

Abstract
We previously demonstrated that plasmid-deficient Chlamydia muridarum retains the ability to infect the murine genital tract but does not elicit oviduct pathology because it fails to activate Toll-like receptor 2 (TLR2). We derived a plasmid-cured derivative of the human genital isolate Chlamydia trachomatis D/UW-3/Cx, strain CTD153, which also fails to activate TLR2, indicating this virulence phenotype is associated with plasmid loss in both C. trachomatis and C. muridarum. As observed with plasmid-deficient C. muridarum, CTD153 displayed impaired accumulation of glycogen within inclusions. Transcriptional profiling of the plasmid-deficient strains by using custom microarrays identified a conserved group of chromosomal loci, the expression of which was similarly controlled in plasmid-deficient C. muridarum strains CM972 and CM3.1 and plasmid-deficient C. trachomatis CTD153. However, although expression of glycogen synthase, encoded by glgA, was greatly reduced in CTD153, it was unaltered in plasmid-deficient C. muridarum strains. Thus, additional plasmid-associated factors are required for glycogen accumulation by this chlamydial species. Furthermore, in C. trachomatis, glgA and other plasmid-responsive chromosomal loci (PRCLs) were transcriptionally responsive to glucose limitation, indicating that additional regulatory elements may be involved in the coordinated expression of these candidate virulence effectors. Glucose-limited C. trachomatis displayed reduced TLR2 stimulation in an in vitro assay. During human chlamydial infection, glucose limitation may decrease chlamydial virulence through its effects on plasmid-responsive chromosomal genes.
Chlamydia trachomatis, Chlamydia muridarum, and Chlamydia psittaci carry a highly conserved plasmid of approximately 7.5 kb, with copy number estimates ranging from 4 to 10 (34) copies per cell. This plasmid encodes eight open reading frames (ORFs). Evidence for the translation of plasmid-encoded ORFs during infection has been obtained by Comanducci et al., who demonstrated that a 28-kDa plasmid-encoded protein, Pgp3, elicits both humoral (7) and mucosal (12) immune responses in convalescent patients. The function of this protein is unknown, as with the remaining plasmid-encoded ORFs. No homology with any other proteins has been observed, with the exception of the tentative identification of a DnaB helicase (17). Two possible recombinases have also been identified (8, 39). Detailed analysis of the plasmid has been severely limited by the lack of a gene transfer system for Chlamydia spp. Naturally occurring plasmid-deficient clinical isolates of C. trachomatis are extremely rare; only three strains have been described (14, 33, 38). Matsumoto et al. described the isolation and characterization of plasmid-deficient C. trachomatis strains in tissue culture-propagated populations (26) and determined that these derivatives were unable to accumulate glycogen within inclusions.
Recently, we derived a plasmid-cured derivative of C. muridarum, strain CM972, which displays altered plaque morphology and reduced infectivity in cell culture and fails to accumulate glycogen within the inclusion (31). Both CM972 and CM3.1, a derivative of CM972 that has recovered wild-type infectivity, were attenuated in the mouse genital tract infection model. Infection of immunologically normal mice with CM972 or CM3.1 resulted in reduced cytokine expression and limited or absent oviduct pathology (29), most likely because of their inability to activate the innate immune receptor, Toll-like receptor 2 (TLR2) (10). Thus, loss of the plasmid from C. muridarum impacts two virulence-associated phenotypes, infectivity and TLR2 activation, as well as the ability of chlamydiae to accumulate glycogen (26, 30).
Using the same novobiocin treatment and screening methods that were used for isolation of plasmid-deficient derivatives of C. muridarum, we isolated plasmid-deficient derivatives of C. trachomatis D/UW-3/Cx, a human genital tract isolate, and one derivative, designated CTD153, was chosen for further characterization. In vitro and in vivo experiments examined the effects of plasmid loss on plaque size, infectivity, glycogen accumulation, and TLR2 activation and confirmed that CTD153 exhibited the same plasmid-associated phenotypic changes previously described for C. muridarum CM972.
Using custom chlamydial genome microarrays, we detected transcriptional differences between CM972 and CM3.1 and the parental C. muridarum Nigg strain and between CTD153 and the parental C. trachomatis D/UW-3/Cx strain. A conserved group of plasmid-responsive chromosomal loci (PRCLs) was identified that may include effectors of the plasmid-associated phenotypes we have described. Quantitative reverse transcription-PCR (RT-PCR) confirmed that transcription of these loci was altered similarly in plasmid-cured strains and in plasmid-deficient clinical isolates of C. trachomatis. Since lack of glycogen accumulation was associated with plasmid loss, the effect of glucose limitation on PRCL transcription was examined in both C. trachomatis and C. muridarum. PRCL transcription was inhibited in response to glucose limitation in C. trachomatis, and TLR2 activation by glucose-limited C. trachomatis was reduced but was unaltered in glucose-limited C. muridarum. Thus, PRCL transcription may be environmentally responsive during C. trachomatis infection.
MATERIALS AND METHODS
Strains, cell lines, and culture conditions.
The C. muridarum strains Nigg (provided by Roger Rank), CM972, and CM3.1 used in this study were previously described (29, 31). C. trachomatis D/UW-3Cx (37) was obtained from the American Type Culture Collection (Manassas, VA) and plaque purified before use. C. trachomatis L2/434/Bu, C. trachomatis 25667R, an L2 serovar strain that lacks the cryptic plasmid (33), C. trachomatis E/Bour (16), and C. trachomatis CT599, a serovar E clinical isolate (38), were also used in this study. Neither 25667R nor CT599 stain positively with iodine, indicating that they are unable to accumulate glycogen within their inclusions (31).
Chlamydiae were routinely cultured in L929 or McCoy cells. Cell culture media and reagents were purchased from Mediatech (Herndon, VA). Cells were infected at an approximate multiplicity of infection (MOI) of 0.5 to 1, before being centrifuged for 1 h at 37°C. The cell culture medium was then removed and replaced with 1× Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), gentamicin (20 μg ml−1), and 0.1 μg ml−1 cycloheximide. Infected cells were harvested into sucrose-phosphate glutamate (SPG) buffer at 40 h postinfection, sonicated, and maintained at −80°C. Bacteria were subsequently titrated by either plaque assay (31) or as inclusion-forming units (IFU) by using a genus-specific fluorescently tagged anti-chlamydial lipopolysaccharide monoclonal antibody (Bio-Rad, Hercules, CA). All MOI calculations were performed using IFU titers unless otherwise stated. Chlamydiae cultured for the purpose of generating cytoplasmic extracts for enzyme assays were infected at an MOI of 2 to 3 into L929 cells grown in suspension in RPMI 1640 medium supplemented with 10% FBS, 2.5 mM HEPES, once they reached a cell density of ∼5 × 105 cells ml−1. Immunoblotting of chlamydial strains was performed using anti-Pls1 antibody (20), provided by Raphael Valdivia, and anti-Mip antibody (4), provided by Sylvette Bas. Serovar D-specific anti-Momp monoclonal antibody used to confirm the lineage of CTD153 was a generous gift of Byron E. Batteiger.
Infectivity assays. (i) In vitro infectivity.
In vitro infectivity of CTD153 was evaluated by comparing the plaquing efficiency and competitiveness of CTD153 with D/UW-3Cx. Plaquing efficiency was determined by calculating the efficiency of plaquing (EOP) for strains titrated in a modified plaque assay without centrifugation versus the titer obtained via the standard assay (with centrifugation) (31). An in vitro competition assay was performed as follows: suspensions comprising C. trachomatis D/UW-3/Cx and CTD153 elementary bodies (EB) were inoculated onto L929 monolayers at fixed ratios ranging from 1:10 to 1:106 under culture conditions favoring entry of plasmid-containing chlamydiae (i.e., without centrifugation) and harvested after 40 h of incubation by lysing the infected cells in 1 ml SPG buffer. A 0.1-ml aliquot of the resulting chlamydial suspension was then applied to a fresh L929 monolayer for a total of five sequential passages. The remaining suspension was frozen at −80°C for subsequent analysis by using iodine staining (36) to detect glycogen within chlamydial inclusions and to distinguish inclusions formed by plasmid-deficient chlamydiae from wild-type inclusions.
In vivo infectivity.
For in vivo infectivity determinations, groups of five female C3H/HeouJ mice were obtained from Jackson Laboratories (Bar Harbor, ME) and inoculated vaginally with 5 × 105 IFU of D/UW-3Cx or CTD153 in 30 μl of SPG buffer. Mice were injected with medroxyprogesterone acetate 7 days prior to infection as previously described (9). Vaginal swabs obtained daily from days 2 through 10 postinoculation were cultured on monolayers of L929 cells to titrate lower genital tract shedding of chlamydiae, and genital tract lavage fluids were obtained to determine cytokine production in response to infection. Gamma interferon (IFN-γ), interleukin-6 (IL-6), macrophage inflammatory protein 2 (MIP-2 [CXCL2]), and tumor necrosis factor alpha (TNF-α) were assayed by using a multiplex cytometric bead array (Millipore, Billerica, MA). All data points were assayed in triplicate and are reported as the means ± standard deviations (SD). The analysis of variance (ANOVA) plus post hoc test was used to analyze differences in bacterial burden and cytokine production between the groups, and P values of <0.05 were considered significant. The Institutional Animal Care and Use Committee of the University of Pittsburgh approved all of the animal experiments.
In vitro analysis of TLR2 activation.
In vitro infection with chlamydiae was performed using murine bone marrow-derived dendritic cells (BMDDCs) grown from bone marrow cultures, following the procedure of Inaba et al. (19). The TLR2 agonist Pam3Cys-Ser-(Lys)4 (Axxora LLC, San Diego, CA) was used as a positive-control stimulant. Dendritic cells were plated in 24-well tissue culture dishes at a density of 105 cells per well. Infections were carried out by overlaying cells with an MOI of 1 or 3 IFU/cell. The inoculated cells were centrifuged at 1,600 × g for 1 h before being incubated for 24 h at 37°C, 5% CO2. Supernatants were harvested and assayed for granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-1β, IL-6, IFN-γ, and TNF-α by using the multiplex cytometric bead array (Millipore).
TLR2 activation by chlamydiae was also assayed using HEK-Blue-2 cells obtained from Invivogen (San Diego, CA). These HEK293 cells, which express TLR1 and TLR6 endogenously, are cotransfected with TLR2 and CD14 genes and an NF-κB promoter fused to a secreted alkaline phosphatase (SEAP) reporter. Similarly processed D/UW-3/Cx, CTD153, Nigg, and CM3.1 were harvested from untreated cells. Each preparation was then assayed via quantitative PCR using 16S primers and normalized to suspensions containing 1.25 × 107 genome equivalents ml−1 prior to inactivation via X-ray irradiation (383.4 Gy) in an IsoVolt Titan E irradiator (GE Inspection Technologies, Ahrensburgh, Germany). We had previously determined that this dose results in a minimum 4-log reduction in IFU titer. TLR2 activation in these cells was measured 24 h after incubation with X-ray-inactivated chlamydial suspensions via a colorimetric enzyme assay of cell supernatants by using QUANTI-Blue (Invivogen). All data points were assayed in triplicate and are reported as the means ± SD.
Microarray screening for transcriptional differences.
Total RNA was purified from infected HeLa 229 cultures (9 × 105 cells per well cultivated as monolayers in six-well culture plates) at 30 h postinfection. The culture medium was discarded, cells were lysed in 1 ml per well lysis buffer containing proteinase K (0.167 μg/μl; MasterPure; Epicentre, Madison, WI), and total nucleic acid was isolated. The lysate was treated with DNase I and used for total RNA preparation according to the manufacturer's instructions. RNA intended for microarray analysis was further purified from residual contaminating DNA by treatment with DNase I (TurboDNAFree; 0.3U per 1 μg RNA; Ambion) at 37°C for 1 h before being precipitated and resuspended in diethylpyrocarbonate (DEPC) water (1 μg/μl). The RNA (250 μg) was then enriched using MICROBEnrich (Ambion, Austin, TX) and MICROBExpress (Ambion) protocols, which resulted in the removal of a significant portion of HeLa cell polyadenylated mRNA and 18S and 28S rRNA in addition to bacterial 16S and 23S rRNA. Once purified, the RNA was reverse transcribed, hybridized, washed, stained, and scanned according to the Prokaryotic Sample and Array Processing section in the GeneChip Expression Analysis technical manual (Affymetrix). The hybridization cocktail, consisting of labeled cDNA, control B2 oligo (Affymetrix), herring sperm DNA (Promega), and bovine serum albumin (BSA; Invitrogen, CA) was then hybridized to a MPAUT-1 GeneChip and incubated for ∼16 h in an Affymetrix hybridization oven 640 (Affymetrix) at 45°C rotating at 60 rpm. Samples were then washed and stained on an Affymetrix fluidics station 450 (Affymetrix) using streptavidin (Pierce Chemical), antistreptavidin antibody (Vector Labratories), and R-streptavidin-phycoerythrin (Molecular Probes), using fluidics protocol ProKGE-WS2_450 in GCOS 1.1 (Affymetrix). After processing, the chips were scanned using a Genechip scanner 3000 (Affymetrix). Data were scaled using GCOS 1.1 (Affymetrix) to a mean intensity of 1,000 by using the C. trachomatis ORFs as a scaling mask. The pivot file for the sample was then saved in GCOS as a text tab-delimited file, and the data were imported into GeneSpring 7.2 (Agilent) for data analysis.
Quantitative RT-PCR.
Total RNA was isolated from chlamydia-infected L929 cells 24 h postinfection (MOI, ∼1) by using an RNeasy RNA isolation kit obtained from Qiagen (Valencia, CA). For quantitative RT-PCR analysis, the RNA (2 μg) was pretreated with DNase I (Ambion), and cDNA was prepared using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. Primer pairs were selected for amplification of the genes identified in this study and for 16S rRNA (Table 1) by using the Beacon Designer 2.13 program (Premier Biosoft International, Palo Alto, CA). Primers were purchased from IDT (Coralville, IA). Real-time PCRs were carried out using iQ Syber green supermix (Bio-Rad, Hercules, CA) in a Bio-Rad iCycler using a two-step reaction, 95°C for 10 s and 55°C for 1 min, for a total of 40 cycles. Melting curve analysis showed that the accumulation of SYBR green-bound DNA was gene specific and not due to primer dimers. Transcripts from the 16S rRNA genes of C. trachomatis and C. muridarum served as endogenous references, and data were analyzed by the 2−ΔΔCT method (25) using Bio-Rad proprietary software. Each sample was assayed in triplicate, and each experiment was performed at least twice. Statistical significance was determined by using Student's t test. A P value of <0.05 was considered significant.
TABLE 1.
PCR primer sets used in this study
| Strain | Locusa | Sense primer | Antisense primer |
|---|---|---|---|
| C. muridarum | TC0319 | GCGGCAATCCCTCCTACC | TGACAACACGACAACATCCTG |
| Nigg | TC0357 | CATCTCCACAACTCACTCTG | AGGTTTGGGTCTTAATGAAATG |
| TC0419 | CTGTAGGAAGAGATGTTGAC | GGAGTTCGGATGTAATTCG | |
| 382.1b | GGCGTTATGTGTTTGACCTGTAG | GCGTCGTAAGAAAGGCTTGAAAG | |
| TC075 | TACGAACTATACGATGAATCC | TGTAGAATTGACGAGTAACG | |
| TC0181 (glgA) | TGTTTCAGGGTCCAAAATCG | CCTTCAAGACTATTCGGACTATG | |
| TC0831 (glpT) | TTGTAAGCGGTGTGATGTC | TTAGAATACCAGTGAGTCAGTAG | |
| TC0828 (mip) | CGGGTAAACCTAACGCTTCTATTG | AACTTCTCCTGTCTCGCATTCC | |
| TCA03 | ACTTACAACTTATTAACTCATCTG | TTGACCGCTATCTCTTAGG | |
| ORF 8b | GATGGGAAGGTCTAACTGATGAAG | GTTCGCAGGCTTGACTACG | |
| TCA04 | GCTATGGCTATTCTTCTGGTGTG | CCGCTCTCTAATCCTCCTATTCG | |
| TCA05 | CCCGAGCTAGACTTGAAAAT | CTCTTGTGGTAGAGTTCTAAGG | |
| TCA06 | CATCCCAAGCAGCAATTTACG | TGGCTCAGGAATCAAACATACG | |
| TCA07 | GGGAAAGTTTGTGGAATGTC | GCGAAGCAGTTCTACGAG | |
| TCA01 | GGCGTGTGTTTATTTCTGAG | TCTTCATCTCTTAATCCCATTTG | |
| TCA02 | CAAGCAACTATCTCTTTCCTC | CGGGCAATTTGTCTTAACC | |
| C. trachomatis | 16S rRNAc | CGTTAATACCCGCTGGATTTGAG | GCCCCGATCTTTGACAACTAAC |
| D/UW3-Cx | CT049 | GACAACCACTCCAGAATCAG | CCATCCGAAGAGCCTACC |
| CT084 | AAGTGTCCGCTCGCATTCC | TGTTCGTCCCAAGGTAAGATAGG | |
| CT142 | GCTTGGTGTATCAATCGTCTC | AATCTCCTCCATATCCATTATTCC | |
| CT382.1 | GGCGTTATGTGTTTAACTTGTAG | ACATCTATCAGCGTCGTAAG | |
| CT702 | AAGGTCTGTTGGGAAGGG | ATGATGATATTGTCTAAGTTGGC | |
| CT798 (glgA) | GCAGGGCTGTTGTTTGAGTAGT | AAGCGATGATGCCTTCCGTTT | |
| CT544 (uhpC) | GAGCGGCTTTACAGGATGGT | TCCTGAGCCACCTTCCCTAAA | |
| CT600 (pal) | GATAACCTCACGATTCTTGC | AGTGAATAAGCGGTCTGC | |
| pGP1 | GCCTCAGAGCTAAACATTCCT | TTGATAAACAAAATCACATCTGCG | |
| pGP2 | AAGGATGAATCGGTACATTTGTA | AAATTCAATGCGTTTTCTTCTAGA | |
| pGP3 | TCACCTTCTCGTACCAAAGC | TCTGGGAGCATGTTCTTAGTC | |
| pGP4 | CTGCTTCAACAAATAGAAAACTCT | AGACAAGATAAGCATAATCAAAGC | |
| pGP5 | TAGACCCGCAATCCAATTTATC | TTTGATGTGTATACTATGTCGTGT | |
| pGP6 | CGATGTGATAGGGAAAGTATGTG | TCTTTGTTTCTAGATGAAGGAAGA | |
| pGP7 | CTATCAGCTTAATGGAGGAGTTG | ACAGGAATCCCTATTTTAGAAACA | |
| pGP8 | CCATCTTCTTTGAAGCGTTGT | GTGCTCAGACTCCGACATAAT | |
| C. trachomatis | CTL0305 | ATGTTGTCGGAGAGTATAGAG | AACCACTTAGCATCTGTAGG |
| L2/434/Bu | CTL0339 | AAGTGTCCGCTCGCATTCC | TGTTCGTCCCAAGGTAAGATAGG |
| CTL071 | AAGGTCTGTTGGGAAGGG | ATGATGATATTGTCTAAGTTGGC | |
| CTL0803 (mip) | GTCGCTCTGCCTATCGTAG | GCATCCCTTTAATCACTTCAAC |
Chromosomal loci are identified by their annotated designations: C. muridarum Nigg, AE002160.2; C. trachomatis D/UW-3/Cx, AE001273.1; L2/434/Bu, AM884176.1. Plasmid ORFs are named as per the corresponding GenBank entry: pMoPn, AE002162; pCHL1 (C. trachomatis D/G0/86), J03321.
An unannotated ORF present in C. muridarum Nigg. Consequently, no formal locus designation was available.
The same 16S primer pair was used for all strains and contains a single mismatch with the C. muridarum gene that did not impair efficiency. Unless otherwise specified, the primer pair designed to amplify C. trachomatis D/UW-3/Cx was also used to assay L2/434/Bu, 25667R, E/Bour, and CT599 homologs.
Preparation of chlamydial cytoplasmic extracts and glycogen synthase assay.
Infected L929 cells were separated from the culture medium after 12 and 24 h of incubation by centrifugation at 450 × g and washed with once in Hanks balanced salt solution (HBSS) before being gently sonicated to release the bacteria. The sonicated cell suspension was then centrifuged at 1,900 × g to pellet unlysed cells and associated debris. The supernatant was then centrifuged at 20,000 × g for 30 min to concentrate the bacteria. The resultant bacterial pellet was washed twice with 1× HBSS and then resuspended in extract buffer (10% glycerol, 50 mM HEPES, 1 mM EDTA [pH 8.0]). The nonionic detergent Triton X-100 was then added to a final concentration of 1%, and the bacteria were disrupted by repetitive shearing through an 18-gauge needle. The resulting chlamydial lysate was immediately frozen and stored at −80°C until assayed. Enzyme assays to measure the incorporation of ADP-Glc into glycogen (15) included 50 mM HEPES (pH 8), 2.5 mg ml−1 rabbit liver glycogen (Sigma), 5 mM magnesium acetate, 0.5 mg ml−1 BSA, and 1 mM ADP-Glc (14C-labeled glucose; ∼500 cpm/nmol; Sigma) and enzyme extract in a total volume of 200 μl. The assays were initiated by the addition of enzyme, and the reactions were terminated after 10 min by boiling for 1 min; the generated labeled glycogen was precipitated (and thus separated from unincorporated ADP-Glc) by the addition of 2 ml of a 75% methanol-1% KCl solution. After incubation on ice for 10 min followed by centrifugation, the resulting precipitate was resuspended in distilled deionized water and subjected to two more rounds of precipitation prior to resuspension and quantitation by liquid scintillation counting. A unit of activity is defined as the amount of enzyme catalyzing the incorporation of 1 μmol of [14C]ADP-Glc per minute at 37°C. Protein concentrations were determined by the method of Bradford (5).
RESULTS
Generation of a plasmid-cured derivative of C. trachomatis causing human genital tract infections.
C. trachomatis D/UW-3/Cx, a representative serovar D strain associated with human genital tract infections, was treated with novobiocin and plated in a plaque assay as previously described (31). A total of 48 plaques were randomly selected, picked from the monolayer, and passaged twice in L929 cells cultured in 24-well dishes. Each isolate was screened by PCR using primers directed against the plasmid and by iodine staining to detect derivatives that failed to accumulate glycogen within their inclusions. Three PCR-negative, iodine-negative isolates were identified via these screens and were plaque purified once more to ensure clonality. Amplification and sequencing of the ompA gene (Fig. 1A) from the plasmid-deficient derivatives confirmed their genetic lineage, and intracellular inclusions formed by this strain stained positively with a mouse monoclonal antibody directed against the serovar D major outer membrane protein (Fig. 1B). A single isolate was selected for further study and designated C. trachomatis CTD153. Consistent with our prior observations of plasmid-cured C. muridarum, CTD153 failed to accumulate glycogen within its inclusions (Fig. 1C).
FIG. 1.

C. trachomatis CTD153 is plasmid deficient and is unable to accumulate glycogen within its inclusions. (A) Equal amounts of template were added to PCR mixtures with primers directed against the cryptic plasmid (pL2F1/R1) or the chlamydial genome (ompA). The first lane contains a standard 10-kb to 100-bp ladder, and differences in resolution reflect different agarose percentages (top, 0.8%; lower, 2%). Purified PCR products (+PCR) were used as positive controls, and the no-template control lanes are indicated (−PCR). (B) L929 cells infected with either D/UW-3/Cx or CTD153, the plasmid-deficient derivative, were fixed and stained with a strain-specific mouse monoclonal antibody 24 h after infection. The monoclonal antibody was detected by using a goat anti-mouse Alexa 488-conjugated antibody with Evans blue stain (1:1,000) of cytoplasm before being photographed at 1,000× magnification. (C) In a parallel experiment, L929 cells infected with either D/UW-3/Cx or CTD153 were fixed and stained with iodine 40 h postinfection before being photographed at 1,000× magnification.
Plasmid-deficient CTD153 replicates normally but displays reduced in vitro infectivity.
Reduced in vitro infectivity of CTD153 relative to D/UW-3/Cx was determined via analysis of plaquing efficiency and by the performance of an in vitro competition assay. CTD153 displayed an ∼85-fold-reduced plaquing efficiency in vitro (EOP for C. trachomatis D/UW-3/Cx, 2.23 × 10−1 ± 2.41 × 10−1; EOP for C. trachomatis CTD153, 2.61 × 10−3 ± 2.20 × 10−3) as well as reduced plaque size compared with its parent (Fig. 2A), suggesting an infectivity defect similar to what we previously described for C. muridarum CM972 (29). The reductions in plaquing efficiency and plaque size observed were not due to a replication defect, because we were unable to detect any differences in growth rate or yield for CTD153 during synchronous infection of L929 cells compared with D/UW-3/Cx (P = 0.73) (Fig. 2B).
FIG. 2.

C. trachomatis CTD153 replicates normally but displays reduced infectivity in vitro and in vivo. (A) Plaques formed by CTD153 are smaller than those of D/UW-3/Cx. (B) CTD153 replicates normally during synchronous infection of L929 cells. D/UW-3/Cx and CTD153 were inoculated at an MOI of ∼1 into L929 cells growing in 24-well dishes, and then samples were harvested at intervals and titrated to determine the IFU/well (P = 0.73). (C) D/UW-3/Cx outcompetes CTD153 over three serial passages when inoculated at a ratio of 1:100. Iodine staining was used to distinguish between inclusions formed by D/UW-3/Cx (iodine+) and those formed by CTD153 (iodine−). Data presented were obtained from the simultaneous counting and iodine scoring of 150 to 200 inclusions/well, with two replicates per passage. Each experiment was performed twice. (D) Lower genital tract shedding of chlamydiae by mice infected with CTD153 is significantly reduced compared to shedding by mice infected with D/UW-3/Cx. Each group of mice (n = 5/group) was inoculated with 5 × 105 IFU chlamydiae and swabbed daily from days 2 to 10 after infection (P = 0.003 by two-way repeated-measures [RM] ANOVA), and this experiment was performed twice.
We also compared the relative fitness of strains D/UW-3/Cx and CTD153 in mixed inocula in which plasmid-deficient chlamydiae competed with wild-type microorganisms to establish infection. Suspensions comprising C. trachomatis D/UW-3/Cx and CTD153 were passaged three times sequentially by inoculation onto L929 monolayers at fixed ratios ranging from 1:10 to 1:106 under conditions that favored entry of plasmid-containing chlamydiae (i.e., without centrifugation). Samples from each passage were cultured in L929 cells under conditions that equally favored infection by both strains, before being fixed and stained with iodine to detect D/UW-3/Cx (iodine-positive) and CTD153 (iodine-negative) inclusions. The ratio of iodine positivity [(iodine+ inclusions/(iodine − + iodine+ inclusions)] for each sample was determined and plotted against passage number (Fig. 2C). Each strain was passaged alone as a control. At least 87% of D/UW-3/Cx inclusions in monoculture stained positively with iodine, while inclusions formed by CTD153 always stained negatively. When mixed inocula comprising D/UW-3/Cx and CTD153 at ratios of 1:10, 1:100, and 1:1,000 were passaged, the population of iodine+ inclusions in each harvested passage increased. In mixed infections where D/UW-3/Cx was inoculated at a ratio of 1:10, it expanded to ∼50% of the population by the second passage, although we did not observe continued expansion in the subsequent passage. Where D/UW-3/Cx was inoculated at ratios of 1:100 and 1:1,000, it expanded to between 13 and 20% of the infecting population by passage 3. In mixed infections where D/UW-3/Cx was present at ratios of ≥1:104, iodine+ inclusions were not detected (data not shown). Plating of CTD153 in the plaque assay, even with prolonged incubation, did not yield secondary mutants in which the infectivity defect had reverted, as we previously described for C. muridarum CM972.
The in vivo consequence of the reduced in vitro infectivity of plasmid-deficient CTD153 was detected by evaluation of the course of lower genital tract infections by CTD153 and D/UW-3/Cx in C3H/HeouJ mice. Cervical swabs obtained from days 2 through 10 after inoculation revealed a significant reduction in the extent of chlamydial shedding in mice infected with CTD153 compared with those infected with D/UW-3/Cx (P = 0.003) (Fig. 2D).
Plasmid-regulation of TLR2-dependent signaling is conserved in C. trachomatis.
We previously demonstrated that plasmid-deficient derivatives of C. muridarum do not cause oviduct disease in the mouse genital tract infection model, most likely because they are unable to elicit TLR2-dependent cytokine expression by the host (29). To determine if this association between the presence of the plasmid and TLR2 activation is preserved in C. trachomatis strains that infect humans, we examined cytokine production from murine BMDDCs incubated with D/UW-3/Cx or CTD153 at MOIs of 1 and 3 for 24 h. Supernatants from murine BMDDCs inoculated with CTD153 contained strikingly lower amounts of TNF-α and IL-6 (Fig. 3A and B) than supernatants from cells incubated with D/UW-3/Cx (MOI, 3). GM-CSF, IL-1β, and IFN-γ levels were extremely low (for GM-CSF) or were undetectable in supernatants of cells cultured with either D/UW-3/Cx or CTD153 (data not shown). Consistent with these observations, vaginal lavage fluids obtained from mice infected with CTD153 contained TNF-α, IL-6 (Fig. 3C and D), and MIP-2 (data not shown) at significantly lower levels than those obtained from D/UW-3/Cx-infected mice. Significantly lower levels of these mediators were noted in genital secretions of CTD153-infected mice on day 8, when the difference in bacterial burden between CTD153- and D/UW-3/Cx-infected mice was slight. These data demonstrate that TLR2 activation by plasmid-cured C. trachomatis is reduced in vitro and in vivo and indicate that the plasmid regulates TLR2-dependent signaling by C. trachomatis, as observed previously for C. muridarum (29).
FIG. 3.

Plasmid-cured C. trachomatis fails to elicit TLR2-dependent cytokine secretion. (A and B) C. trachomatis CTD153 induces weak or absent expression of TNF-α (A) or IL-6 (B) by DCs. Supernatants from murine BMDDCs cultured for 24 h at an MOI of 1 or 3 with D/UW-3/Cx or CTD153, or with the positive control TLR2 ligand, Pam3Cys, were assayed for TNF-α and IL-6 by bead array. Bars indicate means ± SD, calculated from triplicate wells in a single representative experiment (P < 0.001 at MOI of 3). (C and D) Vaginal secretions from mice infected with CTD153 also contained lower levels of TNF-α (P = 0.043 days 3 to 8) (C) or IL-6 (P = 0.013) (D) than those collected from mice infected with D/UW-3/Cx. This experiment was performed twice. Cytokines were assayed by bead array, and data points represent the means ± SD for each group of five mice.
Transcriptional analysis of C. muridarum CM972 and C. trachomatis CTD153 reveals a conserved group of plasmid-responsive chromosomal genes.
The association between the presence of the chlamydial plasmid and the phenotypes of TLR2 signaling and the ability to accumulate glycogen within inclusions was conserved in both C. muridarum and C. trachomatis, although the pathogenic TLR2 ligand expressed by these chlamydiae remains unknown. We hypothesized that comparison of the transcriptional profiles of the plasmid-cured strains with their parent might identify genes encoding effectors that are involved in the expression of these phenotypes. A screening microarray analysis comparing the transcriptional profiles of CM972 with that of C. muridarum Nigg was performed using a custom microarray based on the published C. muridarum genome. Total RNA was prepared from McCoy cells infected with C. muridarum Nigg or CM972 (MOI, ∼1) at 30 h postinfection. The RNA was processed for analysis as previously described (32). The transcriptional profiles obtained from microarray analyses of CM972 (Fig. 4A) and CM3.1 (data not shown) strongly resembled that of strain Nigg, although, as was expected, transcripts derived from the chlamydial cryptic plasmid were not detected in either of the plasmid-deficient C. muridarum strains. Nevertheless, a small number of chromosomal loci were differentially transcribed in the absence of the plasmid. Similar results were obtained when an analysis was performed comparing the transcriptional profile of C. trachomatis CTD153 with its parent D/UW-3/Cx (Fig. 4B). Table 2 lists genes whose transcription levels were altered at least 2-fold based on microarray screening in both C. trachomatis and C. muridarum. The transcription of either mip or pal was also evaluated by quantitative PCR because no difference in the transcription of these genes was noted in the microarray screenings, and thus these genes served as controls. We confirmed the transcriptional changes detected via microarray between D/UW-3Cx and CTD153 and between Nigg and CM972, with the exception of TC0357, the transcription of which did not appear significantly altered in CM972 compared with the parental C. muridarum Nigg when assayed by PCR 24 h after infection (Fig. 4C and D). However, transcription of this gene was 0.12-fold ± 0.11-fold reduced (P < 0.001; mean ± SD of three independent experiments) when assayed 36 h after infection.
FIG. 4.

Transcriptional profiling via microarray screening and quantitative RT-PCR of plasmid-deficient C. muridarum and C. trachomatis reveals a subpopulation of plasmid-responsive genes. (A and B) Scatter plot illustrations of microarray comparisons of C. muridarum Nigg with strain CM972 (A) and C. trachomatis D/UW3-Cx with CTD153 (B). (C and D) RNA was isolated from infected cells 30 h after infection. Quantitative RT-PCR confirmed reduced transcription of candidate PRCL in C. muridarum CM972 (C) and C. trachomatis CTD153 (D). Total RNA was isolated from infected cells 24 h after infection, and PRCL transcripts were measured by quantitative RT-PCR. The transcriptional differences are presented as the fold change in expression for each gene in a single representative experiment, although each experiment was performed independently at least twice and samples were assayed in triplicate. *, P ≤ 0.013 (Nigg versus CM972) or P ≤ 0.025 (D/UW-3Cx versus CTD153). TC0828 (mip) or CT600 (pal) are included as examples of non-plasmid-responsive controls, because we did not detect a difference in the transcription of these genes by microarray screen.
TABLE 2.
Genes demonstrating a 2-fold or greater reduction in transcription via microarray screena in plasmid-deficient derivatives of C. muridarum Nigg (CM972 and CM3.1) and C. trachomatis D/UW-3/Cx
| ORF(s) designation in species |
Function (where known) | |
|---|---|---|
| C. trachomatis | C. muridarum | |
| CT049 | TC0319 | Located downstream of CT048 (SAM-dependent methytransferase); encodes pmp-like protein Pls1 (19) |
| CT084 | TC0357 | Phosphatidylcholine-hydrolyzing phopospholipase, possibly secreted; similar to CT157 |
| CT142 to CT144 | TC0419 to TC0421 | Conserved hypothetical three-gene operon with limited homology to transport systems, upstream of CT145 (serine/threonine protein kinase) |
| CT382.1 | 382.1b | Conserved hypothetical, small protein annotated as membrane or cell envelope associated |
| CT702 | TC075 | Conserved hypothetical protein downstream of GTPase and upstream of CT701 (secA) |
A single microarray analysis was performed for each strain pairing, using mRNA recovered 30 h after infection.
ORF recognized and mapped but unannotated.
We also extended these studies to plasmid-deficient clinical isolates C. trachomatis 25667R and CT599 (Fig. 5A and B). Conservation of plasmid responsiveness of these loci was observed in 25667R and, for glgA, CT382.1 and CT702 in CT599. Data were not obtained for CT049, CT084, or CT142 transcription by either CT599 or E/Bour because the primer sets used did not amplify a product for these genes, most likely reflecting sequence variations in these strains at those sites. Additional confirmation that the transcriptional changes observed reflected altered gene expression was obtained by immunoblotting of purified EB extracts of D/UW-3/Cx and CTD153 using a polyclonal antibody directed against the protein Pls1 encoded by CT049 (Fig. 6). Expression of Pls1 was clearly reduced in CTD153 compared with D/UW-3/Cx, even when overloaded with respect to the Mip protein control.
FIG. 5.

PRCL transcription is reduced in plasmid-deficient C. trachomatis clinical isolates. C. trachomatis 25667R genes are identified according to the annotation for strain L2/434/Bu; thus, CTL0305 is homologous to CT049, CTL0339 to CT084, CTL0397 to CT142, CTL0638 to CT382.1, and CTL071 to CT702. C. trachomatis E/Bour and CT599 genes were assayed with the primers directed against strain D/UW-3/Cx and are identified according to that annotation, because neither serovar E strain has been sequenced. Total RNA was isolated from infected cells 24 h after infection, and PRCL transcripts were measured by quantitative RT-PCR. The transcriptional differences are presented as the fold change in expression for each gene from a single representative experiment. Each experiment was performed independently at least twice, and samples were assayed in triplicate. *, P ≤ 0.011 (L2/434/Bu versus 25667R) or P ≤ 0.006 or less (E/Bour versus CT599).
FIG. 6.
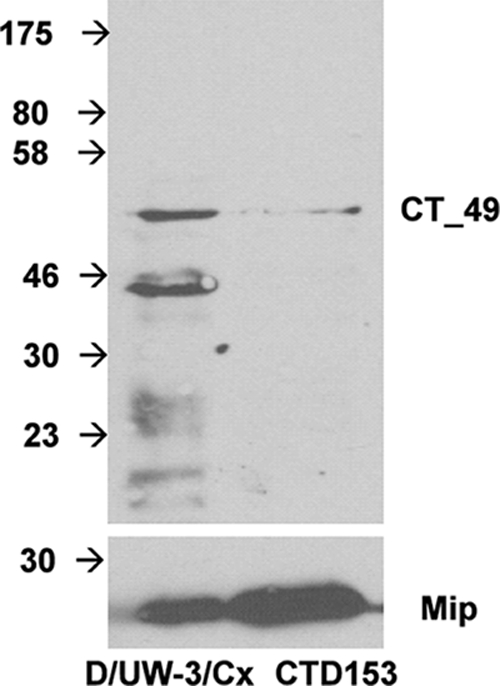
Expression of Pls1 is reduced in C. trachomatis CTD153 EBs. EBs of C. trachomatis D/UW-3/Cx and CTD153 were harvested from infected L929 cells 40 h after infection, gradient purified, and then lyzed by sonication. Immunoblotting was performed by using rabbit polyclonal anti-Pls1 antibody (1:10). As a loading control, Mip, the transcription of which is not altered in the absence of the chlamydial plasmid, was detected by using a rabbit anti-MIP antibody (1:100) (lower panel).
Differential regulation of glgA transcription in C. muridarum and C. trachomatis.
Surprisingly, transcription of glgA was only minimally altered (∼0.6-fold) in plasmid-deficient CM972 (Fig. 4C) and CM3.1 (data not shown), although the strains are unable to accumulate glycogen within intracytoplasmic inclusions. Glycogen synthase, the product of the glgA gene, is required for the formation of the α-1-4-glucosidic linkage characteristic of the main polyglucose chains of glycogen, and bacteria with glgA deleted or mutated are unable to synthesize glycogen (23). Transcription of this gene was ∼6-fold reduced in strain CTD153 relative to its parent (Fig. 4D) and also in 25667R (Fig. 5A) and CT599 (Fig. 5B) compared to plasmid-containing strains of the same serovar. A functional assay of glycogen synthase activity of strains L2/434/Bu, 25667R, Nigg, and CM3.1 at the peak of glgA transcription, 24 h after infection (Fig. 7A and B), confirmed the presence of similar levels of active enzyme in C. muridarum Nigg and CM3.1. In contrast, glycogen synthase remained almost undetectable in the plasmid-deficient C. trachomatis 25667R, while the wild-type, plasmid-containing L2/434/Bu expressed high levels of activity. ADP glucose pyrophosphorylase was detected in chlamydial lysates of all plasmid-deficient and wild-type strains examined 12 and 24 h after infection (data not shown). This enzyme, encoded by glgC, is the first and rate-limiting step of glycogen biosynthesis by bacteria (3). These data suggested that the minor transcriptional difference in glgA transcription detected in plasmid-cured C. muridarum was unlikely to be biologically significant. Furthermore, expression of glycogen synthase was insufficient for glycogen accumulation within inclusions of C. muridarum.
FIG. 7.

Glycogen synthase expression is not sufficient for accumulation of glycogen by C. muridarum. Glycogen synthase expression is reduced in cytoplasmic extracts of plasmid-deficient C. trachomatis 25667R harvested 24 h after infection compared to extracts of L2/434/Bu, a representative plasmid-containing strain of the same serovar (A), but it is normally expressed in extracts of C. muridarum Nigg and CM3.1 (B), although neither plasmid-deficient strain accumulates glycogen within its inclusions (31). Enzyme assays were performed in a linear range (enzyme concentration versus rate, under steady-state conditions) during the time course of the assay. Data points represent the averages of at least two determinations that differed by less than 5% (error bars) in a single experiment.
Transcription of plasmid-responsive genes in C. trachomatis is coordinately regulated in response to glucose limitation.
Glycolysis is the pathway by which chlamydiae utilize glucose as an energy source (reviewed by McClarty and Stephens [27]). Thus, expression of glgA and glycogen biosynthesis are not essential for chlamydial replication or differentiation but likely function as a secondary metabolic pathway. Chlamydiae are unable to utilize glucose directly. C. trachomatis lacks the sugar-specific components of the phosphoenolpyruvate-dependent sugar transferase system (PTS) (27) and does not express a hexokinase (40). Sequencing of the chlamydial genome has confirmed the absence of hexokinase, although the remaining steps of glycolysis appear intact. Instead, C. trachomatis is believed to use glucose-6-phosphate (G6P) obtained from the host cell. When chlamydia-infected cells are treated with the glucose analog 2-deoxyglucose (2DG), it is transported into the cytoplasm via the ubiquitous glucose transporter GLUT1 and phosphorylated by the eukaryotic hexokinase (21). The phosphorylated reaction product, 2DG-6-phosphate, inhibits hexokinase and blocks any further synthesis of G6P, specifically interrupting the flow of this substrate to chlamydiae. We observed that C. trachomatis L2/434/Bu fails to accumulate glycogen within the inclusion when 2DG is added (Fig. 8A). Similarly, glycogen accumulation within inclusions was inhibited in cells infected with either C. trachomatis D/UW-3/Cx or C. muridarum Nigg treated with 2DG (data not shown). Control experiments were performed in which the infected cells were treated with equivalent molarities of 3-o-methyl glucopyranoside, an unmetabolized analog of glucose that has no impact on host glycolysis, and we observed that glycogen accumulation by L2/434/Bu was unaffected (data not shown).
FIG. 8.

2DG-treated C. trachomatis does not accumulate glycogen within inclusions or downregulate PRCL transcription. (A) L929 cells infected with C. trachomatis L2/434/Bu were supplemented with 0.45 mg ml−1 glucose and treated with 2DG as indicated. The cells were fixed and stained with iodine at ∼40 h postinfection and photographed at 1,000× magnification. Arrows indicate chlamydial inclusions. (B to D) Plasmid-responsive gene transcription was differentially transcribed by C. trachomatis L2/434/Bu and D/UW-3/Cx (B and C) but not C. muridarum (D) in response to treatment with 10 mM 2DG. Total RNA was isolated from infected cells 24 h after infection, and PRCL transcripts were measured by quantitative RT-PCR. Transcription of uhpC, the gene encoding the chlamydial hexose phosphate transporter, was unaltered in response to 2DG treatment and served as a control. The transcriptional differences are presented as the fold change in expression for each gene in a single representative experiment, although each experiment was performed independently at least twice and samples were assayed in triplicate. *, P ≤ 0.005 (L2 versus L2 + 2DG) or P ≤ 0.016 (D/UW-3/Cx versus D/UW-3/Cx + 2DG). Values obtained for C. muridarum Nigg transcripts treated with 2DG did not differ significantly from the untreated control.
To determine if limiting the availability of substrate for the glycogen biosynthetic pathway influences glgA transcription by chlamydiae, we cultured L929 cells infected with plasmid-containing strains D/UW-3/Cx, L2/434/Bu, and Nigg in the presence or absence of 10 mM 2DG and harvested total RNA from the infected cells 24 h after infection. Quantitative PCR performed on the cDNAs synthesized from these preparations revealed transcription of glgA was altered in response to 2DG treatment in both C. trachomatis D/UW-3/Cx and L2/434/Bu, while expression of uhpC, the gene encoding the chlamydial hexose phosphate transporter, remained unaffected. Furthermore, PRCL transcription appeared reduced to levels comparable with those we observed in plasmid-deficient strains (Fig. 8B and C). In striking contrast, transcription of glgA by C. muridarum Nigg remained unaltered in response to 2DG treatment, and transcription of PRCLs was unchanged or increased (Fig. 8D).
It is not possible to directly investigate the effects of 2DG-mediated G6P limitation on TLR2 activation in epithelial cells, because it limits cellular metabolism and thus suppresses inflammatory responses (data not shown). Therefore, we examined the effect of G6P restriction on the ability of C. trachomatis and C. muridarum to activate TLR by using an in vitro signaling assay with D/UW-3/Cx and Nigg cultured in 2DG-treated L929 cells. Chlamydiae were harvested 40 h postinfection, washed, and concentrated via centrifugation before being inactivated via X-ray irradiation. Similarly processed D/UW-3/Cx, Nigg, CTD153, and CM3.1 organisms were harvested from untreated cells. The inactivated chlamydiae were incubated with HEK-Blue-2 cells for 24 h. The culture supernatant from the cells was then subjected to a colorimetric assay of phosphatase activity from the NF-κB reporter fusion expressed by the HEK cells. X-ray irradiation of the bacterial suspensions prevented chlamydial replication and thus possible de novo TLR2 ligand expression within glucose-sufficient HEK cells during incubation. By using this assay we observed that when D/UW-3/Cx was cultured in cells treated with 2DG, its ability to stimulate TLR2 was significantly reduced at MOIs of 0.5 and 3.0 (P < 0.05) (Fig. 9A). Indeed, at an MOI of 0.5, the degree of activation observed was not statistically different from that of cells incubated with CTD153. In contrast, TLR2 activation by Nigg cultured in 2DG-treated cells was not reduced but appeared enhanced (Fig. 9B). These data indicate that regulation of PRCL transcription in response to glucose availability occurs in C. trachomatis but not in C. muridarum and suggest that glucose limitation is an environmental signal that could result in modulation of chlamydial virulence during human infection.
FIG. 9.

C. trachomatis displays reduced TLR2 activation when cultured within 2DG-treated cells. (A) HEK-Blue-2 cells stimulated with D/UW-3/Cx and CTD153 or with D/UW-3Cx harvested from 2DG-treated L929 cells at an MOI of 0.5 or 3.0. After 24 h of incubation, NF-κB-induced SEAP activity was assessed using QUANTI-Blue. *, P < 0.05 for D/UW-3/Cx versus CTD153 or D + 2DG at an MOI of 0.5 or 3.0; @, P < 0.05 for CTD153 versus D + 2DG at an MOI of 3.0 by one-way ANOVA. (B) HEK-Blue-2 cells stimulated with Nigg and CM3.1 or with Nigg harvested from 2DG-treated L929 cells at an MOI of 3.0. *, P < 0.05 for Nigg versus CM3.1 at an MOI of 3.0 by one-way ANOVA. The TLR2 agonist Pam3Cys was used as a positive control. The data presented are from a single representative experiment, although each experiment was performed independently at least twice and samples were assayed in triplicate.
DISCUSSION
Despite the availability of suitable animal models for the study of genital tract infection by C. trachomatis, an understanding of the mechanisms by which the bacterium causes disease has been limited by the lack of defined genetic mutants with characterized virulence defects. We described plasmid-cured C. muridarum mutants that are attenuated for disease because of their inability to stimulate TLR2 during infection and, in the case of CM972, by an infectivity defect that results in reduced infection of the murine oviduct (29). In this study our derivation of C. trachomatis CTD153, a novel, plasmid-cured strain of the human genital tract isolate C. trachomatis D/UW-3/Cx, confirmed the plasmid-associated phenotypes that we had associated with chlamydial virulence and enabled us to screen for candidate effectors of these phenotypes via microarray analyses.
Our in vitro and in vivo studies demonstrated a reduction in infectivity in CTD153, similar to that which we have described for C. muridarum CM972. This was quantified as a reduction in plaquing efficiency of ∼85-fold and was confirmed by an in vitro competition assay where D/UW-3Cx competed successfully with CTD153 over three successive passages, even when inoculated at a ratio of 1:100. In vivo, we observed significantly reduced shedding of chlamydiae from the lower genital tracts of mice infected with the plasmid-cured derivative than in mice infected with the parental strain. Importantly, CTD153 also failed to elicit the expression of TLR2-dependent cytokines in vitro and in vivo. TLR2-dependent signaling in response to chlamydial infection was linked to the development of oviduct disease in an in vivo study of genital tract infection of TLR2-deficient mice infected with C. muridarum (10). In TLR2-deficient mice, significantly lower levels of TNF-α and MIP-2 were detected in genital tract secretions during the first week of infection, and there was a striking reduction in oviduct pathology after resolution of infection. A similar response with reduced cytokine expression over the course of infection and limited or absent oviduct pathology was observed in immunologically normal mice infected intravaginally with plasmid-deficient C. muridarum (29). In this study we determined that CTD153 failed to elicit the production of TLR2-dependent cytokines by dendritic cells and observed reduced cytokine expression in mice infected with CTD153 compared with those infected with D/UW-3/Cx, suggesting that this plasmid-cured derivative of C. trachomatis resembles CM972 with respect to these important virulence phenotypes and indicating that the chlamydial plasmid plays an important role in their expression.
Screening via microarray of the transcriptional profiles of CM972, CM3.1, and CTD153 revealed a conserved group of PRCL. This was further confirmed at the transcriptional level by quantitative PCR in these plasmid-cured strains and in plasmid-deficient clinical isolates and at the level of protein expression for Pls1 encoded by CT049 (20) and for glycogen synthase. We note that of the chromosomal loci previously described as differentially transcribed in strain 25667R in the investigation of Carlson et al. (6), four loci, including CTL0071, CTL0397, CTL0638, and glgA, showed similar expression in CTD153, while CTL0305 and CTL0339 proved to be differentially transcribed when investigated as orthologs of the candidate PRCLs, CT049 and CT084. These differences most likely reflect variations resulting from the experimental procedures and time points employed. However, although differentially transcribed in microarray analysis of both CM972 and CM3.1 (data not shown) and via quantitative RT-PCR 36 h after infection, we did not observe differential transcription of TC0357 by RT-PCR at 24 h postinfection, indicating that the screening approach we employed as a survey for conserved changes in global gene transcription was not exhaustive, having been performed using RNA obtained at a single time point. Thus, additional PRCL may yet be identified that are more profoundly differentially transcribed at other times during the chlamydial developmental cycle. These chromosomal loci represent additional candidate effectors or regulators of chlamydial infectivity and TLR2 stimulation if these are not encoded by the chlamydial plasmid directly. Recently, immunostimulatory properties have been ascribed to the plasmid-encoded protein Pgp3 (24). However, this contrasts with the work of Bas et al., who failed to observe any response to Pgp3 that significantly exceeded background levels in a study demonstrating TLR2-activating properties for the chlamydial Mip protein in human macrophages (4). Interestingly, mip was normally transcribed by the plasmid-deficient strains that we examined. Further studies are needed to identify the pathological TLR2 ligand(s) of chlamydiae and to determine the role played by the plasmid in chlamydia-induced TLR2 stimulation. The mechanism(s) regulating transcription of these PRCL remains unknown and may involve regulatory activities of the proteins encoded by the plasmid or the lack of expression of two noncoding RNAs expressed at high levels by the plasmid (1, 13), and both of these mechanisms are under investigation at this time.
One striking exception to the conservation of PRCL was our observation that expression of glycogen synthase appeared plasmid independent in C. muridarum. We were surprised to observe only minimal alteration of glgA transcription in this strain, because glgA was differentially transcribed in the other plasmid-deficient strains that we examined, including plasmid-cured CTD153 and plasmid-deficient clinical isolates C. trachomatis 25667R and CT599. We confirmed that functional glycogen synthase was expressed by CM3.1, indicating that the presence of active enzyme was insufficient for glycogen accumulation within inclusions. Thus, even when the enzymes of the biosynthetic pathway are active during infection of cells cultured under conditions of glucose excess, glycogen accumulation remains plasmid dependent. Plasmid-encoded proteins and/or the proteins expressed by the subset of PRCL that we have identified may be candidate chlamydial effectors of substrate uptake and consequently required for glycogen accumulation within the inclusion.
Even more surprising was our observation that glgA and PRCL transcription was depressed when G6P was limiting for strains of C. trachomatis and that this resulted in reduced TLR2-activation in an in vitro assay. Chlamydiae are not considered to be transcriptionally responsive to carbon limitation. A comprehensive analysis undertaken by Iliffe-Lee and McClarty (18) led them to conclude that although production of infectious organisms required that the host cells be supplemented with glucose, expression of a subset of core carbon metabolism genes was not responsive to either the source or amount of carbon present in the culture medium. More recently, Nicholson et al. (28) expanded this analysis to cover global gene families, and they obtained data which appeared to confirm these observations, finding no evidence of a chlamydial transcriptional response to carbon availability. It is possible that our use of 2DG to inhibit G6P biosynthesis by the host cell resulted in a very specific signal not usually transduced if the host cell is globally limited for glucose. Under these conditions, the host's metabolic response may prioritize energy pathways such as glycolysis and thus blunt the amplitude of the signal. It is likely advantageous for chlamydiae to limit the expression of PRCL should they encounter an energy stress. We have been unable to demonstrate any growth disadvantage associated with plasmid deficiency either in CTD153 or in plasmid-cured C. muridarum (29), indicating that expression of these genes is not essential for chlamydial replication. Importantly, glucose-restricted C. muridarum did not downregulate PRCL transcription or its ability to stimulate TLR2 signaling in response to 2DG treatment, consistent with our observation that glgA transcription was not plasmid responsive in this strain. This raises the possibility that PRCL transcribed by C. trachomatis strains infecting humans are subject to additional regulators unrelated to the plasmid and that this pathway is not active in C. muridarum. Of possible clinical significance, we speculate that if expression of plasmid-associated phenotypes, e.g., TLR2 activation, is modulated by G6P availability during human infection, this could contribute to the spectrum of disease outcome that is observed. Oviduct epithelial cells are the primary source of glucose in follicular fluid, the concentration of which varies over the course of the menstrual cycle (reviewed by Leese et al. [22]). Consequently, the cytoplasmic availability of G6P in these cells may vary in response to hormonal signaling, etc., and become potentially limiting for infecting chlamydiae, leading to reduced TLR2 activation and milder disease.
Is there a role for chlamydial glycogen in pathogenesis? Carlson et al. previously noted plasmid responsiveness of glgA transcription in plasmid-deficient C. trachomatis (6). That study compared the infection kinetics of 25667R with C. trachomatis L2/434/Bu, a wild-type strain of the same serovar, in the murine genital tract. They determined that the duration of infection and bacterial burden were reduced with 25667R, although they observed no infectivity defect in vitro. Their observations contrast with our previous findings with regard to C. muridarum CM972 infectivity (29, 31) and the in vitro and in vivo infectivity deficiency we detected in C. trachomatis CTD153. This may reflect procedural differences in the in vitro assays performed, e.g., determination of plaquing efficiency versus plating efficiency (6). Most recently, we observed that coculture competition assays in vitro are more reflective of in vivo differences than differences in plaquing efficiency (35) and could facilitate further analysis of 25667R's infectivity. An additional confounding factor is the differences in cell attachment/entry associated with strains of the C. trachomatis LGV serogroup, including L2/434/Bu and 25667R (11), that might mask or override plasmid-associated infectivity differences. Carlson et al. (6) speculated that glycogen may enhance chlamydial attachment or invasion and that loss of glycogen biosynthesis, as a consequence of the failure to transcribe glgA, might impair infectivity in vivo. Alternatively, the lack of glycogen as a carbon source might compromise the in vivo growth potential of the plasmid-deficient strain. Although we were unable to investigate the contribution of glycogen to infectivity in CTD153 because we did not recover spontaneous “revertants” of the infectivity defect, we were also unable to demonstrate a role for glycogen in infectivity in C. muridarum, because plasmid-deficient CM3.1, which displays wild-type infectivity in vitro, had a normal course of infection when inoculated intravaginally into mice and established infection in the oviducts at levels that were not significantly different from the parental Nigg strain (29) despite its inability to accumulate glycogen. Recently, genetic characterization of both C. muridarum strains CM972 and CM3.1 via comparative genome hybridization revealed no additional mutations other than loss of the plasmid to account for the phenotypic changes in CM972, and a single base substitution in TC0236 was detected in CM3.1 that may be responsible for restored infectivity (35).
Recognizing that the biosynthesis of glycogen is a secondary metabolic pathway, utilized only when substrate is available in excess, we hypothesize that the sensing/regulatory components required for the control of glgA have evolved to coregulate chlamydial virulence genes, at least in strains of C. trachomatis that infect humans. Regulation of glycogen biosynthesis at the level of transcription is rare in bacteria and is more often observed as allosteric regulation of the gene product of glgC, ADP glucose pyrophosphorylase, which controls the first, rate-limiting step of glycogen biosynthesis (3). However, we have yet to determine if G6P restriction is sensed directly at the glgA promoter or if the regulation we observed is indirect. This pathway is one of the general housekeeping regulons transcribed by σ70 in Escherichia coli, but whether the chlamydial homolog σ 66 is the dominant sigma factor for glycogen gene expression in this microorganism is not known. Recognized regulators of glycogen metabolism in Gram-negative bacteria, such as adenylate cyclase or the cyclic AMP receptor protein, positive regulators of glycogen metabolism in E. coli, and csrA, a negative regulator of the pathway that acts posttranscriptionally by manipulating mRNA decay rates (2), appear absent from the chlamydial genome.
In summary, we have demonstrated a central role for the C. trachomatis plasmid in the expression of at least two virulence phenotypes, infectivity and TLR2 activation, consistent with our previous observations for C. muridarum. We have identified candidate effectors of these phenotypes via transcriptional profiling by using plasmid-cured derivatives of both biovars, although we have yet to identify the specific genes involved in the expression of these pathogenic mechanisms. Interestingly, we have also identified a role for the chlamydial plasmid in the accumulation of glycogen within inclusions by using plasmid-cured C. muridarum, which is independent of differential glgA transcription. Furthermore, our data suggest that chlamydiae infecting humans can sense and respond to environmental signals by limiting expression of virulence genes.
Acknowledgments
We thank Andrew Nowalk for helpful discussions during the preparation of the manuscript.
This work was supported by NIH NIAID via grants R21 AI083657 to C.M.O. and R01 AI070693 to R.J.B. and by the NSF via grant 0448676 to C.R.M.
Editor: S. R. Blanke
Footnotes
Published ahead of print on 3 January 2011.
REFERENCES
- 1.Albrecht, M., C. M. Sharma, R. Reinhardt, J. Vogel, and T. Rudel. 2010. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 38:868-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker, C. S., I. Morozov, K. Suzuki, T. Romeo, and P. Babitzke. 2002. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol. Microbiol. 44:1599-1610. [DOI] [PubMed] [Google Scholar]
- 3.Ballicora, M. A., A. A. Iglesias, and J. Preiss. 2003. ADP-glucose pyrophosphorylase, a regulatory enzyme for bacterial glycogen synthesis. Microbiol. Mol. Biol. Rev. 67:213-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bas, S., et al. 2008. The proinflammatory cytokine response to Chlamydia trachomatis elementary bodies in human macrophages is partly mediated by a lipoprotein, the macrophage infectivity potentiator, through TLR2/TLR1/TLR6 and CD14. J. Immunol. 180:1158-1168. [DOI] [PubMed] [Google Scholar]
- 5.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 6.Carlson, J. H., et al. 2008. The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect. Immun. 76:2273-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Comanducci, M., et al. 1994. Humoral immune response to plasmid protein Pgp3 in patients with Chlamydia trachomatis infection. Infect. Immun. 62:5491-5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Comanducci, M., S. Ricci, R. Cevenini, and G. Ratti. 1990. Diversity of the Chlamydia trachomatis common plasmid in biovars with different pathogenicity. Plasmid 23:149-154. [DOI] [PubMed] [Google Scholar]
- 9.Darville, T., et al. 1997. Mouse strain-dependent variation in the course and outcome of chlamydial genital tract infection is associated with differences in host response. Infect. Immun. 65:3065-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darville, T., et al. 2003. Toll-like receptor-2, but not toll-like receptor-4, is essential for development of oviduct pathology in chlamydial genital tract infection. J. Immunol. 171:6187-6197. [DOI] [PubMed] [Google Scholar]
- 11.Davis, C. H., and P. B. Wyrick. 1997. Differences in the association of Chlamydia trachomatis serovar E and serovar L2 with epithelial cells in vitro may reflect biological differences in vivo. Infect. Immun. 65:2914-2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donati, M., et al. 2003. DNA immunization with pgp3 gene of Chlamydia trachomatis inhibits the spread of chlamydial infection from the lower to the upper genital tract in C3H/HeN mice. Vaccine 21:1089-1093. [DOI] [PubMed] [Google Scholar]
- 13.Fahr, M. J., K. S. Sriprakash, and T. P. Hatch. 1992. Convergent and overlapping transcripts of the Chlamydia trachomatis 7.5-kb plasmid. Plasmid 28:247-257. [DOI] [PubMed] [Google Scholar]
- 14.Farencena, A., M. Comanducci, M. Donati, G. Ratti, and R. Cevenini. 1997. Characterization of a new isolate of Chlamydia trachomatis which lacks the common plasmid and has properties of biovar trachoma. Infect. Immun. 65:2965-2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox, J., K. Kawaguchi, E. Greenberg, and J. Preiss. 1976. Biosynthesis of bacterial glycogen. Purification and properties of the Escherichia coli B ADPglucose:1,4-alpha-D-glucan 4-alpha-glucosyltransferase. Biochemistry 15:849-857. [DOI] [PubMed] [Google Scholar]
- 16.Hanna, L., P. Thygeson, E. Jawetz, and C. Dawson. 1959. Elementary-body virus isolated from clinical trachoma in California. Science 130:1339-1340. [DOI] [PubMed] [Google Scholar]
- 17.Hatt, C., M. E. Ward, and I. N. Clarke. 1988. Analysis of the entire nucleotide sequence of the cryptic plasmid of Chlamydia trachomatis serovar L1. Evidence for involvement in DNA replication. Nucleic Acids Res. 16:4053-4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iliffe-Lee, E. R., and G. McClarty. 1999. Glucose metabolism in Chlamydia trachomatis: the ‘energy parasite’ hypothesis revisited. Mol. Microbiol. 33:177-187. [DOI] [PubMed] [Google Scholar]
- 19.Inaba, K., et al. 1992. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176:1693-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jorgensen, I., and R. H. Valdivia. 2008. Pmp-like proteins Pls1 and Pls2 are secreted into the lumen of the Chlamydia trachomatis inclusion. Infect. Immun. 76:3940-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kletzien, R. F., and J. F. Perdue. 1975. Induction of sugar transport in chick embryo fibroblasts by hexose starvation. Evidence for transcriptional regulation of transport. J. Biol. Chem. 250:593-600. [PubMed] [Google Scholar]
- 22.Leese, H. J., J. I. Tay, J. Reischl, and S. J. Downing. 2001. Formation of fallopian tubal fluid: role of a neglected epithelium. Reproduction 121:339-346. [DOI] [PubMed] [Google Scholar]
- 23.Leung, P. S., and J. Preiss. 1987. Cloning of the ADP glucose pyrophosphorylase (glgC) and glycogen synthase (glgA) structural genes from Salmonella typhimurium LT2. J. Bacteriol. 169:4349-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, Z., D. Chen, Y. Zhong, S. Wang, and G. Zhong. 2008. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect. Immun. 76:3415-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 26.Matsumoto, A., H. Izutsu, N. Miyashita, and M. Ohuchi. 1998. Plaque formation by and plaque cloning of Chlamydia trachomatis biovar trachoma. J. Clin. Microbiol. 36:3013-3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McClarty, G., and R. S. Stephens. 1999. Chlamydial metabolism as inferred from the complete genome sequence, p. 69-100. In R. S. Stephens (ed.), Chlamydia: intracellular biology, pathogenesis, and immunity. American Society for Microbiology, Washington, DC.
- 28.Nicholson, T. L., K. Chiu, and R. S. Stephens. 2004. Chlamydia trachomatis lacks an adaptive response to changes in carbon source availability. Infect. Immun. 72:4286-4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Connell, C. M., R. R. Ingalls, C. W. Andrews, Jr., A. M. Skurlock, and T. Darville. 2007. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J. Immunol. 179:4027-4034. [DOI] [PubMed] [Google Scholar]
- 30.O'Connell, C. M., I. A. Ionova, A. J. Quayle, A. Visintin, and R. R. Ingalls. 2006. Localization of TLR2 and MyD88 to Chlamydia trachomatis inclusions. Evidence for signaling by intracellular TLR2 during infection with an obligate intracellular pathogen. J. Biol. Chem. 281:1652-1659. [DOI] [PubMed] [Google Scholar]
- 31.O'Connell, C. M., and K. M. Nicks. 2006. A plasmid-cured Chlamydia muridarum strain displays altered plaque morphology and reduced infectivity in cell culture. Microbiology 152:1601-1607. [DOI] [PubMed] [Google Scholar]
- 32.Ouellette, S. P., et al. 2006. Global transcriptional upregulation in the absence of increased translation in Chlamydia during IFNγ-mediated host cell tryptophan starvation. Mol. Microbiol. 62:1387-1401. [DOI] [PubMed] [Google Scholar]
- 33.Peterson, E. M., B. A. Markoff, J. Schachter, and L. M. De La Maza. 1990. The 7.5-kb plasmid present in Chlamydia trachomatis is not essential for the growth of this microorganism. Plasmid 23:144-148. [DOI] [PubMed] [Google Scholar]
- 34.Pickett, M. A., J. S. Everson, P. J. Pead, and I. N. Clarke. 2005. The plasmids of Chlamydia trachomatis and Chlamydophila pneumoniae (N16): accurate determination of copy number and the paradoxical effect of plasmid-curing agents. Microbiology 151:893-903. [DOI] [PubMed] [Google Scholar]
- 35.Russell, M., et al. 2010. Infectivity acts as in vivo selection for maintenance of the chlamydial “cryptic” plasmid. Infect. Immun. 79:98-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schachter, J., and C. R. Dawson. 1978. Human chlamydial infections, p. 181-220. PSG Publishing Company Inc., Littleton, MA.
- 37.Stephens, R. S., et al. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282:754-759. [DOI] [PubMed] [Google Scholar]
- 38.Stothard, D. R., J. A. Williams, P. B. Van Der Pol, and R. B. Jones. 1998. Identification of a Chlamydia trachomatis serovar E urogenital isolate which lacks the cryptic plasmid. Infect. Immun. 66:6010-6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas, N. S., M. Lusher, C. C. Storey, and I. N. Clarke. 1997. Plasmid diversity in Chlamydia. Microbiology 143:1847-1854. [DOI] [PubMed] [Google Scholar]
- 40.Vender, J., and J. W. Moulder. 1967. Initial step in catabolism of glucose by the meningopneumonitis agent. J. Bacteriol. 94:867-869. [DOI] [PMC free article] [PubMed] [Google Scholar]