

Abstract
The pathogenesis of a Citrobacter rodentium infection was evaluated in mice fed diets with a single deficiency in either selenium or vitamin E or with a double deficiency in both selenium and vitamin E compared to mice on nutritionally adequate diets. Mice fed the selenium- and vitamin E-deficient diet for 6 weeks had increased loads of C. rodentium in the colon and spleen, which were not observed in mice fed either of the singly deficient diets or the adequate diet. Infected mice fed the doubly deficient diet had increased colon crypt hyperplasia and an influx of infiltrating cells along with gross changes to crypt architecture, including ulceration and denuding of the epithelial layer. Cytokine and chemokine mRNA levels in the colon were measured by real-time PCR. Expression of proinflammatory cytokines and chemokines was upregulated on day 12 after infection with C. rodentium in mice fed the doubly deficient diet compared to mice fed the control diet. Heme oxygenase 1, an enzyme upregulated by oxidative stress, also was more highly induced in infected mice fed the doubly deficient diet. Production of C. rodentium antigen-specific IgM and IgG antibodies was not affected by feeding the doubly deficient diet. The results indicated that selenium and vitamin E play an important role in host resistance and in the pathology induced by C. rodentium, an infection that mimics disease caused by common food-borne bacterial pathogens in humans.
Citrobacter rodentium is a mouse pathogen that shares many characteristics with human enteropathogenic Escherichia coli (EPEC) and enterohemorrhagic E. coli (EHEC) (19). EPEC and EHEC (e.g., E. coli O157:H7) are food-borne pathogens that are the causative agents for human diseases ranging from diarrhea to hemorrhagic colitis and hemolytic-uremic syndrome. Infection of mice with C. rodentium induces lesions in the mouse colon that are indistinguishable from those observed in humans infected with EPEC and EHEC (32). Citrobacter rodentium-induced changes to the colon include crypt hyperplasia, epithelial cell proliferation, crypt dilation, mucosal thickening, and an uneven apical enterocyte surface (32). Citrobacter rodentium growth is usually restricted to the colon, with little bacterial translocation except in highly susceptible mouse strains such as C3H (57).
The distal colon is preferentially colonized by C. rodentium, with a high bacterial burden by day 7 after infection that persists through day 14 and is typically cleared by day 21 (60). Oral infection with C. rodentium induces a robust immune response characterized by a mixed Th1/Th17 response with increased gene expression of interleukin-17 (IL-17), IL-22, IL-23, IL-12, gamma interferon (IFN-γ), IL-1β, and tumor necrosis factor alpha (TNF-α) (20, 64). Both IL-23 and IL-22 have been shown to be critical for resistance to C. rodentium (64). In contrast, mice deficient in IFN-γ, IL-17A, or IL-17F survived infection with C. rodentium but exhibited delayed clearance or increased bacterial load and increased colonic pathology (21, 48). CD4+, but not CD8+, T cells are critical for controlling infection with C. rodentium, as are B cells and antigen-specific antibody responses (7, 47); indicating that a T-cell-dependent systemic antibody response is required for clearance of C. rodentium (6).
Selenium and vitamin E are important in host antioxidant defense and immune function. Vitamin E is a potent peroxyl radical scavenger that prevents lipid peroxidation (8) and is found in high concentrations in immune cells (10). Deficiency in vitamin E is associated with increased oxidative stress (39) and impaired immune function, including both humoral and cell-mediated immunity, phagocyte function, and lymphocyte proliferation (37). Age-related declines in immune function can be restored by vitamin E supplementation (61).
Selenium also has an important role in antioxidant defense and immune function. Due to its incorporation as selenocysteine into glutathione peroxidase (GPX) (18) and thioredoxin reductase (55), selenium is important for the control of oxidative stress and, therefore, the redox tone of the cell. In total, there are 25 identified selenoproteins (24 in rodents), many with unknown function (38). Selenium is important for cytotoxic T-lymphocyte and natural killer cell activity (26), respiratory burst (2), and protection against endotoxin-induced oxidative stress (42). Multiple studies have shown that NF-κB activation can be affected by selenium status (23, 33), and selenium deficiency can alter chemokine and cytokine expression during viral infections (4).
Previous work with mice established that deficiencies in selenium and/or vitamin E altered the normal physiological response and intestinal clearance of a parasitic nematode, Heligmosomoides polygyrus (1, 49). A secondary H. polygyrus infection induced a potent T-helper cell 2 (Th2) immune (memory) response, and both single and double deficiencies in selenium and vitamin E delayed adult worm expulsion, increased fecundity (egg production), and impaired some, but not all, IL-4 receptor-dependent changes to the small intestine. These results suggested that both selenium and vitamin E are required for changes in intestinal physiology that promote host protection against H. polygyrus. To further extend our knowledge of the roles of selenium and vitamin E in intestinal function and mucosal immunity, we characterized the host Th1/Th17-related response to C. rodentium infection in mice fed a diet deficient in selenium and vitamin E. The study demonstrated that a combined selenium and vitamin E deficiency altered resistance to C. rodentium and enhanced the pathology associated with infection of the colon.
MATERIALS AND METHODS
Mice.
Three-week-old male C57BL/6 mice were obtained from the Small Animals Division of the National Cancer Institute (Frederick, MD). Mice received one of five isocaloric torula yeast-based diets (prepared by Harlan Teklad, Madison, WI) that were adequate in all nutrients except those specified below (3). The first control diet contained 4% menhaden oil plus 1% corn oil as the fat source, 0.2 μg/g of sodium selenite, and 50 mg/kg of d-α-tocopherol acetate. A second diet, which was doubly deficient in vitamin E and selenium, was prepared by omitting the sodium selenite and d-α-tocopherol acetate from the control diet, while a third diet, which was deficient in vitamin E, included 0.2 μg/g of sodium selenite but no d-α-tocopherol acetate. A fourth diet, a control for studies examining only selenium deficiency, was identical to the first control diet described above except that lard was substituted for menhaden oil, and a fifth diet, which was deficient in selenium, was prepared by omitting the sodium selenite from this control diet. Menhaden oil is generally used as the fat source for studies on vitamin E deficiency because it is known to increase the requirement for vitamin E, thereby hastening the onset of a vitamin E deficiency (11). Mice were fed the diets for 5 to 6 weeks prior to infection with C. rodentium and were maintained on the respective diets for the remainder of the experiment. Body weight was monitored throughout the study. All animal studies were approved by the USDA Beltsville Area Animal Care and Use Committee.
Citrobacter rodentium infection and antigen preparation.
The C. rodentium strain used was a nalidixic acid-resistant mutant of strain DBS100 (ATCC 51459). Citrobacter rodentium was grown in Luria-Bertani (LB) medium containing 20 μg/ml nalidixic acid to stationary phase, collected by centrifugation, and resuspended in LB medium. Mice were infected by oral gavage with 0.2 ml of the C. rodentium suspension, which contained ca. 1.0 × 1010 CFU. The dose was confirmed by retrospective plating on LB agar plates containing 50 μg/ml nalidixic acid. For enzyme-linked immunosorbent assays (ELISAs), a B-PER extract of C. rodentium was prepared and the protein content determined using the bicinchoninic acid (BCA) protein reagent (Pierce, Chicago, IL).
Measurement of liver vitamin E and GPX activity.
To confirm the vitamin E and selenium status of mice fed the experimental diets, the liver vitamin E concentration and the enzyme activity of glutathione peroxidase (GPX), a selenium-dependent enzyme sensitive to dietary selenium levels, were determined in samples obtained from mice fed control or deficient diets (9, 50). Data were expressed as the mean ± standard error of the mean (SEM).
Sample collection.
Mice were weighed and sacrificed at various time points after infection. Blood samples were collected and liver samples removed and frozen for subsequent biochemical analyses. The distal 5 cm of colon and the spleen were removed aseptically. The colon was weighed after removal of the fecal pellets, and 1-cm portions of the colon were fixed in 4% formalin for histology or snap-frozen in liquid nitrogen for gene expression analysis. The extent of colonization of the spleen and colon by C. rodentium was determined by homogenizing the tissues in LB medium, and then viable counts were determined on LB agar plates with (colon) or without (spleen) 50 μg/ml nalidixic acid. Bacterial colonies were enumerated the following day. The presence of C. rodentium also was confirmed by plating on MacConkey agar plates. Colonies gave the characteristic morphology, a red center with a white rim, as previously described (43). The clearance of C. rodentium from the host was based on a colony count of 0 obtained from duplicate plates inoculated with samples of undiluted colon and spleen homogenate at different times during the infection. The limit of detection was assigned a colony count of 0.5 for statistics and graphing.
Spleens were removed and pressed through 70-μm nylon cell strainers to prepare single-cell suspensions (Becton Dickinson, Franklin Lakes, NJ); these were centrifuged and the resulting cell pellet resuspended in 0.15 M NH4Cl-1 mM KHCO3-0.1 mM EDTA (pH 7.3) to lyse red blood cells. The final suspension was filtered through 100-μm mesh to remove cellular debris and resuspended in culture medium, and cells were counted.
Measurement of colonic cytokine gene expression.
cDNA for gene expression studies was prepared from colonic tissue (13). Briefly, 50 ng cDNA per reaction was used for quantitative PCR (qPCR) amplification with a commercially available kit (Abgene, Rochester, NY), and amplification was measured on an ABI7700 instrument (Applied Biosystems, Foster City, CA). The probes were 5′-tetrachloro-6-carboxyfluoroscein labeled and 3′-Black Hole Quencher 1 labeled and were synthesized by Biosource International (Camarillo, CA). Fluorescence signals measured during amplification were processed postamplification to determine the threshold cycle (CT) value using the manufacturer's software. Data obtained from three independent experiments were merged to increase the number of replicates and the power of the analysis. To evaluate the effects of treatment, genes of interest were normalized to the housekeeping gene RPL32 (ΔCT) and then analyzed using the ΔΔCT method (30).
Measurement of serum antibody and total nitrite levels.
A C. rodentium protein extract was used for coating Maxisorb (Nunc, Rochester, NY) ELISA plates. Plates were coated overnight with 10 μg/ml of bacterial antigen in phosphate-buffered saline (PBS) and blocked with 3% nonfat milk (Bio-Rad, Hercules, CA) in PBS. Mouse serum was then serially diluted into PBS-0.05% Tween 20 (TPBS) containing 1% nonfat milk. Plates were washed with TPBS between all steps. Fifty microliters of each sample was then placed in duplicate into wells of the ELISA plate and incubated for 30 min at 37°C. Plates were then incubated with biotinylated class-specific antibodies (anti-IgG [Vector Laboratories, Burlingame, CA] or anti-IgM [Antibodies Incorporated, Davis, CA]) for 30 min at 37°C, followed by a 30-min incubation with a streptavidin-horseradish peroxidase complex (Vector Laboratories, Burlingame, CA). After the final wash, wells were incubated with 3,3′,5,5′-tetramethylbenzidine color substrate (BD Biosciences, San Jose, CA). The color reaction was terminated by addition of 4 M sulfuric acid and read at 450 nm.
Total serum nitrite levels were measured using an R&D Systems total nitric oxide assay (catalog no. DE1600; R&D Systems, Minneapolis, MN) following the manufacturer's protocol. Briefly, serum samples were deproteinated by ultrafiltration through 10,000-molecular-weight-cutoff Microcon filters (Millipore, Billerica, MA). Nitrate present in the samples was converted to nitrite by nitrate reductase treatment followed by reaction with Griess reagent. Samples were read on a SpectoMax Plus microplate reader (Molecular Devices, Sunnyvale, CA) at 540 nm. Total serum nitrite concentrations were extrapolated from a standard curve generated using a nitrate standard provided in the kit. Data from three separate experiments were merged to increase the number of replicates.
Histopathological evaluation.
Approximately 1-cm sections from equivalent positions in the distal colon were obtained from mice fed adequate or deficient diets on days 12 and 17 postinfection. The colon sections were cut longitudinally and pinned out flat with the mucosal side up in a buffered formalin solution. After fixation, the sections were paraffin embedded, sectioned (5 μm), and stained with Giemsa stain. Sections were evaluated for changes in the mucosa architecture and the presence of an inflammatory infiltrate using a modification of a previously described scoring system (46). Sections were scored for damage to the surface epithelial cells (0 to 4), edema (0 to 2), hemorrhage (0 to 2), crypt dilation (0 to 1), and inflammatory infiltrate (0 to 3), and the scores were summed. Mucosa height was measured using an Arcturus XT microdissection system. Only well-oriented crypts were measured, and seven or more individual measurements were averaged for each mouse.
Data analysis.
Data in the table and figures were expressed as the mean ± standard deviation (SD) or SEM. Statistical analyses were performed using a t test, one-way analysis of variance (ANOVA), or two-way ANOVA with post hoc analysis for multiple comparisons using the Holm-Sidak method (SigmaStat 3.1 or SigmaPlot 11; Systat Inc., Richmond, CA). Data were transformed, if necessary, to generate normality and equal-variance parameters, but a Mann-Whitney rank sum test was run and the median value reported if this could not be achieved. A P value of <0.05 was considered significant.
RESULTS
Biochemical parameters.
Vitamin E deficiency was confirmed by liver analysis, which showed 16.3 ± 1.0 μg vitamin E/g of tissue in mice fed the control diet versus 2.2 ± 0.1 μg vitamin E/g of tissue in mice fed the doubly selenium- and vitamin E-deficient diet (mean ± SEM). Selenium deficiency was confirmed by determining the activity of GPX, a selenium-dependent enzyme sensitive to dietary selenium levels (29), in the liver. Liver GPX activity (mean ± SEM) was 801 ± 22 and 6.3 ± 0.4 mU/mg of protein in mice fed the diets adequate and deficient in selenium, respectively. Thus, selenium and vitamin E levels were reduced by approximately 99 and 85%, respectively, in mice fed the doubly deficient diet. Similar reductions in liver vitamin E concentration and GPX activity were observed in mice fed diets deficient in vitamin E or selenium, respectively.
Effect of diet and infection on growth.
Mice fed the doubly deficient diet were within 4% of the weight of mice fed the control adequate diets after approximately 6 weeks on the diet prior to infection (data not shown). Weights of mice on the control adequate diets were not affected by infection with C. rodentium. Some mice on the doubly deficient diet that were infected with C. rodentium exhibited weight loss ranging from 3 to 24% between days 4 and 17. In addition, infected mice fed the doubly deficient diet had stools that were soft or liquid compared to those of infected mice on the control adequate diet.
Doubly deficient, but not singly deficient, diets increase C. rodentium colonization of the colon.
Colonization of the colon by C. rodentium was assessed at days 7, 12, and 17 postinfection. On day 7 postinfection the colonic C. rodentium loads were the same in mice on both dietary regimens (Fig. 1 A). By day 12, however, mice fed the control adequate diet had a slightly reduced C. rodentium load compared to that on day 7, which was decreased substantially by day 17. In contrast, mice fed the doubly deficient diet had a colonic C. rodentium load that was significantly higher on days 12 and 17 postinfection than that in infected mice fed the control adequate diet (P < 0.001). Mice fed a diet deficient in only selenium or vitamin E did not have increased C. rodentium colonization (data not shown). Additional studies demonstrated that the infection is essentially cleared in mice around day 20 regardless of the diet fed (data not shown).
FIG. 1.

Mice fed a doubly selenium- and vitamin E-deficient diet have increased C. rodentium colonization of the colon and spleen. Mice (n = 7 or 8) were infected orally with approximately 1.0 × 1010 CFU of C. rodentium and sacrificed at 7, 12, and 17 days postinfection. The C. rodentium colon (A) and spleen (B) burdens were determined by plating serial dilutions of the tissue homogenates. Only within-day comparisons were made, using a t test (panel A and day 7 in panel B) or Mann-Whitney rank sum test (days 12 and 17 in panel B). The dashed line indicates the limit of detection. Within each day the mean (panel A and day 7 in panel B) or the median (days 12 and 17 in panel B) is indicated by a dash.
Mice fed the doubly deficient diet have increased translocation of C. rodentium into systemic compartments.
Total viable bacteria in the spleen were enumerated to assess whether a deficiency in selenium and vitamin E could alter epithelial barrier function, resulting in increased translocation of luminal bacteria. Colonization of the spleen was observed on day 7 in infected mice fed both the control adequate and doubly deficient diets, but the levels were not significantly different (Fig. 1B). On day 12 and again on day 17, splenic colonization was higher in infected mice fed the doubly deficient diet than in infected mice on the control adequate diet. The data shown in Fig. 1B are representative of three experiments, but colonization was more variable in other experiments and the trend toward increased splenic colonization in doubly deficient mice did not always achieve significance. Mice fed a diet deficient in only selenium or vitamin E did not have increased splenic colonization (data not shown).
The spleens from infected mice fed the doubly deficient diet were also enlarged compared to those of infected mice fed the control adequate diet. The spleen-to-body weight ratios of infected mice fed the control adequate diet (Fig. 2 A) and mice fed diets deficient in only selenium or vitamin E (data not shown) remained relatively constant from day 4 to 17, while the ratio increased in infected mice fed the doubly deficient diet, with the difference being significant on days 12 and 17 after infection (Fig. 2A). Spleen size correlated with increased cell counts on day 12 postinfection, indicating that more cells were present in spleens from infected mice fed the doubly deficient diet (11.1 × 107 ± 1.3 × 107) than in spleens from uninfected mice fed the control adequate (5.4 × 107 ± 0.6 × 107) or deficient (5.7 × 107 ± 0.8 × 107) diet or in spleens from infected mice fed the control adequate diet (7.7 × 107 ± 1.2 × 107).
FIG. 2.
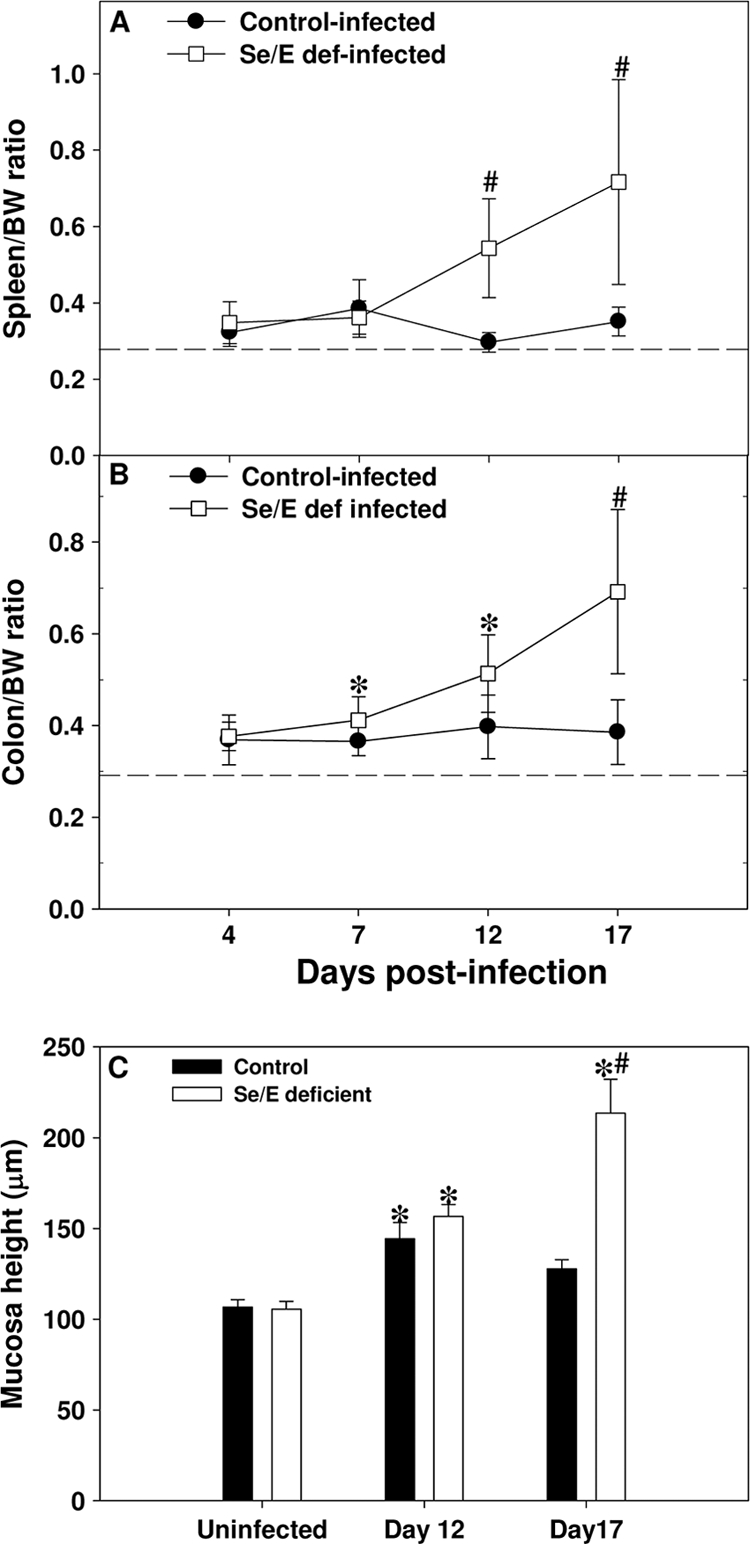
C. rodentium-infected mice fed a doubly selenium- and vitamin E-deficient diet have higher organ/body weight ratios and mucosa heights. Mice (n = 7 or 8) were infected orally with approximately 1.0 × 1010 CFU of C. rodentium and sacrificed at 4, 7, 12, and 17 days postinfection. Mice were weighed just prior to euthanasia. (A and B) The spleen and first 5 cm of the distal colon from the anus were removed. Any fecal pellets present in the colon section were removed and both the spleen (A) and colon (B) weighed. Data are expressed as the mean ± SD percent organ/body weight (BW) ratio. *, P < 0.05; #, P < 0.001 (for comparison with infected mice fed the control adequate diet). Only within-day comparisons were made. The dashed line indicates the average organ weight in uninfected mice fed the control adequate diet. (C) Mucosa height was measured using an Arcturus XT microdissection system. Only well-oriented crypts were measured and averaged for each mouse. Data are expressed as the group mean ± SEM. *, significant different (P < 0.05) from uninfected mice fed the respective diet; #, the doubly selenium- and vitamin E-deficient infected group was significantly different from the infected control group on day 17.
Infected mice fed a doubly deficient diet have increased colonic pathology.
In agreement with previously published results (57), infection with C. rodentium produced a hyperproliferative response in the colonic crypts that resulted in an increase in the thickness and rigidity of the colon. There were no obvious visual differences at day 4 postinfection in colons from infected mice fed the control adequate or doubly deficient diet, but the colons from infected mice fed the doubly deficient diet became thicker and more rigid and had a higher percent colon/body weight ratio than those of infected mice fed the control adequate diet as the infection progressed through day 17 (Fig. 2B). This result was further supported by an increase in the mucosal height in mice fed a doubly deficient diet on day 17 after infection, which paralleled the increase in the colon/body weight ratio (Fig. 2C) (P < 0.001). No difference in mucosal height was observed between uninfected mice fed either diet. Infected mice fed diets deficient only in selenium or vitamin E did not have increased colon/body weight ratios (data not shown) compared to infected mice fed the control adequate diet, indicating an exaggerated hyperplasia in only the infected mice fed the doubly deficient diet.
Infection of mice with C. rodentium, in general, caused increased crypt expansion, hyperplasia of the entire mucosa, and an increased inflammatory infiltrate. On day 12, infected mice fed the control adequate diet displayed colonic hyperplasia, but the epithelial cell border was largely intact with only a mild to moderate cellular infiltrate, and goblet cells were plentiful with a large amount of mucus (Fig. 3 C). On day 17, infected mice fed the control adequate diet still exhibited signs of hyperplasia, but the cellular infiltrate was reduced, and both the epithelial cell border and mucus production were intact (Fig. 3E). Mice fed the doubly deficient diet, on day 12 after infection (Fig. 3D) and even more so on day 17 (Fig. 3F), had disruption of the crypt architecture with a large increase in the amount of cellular infiltration, frank ulceration, and denuding of the mucosal epithelial surface. This was reflected by an increase in the pathology score (mean ± SEM) on days 12 (control adequate, 3.1 ± 0.2; deficient, 6.6 ± 0.7 [P < 0.001]) and 17 (control adequate, 1.9 ± 0.4; deficient, 6.3 ± 0.7 [P < 0.001]) postinfection in infected doubly deficient mice. No differences in histology between uninfected mice on either diet were noted (Fig. 3A and B).
FIG. 3.

C. rodentium-infected mice fed doubly selenium- and vitamin E-deficient diet have increased colon pathology. (A, C, and E) Mice fed control adequate diet; (B, D, and E) mice fed doubly selenium- and vitamin E-deficient diet. (A and B) Uninfected mice. (C to F) Citrobacter rodentium-infected mice killed on day 12 (C and D) or day 17 (E and F). An equivalent 1-cm section of colon from each mouse was removed, opened longitudinally, pinned out mucosal side up, and fixed in buffered formalin. Samples were then processed for Giemsa staining. All photomicrographs were taken under identical conditions, and the bars indicate 100 μm.
Doubly deficient diets do not affect antigen-specific serum IgG and IgM levels but increase serum total nitrite levels.
Previous work demonstrated that an antigen-specific antibody response was necessary for clearance of C. rodentium (7, 47). Citrobacter rodentium-specific serum antibody levels were measured by ELISA. Considerable variation in antigen-specific antibody production was noted among mice in each dietary group at 16 days postinfection. There was, however, no significant difference in either antigen-specific IgG or IgM production between infected mice fed either the control adequate or doubly deficient diet (data not shown).
The gene for inducible nitric oxide synthase (NOS2) was upregulated in colon by infection with C. rodentium in mice fed either the control adequate or doubly deficient diet (Table 1) but was more highly expressed in the latter group. To evaluate whether there also were systemic changes in nitric oxide production, total serum nitrite levels were measured. Nitrite levels in uninfected mice fed either the control adequate or the doubly deficient diet were similar and not statistically different (Fig. 4). Infection caused a significant increase in serum nitrite levels in mice fed the doubly deficient diet (P < 0.001) (Fig. 4), which correlated with increased NOS2 gene expression.
TABLE 1.
Colonic gene expression is altered in C. rodentium-infected mice fed a doubly selenium- and vitamin E-deficient diet
| Gene product | Gene expressiona in: |
|||||||
|---|---|---|---|---|---|---|---|---|
| Uninfected mice fed adequate diet |
Uninfected mice fed deficient diet |
Infected mice fed adequate diet |
Infected mice fed deficient diet |
|||||
| CT | Fold change | CT | Fold change | CT | Fold change | CT | Fold change | |
| IL-12p40 | 12.7 ± 1.1 | 1.0 | 13.5 ± 0.9 | 0.5 | 11.4 ± 0.7 | 2.4* | 12.5 ± 1.3 | 1.1*# |
| IL-17A | 16.2 ± 1.2 | 1.0 | 15.6 ± 1.1 | 1.5 | 11.0 ± 0.8 | 34.8* | 10.2 ± 1.0 | 61.7*# |
| IL-22 | 17.0 ± 2.0 | 1.0 | 19.0 ± 1.7 | 0.3† | 12.8 ± 2.0 | 18.4* | 11.9 ± 1.1 | 34.9* |
| TNF-α | 9.3 ± 0.5 | 1.0 | 9.5 ± 0.8 | 0.8 | 7.7 ± 0.8 | 3.0* | 6.7 ± 0.8 | 6.0*# |
| iNOS | 10.6 ± 0.6 | 1.0 | 11.0 ± 1.1 | 0.8 | 6.2 ± 1.2 | 21.2* | 5.1 ± 1.3 | 46.2*# |
| IFN-γ | 13.9 ± 0.9 | 1.0 | 14.9 ± 0.6 | 0.5† | 10.6 ± 1.0 | 10.1* | 9.1 ± 0.8 | 27.7*# |
| IL-6 | 14.1 ± 1.3 | 1.0 | 14.1 ± 1.7 | 1.0 | 12.9 ± 1.2 | 2.3* | 10.2 ± 2.2 | 15.1*# |
| CCL2 | 10.1 ± 0.6 | 1.0 | 10.5 ± 0.6 | 0.7 | 7.9 ± 0.9 | 4.7* | 6.4 ± 1.0 | 13.5*# |
| CXCL10 | 7.6 ± 0.8 | 1.0 | 8.5 ± 0.8 | 0.5† | 5.5 ± 1.1 | 4.1* | 3.7 ± 0.9 | 14.7*# |
| GPX1 | 2.9 ± 0.5 | 1.0 | 5.0 ± 0.4 | 0.2† | 2.8 ± 0.5 | 1.0 | 4.9 ± 0.7 | 0.2# |
| GPX2 | 1.1 ± 0.4 | 1.0 | 1.1 ± 0.4 | 1.0 | 0.4 ± 0.5 | 1.7* | −0.4 ± 0.6 | 2.9*# |
| HMOX1 | 8.8 ± 0.7 | 1.0 | 8.1 ± 0.8 | 1.5 | 7.9 ± 0.8 | 1.9* | 5.8 ± 0.9 | 7.8*# |
CT (mean ± SD) is defined as the threshold cycle; the lower the number, the higher the expression. The fold change in colonic gene expression at 12 days postinfection is in comparison to expression in uninfected mice fed a diet adequate in selenium and vitamin E. *, the value is significantly different (P < 0.05) from that for the corresponding uninfected group. #, the value is significantly different (P < 0.05) from that for the infected group fed an adequate diet. †, the value is significantly different (P < 0.05) from that for the uninfected group fed an adequate diet. The results were obtained by merging data from three separate experiments (n = 7 to 21).
FIG. 4.

Serum total nitrite levels are elevated in infected mice fed the doubly deficient diet. Serum total nitrite levels were measured using the Griess reagent. Data are the mean ± SEM serum nitrite concentrations (μM) for each group (n = 13 to 23). Results were obtained by merging data from three separate experiments.
Mice fed the doubly deficient diet have altered immune-related gene expression.
In agreement with previous studies, mice infected with C. rodentium for 12 days had increased expression of multiple proinflammatory cytokines, including IL-12p40, IL-17, IL-22, TNF-α, IFN-γ, and IL-6 (Table 1). Furthermore, with the exception of IL-12p40 and IL-22, the expression levels of these genes, as well as the those for chemokines CCL2 and CXCL10, and of NOS2 were significantly higher in infected mice fed the doubly deficient diet than in infected mice fed control adequate diet (Table 1). The higher chemokine expression after infection in mice fed the doubly deficient diet versus the control adequate diet correlated with the increased cellular infiltrate (Fig. 3). Together these results suggested that infected mice fed the doubly deficient diet had a greater proinflammatory response to C. rodentium.
Expression levels of several other nonimmune genes were also determined (Table 1). Heme oxygenase 1 (HMOX1), whose expression is upregulated by oxidative stress (reviewed in reference 40), was induced by infection in mice fed either diet, but expression was significantly higher in infected mice fed the doubly deficient diet. The expression levels of two other antioxidant genes, those for glutathione peroxidase 1 (GPX1) and glutathione peroxidase 2 (GPX2) were also determined (Table 1). Both enzymes contain selenocysteine residues at their active sites, and their enzymatic activity is sensitive to dietary selenium levels (59), as is the stability of the message for GPX1 (41). Consistent with these results, GPX1 expression was significantly decreased in mice fed a doubly deficient diet compared to mice fed the control adequate diet, and infection did not induce expression in mice fed either diet. In contrast, and in agreement with results reported by others, GPX2 expression was not affected by diet (25) but was significantly increased by infection.
DISCUSSION
Mice fed a diet deficient in both selenium and vitamin E had 99% less selenium-dependent GPX activity and 85% less vitamin E in their livers than mice on an control adequate diet, and these mice had increased pathology in response to infection with C. rodentium. In addition, mice fed the doubly deficient diet had higher colonic C. rodentium burdens and increased bacterial translocation to the spleen compared to infected mice fed the control adequate diet. The increased bacterial translocation may be a reflection of the increased damage to the colonic crypts observed in the doubly selenium- and vitamin E-deficient mice.
Single deficiencies in selenium or vitamin E, however, did not result in increased levels of C. rodentium in the colon or alter the tissue/body weight ratios as observed in mice fed the doubly deficient diet. The reasons for this are unclear, but it is known that vitamin E deficiency enhances the effects of a selenium deficiency (28), and therefore, the combined deficiency may produce a more dramatic effect than selenium deficiency alone. The length of time that mice were on deficient diet prior to infection (5 to 6 weeks), while sufficient to deplete the selenocysteine-containing enzyme GPX1, which is known to be very sensitive to dietary selenium levels, may not be sufficient to lower other selenium pools within the host to a functionally critical level. Furthermore, the functions of many of the 25 known selenocysteine-containing proteins are poorly defined, and any role in protection against infectious colitis needs to be determined. The dose of C. rodentium administered in our study was experimentally found to ensure a uniform infection in mice on a control adequate diet, but a wider range of doses could produce different results in mice fed deficient diets. Additional studies will be needed to determine if longer-term single selenium or vitamin E deficiency can produce results similar to those obtained more rapidly with a double deficiency.
Increased bacterial translocation was associated with a large increase in the size of spleens obtained from infected mice fed the doubly deficient diet at both days 12 and 17 and was associated with increased cellularity. Diet itself had no effect on spleen size or cellularity in uninfected mice, so the increase in spleen size and cellularity is likely the result of a lymphocyte proliferative response to the higher splenic bacterial burden present in deficient mice.
Infection-induced hyperplasia was observed in mice fed either the control adequate or doubly deficient diet but was greater in mice fed the doubly deficient diet as reflected by the increase in colon/body weight ratio and increased mucosal height. The mechanism responsible for the increased level of hyperplasia in the mice fed the doubly deficient diet is not clear, but it could result from the higher bacterial burden in mice fed the doubly deficient diet, altered cell-mediated immunity, or increased oxidative stress that activates cell growth pathways. Hyperplasia was observed with higher bacterial burden in IFN-γ−/− and IL-12−/− mice (48). Reactive oxygen species can act as signaling molecules that can activate transcription factors (45). Decreased selenium levels translate into decreased GPX activity (5) and increased concentrations of intracellular hydrogen peroxide that can activate NF-κB (35). Vitamin E, along with selenium, blocks activation of NF-κB and AP-1 (35, 54). Furthermore, signaling through Toll-like receptor 4 has been implicated in much of the inflammation and pathology seen during infection with C. rodentium (22) and results in rapid induction of NF-κB that parallels increased proliferation of colonic epithelium (58). Thus, a deficiency in both selenium and vitamin E may increase oxidative stress, altering signal transduction and transcription factor activation, and impact tissue damage and the hyperproliferative response of the host to a gastrointestinal tract infection. Further studies are needed to determine which of these factors contributed to the increased tissue damage and hyperplasia observed in the mice fed the doubly deficient diet.
Of interest is the increased expression of GPX2 in response to infection in mice on either the control adequate or doubly deficient diet (Table 1). Expression of GPX2 has been shown to increase the response to initial colonization of the colon by commensal bacteria (15) and in three different models of experimental colitis (56). It was also shown that trace amounts of GPX2 were protective against bacterially induced ileocolitis (16). Further evidence suggested a role for GPX2 in growth and maintenance of epithelial cells (24). Our results indicated that increased expression of GPX2 is also part of the host response to bacterial pathogens. GPX2 gene expression was not affected by dietary selenium levels, confirming previous results (25). Higher gene expression was seen in mice fed the doubly deficient diet and correlated with the increased pathology, thus providing further evidence that GPX2 acts as stress response protein.
Citrobacter rodentium is a potent inducer of a Th1/Th17-type response. The increase in cytokine and chemokine expression in infected mice fed the doubly deficient diet compared to those fed the control adequate diet suggested that the deficiency resulted in a greater proinflammatory response to infection that correlated with the increased colonic pathology. NOS2 expression and serum nitrite levels were also elevated in mice fed the doubly deficient diet. Nitric oxide has antimicrobial functions (reviewed in reference 17), but overexpression has been implicated in the pathology associated with colitis (52). Thus, elevated levels of nitric oxide at the site of infection also may enhance pathology in infected mice fed the doubly deficient diet.
Whether expression of IL-17A and IL-22 produces a net beneficial or deleterious immune response is not entirely clear. IL-17A contributes to the development of experimental autoimmune encephalitis (14) but is important for controlling bacterial infections, including C. rodentium infection (63). IL-22 is critical for host defense against C. rodentium (64), and increased serum levels correlated with Crohn's disease activity in humans (44). In contrast, IL-22 ameliorated intestinal inflammation in a Th2-related mouse model of ulcerative colitis (53). The conflicting effects of IL-22 may be due to the cytokine environment. Sonnenberg et al. (51) demonstrated that IL-22 in the absence of IL-17A was protective against bleomycin-induced lung pathology but was proinflammatory in the presence of IL-17A. The doubly selenium- and vitamin E-deficient mice described here had increased expression of IL-17A and IFN-γ and a trend toward higher expression of IL-22 in response to infection. Thus, it is not clear whether IL-22 expression is a response to or a mediator of the increased pathology observed in mice fed the doubly deficient diet.
Interestingly, expression of both IL-6 and CCL2 was upregulated in mice fed the doubly deficient diet compared to infected mice fed the adequate diet. Interleukin-6 was shown to be important for host defense against C. rodentium, as IL-6-deficient mice had increased mortality, delayed bacterial clearance, and increased damage to the colonic epithelium (12). However, IL-6 has also been implicated in intestinal pathology and is elevated in Crohn's disease patients (34), and IL-6 mRNA expression correlated with disease progression in a murine T-cell transfer model of colitis (27). Treatment with anti-IL-6 receptor (anti-IL-6R) antibodies reduced colitis in this model and also resulted in reduced expression of IFN-γ, TNF-α, and IL-1β (62), indicating that IL-6 is a key cytokine involved in controlling intestinal inflammation. However, IL-6 expression decreased apoptosis of leukocytes (36), increasing the length of time that proinflammatory cells remained at the infection site. Thus, while IL-6 can exhibit anti-inflammatory effects, elevated levels of IL-6 are likely to be associated with increased inflammation and may contribute to the increased pathology observed in mice fed the doubly selenium- and vitamin E-deficient diet.
Interleukin-6 can also influence expression of chemokines, including CCL2 (36), which was more highly upregulated in C. rodentium-infected mice fed the doubly deficient diet (Table 1). Higher expression of CCL2 and CXCL10 in infected mice fed the doubly deficient diet may be related causally to the increased cellular infiltrate observed in colonic tissue on days 12 and 17 compared to that in infected mice fed the control adequate diet and may be responsible for the trend toward higher proinflammatory cytokine mRNA expression. Further studies are needed to identify the specific cell types in the cellular infiltrate and the contribution to the increased hyperplasia and histopathology observed in the colons of doubly deficient mice.
B cells also are important for controlling C. rodentium infections, as uMT mice (which have normal T cells and no B cells) are highly susceptible to C. rodentium (47) and CD4−/− mice receiving immune serum Ig from convalescing mice were able to clear the infection (6), indicating an antibody-dependent protective response against C. rodentium. IgA antibody production, however, does not appear to be required for bacterial clearance (31). Diminished IgG or IgM responses cannot be responsible for the increased C. rodentium load in mice fed the doubly deficient diet, as no differences in C. rodentium-specific IgM or IgG between infected mice fed either the control adequate or the doubly deficient diet were noted.
In conclusion, these results demonstrate that a dual deficiency in selenium and vitamin E altered the host response to infection with C. rodentium and resulted in enhanced pathology. Increased bacterial burden, increased cellular infiltrate, and oxidant-induced tissue injury in response to infection appear to contribute to increased pathology. These results highlight the need to consider the role that diet and oxidative stress play in the exacerbation of food-borne infections, which are reported in increasing numbers today.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 18 January 2011.
REFERENCES
- 1.Au Yeung, K. J., et al. 2005. Impact of vitamin E or selenium deficiency on nematode-induced alterations in murine intestinal function. Exp. Parasitol. 109:201-208. [DOI] [PubMed] [Google Scholar]
- 2.Baker, S. S., and H. J. Cohen. 1983. Altered oxidative metabolism in selenium-deficient rat granulocytes. J. Immunol. 130:2856-2860. [PubMed] [Google Scholar]
- 3.Beck, M. A., et al. 1994. Vitamin E deficiency intensifies the myocardial injury of coxsackievirus B3 infection of mice. J. Nutr. 124:345-358. [DOI] [PubMed] [Google Scholar]
- 4.Beck, M. A., and C. C. Matthews. 2000. Micronutrients and host resistance to viral infection. Proc. Nutr. Soc. 59:581-585. [DOI] [PubMed] [Google Scholar]
- 5.Bermano, G., et al. 1995. Tissue-specific regulation of selenoenzyme gene expression during selenium deficiency in rats. Biochem. J. 311:425-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bry, L., and M. B. Brenner. 2004. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J. Immunol. 172:433-441. [DOI] [PubMed] [Google Scholar]
- 7.Bry, L., M. Brigl, and M. B. Brenner. 2006. CD4+-T-cell effector functions and costimulatory requirements essential for surviving mucosal infection with Citrobacter rodentium. Infect. Immun. 74:673-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burlakova, E. B., S. A. Krashakov, and N. G. Khrapova. 1998. The role of tocopherols in biomembrane lipid peroxidation. Membr. Cell Biol. 12:173-211. [PubMed] [Google Scholar]
- 9.Catignani, G. L., and J. G. Bieri. 1983. Simultaneous determination of retinol and alpha-tocopherol in serum or plasma by liquid chromatography. Clin. Chem. 29:708-712. [PubMed] [Google Scholar]
- 10.Coquette, A., B. Vray, and J. Vanderpas. 1986. Role of vitamin E in the protection of the resident macrophage membrane against oxidative damage. Arch. Int. Physiol. Biochim. 94:S29-34. [PubMed] [Google Scholar]
- 11.Dam, H. (ed.). 1962. Interaction between vitamin E and polyunsaturated fatty acids in animals. Academic Press, Orlando, FL.
- 12.Dann, S. M., et al. 2008. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J. Immunol. 180:6816-6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dawson, H., et al. 2009. Localized Th1-, Th2-, T regulatory cell-, and inflammation-associated hepatic and pulmonary immune responses in Ascaris suum-infected swine are increased by retinoic acid. Infect. Immun. 77:2576-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El-Behi, M., A. Rostami, and B. Ciric. 2010. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 5:189-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esworthy, R. S., S. W. Binder, J. H. Doroshow, and F. F. Chu. 2003. Microflora trigger colitis in mice deficient in selenium-dependent glutathione peroxidase and induce Gpx2 gene expression. Biol. Chem. 384:597-607. [DOI] [PubMed] [Google Scholar]
- 16.Esworthy, R. S., L. Yang, P. H. Frankel, and F. F. Chu. 2005. Epithelium-specific glutathione peroxidase, Gpx2, is involved in the prevention of intestinal inflammation in selenium-deficient mice. J. Nutr. 135:740-745. [DOI] [PubMed] [Google Scholar]
- 17.Fang, F. C. 2004. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2:820-832. [DOI] [PubMed] [Google Scholar]
- 18.Forstrom, J. W., J. J. Zakowski, and A. L. Tappel. 1978. Identification of the catalytic site of rat liver glutathione peroxidase as selenocysteine. Biochemistry 17:2639-2644. [DOI] [PubMed] [Google Scholar]
- 19.Gobert, A. P., K. T. Wilson, and C. Martin. 2005. Cellular responses to attaching and effacing bacteria: activation and implication of the innate immune system. Arch. Immunol. Ther. Exp. (Warsz.) 53:234-244. [PubMed] [Google Scholar]
- 20.Higgins, L. M., G. Frankel, G. Douce, G. Dougan, and T. T. MacDonald. 1999. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect. Immun. 67:3031-3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishigame, H., et al. 2009. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30:108-119. [DOI] [PubMed] [Google Scholar]
- 22.Khan, M. A., et al. 2006. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect. Immun. 74:2522-2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim, S. H., V. J. Johnson, T. Y. Shin, and R. P. Sharma. 2004. Selenium attenuates lipopolysaccharide-induced oxidative stress responses through modulation of p38 MAPK and NF-kappaB signaling pathways. Exp. Biol. Med. 229:203-213. [DOI] [PubMed] [Google Scholar]
- 24.Kipp, A., A. Banning, and R. Brigelius-Flohe. 2007. Activation of the glutathione peroxidase 2 (GPx2) promoter by beta-catenin. Biol. Chem. 388:1027-1033. [DOI] [PubMed] [Google Scholar]
- 25.Kipp, A., et al. 2009. Four selenoproteins, protein biosynthesis, and Wnt signalling are particularly sensitive to limited selenium intake in mouse colon. Mol. Nutr. Food Res. 53:1561-1572. [DOI] [PubMed] [Google Scholar]
- 26.Kiremidjian-Schumacher, L., M. Roy, H. I. Wishe, M. W. Cohen, and G. Stotzky. 1994. Supplementation with selenium and human immune cell functions. II. Effect on cytotoxic lymphocytes and natural killer cells. Biol. Trace Elem Res. 41:115-127. [DOI] [PubMed] [Google Scholar]
- 27.Kitamura, K., Y. Nakamoto, S. Kaneko, and N. Mukaida. 2004. Pivotal roles of interleukin-6 in transmural inflammation in murine T cell transfer colitis. J. Leukoc. Biol. 76:1111-1117. [DOI] [PubMed] [Google Scholar]
- 28.Lee, C. Y., and J. M. Wan. 2002. Immunoregulatory and antioxidant performance of alpha-tocopherol and selenium on human lymphocytes. Biol. Trace Elem. Res. 86:123-136. [DOI] [PubMed] [Google Scholar]
- 29.Levander, O. A. 1985. Considerations on the assessment of selenium status. Fed. Proc. 44:2579-2583. [PubMed] [Google Scholar]
- 30.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 31.Maaser, C., et al. 2004. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect. Immun. 72:3315-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacDonald, T. T., G. Frankel, G. Dougan, N. S. Goncalves, and C. Simmons. 2003. Host defences to Citrobacter rodentium. Int. J. Med. Microbiol. 293:87-93. [DOI] [PubMed] [Google Scholar]
- 33.Maehira, F., I. Miyagi, and Y. Eguchi. 2003. Selenium regulates transcription factor NF-kappaB activation during the acute phase reaction. Clin. Chim. Acta 334:163-171. [DOI] [PubMed] [Google Scholar]
- 34.Mahida, Y. R., L. Kurlac, A. Gallagher, and C. J. Hawkey. 1991. High circulating concentrations of interleukin-6 in active Crohn's disease but not ulcerative colitis. Gut 32:1531-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyer, M., R. Schreck, and P. A. Baeuerle. 1993. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 12:2005-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitsuyama, K., M. Sata, and S. Rose-John. 2006. Interleukin-6 trans-signaling in inflammatory bowel disease. Cytokine Growth Factor Rev. 17:451-461. [DOI] [PubMed] [Google Scholar]
- 37.Moriguchi, S., and M. Muraga. 2000. Vitamin E and immunity. Vitam. Horm. 59:305-336. [DOI] [PubMed] [Google Scholar]
- 38.Papp, L. V., A. Holmgren, and K. K. Khanna. 2010. Selenium and selenoproteins in health and disease. Antioxid. Redox. Signal 12:793-795. [DOI] [PubMed] [Google Scholar]
- 39.Pratico, D., R. K. Tangirala, D. J. Rader, J. Rokach, and G. A. FitzGerald. 1998. Vitamin E suppresses isoprostane generation in vivo and reduces atherosclerosis in ApoE-deficient mice. Nat. Med. 4:1189-1192. [DOI] [PubMed] [Google Scholar]
- 40.Ryter, S. W., and A. M. Choi. 2002. Heme oxygenase-1: molecular mechanisms of gene expression in oxygen-related stress. Antioxid Redox Signal. 4:625-632. [DOI] [PubMed] [Google Scholar]
- 41.Saedi, M. S., et al. 1988. Effect of selenium status on mRNA levels for glutathione peroxidase in rat liver. Biochem. Biophys. Res. Commun. 153:855-861. [DOI] [PubMed] [Google Scholar]
- 42.Sakaguchi, S., et al. 2000. Roles of selenium in endotoxin-induced lipid peroxidation in the rats liver and in nitric oxide production in J774A. 1 cells. Toxicol. Lett. 118:69-77. [DOI] [PubMed] [Google Scholar]
- 43.Schauer, D. B., and S. Falkow. 1993. The eae gene of Citrobacter freundii biotype 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect. Immun. 61:4654-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmechel, S., et al. 2008. Linking genetic susceptibility to Crohn's disease with Th17 cell function: IL-22 serum levels are increased in Crohn's disease and correlate with disease activity and IL23R genotype status. Inflamm. Bowel Dis. 14:204-212. [DOI] [PubMed] [Google Scholar]
- 45.Schreck, R., P. Rieber, and P. A. Baeuerle. 1991. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 10:2247-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shea-Donohue, T., et al. 2008. Mice deficient in the CXCR2 ligand, CXCL1 (KC/GRO-alpha), exhibit increased susceptibility to dextran sodium sulfate (DSS)-induced colitis. Innate Immun. 14:117-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simmons, C. P., et al. 2003. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect. Immun. 71:5077-5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simmons, C. P., et al. 2002. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J. Immunol. 168:1804-1812. [DOI] [PubMed] [Google Scholar]
- 49.Smith, A., et al. 2005. Deficiencies in selenium and/or vitamin E lower the resistance of mice to Heligmosomoides polygyrus infections. J. Nutr. 135:830-836. [DOI] [PubMed] [Google Scholar]
- 50.Smith, A. D., V. C. Morris, and O. A. Levander. 2001. Rapid determination of glutathione peroxidase and thioredoxin reductase activities using a 96-well microplate format: comparison to standard cuvette-based assays. Int. J. Vitam. Nutr. Res. 71:87-92. [DOI] [PubMed] [Google Scholar]
- 51.Sonnenberg, G. F., M. G. Nair, T. J. Kirn, C. Zaph, L. A. Fouser, and D. Artis. 2010. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J. Exp. Med. 207:1293-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Southey, A., et al. 1997. Pathophysiological role of nitric oxide in rat experimental colitis. Int. J. Immunopharmacol. 19:669-676. [DOI] [PubMed] [Google Scholar]
- 53.Sugimoto, K., et al. 2008. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118:534-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki, Y. J., and L. Packer. 1993. Inhibition of NF-kappa B activation by vitamin E derivatives. Biochem. Biophys. Res. Commun. 193:277-283. [DOI] [PubMed] [Google Scholar]
- 55.Tamura, T., and T. C. Stadtman. 1996. A new selenoprotein from human lung adenocarcinoma cells: purification, properties, and thioredoxin reductase activity. Proc. Natl. Acad. Sci. U. S. A. 93:1006-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Te Velde, A. A., I. Pronk, F. de Kort, and P. C. Stokkers. 2008. Glutathione peroxidase 2 and aquaporin 8 as new markers for colonic inflammation in experimental colitis and inflammatory bowel diseases: an important role for H2O2? Eur. J. Gastroenterol. Hepatol. 20:555-560. [DOI] [PubMed] [Google Scholar]
- 57.Vallance, B. A., W. Deng, K. Jacobson, and B. B. Finlay. 2003. Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infect. Immun. 71:3443-3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang, Y., G. S. Xiang, F. Kourouma, and S. Umar. 2006. Citrobacter rodentium-induced NF-kappaB activation in hyperproliferating colonic epithelia: role of p65 (Ser536) phosphorylation. Br. J. Pharmacol. 148:814-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whanger, P. D., and J. A. Butler. 1988. Effects of various dietary levels of selenium as selenite or selenomethionine on tissue selenium levels and glutathione peroxidase activity in rats. J. Nutr. 118:846-852. [DOI] [PubMed] [Google Scholar]
- 60.Wiles, S., et al. 2004. Organ specificity, colonization and clearance dynamics in vivo following oral challenges with the murine pathogen Citrobacter rodentium. Cell. Microbiol. 6:963-972. [DOI] [PubMed] [Google Scholar]
- 61.Wu, D., et al. 2000. In vitro supplementation with different tocopherol homologues can affect the function of immune cells in old mice. Free Radic. Biol. Med. 28:643-651. [DOI] [PubMed] [Google Scholar]
- 62.Yamamoto, M., K. Yoshizaki, T. Kishimoto, and H. Ito. 2000. IL-6 is required for the development of Th1 cell-mediated murine colitis. J. Immunol. 164:4878-4882. [DOI] [PubMed] [Google Scholar]
- 63.Ye, P., et al. 2001. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am. J. Respir. Cell Mol. Biol. 25:335-340. [DOI] [PubMed] [Google Scholar]
- 64.Zheng, Y., et al. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14:282-289. [DOI] [PubMed] [Google Scholar]