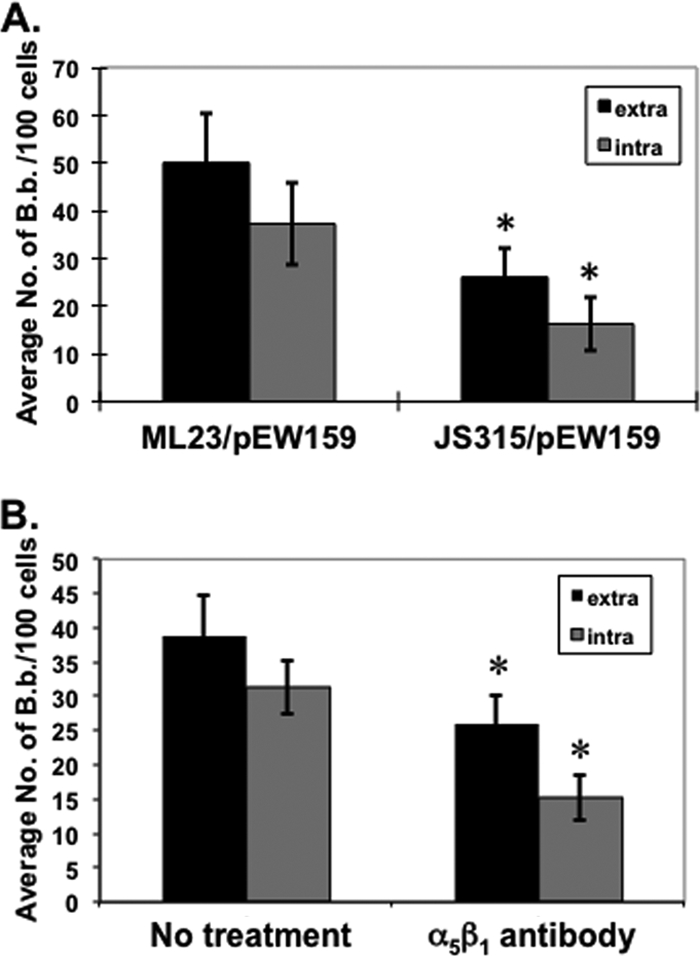
The borrelial Fn binding protein BBK32 does not directly contribute to invasion of B. burgdorferi. (A) Fibroblasts were infected with infectious gfp-expressing B. burgdorferi (ML23/pEW159) or a bbk32::Strr mutant (JS315/pEW159) at an MOI of 5 for 18 h. Borrelial cells were enumerated as described in the legends to Fig. 2 to 4. The data shown for extracellular and intracellular B. burgdorferi are the averages from 3 independent assays from >3,800 host cells analyzed per group per assay. Error bars depict standard deviation. Asterisks indicate a statistically significant difference of both attachment and internalization between ML23/pEW159 and JS315/pEW159 (P < 0.01). (B) Same experiment as described in the legend to panel A except that gfp-expressing B. burgdorferi (ML23/pEW159) cells were incubated with antibody to the α5β1 integrin (10 μg/ml) for 1 h prior to infection. Data shown are the averages from 3 independent assays from >4,000 host cells analyzed per group per assay (total of >12,000 analyzed). Error bars depict standard deviation. Asterisks indicate a statistically significant difference of attachment and internalization of untreated B. burgdorferi relative to cells incubated with α5β1 antibody (P < 0.01).
To provide evidence that the decreased binding seen for the bbk32 mutant was due to its inability to bind the α5β1 integrin, we asked how the addition of an antibody specific for the α5β1 integrin heterodimer affected borrelia-host cell interactions. The data obtained indicated that the addition of this antibody inhibited the attachment of wild-type B. burgdorferi to levels comparable to that observed for the bbk32 mutant strain JS315, suggesting that the reduction of binding seen in JS315 was due, in part, to the loss of binding to α5β1 integrins (Fig. 5B). The difference in internalized B. burgdorferi was also significantly decreased following anti-α5β1 treatment, but in a manner that was directly proportional to their decreased attachment. Based on these observations, the α5β1 integrin participates in the attachment/adherence of B. burgdorferi to fibroblasts but is not required for invasion of these in vitro-cultivated cells.
Invasion of B. burgdorferi requires β1 integrins.
Despite the observation that the α5β1 integrin does not seem to be required for the internalization of B. burgdorferi, it was possible that other β1 integrins might be involved in this process. To test this hypothesis, we used a fibroblast cell line that is devoid of the β1 integrin subunit, designated GD25, and a derivative line that has been transfected to now synthesize β1 and denoted GD25β1 (27, 32). Subsequent infectivity studies with these fibroblasts indicated that the β1 subunit is important for borrelial invasion, given that fibroblasts containing β1 exhibit 3.4-fold-greater internalization and a 2.2-fold-greater attachment-to-invasion ratio relative to fibroblasts lacking the β1 subunit (Fig. 6). These observations suggest that β1 integrins are involved in the internalization of B. burgdorferi into fibroblasts. Furthermore, the absence of a demonstrable change in borrelial invasion following incubation with antibody against α5β1 (Fig. 5B) indicates that a β1-containing integrin heterodimer other than α5β1 is involved in the internalization of B. burgdorferi.
FIG. 6.
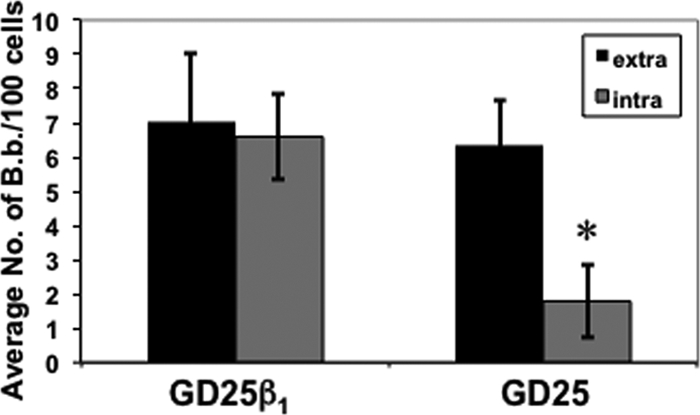
The integrin β1 subunit is required for borrelial invasion. Fibroblasts lacking the integrin β1 subunit were exposed to infectious B. burgdorferi expressing gfp (ML23/pEW159), and attachment and invasion were assessed as indicated in the legends to Fig. 2 to 5. The data shown for extracellular and intracellular B. burgdorferi are the averages from 3 independent assays from >2,000 fibroblasts analyzed per group per assay (total of >6,000 analyzed). Error bars depict standard deviation. The asterisk indicates a statistically significant P value of <0.001 between the levels of invasion observed in cells with or without the β1 integrin subunit.
B. burgdorferi remains viable once it is internalized.
Since we hypothesize that the internalization of B. burgdorferi is involved in long-term survival, it follows that internalized B. burgdorferi would need to retain viability. To test this hypothesis, we applied a variation of the gentamicin protection assay to B. burgdorferi-infected cells. First, we tested the ability of gentamicin to kill B. burgdorferi in the culture media used. Consistent with the known susceptibility of B. burgdorferi to gentamicin (19, 25), it was not surprising that the exposure of B. burgdorferi to 100 μg/ml or 200 μg/ml gentamicin for 5 h in DMEM completely killed all spirochetes (Fig. 7 A). When L929 cells were infected with B. burgdorferi and then exposed to gentamicin for 5 h, live spirochetes were recovered from pelleted cells but never from culture supernatants, suggesting that the internalization or association of B. burgdorferi cells with the L929 cells protected them from the bactericidal effect of gentamicin (Fig. 7A).
FIG. 7.
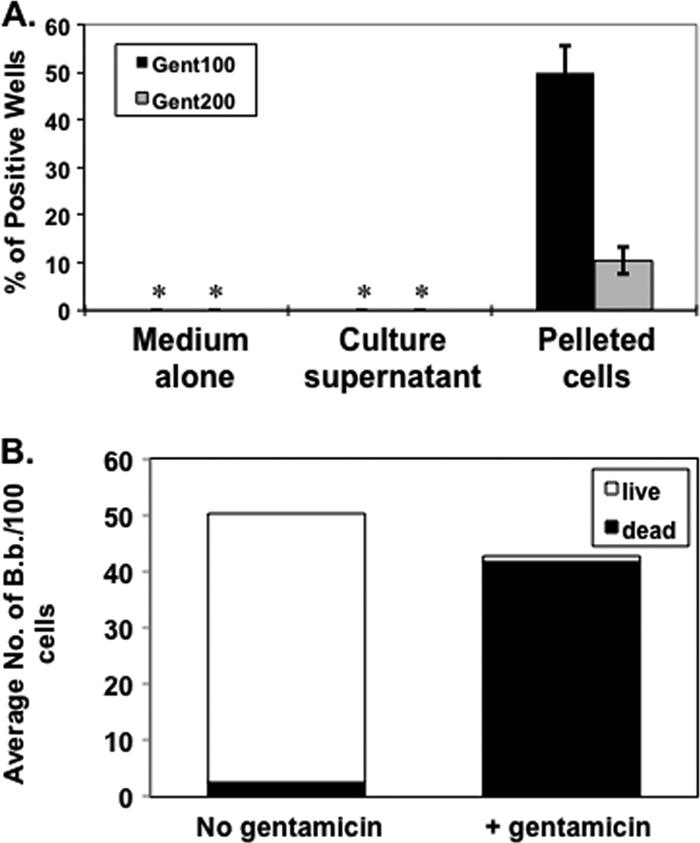
Borrelial invasion is required for survival following antibiotic treatment. (A) Viable B. burgdorferi strain MSK5 bacteria are recovered only from pelleted fibroblasts following exposure to either 100 or 200 μg/ml of gentamicin (Gent100 or Gent200, respectively). Infection of L929 fibroblasts was as described in the legend to Fig. 2. Growth in medium alone and the culture supernatant with added gentamicin did not support growth of B. burgdorferi as indicated by the asterisks. (B) Inhibition by added PP2 and subsequent addition of gentamicin results in the near elimination of extracellular B. burgdorferi strain MSK5. The MSK5 cells represented in panel B were pretreated with CFDA SE prior to their addition to the fibroblasts.
Previously we found that the addition of PP2 greatly inhibited the internalization of B. burgdorferi but did not alter attachment (Fig. 4A). To independently determine the fate of borrelial cells exposed to gentamicin, we labeled B. burgdorferi strain MSK5 with CFSE and then tested how these cells fared when PP2 was added with or without gentamicin, using the LIVE/DEAD fluorescent viability stain. The assay indicated that 97.7% of the extracellular B. burgdorferi cells were killed following the subsequent 5-h exposure to gentamicin (Fig. 7B), relative to controls with PP2 alone, of which only 5.1% were killed (Fig. 7B), suggesting that the live spirochetes observed following gentamicin treatment alone, shown in Fig. 7A, were internalized and protected from the bactericidal action of the added antibiotic.
We next needed an indication that B. burgdorferi cells were metabolically viable following internalization. To assess this, we used real-time RT-PCR to quantify borrelial transcripts following the infection of fibroblasts and treatment with gentamicin. All samples were normalized to total DNA content for both B. burgdorferi and the infected fibroblasts and conducted independently in triplicate. The results shown are the ratio of borrelial recA transcripts to the abundant β-actin host cell transcript, both normalized to their DNA targets. The analysis indicated that borrelial transcripts detected from pelleted fibroblasts were reduced 2.7-fold following incubation with gentamicin, relative to those from parallel samples without added gentamicin (Fig. 8 A). It is important to note that although the decreased level of recA transcripts per borrelial cell relative to β-actin transcripts per fibroblast implies that the assay was insensitive for B. burgdorferi transcripts, the CT values for all RNA targets averaged 17.7 and 17.2 for both recA and β-actin, respectively (data not shown), indicating the high-level expression of both targeted genes. Controls with no added reverse transcriptase had nearly undetectable CT values (data not shown), indicating that the signals detected were due to cDNA converted from RNA in all samples tested. As shown in Fig. 7B, nearly all of the spirochetes were killed by gentamicin treatment if invasion was abrogated by added PP2. As such, these results strongly suggest that the majority of the transcripts detected are from borrelial cells that reside intracellularly. In support of this contention, supernatants from the infected fibroblast cultures treated with gentamicin exhibited a 66.7-fold reduction in borrelial transcripts relative to samples lacking any added antibiotic (Fig. 8B). This is in stark contrast to the pelleted cells, for which a 2.3-fold decrease in borrelial transcript was observed following exposure to gentamicin (Fig. 8B), presumably due to the killing of borrelial cells that are attached but not internalized. The vast difference in borrelial transcript levels, based on extracellular versus intracellular locales (e.g., not as reduced following antibiotic treatment in pelleted cells relative to culture supernatants) support the notion that B. burgdorferi cells are metabolically active following internalization into cultured fibroblasts.
FIG. 8.
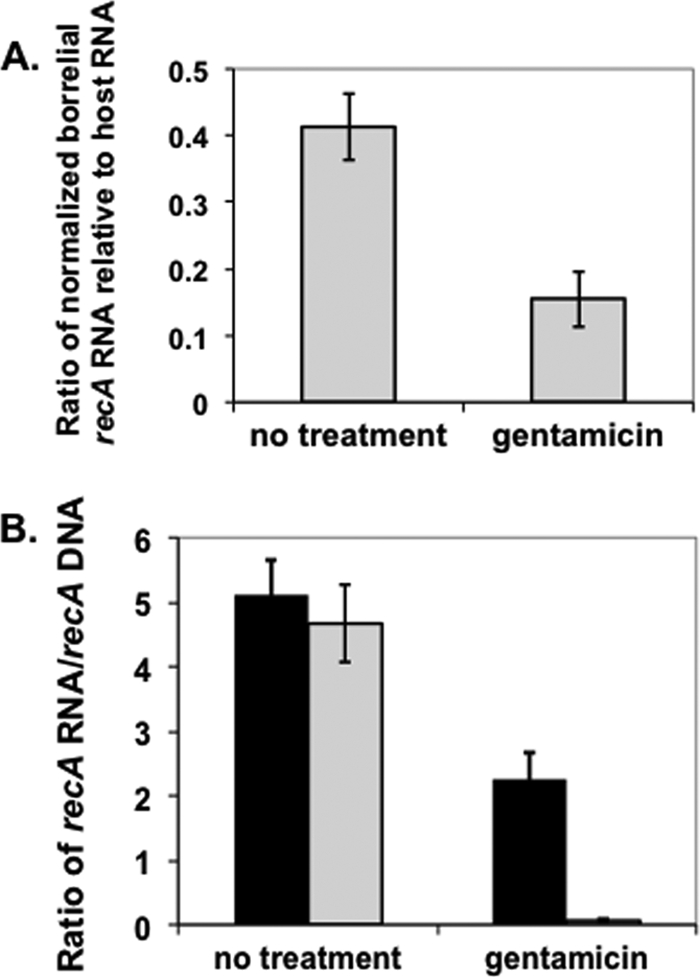
Intracellular B. burgdorferi cells are metabolically active. (A) Transcripts of borrelial recA were detected following infection with B. burgdorferi MSK5 of pelleted L929 fibroblasts (18-h infection at an MOI of 1), in the absence and presence gentamicin (at 200 μg/ml for 5 h postinfection) as indicated and normalized to their respective DNA targets. The values shown are the ratio of borrelial recA transcripts per borrelial genome equivalents relative to β-actin transcripts per β-actin genomic copies (divided by two). Error bars represent standard deviation. (B) Added gentamicin dramatically reduces the amount of transcripts from extracellular B. burgdorferi. Pelleted fibroblasts (black bars) and the resulting supernatant (gray bars) were tested for their levels of recA transcripts relative to recA genomic copies following standard cultivation (no treatment) and incubation with gentamicin. The values shown indicate the ratio of the recA transcripts to recA genomic equivalents within the samples tested. Error bars indicate standard deviation.
B. burgdorferi cells survive following long-term coculture with primary fibroblasts.
Although the initial short-term infection of the L929 cells provided support to our hypothesis that B. burgdorferi can survive intracellularly in nonimmune cells, we were interested in determining if primary cells exhibited this same protection following infection. To assess this, we used NHDFs (primary cells) infected with B. burgdorferi strain MSK5. NHDFs are suited for this type of study since they can be cultivated for several weeks following infection and various treatments. Following infection, NHDFs afforded similar protection to B. burgdorferi following subsequent treatment with gentamicin following a 4-week coinfection (Fig. 9). If invasion was inhibited by the addition of PP2, spirochete levels were 4-fold less than those in cells treated with gentamicin alone (Fig. 9), implying that some B. burgdorferi cells may be able to survive long term or within the secreted extracellular matrix produced by the primary fibroblasts or may escape from infected NHDFs over time. Alternatively, the concentration of PP2 used may not have inhibited invasion for the duration of the experiment, and we do not know whether the cells detected in this assay are extracellular or intracellular. Nevertheless, the enhanced recovery of B. burgdorferi in primary fibroblasts that were treated with gentamicin relative to PP2 alone suggested that the majority of recovered B. burgdorferi cells were the result of invasion and subsequent protection from the bactericidal action of the antibiotic (Fig. 9). The concurrent addition of gentamicin and PP2 completely abrogated the recovery of B. burgdorferi (Fig. 9). As controls, spirochetes that were cultured in either tissue culture medium alone (DMEM, 10% FBS) without NHDFs or in a transwell plate sharing the cell culture supernatant with NHDF host cells, and thus unable to contact the NHDF cells, were not viable (data not shown). These results indicated that neither the tissue culture media nor soluble factors released from cultured primary fibroblasts could support the growth of B. burgdorferi, providing further support that the majority of the long-term borrelial survivors observed were due to the internalization of the spirochete within the NHDF cells.
FIG. 9.

B. burgdorferi survives following long-term coculture with primary dermal fibroblasts (NHDFs) following gentamicin exposure. NHDFs were infected with B. burgdorferi at an MOI of 10 for 18 h and either left untreated, incubated with gentamicin for 1 h, exposed to PP2 for 1 h, or treated with both gentamicin and PP2 for 1 h as indicated above. Subsequently the samples were propagated for 28 days in DMEM, pelleted, washed, processed to release B. burgdorferi, and then scored for growth as described in Materials and Methods. The overall growth (per well) from 3 independent assays was compiled and converted to the percentage of total wells tested, and the untreated sample was normalized to a value of 1.0. The errors listed and the error bars shown represent the standard deviations of the three independent assays conducted. Based on their observed growth, the other samples were adjusted and compared accordingly.
DISCUSSION
In order to establish a persistent infection, pathogens must evade absolute clearance from the host innate and adaptive immune responses and/or the bactericidal or bacteriostatic effect of administered antibiotics. During late-stage B. burgdorferi infection, high-titered antibodies that efficiently kill B. burgdorferi in vitro are ineffective at clearance (3, 5, 28, 80, 81). This is due presumably to the ability of B. burgdorferi to occupy unknown immunoprivileged niches during experimental infection where it may avoid innate and adaptive immune-mediated killing. To circumvent host clearance, some pathogens invade immune cells and alter the ability of these cells to kill them by quelling their antimicrobial activity and/or modifying normal host cell trafficking (reviewed recently in references 23 and 31). However, in vitro-cultivated B. burgdorferi cells are not known to override the killing mediated by innate immune cells and, as a consequence, for the most part are cleared by these cells in vitro (9, 20, 29, 56-58, 69, 81).
Historically B. burgdorferi has been categorized as an extracellular pathogen, but this is due primarily to the inability to detect B. burgdorferi in appreciable numbers during the infectious process. Although the ability of B. burgdorferi to survive within connective tissue has been observed (6, 36, 38), it is also possible that these spirochetes may invade and replicate at low levels inside nonimmune cells, such as fibroblasts, endothelial cells, and neuronal and neuroglial cells (24, 46, 52, 53). Along these lines, previous studies demonstrated that B. burgdorferi invades human umbilical vein endothelial cells (HUVECs), human-foreskin fibroblasts, and an immortalized B cell line (24, 46, 53). The major limitation of these studies was the high multiplicity of infection (MOI) employed, ranging from 100 to 5,000, since this bacterial load is unlikely to be physiologically relevant, and the lack of information provided regarding the fate of the borrelial cells. More recently Livengood and Gilmore demonstrated that B. burgdorferi is taken up by both neuronal and neuroglial cells at a lower MOI and appear to be unaffected, at least morphologically, following this process (52). In our studies, we infected immortalized L929 fibroblasts, as well as primary normal human dermal fibroblasts (NHDFs) and HUVECs, with gfp-expressing B. burgdorferi at an MOI of 1 to 5, which is significantly lower and, by our estimation, more in agreement with what might occur initially in the infectious process. While the extracellular B. burgdorferi spirochetes kept their linear coiled shape, over time the intracellular spirochetes changed their morphology (Fig. 1). Because of the length of linear B. burgdorferi, we speculate that this anomalous configuration may be either due to volume constraints of the host cells or reminiscent of nutritionally restricted quiescent cells having a cyst-like morphology (4). Although unproven, it is tempting to speculate that some of the intracellular forms observed may represent the cyst morphology and can be recovered when they are provided with a nutritionally replete environment.
The enhanced internalization in L929 cells relative to human primary fibroblasts (Fig. 2) suggests that murine cells are either more adept at internalizing B. burgdorferi or less capable of eliminating the spirochetes relative to primary human cells due to the different densities of host receptors for B. burgdorferi or significant differences in intracellular trafficking of internalized spirochetes. These differences also imply that internalization of B. burgdorferi is a rare event in vivo and may explain why such localized niches are not well characterized. Further insight into the trafficking mechanisms of internalized B. burgdorferi may delineate the differences observed between the murine L929 and human primary dermal fibroblast cells.
Given that integrin binding of pathogenic bacteria, in concert with fibronectin binding, is known to mediate adherence and invasion of eukaryotic cells and contribute to their ability to infect susceptible hosts (reviewed recently in references 12, 34, and 43), we asked whether a similar activity could be attributable to B. burgdorferi. Previously we determined that BBK32 was required for maximal binding to fibroblasts (74), and while the studies herein confirmed this prior observation, the degree of invasion was commensurate with binding (Fig. 5A). The results indicate that, although BBK32 was not required for invasion (Fig. 5A), some β1 integrin structure was needed (Fig. 6). The alternative α subunit that engages the β1 subunit for borrelial invasion is not known but, based on known expression profiles of fibroblasts, may be the α1, α2, α3, α8, or α11 subunit (11, 15, 42, 48, 83, 85). Recently α3β1-B. burgdorferi interactions were reported for chondrocytes (7, 8), and given the abundance of α3β1 in a number of cell types (21, 45, 49, 82), as well as the ability of α3β1 to bind a large number of extracellular matrix proteins, including fibronectin, laminin, and collagen (22, 26, 88), it is possible that α3β1 may mediate the B. burgdorferi-host cell interaction observed.
One possibility for the difference in invasive activity associated with B. burgdorferi fibronectin binding could be due to the different stoichiometry of BBK32 and fibronectin binding relative to that of other fibronectin binding proteins from Gram-positive pathogens, even though Borrelia and the Gram-positive pathogens all use the tandem β-zipper mechanism for binding host fibronectin (17, 37, 44, 65, 68, 71, 72). Specifically, SfbI and the FnBPs from Streptococcus and Staphylococcus, respectively, each contain multiple amino terminal domain (NTD) binding sites that recognize F1 modules on fibronectin and promote integrin clustering and the subsequent invasion of these pathogens (17, 37, 44, 65, 68, 71, 72). In contrast, BBK32 contains a sole NTD binding site and, as such, may not be able to attract enough integrins to cluster in order to trigger the downstream signaling pathway needed for optimized internalization. Despite these limitations, it is clear that B. burgdorferi is capable of invasion in vitro.
The observation that exposure to cytochalasin D, a potent inhibitor of actin polymerization, inhibited the internalization of B. burgdorferi indicated that actin microfilament reorganization is also required for borrelial invasion (Fig. 3). Actin reorganization is triggered by integrin signaling pathways and plays an important role in the invasion of several intracellular pathogens (reviewed recently in references 12, 18, 35, and 43). Furthermore, integrin signaling pathways are activated by interactions with extracellular matrix components, including fibronectin (1, 2, 32, 33, 35, 54, 62, 77), resulting in the aforementioned integrin clustering and characteristic activation of kinases, including the Src family protein tyrosine kinases (PTKs), focal adhesion kinase (FAK), and several actin-binding adaptor molecules (1, 2, 32, 63). As such, B. burgdorferi utilizes common mechanisms exploited by other pathogens to invade eukaryotic cells. As indicated above, how borrelial cells ultimately traffic once inside host cells and how this factors into long-term survival are not known.
Given that several intracellular pathogens utilize Src kinase for internalization and dissemination (16, 32, 51, 61, 78, 90), we tested whether specific Src family kinase (SFK) inhibitors inhibited B. burgdorferi internalization in vitro. Specifically, Src kinase activity is required for the internalization of Neisseria spp. and Staphylococcus aureus and is needed for both Streptococcus pneumoniae and Bacillus anthracis dissemination (albeit via different mechanisms), given that translocation through lung epithelial cells is inhibited when SFK inhibitors are added in vitro to experiments with the last two pathogens (1, 2, 32, 63, 78, 89). In a comparable manner, the addition of PP2 clearly inhibits B. burgdorferi invasion (Fig. 4A); however, it is still not known which of the SFK members are involved in this process. Since SU6656 has been shown to alter the dissemination of B. anthracis during infection (89), it is possible that B. burgdorferi dissemination and/or long-term survival may be adversely affected in existing SFK knockout mice or in mice that are treated over time with SU6656. Additional experimentation is required to address this possibility.
Our intent with the modified gentamicin recovery experiment was to determine whether B. burgdorferi could be protected from the bactericidal effect of the antibiotic, thereby providing evidence that the spirochetes resided intracellularly. Typically, colony formation is used as a readout for survival but requires detergent treatment or osmotic lysis to release intracellular bacteria; however, the fragile nature of B. burgdorferi made these techniques untenable (i.e., no viable spirochetes were recovered). To circumvent this dilemma, we lysed cells mechanically and found that B. burgdorferi spirochetes were recovered from host cell pellets, but not from the cell culture supernatant, following 5 h of treatment with gentamicin, suggesting that the recovery of live B. burgdorferi spirochetes was due to their intracellular locale (Fig. 7A). This result is consistent with the observation that antibiotics with increased intracellular penetration, i.e., doxycycline and erythromycin, are more efficient at clearing B. burgdorferi when cocultured with eukaryotic cells (13) and given to treat infection (3, 80, 81). Furthermore, our quantitative RT-PCR results that quantify recA transcripts strongly support the contention that B. burgdorferi cells are internalized and are metabolically active following gentamicin exposure under conditions that clear nearly all spirochetes (Fig. 7 and 8). Whether the internalized borrelial cells are capable of replication is not clear, but our observations, as well as those by Livengood and Gilmore, of B. burgdorferi inside neuroglial cells (52), suggest that the spirochetes do not attain high numbers but instead appear to be in a quiescent but viable state. Despite these limitations, viable spirochetes are recovered from both neural cells and fibroblasts and may reflect differential processing and or trafficking mechanisms that factor into the ability of B. burgdorferi to survive in distinct niches within the infected host.
Finally, we asked whether the intracellular survival of B. burgdorferi observed was limited to the short time frame of our and others' analyses. The results indicated that, while no live spirochetes were recovered in the cell culture supernatant, viable B. burgdorferi cells were obtained at a higher rate and preferentially from primary fibroblasts that were treated with gentamicin alone, suggesting that the ability to invade cells prior to the addition of antibiotic protected them and provided the best chance for survival. It is true that a small proportion of cells survived the treatment with the potent SFK inhibitor PP2 alone (Fig. 9), but even if these cells were capable of residing extracellularly, they are still at a significant disadvantage relative to cells that are internalized, based on the data shown in Fig. 7A and B. One limitation of these studies is the inability to track whether B. burgdorferi can be released from infected cells and repopulate the extracellular milieu of the cultured primary fibroblasts. Given the sensitivity of B. burgdorferi within the cell culture medium (Fig. 7A), it seems unlikely that the spirochetes would be viable in this environment. The studies described herein provide the foundation to further evaluate the importance of the intracellular localization and trafficking of B. burgdorferi following long-term infection.
Supplementary Material
Acknowledgments
We thank Steve Norris for generously providing the borrelial shuttle vector pBBE22 and Tara Moriarty and George Chaconas for supplying us with pTM61. We are grateful to James Carroll for sending us the B. burgdorferi shuttle vector pBSVφ. We also thank Yi Xu and Dana Shaw for helpful conversations and construction of the B. burgdorferi recA plasmid construct pCR2.1/BbrecA, respectively. We also acknowledge the Biomedical Sciences Image Analysis Laboratory at Texas A&M University, College of Veterinary Medicine, for assistance with the confocal imaging shown.
This work was supported by Public Health Service grant R01-AI058086 (to J.T.S. and M.H.) from the National Institute of Allergy and Infectious Diseases. J.W. and E.H.W. were both recipients of Training Grant matching funds from the College of Medicine, Texas A&M Health Science Center.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 20 December 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Agerer, F., et al. 2005. Cellular invasion by Staphylococcus aureus reveals a functional link between focal adhesion kinase and cortactin in integrin-mediated internalisation. J. Cell Sci. 118:2189-2200. [DOI] [PubMed] [Google Scholar]
- 2.Agerer, F., A. Michel, K. Ohlsen, and C. R. Hauck. 2003. Integrin-mediated invasion of Staphylococcus aureus into human cells requires Src family protein-tyrosine kinases. J. Biol. Chem. 278:42524-42531. [DOI] [PubMed] [Google Scholar]
- 3.Aguero-Rosenfeld, M. E., G. Wang, I. Schwartz, and G. P. Wormser. 2005. Diagnosis of Lyme borreliosis. Clin. Microbiol. Rev. 18:484-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alban, P. S., P. W. Johnson, and D. R. Nelson. 2000. Serum-starvation-induced changes in protein synthesis and morphology of Borrelia burgdorferi. Microbiology 146:119-127. [DOI] [PubMed] [Google Scholar]
- 5.Barthold, S. W., M. S. de Souza, J. L. Janotka, A. L. Smith, and D. H. Persing. 1993. Chronic Lyme borreliosis in the laboratory mouse. Am. J. Pathol. 143:959-971. [PMC free article] [PubMed] [Google Scholar]
- 6.Barthold, S. W., E. Hodzic, S. Tunev, and S. Feng. 2006. Antibody-mediated disease remission in the mouse model of Lyme borreliosis. Infect. Immun. 74:4817-4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behera, A. K., et al. 2008. Borrelia burgdorferi BBB07 interaction with integrin a3b1 stimulates production of pro-inflammatory mediators in primary human chondrocytes. Cell. Microbiol. 10:320-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Behera, A. K., et al. 2006. Identification of a TLR-independent pathway for Borrelia burgdorferi-induced expression of matrix metalloproteinases and inflammatory mediators through binding to integrin a3b1. J. Immunol. 177:657-664. [DOI] [PubMed] [Google Scholar]
- 9.Benach, J. L., G. S. Habicht, B. L. Gocinski, and J. L. Coleman. 1984. Phagocytic cell responses to in vivo and in vitro exposure to the Lyme disease spirochete. Yale J. Biol. Med. 57:599-605. [PMC free article] [PubMed] [Google Scholar]
- 10.Blake, R. A., et al. 2000. SU6656, a selective Src family kinase inhibitor, used to probe growth factor signaling. Mol. Cell. Biol. 20:9018-9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouzeghrane, F., C. Mercure, T. L. Reudelhuber, and G. Thibault. 2004. a8b1 integrin is upregulated in myofibroblasts of fibrotic and scarring myocardium. J. Mol. Cell. Cardiol. 36:343-353. [DOI] [PubMed] [Google Scholar]
- 12.Boyle, E. C., and B. B. Finlay. 2003. Bacterial pathogenesis: exploiting cellular adherence. Curr. Opin. Cell Biol. 15:633-639. [DOI] [PubMed] [Google Scholar]
- 13.Brouqui, P., S. Badiaga, and D. Raoult. 1996. Eucaryotic cells protect Borrelia burgdorferi from the action of penicillin and ceftriaxone but not from the action of doxycycline and erythromycin. Antimicrob. Agents Chemother. 40:1552-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carroll, J. A., P. E. Stewart, P. Rosa, A. F. Elias, and C. F. Garon. 2003. An enhanced GFP reporter system to monitor gene expression in Borrelia burgdorferi. Microbiology 149:1819-1828. [DOI] [PubMed] [Google Scholar]
- 15.Caswell, C. C., et al. 2008. Identification of the first prokaryotic collagen sequence motif that mediates binding to human collagen receptors, integrins a2b1 and a11b1. J. Biol. Chem. 283:36168-36175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho, B., N. Cho, S. Seong, M. Choi, and I. Kim. 2010. Intracellular invasion by Orientia tsutsugamushi is mediated by integrin signaling and actin cytoskeleton rearrangements. Infect. Immun. 78:1915-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coburn, J., J. R. Fischer, and J. M. Leong. 2005. Solving a sticky problem: new genetic approaches to host cell adhesion by the Lyme disease spirochete. Mol. Microbiol. 57:1182-1195. [DOI] [PubMed] [Google Scholar]
- 18.Cossart, P., and P. J. Sansonetti. 2004. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304:242-248. [DOI] [PubMed] [Google Scholar]
- 19.Criswell, D., V. L. Tobiason, J. S. Lodmell, and D. S. Samuels. 2006. Mutations conferring aminoglycoside and spectinomycin resistance in Borrelia burgdorferi. Antimicrob. Agents Chemother. 50:445-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cruz, A. R., et al. 2008. Phagocytosis of Borrelia burgdorferi, the Lyme disease spirochete, potentiates innate immune activation and induces apoptosis in human monocytes. Infect. Immun. 76:56-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.da Silva, R. G., et al. 2010. Endothelial a3b1-integrin represses pathological angiogenesis and sustains endothelial-VEGF. Am. J. Pathol. 177:1534-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delwel, G. O., et al. 1994. Distinct and overlapping ligand specificities of the a3Ab1 and a6Ab1 integrins: recognition of laminin isoforms. Mol. Biol. Cell 5:203-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diacovich, L., and J. Gorvel. 2010. Bacterial manipulation of innate immunity to promote infection. Nat. Rev. Microbiol. 8:117-128. [DOI] [PubMed] [Google Scholar]
- 24.Dorward, D. W., E. R. Fischer, and D. M. Brooks. 1997. Invasion and cytopathic killing of human lymphocytes by spirochetes causing Lyme disease. Clin. Infect. Dis. 25(Suppl.1):S2-S8. [DOI] [PubMed] [Google Scholar]
- 25.Elias, A. F., et al. 2003. New antibiotic resistance cassettes suitable for genetic studies in Borrelia burgdorferi. J. Mol. Microbiol. Biotechnol. 6:29-40. [DOI] [PubMed] [Google Scholar]
- 26.Elices, M. J., L. A. Urry, and M. E. Hemler. 1991. Receptor functions for the integrin VLA-3: fibronectin, collagen, and laminin binding are differentially influenced by Arg-Gly-Asp peptide and by divalent cations. J. Cell Biol. 112:169-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fässler, R., et al. 1995. Lack of b1 integrin gene in embryonic stem cells affects morphology, adhesion, and migration but not integration into the inner cell mass of blastocysts. J. Cell Biol. 128:979-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fikrig, E., et al. 1994. Sera from patients with chronic Lyme disease protect mice from Lyme borreliosis. J. Infect. Dis. 169:568-574. [DOI] [PubMed] [Google Scholar]
- 29.Filgueira, L., F. O. Nestle, M. Rittig, H. I. Joller, and P. Groscurth. 1996. Human dendritic cells phagocytose and process Borrelia burgdorferi. J. Immunol. 157:2998-3005. [PubMed] [Google Scholar]
- 30.Fischer J. R., K. T. LeBlanc, and J. M. Leong. 2006. Fibronectin binding protein BBK32 of the Lyme disease spirochete promotes bacterial attachment to glycosaminoglycans. Infect. Immun. 74:435-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flannagan, R. S., G. Cosío, and S. Grinstein. 2009. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 7:355-366. [DOI] [PubMed] [Google Scholar]
- 32.Fowler, T., S. Johansson, K. K. Wary, and M. Höök. 2003. Src kinase has a central role in in vitro cellular internalization of Staphylococcus aureus. Cell. Microbiol. 5:417-426. [DOI] [PubMed] [Google Scholar]
- 33.Fowler, T., et al. 2000. Cellular invasion by Staphylococcus aureus involves a fibronectin bridge between the bacterial fibronectin-binding MSCRAMMs and host cell b1 integrins. Eur. J. Cell Biol. 79:672-679. [DOI] [PubMed] [Google Scholar]
- 34.Hauck, C. R., F. Agerer, P. Muenzner, and T. Schmitter. 2006. Cellular adhesion molecules as targets for bacterial infection. Eur. J. Cell Biol. 85:235-242. [DOI] [PubMed] [Google Scholar]
- 35.Hauck, C. R., and K. Ohlsen. 2006. Sticky connections: extracellular matrix protein recognition and integrin-mediated cellular invasion by Staphylococcus aureus. Curr. Opin. Microbiol. 9:5-11. [DOI] [PubMed] [Google Scholar]
- 36.Hodzic, E., S. Feng, K. Holden, K. J. Freet, and S. W. Barthold. 2008. Persistence of Borrelia burgdorferi following antibiotic treatment in mice. Antimicrob. Agents Chemother. 52:1728-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.House-Pompeo, K., Y. Xu, D. Joh, P. Speziale, and M. Höök. 1996. Conformational changes in the fibronectin binding MSCRAMMs are induced by ligand binding. J. Biol. Chem. 271:1379-1384. [DOI] [PubMed] [Google Scholar]
- 38.Hunfeld, K., E. Ruzić-Sabljić, D. E. Norris, P. Kraiczy, and F. Strle. 2006. Risk of culture-confirmed borrelial persistence in patients treated for erythema migrans and possible mechanisms of resistance. Int. J. Med. Microbiol. 296(Suppl. 40):233-241. [DOI] [PubMed] [Google Scholar]
- 39.Hyde, J. A., J. P. Trzeciakowski, and J. T. Skare. 2007. Borrelia burgdorferi alters its gene expression and antigenic profile in response to CO2 levels. J. Bacteriol. 189:437-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hyde, J. A., D. K. Shaw, R. Smith Iii, J. P. Trzeciakowski, and J. T. Skare. 2009. The BosR regulatory protein of Borrelia burgdorferi interfaces with the RpoS regulatory pathway and modulates both the oxidative stress response and pathogenic properties of the Lyme disease spirochete. Mol. Microbiol. 74:1344-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hyde, J. A., D. K. Shaw, R. Smith, J. P. Trzeciakowski, and J. T. Skare. 2010. Characterization of a conditional bosR mutant in Borrelia burgdorferi. Infect. Immun. 78:265-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hynes, R. O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell 110:673-687. [DOI] [PubMed] [Google Scholar]
- 43.Kerr, J. R. 1999. Cell adhesion molecules in the pathogenesis of and host defence against microbial infection. Mol. Pathol. 52:220-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim, J. H., et al. 2004. BBK32, a fibronectin binding MSCRAMM from Borrelia burgdorferi, contains a disordered region that undergoes a conformational change on ligand binding. J. Biol. Chem. 279:41706-41714. [DOI] [PubMed] [Google Scholar]
- 45.Kim, K. K., et al. 2009. Epithelial cell a3b1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Invest. 119:213-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klempner, M. S., R. Noring, and R. A. Rogers. 1993. Invasion of human skin fibroblasts by the Lyme disease spirochete, Borrelia burgdorferi. J. Infect. Dis. 167:1074-1081. [DOI] [PubMed] [Google Scholar]
- 47.Labandeira-Rey, M., and J. T. Skare. 2001. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect. Immun. 69:446-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.La Linn, M., et al. 2005. An arthritogenic alphavirus uses the a1b1 integrin collagen receptor. Virology 336:229-239. [DOI] [PubMed] [Google Scholar]
- 49.Lemieux, J. M., M. C. Horowitz, and M. A. Kacena. 2010. Involvement of integrins a3b1 and a5b1 and glycoprotein IIb in megakaryocyte-induced osteoblast proliferation. J. Cell. Biochem. 109:927-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li, X., X. Liu, D. S. Beck, F. S. Kantor, and E. Fikrig. 2006. Borrelia burgdorferi lacking BBK32, a fibronectin-binding protein, retains full pathogenicity. Infect. Immun. 74:3305-3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu, W., et al. 2010. Involvement of Src tyrosine kinase in Escherichia coli invasion of human brain microvascular endothelial cells. FEBS Lett. 584:27-32. [DOI] [PubMed] [Google Scholar]
- 52.Livengood, J. A., and R. J. Gilmore. 2006. Invasion of human neuronal and glial cells by an infectious strain of Borrelia burgdorferi. Microbes Infect. 8:2832-2840. [DOI] [PubMed] [Google Scholar]
- 53.Ma, Y., A. Sturrock, and J. J. Weis. 1991. Intracellular localization of Borrelia burgdorferi within human endothelial cells. Infect. Immun. 59:671-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Massey, R. C., et al. 2001. Fibronectin-binding protein A of Staphylococcus aureus has multiple, substituting, binding regions that mediate adherence to fibronectin and invasion of endothelial cells. Cell. Microbiol. 3:839-851. [DOI] [PubMed] [Google Scholar]
- 55.Montgomery, R. R., and S. E. Malawista. 1996. Entry of Borrelia burgdorferi into macrophages is end-on and leads to degradation in lysosomes. Infect. Immun. 64:2867-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montgomery, R. R., S. E. Malawista, K. J. Feen, and L. K. Bockenstedt. 1996. Direct demonstration of antigenic substitution of Borrelia burgdorferi ex vivo: exploration of the paradox of the early immune response to outer surface proteins A and C in Lyme disease. J. Exp. Med. 183:261-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Montgomery, R. R., D. Lusitani, A. D. B. Chevance, and S. E. Malawista. 2002. Human phagocytic cells in the early innate immune response to Borrelia burgdorferi. J. Infect. Dis. 185:1773-1779. [DOI] [PubMed] [Google Scholar]
- 58.Moore, M. W., et al. 2007. Phagocytosis of Borrelia burgdorferi and Treponema pallidum potentiates innate immune activation and induces gamma interferon production. Infect. Immun. 75:2046-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moriarty, T. J., et al. 2008. Real-time high resolution 3D imaging of the Lyme disease spirochete adhering to and escaping from the vasculature of a living host. PLoS Pathog. 4:e1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nadelman, R. B., and G. P. Wormser. 1998. Lyme borreliosis. Lancet 352:557-565. [DOI] [PubMed] [Google Scholar]
- 61.Nerlich, A., et al. 2009. Invasion of endothelial cells by tissue-invasive M3 type group A streptococci requires Src kinase and activation of Rac1 by a phosphatidylinositol 3-kinase-independent mechanism. J. Biol. Chem. 284:20319-20328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nitsche-Schmitz, D. P., M. Rohde, and G. S. Chhatwal. 2007. Invasion mechanisms of Gram-positive pathogenic cocci. Thromb. Haemost. 98:488-496. [PubMed] [Google Scholar]
- 63.Ozeri, V., et al. 2001. De novo formation of focal complex-like structures in host cells by invading streptococci. Mol. Microbiol. 41:561-573. [DOI] [PubMed] [Google Scholar]
- 64.Ozeri, V., I. Rosenshine, D. F. Mosher, R. Fassler, and E. Hanski. 1998. Roles of integrins and fibronectin in the entry of Streptococcus pyogenes into cells via protein F1. Mol. Microbiol. 30:625-637. [DOI] [PubMed] [Google Scholar]
- 65.Prabhakaran, S., X. Liang, J. T. Skare, J. R. Potts, and M. Höök. 2009. A novel fibronectin binding motif in MSCRAMMs targets F3 modules. PLoS One 4:e5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Probert, W. S., and B. J. Johnson. 1998. Identification of a 47 kDa fibronectin-binding protein expressed by Borrelia burgdorferi isolate B31. Mol. Microbiol. 30:1003-1015. [DOI] [PubMed] [Google Scholar]
- 67.Purser, J. E., et al. 2003. A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Mol. Microbiol. 48:753-764. [DOI] [PubMed] [Google Scholar]
- 68.Raibaud, S., et al. 2005. Borrelia burgdorferi binds fibronectin through a tandem b-zipper, a common mechanism of fibronectin binding in staphylococci, streptococci, and spirochetes. J. Biol. Chem. 280:18803-18809. [DOI] [PubMed] [Google Scholar]
- 69.Salazar, J. C., et al. 2009. Activation of human monocytes by live Borrelia burgdorferi generates TLR2-dependent and -independent responses which include induction of IFN-beta. PLoS Pathog. 5:e1000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sambri, V., et al. 1996. Uptake and killing of Lyme disease and relapsing fever borreliae in the perfused rat liver and by isolated Kupffer cells. Infect. Immun. 64:1858-1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwarz-Linek, U., et al. 2004. High affinity streptococcal binding to human fibronectin requires specific recognition of sequential F1 modules. J. Biol. Chem. 279:39017-39025. [DOI] [PubMed] [Google Scholar]
- 72.Schwarz-Linek, U., et al. 2003. Pathogenic bacteria attach to human fibronectin through a tandem b-zipper. Nature 423:177-181. [DOI] [PubMed] [Google Scholar]
- 73.Seshu, J., et al. 2004. A conservative amino acid change alters the function of BosR, the redox regulator of Borrelia burgdorferi. Mol. Microbiol. 54:1352-1363. [DOI] [PubMed] [Google Scholar]
- 74.Seshu, J., et al. 2006. Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Mol. Microbiol. 59:1591-1601. [DOI] [PubMed] [Google Scholar]
- 75.Severgnini, M., et al. 2005. Inhibition of the Src and Jak kinases protects against lipopolysaccharide-induced acute lung injury. Am. J. Respir. Crit. Care Med. 171:858-867. [DOI] [PubMed] [Google Scholar]
- 76.Sinha, B., et al. 2000. Heterologously expressed Staphylococcus aureus fibronectin-binding proteins are sufficient for invasion of host cells. Infect. Immun. 68:6871-6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sinha, B., et al. 1999. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin a5b1. Cell. Microbiol. 1:101-117. [DOI] [PubMed] [Google Scholar]
- 78.Slanina, H., et al. 2010. Entry of Neisseria meningitidis into mammalian cells requires the Src family protein tyrosine kinases. Infect. Immun. 78:1905-1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith, R. P., et al. 2002. Clinical characteristics and treatment outcome of early Lyme disease in patients with microbiologically confirmed erythema migrans. Ann. Intern. Med. 136:421-428. [DOI] [PubMed] [Google Scholar]
- 80.Steere, A. C. 2001. Lyme disease. N. Engl. J. Med. 345:115-125. [DOI] [PubMed] [Google Scholar]
- 81.Steere, A. C., J. Coburn, and L. Glickstein. 2004. The emergence of Lyme disease. J. Clin. Invest. 113:1093-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tavella, S., et al. 1997. Regulated expression of fibronectin, laminin and related integrin receptors during the early chondrocyte differentiation. J. Cell Sci. 110(Pt. 18):2261-2270. [DOI] [PubMed] [Google Scholar]
- 83.Thibault, G., et al. 2001. Upregulation of a8b1-integrin in cardiac fibroblast by angiotensin II and transforming growth factor-beta1. Am. J. Physiol. Cell Physiol. 281:C1457-C1467. [DOI] [PubMed] [Google Scholar]
- 84.Tuchscherr, L., et al. 2010. Staphylococcus aureus small-colony variants are adapted phenotypes for intracellular persistence. J. Infect. Dis. 202:1031-1040. [DOI] [PubMed] [Google Scholar]
- 85.Warren, A. P., C. N. Owens, L. K. Borysiewicz, and K. Patel. 1994. Down-regulation of integrin a1b1 expression and association with cell rounding in human cytomegalovirus-infected fibroblasts. J. Gen. Virol. 75:3319-3325. [DOI] [PubMed] [Google Scholar]
- 86.Wautier, M. P., et al. 1999. Cultured endothelial cells from human arteriovenous malformations have defective growth regulation. Blood 94:2020-2028. [PubMed] [Google Scholar]
- 87.Weening, E. H., et al. 2008. Borrelia burgdorferi lacking DbpBA exhibits an early survival defect during experimental infection. Infect. Immun. 76:5694-5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weitzman, J. B., R. Pasqualini, Y. Takada, and M. E. Hemler. 1993. The function and distinctive regulation of the integrin VLA-3 in cell adhesion, spreading, and homotypic cell aggregation. J. Biol. Chem. 268:8651-8657. [PubMed] [Google Scholar]
- 89.Xue, Q., et al. 2010. Bacillus anthracis spore entry into epithelial cells is an actin-dependent process requiring c-Src and PI3K. PLoS One 5:e11665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zaas, D. W., et al. 2009. Counteracting signaling activities in lipid rafts associated with the invasion of lung epithelial cells by Pseudomonas aeruginosa. J. Biol. Chem. 284:9955-9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.