

Abstract
Cerebral malaria (CM) is a major complication of Plasmodium falciparum infection, particularly in children. The pathogenesis of cerebral malaria involves parasitized red blood cell (RBC)-mediated vascular inflammation, immune stimulation, loss of blood-brain barrier integrity, and obstruction of cerebral capillaries. Therefore, blunting vascular inflammation and immune cell recruitment is crucial in limiting the disease course. Beta interferon (IFN-β) has been used in the treatment of diseases, such as multiple sclerosis (MS) but has not yet been explored in the treatment of CM. Therefore, we sought to determine whether IFN-β also limits disease progression in experimental cerebral malaria (ECM). Plasmodium berghei-infected mice treated with IFN-β died later and showed increased survival, with improved blood-brain barrier function, compared to infected mice. IFN-β did not alter systemic parasitemia. However, we identified multiple action sites that were modified by IFN-β administration. P. berghei infection resulted in increased expression of chemokine (C-X-C motif) ligand 9 (CXCL9) in brain vascular endothelial cells that attract T cells to the brain, as well as increased T-cell chemokine (C-X-C motif) receptor 3 (CXCR3) expression. The infection also increased the cellular content of intercellular adhesion molecule 1 (ICAM-1), a molecule important for attachment of parasitized RBCs to the endothelial cell. In this article, we report that IFN-β treatment leads to reduction of CXCL9 and ICAM-1 in the brain, reduction of T-cell CXCR3 expression, and downregulation of serum tumor necrosis factor alpha (TNF-α). In addition, IFN-β-treated P. berghei-infected mice also had fewer brain T-cell infiltrates, further demonstrating its protective effects. Hence, IFN-β has important anti-inflammatory properties that ameliorate the severity of ECM and prolong mouse survival.
Cerebral malaria (CM) is a major complication of Plasmodium falciparum infection. CM results from a combination of cerebral vascular and immune system dysfunction. Several hypotheses have attempted to explain the pathogenesis of CM. Some emphasize that CM is the result of the mechanical obstruction of microvessels subsequent to an increase in endothelial cell adhesion molecule expression and cell attachment (3, 19, 20). Others stress an immunological basis caused by excessive immune stimulation and proinflammatory cytokine release (22). A combination of these potential mechanisms seems likely with infected red blood cell (RBC) adhesion to the endothelium promoting prothrombotic immune responses, resulting in vascular inflammation, immune stimulation, and cerebral vascular obstruction (22).
CM is particularly prevalent in African children. In one recent study, 33% of children admitted to a hospital in Kenya had malaria, and almost half of these children had neurological symptoms (11). Other field studies have indicated that despite treatment, mortality due to CM can be as high as 30%, while 10% of survivors may experience neurological complications (1). When one considers that there were an estimated 247 million clinical episodes of malaria infection in the world in 2006, many affecting children less than 5 years of age, the social and economic impact of cerebral malaria is staggering (11). The development of a malaria vaccine has proven challenging, heightening the need for continued research focused on the treatment and prevention of malaria-associated cerebral vascular events.
Human studies and mouse models of CM have independently implicated that cytokines, adhesion molecules, and chemokines have important roles in the disease pathogenesis. The tumor necrosis factor alpha (TNF-α) and gamma interferon (IFN-γ) cytokines are two proinflammatory molecules that clearly contribute to CM pathogenesis (7, 8, 13). The ICAM-1 (intercellular adhesion molecule 1) adhesion molecule has long been linked to CM due to its role in parasitized red blood cell (pRBC) engagement and in T-cell adhesion and transendothelial cell migration through the brain endothelial cells. Many chemokines have been noted to exhibit increased expression in cerebral malaria, but the chemokines most strongly implicated to have a role are the chemokine (C-X-C motif) receptor 3 (CXCR3) binding chemokines, including chemokine (C-X-C motif) ligand 4 (CXCL4), CXCL9, and CXCL10 (2, 9, 14, 17, 21).
Studies using the experimental cerebral malaria (ECM) model have demonstrated that the CXCR3 chemokine ligands CXCL10 (interferon-inducible protein of 10 kDa [IP-10]) and CXCL9 (monokine induced by gamma interferon [MIG]) were highly induced in the brains of mice infected with Plasmodium berghei ANKA. It is interesting that mice deficient in their chemokine receptor, CXCR3, were protected against CM and that there was a significant reduction of leukocytes traveling into the brain (2). Luster et al. validated this work by also showing that a number of chemokine mRNAs, including those for CXCR3 and its ligands, are increased in the brain during ECM (14). Moreover, the investigators also demonstrated that CXCL9 and CXCL10 mRNA and protein were localized primarily in the microvasculature and glial cells, respectively (2, 14). These investigations have further suggested that the CXCL9 and CXCL10 CXCR3 ligands have distinct and nonredundant roles in the pathogenesis of ECM (2, 15). The importance of CXCR3 chemokines in the pathogenesis of human infections has also been corroborated in studies demonstrating that the CXCL10 concentrations in plasma are increased in CM. However, to date, CXCL9 has not been studied (1).
Prior work by our group has demonstrated that high levels of IFN-β are produced in human retinal pigment epithelial (RPE) cells through Toll-like receptor 3 (TLR3) signaling (12). Moreover, we demonstrated that IFN-β may in fact be a potent immunosuppressive molecule within the retina, a function that may serve a critical protective mechanism in the ocular microenvironment (4). Also, in a previous study, we documented that IFN-β inhibited gene expression and production of both CXCL9 and ICAM-1 in RPE cells (10), and we suggested that this may be one of the ways in which IFN-β exerts its suppressive activity. This immunosuppressive action is not exclusive to RPE cells. More recently, we observed that the downregulation of CXCL9 by IFN-β treatment occurs in other cell types, including retinal and human umbilical vein endothelial cells (unpublished data).
We now demonstrate using the mouse model of ECM that IFN-β, an immunosuppressive cytokine, potentially has a therapeutic role in the prevention and treatment of cerebral malaria. Furthermore, we show that the mechanism of this protective effect is at least in part due to a downregulation of CXCL9, its receptor, CXCR3, and ICAM-1, TNF-α, and IFN-γ.
MATERIALS AND METHODS
Mouse studies.
C57BL6/J mice were purchased from Jackson Labs. The mice were infected with Plasmodium berghei ANKA by intraperitoneal (i.p.) injection of approximately 0.5 × 106 parasites. The mice were treated with IFN-β (800,000 U/mouse/day) by the i.p. route. Recombinant murine IFN-β was obtained from PBL Interferon Source, Inc. (Piscataway, NJ).
All animal studies were performed at an AAALAC-accredited institute using methods and guidelines approved by The John Hopkins University and University of Rochester Animal Care and Use Committees that comply with United States national guidelines for the use of animals in research (Public Health Service Policy A-3292-01 [16a, 16b]).
The brains were harvested on day 5 because the mice begin to die on day 6, and we sought to obtain data from a complete group of mice at the same time point. The mice typically die overnight and tend to undergo autolysis rapidly, so data from any tissue would be severely compromised. The selection of day 5 for harvest allows for the most complete and accurate data while minimizing the number of animals that must be used in one study.
Brain mononuclear cell isolation.
Brain mononuclear cells were isolated by removing the brains on day 5 postinfection (p.i.), and a single-cell suspension was obtained by grinding the tissue and mincing it with a razor blade in Dulbecco modified Eagle medium with 10% fetal bovine serum while on ice. Cell suspensions were placed in 15-ml conical tubes, and Percoll (Sigma) was added to a final concentration of 30%. One milliliter of Percoll was placed at the bottom of the tube before the cells were added, and the cells were spun at 1,300 × g for 30 min at 4°C. Cells at the interface were isolated, washed twice, resuspended in Tyrode's buffer, and labeled with anti-CD4 and CD8 antibodies for flow cytometry.
This brain cell isolation technique results in the collection of both intravascular and extravascular cells. This allows us to evaluate both already trafficked and recruited cells.
BBB studies.
Blood-brain barrier (BBB) permeability was determined by injecting 200 μl of 2% Evans blue dye intravenously (i.v.) into mice on day 5 p.i. One hour later, the brains were harvested and incubated in 10% buffered formalin for 48 h, and dye extravasation was determined by optical density (6).
Cytokine analysis by EIA.
The levels of CXCL9, CXCL10, soluble ICAM-1 (sICAM-1), TNF-α, and IFN-γ in plasma were determined using commercially available enzyme immunoassay (EIA) kits (R&D Systems, Minneapolis, MN). All plasma samples were tested according to the manufacturer's instructions. The optical density of each sample was determined using the VERSAmax tunable microplate reader (Molecular Devices, Sunnyvale, CA). Results were calculated from a standard curve and reported accordingly in picograms per milliliter or nanograms per milliliter. The minimal detectable dose for each chemokine/cytokine measured is as follows: CXCL9, 3 pg/ml; CXCL10, 2.2 pg/ml; sICAM-1, 0.029 ng/ml; IFN-γ, less than 2.0 pg/ml; and TNF-α, less than 5.1 pg/ml. The unpaired Student t test was used to analyze the plasma cytokine data.
RNA preparation from mouse brain.
The brains were removed from mice, washed in phosphate-buffered saline (PBS), quickly frozen on dry ice, and stored at −70°C. Brain tissue samples were obtained from control, uninfected mice, parasite-infected mice, and parasite-infected mice treated with IFN-β. Total RNA (1 μg) was extracted from brain samples using RNA STAT-60 (Tel-Test Inc., Friendswood, TX) according to the manufacturer's instructions and reverse transcribed into cDNA using StrataScript first-strand synthesis kit (Stratagene). A TaqMan gene expression assay kit containing probe and primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), CXCL4, CXCL9, CXCL10, CXCR3, and ICAM-1 genes was purchased from Applied Biosystems (Carlsbad, CA). Quantitative real-time PCR (qRT-PCR) was performed by heating cDNA samples mixed with primers and TaqMan gene expression master mix (Applied Biosystems) at 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of amplification (one cycle consisting of 15 s at 95°C and 1 min at 60°C), using an Applied Biosystems RT-PCR system 7500 (Applied Biosystems). All infected samples and control uninfected samples were tested at the same time to enable comparisons of GAPDH rRNA-normalized values. Negative-control samples without reverse transcriptase or RNA were included. For data analysis, the amount of reaction product was determined using the analysis software of comparative threshold cycle method (Applied Biosystems). The mRNA levels for each primer pair were normalized to the housekeeping gene GAPDH rRNA level, and fold changes were calculated against the values for control uninfected mice.
IHC.
Harvested transplants were fixed in 60% methanol-30% water-10% acetic acid. Tissue samples were then embedded and sectioned, and immunohistochemistry (IHC) was performed using protocols and procedures described previously (16, 24).
Statistical analysis.
All data were analyzed using the Student paired t test.
RESULTS
IFN-β is protective in experimental cerebral malaria (ECM).
To determine whether IFN-β is protective in cerebral malaria (CM), we first infected mice with P. berghei ANKA and treated mice daily for 10 days with either IFN-β (8 × 105 U in 100 μl given intraperitoneally) or saline (infected mice) and monitored survival for 10 days postinfection (p.i.). Infected mice begin to die on day 6 or 7 p.i. with 20% of mice surviving until day 10 (Fig. 1A). Mice treated with IFN-β died later (day 9) and showed improved survival (60%) compared to infected mice (Fig. 1A) (P < 0.05). However, there was no difference in the parasitemia noted in either group. The data in Fig. 1A are from one experiment but are representative of three separate experiments. These data demonstrate that IFN-β is protective in CM.
FIG. 1.
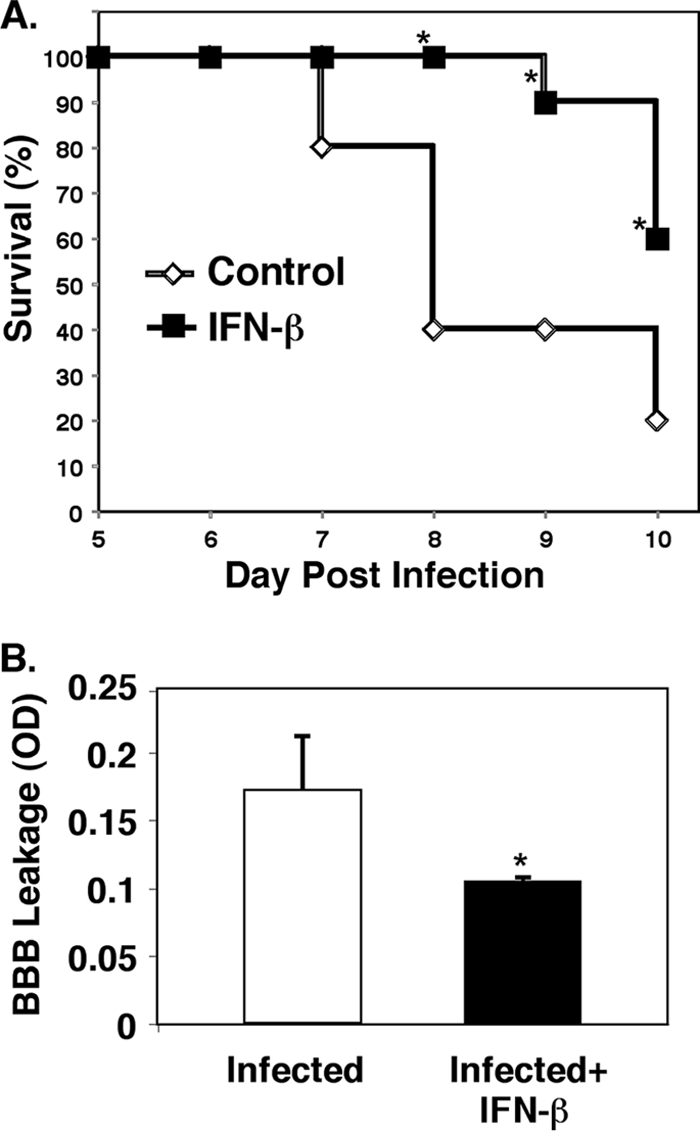
IFN-β is protective in experimental cerebral malaria (ECM). (A) Survival. Mice were infected with Plasmodium berghei, and their survival was monitored. The values that were significantly different (P < 0.05) from the value for control, non-IFN-β-treated mice are indicated by an asterisk. (B) Blood-brain barrier (BBB). On day 5 postinfection, the mice were injected with Evans blue dye, and dye extravasation was measured (n = 5). Values are means plus standard errors of the means (SEMs) (error bars). The value for infected and IFN-β-treated mice was significantly different (P < 0.05) from the value for infected mice and is indicated by an asterisk. OD, optical density.
IFN-β has protective effects on cerebral vascular integrity against multiple sclerosis (MS) (18). To further demonstrate the cerebral vascular protective effects of IFN-β against CM, mice were again infected with P. berghei and treated with IFN-β or saline. On day 5 p.i., blood-brain barrier (BBB) integrity was determined by measuring Evans blue dye extravasation. Brains from IFN-β-treated mice had significantly reduced dye extravasation compared to untreated, infected mice (Fig. 1B) (P < 0.05). This observation indicates that IFN-β treatment improves cerebral vascular integrity in CM. Also, on day 5, brains were harvested for histological examination. P. berghei-infected mice have evidence of numerous blood vessels that are swollen with perivascular inflammatory cells and vasculitis, which is indicated by inflammatory cells within blood vessel walls in the cerebral cortex (Fig. 2). In contrast, infected mice treated with IFN-β had fewer brain inflammatory cell infiltrates and improved vascular integrity compared to infected mice (Fig. 2).
FIG. 2.

Brain pathology in the cerebral cortex. On day 5 postinfection, brains were collected from mice, and hematoxylin and eosin staining was performed. The arrows point to areas of vascular inflammation.
Taken together, the data in Fig. 1 and 2 demonstrate that IFN-β has cerebral vascular protective effects in ECM.
IFN-β suppresses ECM-associated inflammation.
Previous work by our group has demonstrated that IFN-β reduced production of the CXCL9 chemokine and the ICAM-1 adhesion molecule in vitro in retinal pigment epithelial (RPE) cells and endothelial cells (10). Moreover, several investigators have shown that CXCL9 and CXCR3 are critical to the development of ECM (2, 9). To determine whether a protective effect of IFN-β in CM may be due to reduced chemokine and inflammatory cytokine production, we took plasma samples from untreated control mice, infected mice, and infected mice treated with IFN-β. On day 5 p.i., plasma samples were collected and evaluated by EIA for the presence of CXCL9, CXCL10, sICAM-1, TNF-α, and IFN-γ. All infected mice had elevated levels of CXCL9, CXCL10, sICAM-1, TNF-α, and IFN-γ in plasma (Fig. 3A to E). The IFN-β-treated mice demonstrated decreased plasma CXCL9 and sICAM-1 levels (Fig. 3A and C). However, CXCL10 was increased in IFN-β-treated infected mice (Fig. 3B). Numerous studies have also reported the upregulation of TNF-α and IFN-γ in CM. Similarly, we also observed elevated levels of TNF-α and IFN-γ in P. berghei-infected mice that are significantly decreased by IFN-β treatment (Fig. 3D and E) (P < 0.01 and P = 0.02, respectively).
FIG. 3.

IFN-β suppresses ECM-associated inflammation. On day 5 postinfection, mouse plasma was isolated and evaluated by EIA. (A to E) The concentrations of CXCL9 (A), CXCL10 (B), sICAM-1 (C), TNF-α (D), and IFN-γ (E) in the three groups of mice are shown. (A) CXCL9 (n = 5). Values are means plus standard deviation (SDs) (error bars). The value that was significantly different (P < 0.05) from the value for infected mice is indicated by an asterisk. (B) CXCL10 (n = 4 or 5). The value that was significantly different (P < 0.01) from the value for infected mice is indicated by an asterisk. (C) sICAM-1 (n = 5). (D) TNF-α (n = 3). The value that was significantly different (P < 0.01) from the value for infected mice is indicated by an asterisk. (E) IFN-γ (n = 3). The value that was significantly different (P = 0.02) from the value for infected mice is indicated by an asterisk. Values in panels B, D, and E are means plus SEMs (error bars).
Plasma EIA results were also reflected in immunohistochemical (IHC) analysis of brain tissue. On day 5 p.i., the brains from P. berghei-infected mice and infected mice treated with IFN-β were sectioned, and IHC analysis using antibodies specific for CXCL9 and CXCL10 was performed. The cerebral cortex regions of uninfected mice had negligible CXCL9-positive reactivity (not shown). In the infected mice, CXCL9-positive staining was greatly increased and confined to the blood vessels that were primarily small to medium sized (Fig. 4A). Similar to plasma protein results, infected mice treated with IFN-β had greatly reduced CXCL9 expression compared to infected mice, and this staining was also restricted to the vasculature (Fig. 4A). The number of CXCL9-positive vessels per 20× field was counted and demonstrated to be significantly reduced in IFN-β-treated mice (Fig. 4B) (P < 0.01). Infected mice had approximately 14 CXCL9-positive vessels per 20× field. In contrast, IFN-β treatment of infected mice reduced the positive vessel per 20× field to approximately 7 (P < 0.01). These data demonstrate that endothelial cells are a major source of CXCL9 in the brains of P. berghei-infected mice and that IFN-β attenuates this increase in CXCL9. In contrast, CXCL10 was highly expressed in astrocytes and glial cells of infected mice, and the numbers of CXCL10-positive cells were not reduced by IFN-β treatment (Fig. 5A and B). In fact, the number of CXCL10-positive cells was significantly increased (P < 0.01).
FIG. 4.

IFN-β alters brain CXCL9 production. (A) CXCL9 immunohistochemical analysis. The arrows point to blood vessels that are positive for CXCL9. (B) Quantification of CXCL9-positive vessels. Infected mice had approximately 14 CXCL9-positive (+ve) vessels per 20× field, whereas in IFN-β-treated mice, the number of such vessels was reduced to approximately 7 (5 fields per mouse; 5 mice). Values are means plus SDs (error bars). The value for infected and IFN-β-treated mice was significantly different (P < 0.01) from the value for infected mice and is indicated by an asterisk.
FIG. 5.

IFN-β alters brain CXCL10 production. (A) CXCL10 immunohistochemical analysis. The arrows point to CXCL10-positive astrocytes and glial cells. (B) Quantification of CXCL10-positive cells. Infected mice had approximately 40 CXCL10-positive cells per 20× field, whereas in IFN-β-treated mice, this number was increased to greater than 80 (5 fields per mouse; five mice). Values are means plus SDs (error bars). The value for infected and IFN-β-treated mice was significantly different (P < 0.01) from the value for infected mice and is indicated by an asterisk.
To extend and further confirm these important findings, quantitative real-time PCR (qRT-PCR) was performed on brain tissue from untreated control mice, infected mice, and IFN-β-treated infected mice. The qRT-PCR data mirrored the protein expression data. IFN-β decreased total brain CXCL9 mRNA expression and increased CXCL10 expression (Fig. 6A and B) compared to untreated infected mice. The total expression of ICAM-1 mRNA was also decreased by IFN-β (Fig. 6C).
FIG. 6.
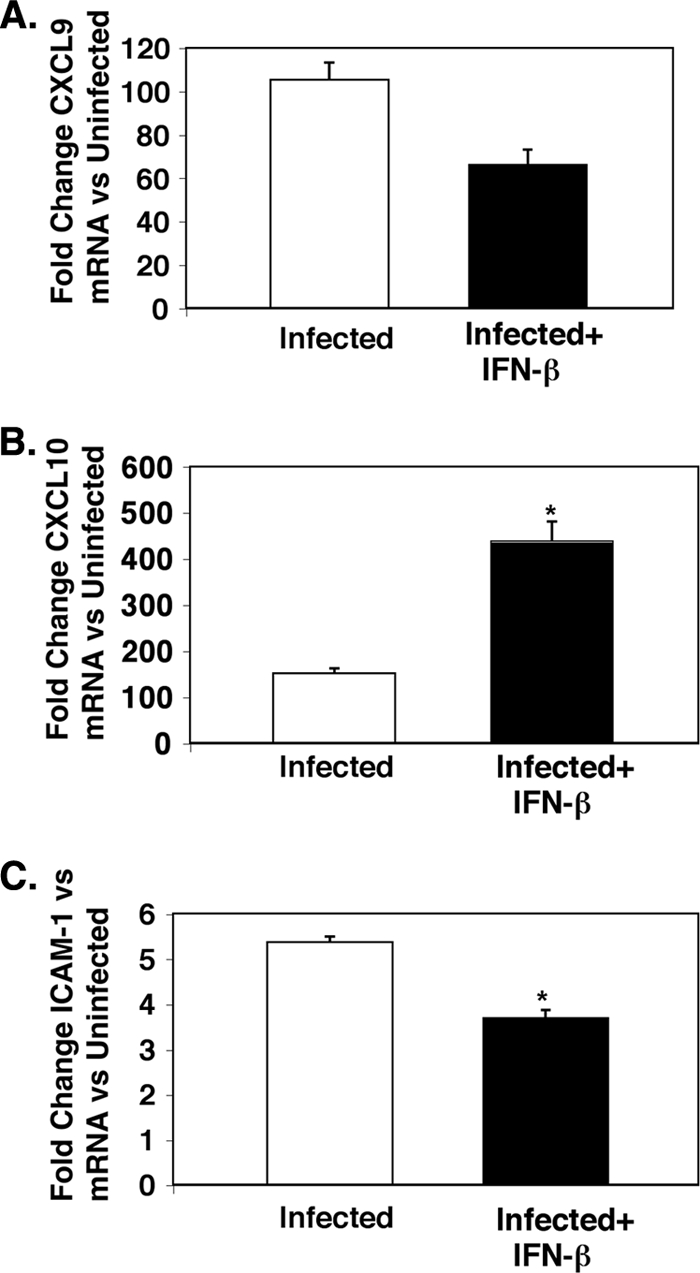
IFN-β alters expression of CXCL9, CXCL10, and ICAM-1 mRNA in the brain. (A) CXCL9 (n = 3 or 4). (B) CXCL10 (n = 3 or 4). The value for infected and IFN-β-treated mice was significantly different (P < 0.05) from the value for infected mice and is indicated by an asterisk. (C) ICAM-1 (n = 3 or 4). The value for infected and IFN-β-treated mice was significantly different (P < 0.02) from the value for infected mice and is indicated by an asterisk. Values are means plus SEMs (error bars). The mRNA levels for each primer pair were normalized to the housekeeping gene GAPDH rRNA level, and fold changes were calculated against control uninfected mice.
IFN-β reduces T-cell trafficking in ECM.
ECM is in part a T-cell-dependent process. To determine whether IFN-β affects reduced T-cell trafficking to the brain, we isolated brains from infected and infected mice treated with IFN-β on day 6 p.i. Brain sections were prepared for IHC analysis with anti-CD3 antibody as a T-cell marker. Minimal CD3 staining was observed in untreated control mice (not shown). Infected mice had numerous sites in the cerebral cortex and meninges where CD3+ cells were noted in vessel walls and adjacent parenchyma (Fig. 7). IFN-β-treated mice, however, had many fewer CD3+ cells (Fig. 7). The number of CD3+ cells per field was counted and demonstrated to be greatly reduced in the IFN-β-treated mice (Fig. 8A) (P < 0.01). Brain T-cell subsets were quantified by isolating mononuclear cells from brain tissue as we have described previously (17), and T-cell numbers were expressed as a percentage of total mononuclear cells using fluorescence-activated cell sorting (FACS) (see File S2 in the supplemental material). As anticipated, both CD4+ and CD8+ T cells were greatly increased in the brains of infected mice (Fig. 8B). Infected mice treated with IFN-β had dramatically reduced CD4+ and CD8+ T-cell infiltrates compared to untreated infected mice (Fig. 8B) (significantly different [P < 0.06] compared to the value for CD8+ T-cell infiltrates from infected mice and significantly different [P < 0.05] compared to the value for CD4+ T-cell infiltrates from infected mice). These data demonstrate that IFN-β reduced the T-cell trafficking to the brain in ECM.
FIG. 7.

IFN-β reduces T-cell trafficking in ECM. T-cell trafficking to the brain in ECM was evaluated by immunohistochemical staining with anti-CD3 antibody.
FIG. 8.

T-cell trafficking in ECM is reduced by IFN-β. (A) Quantification of CD3-positive cells taken from Fig. 7. Infected mice had approximately 25 CD3-positive cells per 20× field, but the number was reduced to less than 10 in IFN-β-treated mice. There were 5 fields per mouse and 5 mice. The value for infected and IFN-β-treated mice was significantly different (P < 0.01) from the value for infected mice and is indicated by an asterisk. In panels A to C, values are means plus SDs (error bars). (B) Isolated T-cell subsets from mouse brains. Mononuclear cells were isolated from the brains of control mice, infected mice, and infected mice treated with IFN-β. T cells were quantified by FACS (n = 5). The value that was significantly different (P < 0.06) from the value for infected, CD8-positive cells is indicated by an asterisk. The value that was significantly different (P < 0.05) from the value for infected, CD4-positive cells is indicated by two asterisks. (C) T-cell CXCR3 expression. Splenocytes were isolated from control, infected, and infected mice treated with IFN-β. T-cell CXCR3 expression was determined by FACS (n = 5). The value that was significantly different (P < 0.05) from the value for infected mice is indicated by an asterisk. MFI, mean fluorescence intensity. (D) CXCR3 mRNA expression in the brain by qRT-PCR (n = 3 or 4). Values are means plus SEMs (error bars). The mRNA levels for each primer pair were normalized to the housekeeping gene GAPDH rRNA level, and fold changes were calculated against control uninfected mice.
Because CXCR3 expression is important for T-cell trafficking in many diseases, including CM (2, 5, 9, 17), and because CXCR3 is a CXCL9 receptor, we examined whether IFN-β treatment decreased T-cell CXCR3 expression as part of the mechanism for reduced T-cell trafficking. Mononuclear cells were isolated from the spleens of untreated, P. berghei-infected, and infected mice treated with IFN-β, and T-cell CXCR3 expression was determined by FACS (see File S1 in the supplemental material). Infected mice had a significant increase in T-cell CXCR3 expression (Fig. 8C), while IFN-β treatment inhibited this increase in T-cell CXCR3 (Fig. 8C) (P < 0.05). Likewise, CXCR3 mRNA expression was increased in the brains of infected mice but reduced in infected mice treated with IFN-β (Fig. 8D).
DISCUSSION
Our results demonstrate that IFN-β delayed the onset of death and improved overall survival in experimental cerebral malaria (ECM). IFN-β exerts its effects in part by suppressing expression of production of all of the following elements: ICAM-1, a critical infected RBC adhesion molecule, serum inflammatory cytokines TNF-α and IFN-γ, and the CXCR3 chemokine ligand (CXCL9). Moreover, administration of IFN-β decreased the expression of T-cell CXCR3 as well as T-cell trafficking (Fig. 9) into the brain. Taken together, this IFN-β-driven multifactorial process can lead to protection of the blood-brain barrier (BBB) and improved ECM survival.
FIG. 9.

Diagram of the hypothesized actions of IFN-β on the pathogenesis of cerebral malaria.
Components of the IFN system are known to be important in the development of ECM (15, 21). As previously reported, high levels of IFN-γ were produced in response to Plasmodium infection, and IFNγ−/− mice were protected against ECM (25). It is known that IFN-γ is responsible for the induction of three IFN-γ-inducible chemokines, CXCL9, CXCL10, and CXCL11 (14). In ECM, these chemokines recruit CD8+ T cells to the brain via CXCR3 on the cell surface. These studies underscore the critical role of IFN-γ in the development of malaria infection.
To date, IFN-β has not been evaluated in ECM. IFN-β is best known for its role as a therapeutic agent in the treatment of MS. In this disease, IFN-β is thought to have its positive effects through multiple actions, including immunosuppressive activities that help maintain BBB integrity. Previously, our laboratory demonstrated in vitro that treatment of retinal cells with IFN-β resulted in the suppression of gene expression and protein production of CXCL9 and ICAM-1 (10). In recent years, interest in the roles of ICAM-1, CXCL9, and CXCL10 in the pathogenesis of ECM has developed (2). Therefore, the aim of our study was to determine whether IFN-β altered development of ECM through these mechanisms. In this report, we demonstrate that murine IFN-β delayed the onset of disease and increased survival in this model system. The effect of IFN-β on ECM was associated with a decrease in the levels of CXCL9 protein in the plasma and CXCL9 expression in brain endothelial cells as well as CXCL9 mRNA in the brain (10). Furthermore, it is of interest to note that in this experimental model the immunosuppressive effect of IFN-β was selective, in that CXCL9 was inhibited, while CXCL10 and CXCL11 were not. In fact, both of these chemokines were elevated.
Although both CXCL9 and CXCL10 signal through CXCR3, IFN-β has different effects on each chemokine that may be critical in the pathogenesis of ECM. This may also imply that perhaps CXCL9 has a more prominent role in accelerating the pathogenesis of ECM. This difference may be due in part to the expression pattern. For example, CXCL9 is expressed primarily in endothelial cells in the brains of infected mice, whereas CXCL10 is found mostly associated with astrocytes and glial cells. The presence of CXCL9 at this vascular interface may therefore have a direct impact on cell recruitment and changes in vascular permeability. Over a longer course of infection, CXCL10 may be very important in neurodegeneration or other cerebral inflammation-mediated processes, but it does not appear to impact disease pathogenesis in this acute-infection setting.
The infiltration of leukocytes represents a critical early event in the development of ECM. Since chemokines and their receptors are essential regulators of leukocyte trafficking, investigators have also examined chemokine receptors in ECM. Three recent studies have reported that CXCR3 is important for the development of ECM. First, the strain-dependent susceptibility to develop ECM is closely related to the expression of CXCR3 on T cells (9, 21). Second, mice deficient in CXCR3 were protected from ECM (2, 9, 17, 21). Moreover, in CXCR3-deficient mice, there was a reduction in the number of CD8+ T cells trafficking to the brain compared to wild-type mice. Our studies provide additional support to explain why T-cell-deficient mice are spared in ECM. We show that IFN-β-treated, Plasmodium-infected mice have reduced expression of CXCR3 in splenic T cells and this is associated with a dramatic decrease in recruitment of T cells to the brains of infected animals. Therefore, one of the multiple mechanisms by which IFN-β is protective is mediated by a reduction in CXCR3 expression and thus results in impaired T-cell recruitment.
ICAM-1 has been reported to play a role in parasitized RBC (pRBC) engagement and mediating many of the early steps in the pathogenesis of ECM (3, 20). Our plasma sICAM-1 data in ECM indicated a trend to IFN-β reduction of ICAM-1 expression that was confirmed by the significant decline in ICAM-1 mRNA by brain qPCR. The plasma data may not be as significant as the mRNA expression data because of tissue bed-mediated effects. ICAM-1 is a membrane protein that sheds from vascular beds, and the plasma expression data may reflect the fact that ICAM-1 is not globally reduced by IFN-β. P. berghei infection can lead to an increase in ICAM-1 expression in other capillary beds, including the lungs. IFN-β has been most studied in the context of the cerebral vascular interface, and its effects on other vascular beds are less well-known. Further study is needed to determine whether this difference in the extent of change mediated by IFN-β between brain and plasma ICAM-1 is a reflection of a regional variability of IFN-β's effects.
One recent study looked at the feasibility of using a human recombinant IFN-α to treat murine ECM (23). The extensive treatment regimen resulted in enhanced survival that was associated with a decrease in parasite replication, decrease in ICAM-1 expression in brain endothelial cells, mild reduction in serum TNF-α levels, and fewer CD8+ T cells trafficking to the brain. There were some distinct differences noted between the human IFN-α in the above report and the mouse IFN-β in our current study. We found that IFN-β treatment produced inhibition of ICAM-1 expression in the brain, decreased the levels of TNF-α and IFN-γ in serum, and reduced T-cell migration to the brain. However, IFN-β did not affect the absolute number of parasitized RBCs in our study. In addition, we show that IFN-β treatment inhibits CXCL9 expression in brain endothelial cells and decreases CXCL9 gene expression in the brain. This chemokine was not evaluated in the IFN-α treatment study. In this study, we also report that the decrease in T-cell trafficking to the brain is associated with decreased levels of CXCR3 on T cells.
The dose of IFN-β used in this study (8 × 105 U/day) is higher than the dose used to treat human MS patients (6 × 106 U twice a day [BID]). It is important to note that mice have a much higher metabolic rate and using standard allometric scaling to account for this, our dose is approximately 4 times higher than the standard human dose. In addition to treatment of MS patients, the type 1 IFNs have been shown to be effective in controlling acute and chronic viral infections. The results of this study in mice may have clinical implications in humans. Including IFN-β in the already established treatment modalities may provide additional immunosuppressive benefits. That is, IFN-β may act on pathogenic mechanisms not controlled by standard therapy.
In summary, we demonstrate that IFN-β ameliorates the severity of ECM and prolongs survival. The proposed mechanisms of action of IFN-β are outlined in Fig. 9. In this scenario, in P. berghei-infected mice, brain endothelial cells express elevated levels of ICAM-1 and CXCL9. Parasitized RBCs bind to ICAM-1 on endothelial cells, and CXCR3-expressing T cells are attracted to CXCL9-positive endothelium. These events lead to a breakdown in cerebral vascular integrity and obstruction of cerebral capillaries. IFN-β treatment interrupts this process and results in a suppression of CXCL9 and ICAM-1 in the brain endothelial cells. In addition, there is a reduction in T-cell CXCR3 expression. The consequence of this IFN-β-mediated combination of events leads to decreased pRBC engagement and entry into the brain, decreased inflammation in the brain, and reinforcement of the blood-brain barrier.
Supplementary Material
Acknowledgments
This work was supported by a National Institutes of Health grant (NIH/NHLBI 1R01HL093179) and grants from The Johns Hopkins University School of Public Health Malaria Research Institute (to C.N.M.) and by the Intramural Research Program, NEI, NIH, to J.J.H.
Editor: J. H. Adams
Footnotes
Published ahead of print on 18 January 2011.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Armah, H. B., et al. 2007. Cerebrospinal fluid and serum biomarkers of cerebral malaria mortality in Ghanaian children. Malar. J. 6:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campanella, G. S., et al. 2008. Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc. Natl. Acad. Sci. U. S. A. 105:4814-4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakravorty, S. J., and A. Craig. 2005. The role of ICAM-1 in Plasmodium falciparum cytoadherence. Eur. J. Cell Biol. 84:15-27. [DOI] [PubMed] [Google Scholar]
- 4.Detrick, B., and J. J. Hooks. 2010. Immune regulation in the retina. Immunol. Res. 47:153-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frangogiannis, N. G. 2007. Chemokines in ischemia and reperfusion. Thromb. Haemost. 97:738-747. [PubMed] [Google Scholar]
- 6.Gramaglia, I., et al. 2006. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat. Med. 12:1417-1422. [DOI] [PubMed] [Google Scholar]
- 7.Grau, G. E., and J. N. Lou. 1995. Experimental cerebral malaria: possible new mechanisms in the TNF-induced microvascular pathology. Soz. Praventivmed. 40:50-57. [DOI] [PubMed] [Google Scholar]
- 8.Grau, G. E., et al. 1993. TNF-induced microvascular pathology: active role for platelets and importance of the LFA-1/ICAM-1 interaction. Eur. Cytokine Netw. 4:415-419. [PubMed] [Google Scholar]
- 9.Hansen, D. S., N. J. Bernard, C. Q. Nie, and L. Schofield. 2007. NK cells stimulate recruitment of CXCR3+ T cells to the brain during Plasmodium berghei-mediated cerebral malaria. J. Immunol. 178:5779-5788. [DOI] [PubMed] [Google Scholar]
- 10.Hooks, J. J., C. N. Nagineni, L. C. Hooper, K. Hayashi, and B. Detrick. 2008. IFN-beta provides immuno-protection in the retina by inhibiting ICAM-1 and CXCL9 in retinal pigment epithelial cells. J. Immunol. 180:3789-3796. [DOI] [PubMed] [Google Scholar]
- 11.Idro, R., et al. 2007. Burden, features, and outcome of neurological involvement in acute falciparum malaria in Kenyan children. JAMA 297:2232-2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar, M. V., C. N. Nagineni, M. S. Chin, J. J. Hooks, and B. Detrick. 2004. Innate immunity in the retina: Toll-like receptor (TLR) signaling in human retinal pigment epithelial cells. J. Neuroimmunol. 153:7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lou, J., et al. 1997. Platelets play an important role in TNF-induced microvascular endothelial cell pathology. Am. J. Pathol. 151:1397-1405. [PMC free article] [PubMed] [Google Scholar]
- 14.Luster, A. D., R. Alon, and U. H. von Andrian. 2005. Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 6:1182-1190. [DOI] [PubMed] [Google Scholar]
- 15.Miu, J., N. H. Hunt, and H. J. Ball. 2008. Predominance of interferon-related responses in the brain during murine malaria, as identified by microarray analysis. Infect. Immun. 76:1812-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murata, K., et al. 2007. Synergistic deposition of C4d by complement-activating and non-activating antibodies in cardiac transplants. Am. J. Transplant. 7:2605-2614. [DOI] [PubMed] [Google Scholar]
- 16a.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC.
- 16b.National Institutes of Health. 2000. Public Health Service policy on humane care and use of laboratory animals. Office of Laboratory Animal Welfare, National Institutes of Health, Bethesda, MD.
- 17.Srivastava, K., et al. 2008. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe 4:179-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanuma, N., H. Sakuma, A. Sasaki, and Y. Matsumoto. 2006. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. (Berl.) 112:195-204. [DOI] [PubMed] [Google Scholar]
- 19.Tripathi, A. K., D. J. Sullivan, and M. F. Stins. 2007. Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J. Infect. Dis. 195:942-950. [DOI] [PubMed] [Google Scholar]
- 20.Tripathi, A. K., D. J. Sullivan, and M. F. Stins. 2006. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect. Immun. 74:3262-3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van den Steen, P. E., et al. 2008. CXCR3 determines strain susceptibility to murine cerebral malaria by mediating T lymphocyte migration toward IFN-gamma-induced chemokines. Eur. J. Immunol. 38:1082-1095. [DOI] [PubMed] [Google Scholar]
- 22.van der Heyde, H. C., J. Nolan, V. Combes, I. Gramaglia, and G. E. Grau. 2006. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 22:503-508. [DOI] [PubMed] [Google Scholar]
- 23.Vigario, A. M., et al. 2007. Recombinant human IFN-alpha inhibits cerebral malaria and reduces parasite burden in mice. J. Immunol. 178:6416-6425. [DOI] [PubMed] [Google Scholar]
- 24.Yamakuchi, M., et al. 2007. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc. Natl. Acad. Sci. U. S. A. 104:1301-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yanez, D. M., D. D. Manning, A. J. Cooley, W. P. Weidanz, and H. C. van der Heyde. 1996. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J. Immunol. 157:1620-1624. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.