

Abstract
Dimethylsulfide (DMS) is a volatile organosulfur compound which has been implicated in the biogeochemical cycling of sulfur and in climate control. Microbial degradation is a major sink for DMS. DMS metabolism in some bacteria involves its oxidation by a DMS monooxygenase in the first step of the degradation pathway; however, this enzyme has remained uncharacterized until now. We have purified a DMS monooxygenase from Hyphomicrobium sulfonivorans, which was previously isolated from garden soil. The enzyme is a member of the flavin-linked monooxygenases of the luciferase family and is most closely related to nitrilotriacetate monooxygenases. It consists of two subunits: DmoA, a 53-kDa FMNH2-dependent monooxygenase, and DmoB, a 19-kDa NAD(P)H-dependent flavin oxidoreductase. Enzyme kinetics were investigated with a range of substrates and inhibitors. The enzyme had a Km of 17.2 (± 0.48) μM for DMS (kcat = 5.45 s−1) and a Vmax of 1.25 (± 0.01) μmol NADH oxidized min−1 (mg protein−1). It was inhibited by umbelliferone, 8-anilinonaphthalenesulfonate, a range of metal-chelating agents, and Hg2+, Cd2+, and Pb2+ ions. The purified enzyme had no activity with the substrates of related enzymes, including alkanesulfonates, aldehydes, nitrilotriacetate, or dibenzothiophenesulfone. The gene encoding the 53-kDa enzyme subunit has been cloned and matched to the enzyme subunit by mass spectrometry. DMS monooxygenase represents a new class of FMNH2-dependent monooxygenases, based on its specificity for dimethylsulfide and the molecular phylogeny of its predicted amino acid sequence. The gene encoding the large subunit of DMS monooxygenase is colocated with genes encoding putative flavin reductases, homologues of enzymes of inorganic and organic sulfur compound metabolism, and enzymes involved in riboflavin synthesis.
Dimethylsulfide (DMS) is a volatile organosulfur compound, important in the biogeochemical cycling of sulfur and global climate regulation (4, 9). Bacterial metabolism of DMS is an important sink of the compound in nature and is thought to account for degradation of over 80% of the DMS produced in the marine environment. Although bacterial pathways of DMS degradation have been studied previously in Hyphomicrobium spp. and in Thiobacillus spp. (12, 36), they remain poorly characterized, and few enzymes of DMS metabolism have been purified (see reference 32). DMS monooxygenase was first reported from an assay of NADH-dependent oxygen uptake in the presence of DMS by cell extracts of Hyphomicrobium S (12), an activity also demonstrated in cell extracts of other Hyphomicrobium, Thiobacillus, and Arthrobacter isolates (6, 7, 34), with specific activities around 30 nmol NADH oxidized min−1 mg protein−1. The enzyme has not previously been purified or characterized.
The aims of this study were to purify and characterize the DMS monooxygenase enzyme from a member of the genus Hyphomicrobium. Since Hyphomicrobium S is no longer available, studies were undertaken using the type strain of H. sulfonivorans. The strain was originally isolated from garden soil and grows on DMS, as well as the related compounds dimethyl sulfoxide (DMSO) and dimethylsulfone (DMSO2). During growth on DMSO2, H. sulfonivorans first reduces DMSO2 to DMSO by a dimethylsulfone reductase, and subsequently a DMSO reductase converts DMSO to DMS, which is further oxidized to methanethiol and formaldehyde by a DMS monooxygenase. Oxidation of methanethiol to formaldehyde by methanethiol oxidase yields another mole of formaldehyde, which is either assimilated into biomass or oxidized to carbon dioxide to provide reducing equivalents (Fig. 1). DMS monooxygenase activity is present in the soluble protein fraction during growth on these compounds (6, 7). A 53-kDa polypeptide was previously observed in organisms grown on DMS, DMSO, and DMSO2 (6, 7), but its significance in the metabolism of these compounds was unknown.
FIG. 1.

Pathway and enzymes of dimethylsulfone degradation in Hyphomicrobium sulfonivorans S1. Reduction of dimethylsulfone [DMSO2; (CH3)2SO2] to dimethyl sulfoxide [DMSO; (CH3)2SO] and further reduction of DMSO to dimethylsulfide provides the substrate for DMS monooxygenase. Formaldehyde is either assimilated (via the serine cycle) or oxidized to CO2 providing reducing equivalents. Sulfide is oxidized to sulfate; see reference 7 for further details.
MATERIALS AND METHODS
Materials.
NADH was obtained from BDH (VWR, Lutterworth, United Kingdom) and was washed with ether before use to remove residual alcohols. Protein purification media were obtained from Pharmacia (Uppsala, Sweden), DMS was from Acros Organics (Geel, Belgium), and alkanesulfonates were purchased as their sodium salts from Aldrich (Gillingham, United Kingdom), Fluka (Buchs, Switzerland), and Sigma (St. Louis, MO) or as free acids from Lancaster Synthesis (Morecombe, United Kingdom) and were neutralized with NaOH. All other chemicals were obtained from Sigma-Aldrich (Gillingham, United Kingdom) unless otherwise stated.
Growth of Hyphomicrobium sulfonivorans.
H. sulfonivorans S1T (DSM 13863, ATCC BAA-113) was grown in DMSO2-limited chemostats ([DMSO2]0 = 40 mM; D = 0.03 h−1) in culture volumes of 4 liters using the medium previously described (6) with the addition of 0.001% (vol/vol) Antifoam 289 (Sigma) in LH Series 210 fermentors (LH Engineering LTD, Stoke Poges, United Kingdom). The cultures were inoculated from a frozen stock, incubated at 30 ± 1°C and maintained at pH 7.4 ± 0.2 by automatic titration with 2 M NaOH or 1 M H2SO4. Cultures were equilibrated by the passage of five culture volumes of medium through the growth vessel and were harvested at steady state by centrifugation at 13,000 × g. Cells were washed in 20 mM PIPES-HCl, pH 7.4, resuspended in the same buffer, snap-frozen in liquid nitrogen, and stored at −80°C until required.
Protein electrophoresis and Western blotting.
Bis-Tricine (10%) NuPAGE gels (Invitrogen, Paisley, United Kingdom) were used with the MOPS (morpholinepropanesulfonic acid) buffering system and denaturing sample buffer according to the manufacturer's instructions and were stained either with Coomassie brilliant blue R-250 or with the PlusOne silver staining kit (Pharmacia) using the mass spectrometry-compatible protocol. For some applications, 12.5% SDS-PAGE slab gels were used. Western blotting onto Hybond-P polyvinylidene difluoride (PVDF) membranes (Amersham) was performed using a Novex XCell electrophoresis cell with the appropriate electroblotting module (Invitrogen) according to the recommendations of the manufacturer. Following transfer, polypeptide bands were visualized on the membrane using a 0.1% (wt/vol) solution of Ponceau S in 1% (vol/vol) acetic acid, before a brief rinse in MilliQ water, air drying, and excision of the target band, which was submitted for N-terminal sequencing to Alta Bioscience Ltd. (University of Birmingham, United Kingdom).
Preparation of soluble protein fractions.
Pooled cell pastes were thawed rapidly at 37°C, and bovine pancreatic DNase (25 μg ml−1) and benzamidine hydrochloride (1 mM) were added. Cells were lysed by three passes through a French pressure cell at 120 MPa. Pooled crude extracts were freed of debris by centrifugation at 13,000 × g before centrifugation at 150,000 × g to remove membranes. Soluble fractions were pooled and concentrated using Amicon Ultra 3500 MWCO centrifugal filter devices (Millipore, Watford, United Kingdom).
Protein quantification.
Protein was quantified using the method of Bradford (8) with the Bio-Rad protein assay reagent (Bio-Rad, Hemel Hempstead, United Kingdom) according to the manufacturer's instructions. Calibration curves were prepared from standard solutions of bovine serum albumin.
Enzyme assays.
DMS monooxygenase activity was assessed in an assay mix comprising 850 μl assay buffer [20 mM PIPES-HCl, pH 7.4, supplemented with 1 mM NADH, 3 μM flavin mononucleotide (FMN), 5 μM dithiothreitol (DTT), and 5 μM (NH4)2Fe(SO4)2] and 50 μl protein solution (containing around 10 mg protein ml−1). The reaction was initiated by the addition of 100 μl of 10 mM DMS solution (or 10×-strength solutions of other test substrates). NADH depletion was monitored as a decrease in absorbance (λ = 340 nm), and the concentration was calculated using the Beer-Lambert law given that ɛ = 6,200 M−1 cm−1. Assays were conducted at 30°C unless otherwise stated using an Ultrospec 3100 Pro UV/Visible spectrophotometer (Amersham) equipped with an 8-cell carousel. All assays were conducted in 7-fold replicate, and the standard error of the mean is given.
Determination of dimethylsulfide and methanethiol.
DMS and methanethiol (MT) were quantified by gas chromatography using a GCD gas chromatograph (PYE Unicam LTD, Cambridge, United Kingdom) as described previously (30).
Determination of formaldehyde.
Formaldehyde was quantified in solution using 4-amino-3-hydrazino-5-mercapto-1,2,4-triazole (Purpald) as previously described (13) with minor modifications. A 0.1-ml sample of 135 mM Purpald solution in 1 M NaOH was allowed to react with 0.2 ml of the solution under test for 15 min at room temperature in a 1-ml volumetric flask before dilution to volume and measurement of absorbance (λ = 550 nm). Standard formaldehyde solutions in the 7 μM to 70 μM range were used to prepare calibration curves. Formaldehyde stock solutions were prepared by the thermal depolymerization of paraformaldehyde (11).
Enzyme purification.
DMS monooxygenase purification was achieved in sequential steps of gel filtration and hydrophobic interaction chromatography. Throughout the purification procedure, Amicon Ultra 3500 MWCO centrifugal filter devices were used to concentrate pooled active fractions from gel filtration and affinity purification steps. HiTrap desalting columns (5 ml bed volume) were used for buffer exchange and to desalt samples. Initially, soluble protein extracts (maximum, 20 ml) were applied at a flow rate of 0.5 ml min−1 to a Superdex S200 HR 26/60 column (Pharmacia) at 4°C that had been equilibrated with three bed volumes of buffer A [20 mM PIPES-HCl, pH 7.4, 150 mM NaCl, 1 mM benzamidine hydrochloride, 10 μM DMS, 50 μM DTT, 50 μM (NH4)2Fe(SO4)2] before use. Fractions were collected at 1-min intervals and assayed for protein concentration and DMS monooxygenase activity before examination of active fractions by SDS-PAGE or NuPAGE. Active fractions were pooled and subjected to a second round of gel filtration using a glass column (30 cm by 2 cm) packed with Sephadex G75 to a total bed volume of 49 ml. The column was equilibrated as described above and run at a flow rate of 0.5 ml min−1. Active fractions from the second round of gel filtration were pooled and subjected to hydrophobic interaction chromatography using phenyl-Sepharose in batch for the removal of putative methanol dehydrogenase from protein preparation after size exclusion chromatography. Buffer B (20 mM PIPES-HCl, pH 7.4, 250 mM NaCl, 1 mM benzamidine, 1.4 M glycerol) was used for binding of methanol dehydrogenase to phenyl-Sepharose, which was then removed by centrifugation at 13,000 × g. In order to separate DMS monooxygenase enzyme subunits, Mimetic Yellow 2 (ProMetic Biosciences LTD, Cambridge, United Kingdom) was used in batch using buffer B. After removal of the resin (and bound small subunit) by centrifugation at 13,000 × g, the large subunit could be recovered from the supernatant. The small subunit was eluted from the resin using buffer C (20 mM PIPES-HCl, pH 7.4, 250 mM NaCl, 1 mM benzamidine, 3 μM FMN).
Determination of molecular mass of enzyme subunits.
The separate subunits were subjected to analytical gel filtration using the Superdex S200 HR 26/60 column after calibration of the column using standards of known molecular mass: bovine hepatic catalase (232 kDa), bovine serum albumin (67 kDa), egg lysozyme (23.1 kDa), equine cardiac cytochrome c (12.4 kDa), and vitamin B12 (1.3 kDa). The void volume was determined with blue dextran (2,000 kDa).
Mass spectrometry analyses.
Bands were excised from PAGE gels using a sterile razor blade and were diced into cubes of approximately 1 mm3. Gel pieces were destained twice in 9.5 M acetonitrile (ACN) containing 50 mM ammonium bicarbonate, washed in ACN, and dried at room temperature. Gel pieces were reduced with 10 mM DTT and alkylated with 55 mM iodoacetamide before washes with ACN and 100 mM ammonium bicarbonate and a further three washes with ACN. Porcine trypsin (150 ng) was added to each sample, along with 25 μl high-performance liquid chromatography (HPLC)-grade water, before incubation at 37°C for 4.5 h. Peptides were extracted from gel pieces first in 30 μl of 380 mM ACN solution containing 265 mM formic acid, followed by a second extraction with 15 μl of 9 M ACN solution containing 123 mM formic acid. Pooled extracts were transferred to a Micromass modular CapLC (Waters, United Kingdom) and autosampler system. A 6.4-μl sample of the pooled extract was mixed with 13.6 μl 26.5 mM formic acid before application to a 0.5-cm LC Packings C18 5-μm 100-Å (inner diameter, 300 μm) precolumn cartridge. The cartridge was flushed with 26.5 mM formic acid to desalt bound peptides before application of a linear gradient of 26.5 mM formic acid in 18 M ACN at a flow rate of 200 nl min−1 to elute peptides for further resolution on a 15-cm LC Packings C18 5-μm 5-Å (inner diameter, 75 μm) PepMap analytical column, with the same gradient.
Metal depletion experiments.
The effect of metal cations on DMS monooxygenase activity in cell-free extracts (CFEs) was assessed. Sequestration of cations from CFE samples was achieved using the EDTA method of Duewel and Woodard (14). Divalent metal cations (e.g., magnesium sulfate, calcium chloride, cupric sulfate, ferrous sulfate, cobaltous chloride, mercuric chloride, manganous sulfate, cadmium chloride, or zinc chloride) were replaced by incubation, at a final concentration of 1 mM, with sequestrated CFE samples for 30 min on ice prior to assessing enzyme activity. Sodium sulfate and sodium chloride were used as controls and did not have a discernible effect on the restoration of DMS monooxygenase activity.
Substrate range.
The substrate range of purified DMS monooxygenase was assessed by assaying enzyme activity as described above using alternative substrates (1 mM final concentration in the assay), including symmetric alkyl/aryl sulfides (C1-C6 and phenyl), asymmetric methyl-alkyl/aryl sulfides (C1-C6 and phenyl), primary alkyl thiols (C1-C6), symmetric alkyl sulfones (C1-C6), symmetric alkyl sulfoxides (C1-C6), primary alkyl sulfonates (C1-C14, as sodium salts), primary alcohols (C1-C6 and phenol), alkanes (C1-C6), sodium nitrilotriacetate, dimethyl disulfide, and dibenzothiophene sulfone. In the case of gaseous compounds, saturated solutions in water were used in the enzyme assay. Thiol solutions were prepared in degassed water under argon to prevent autooxidation to disulfides.
Determination of activity with alternative cofactors.
FMN, FAD, riboflavin, lumiflavin, and lumichrome (all at 3 μM) each with NADH or NADPH (both at 1 mM) were assessed as alternative cofactors with the DmoAB complex and with the individual subunits.
Inhibition experiments.
Purified enzyme samples (20 mg ml−1) were incubated at 30°C for 15 min in the presence of 1 mM inhibitor (i.e., 50 nmol mg−1 protein) which was added from an appropriate stock solution before use in enzyme assays. Inhibitors assessed were CdSO4, PbCl2, HgCl2 and KCN, formaldehyde, methanethiol, ethyl vinyl sulfide (EVS), sodium 8-anilinonaphthalenesulfonate (ANS), coumarin, umbelliferone, methyl-tert-butyl ether (MTBE), chloroform, 3-amino-1,2,4-triazole (amitrole), sodium bathocuproinedisulfonate, EDTA, and EGTA.
DNA extraction.
Genomic DNA of H. sulfonivorans was extracted from the biomass of a 50-ml culture grown on 25 mM methanol, harvested in late exponential phase by centrifugation at 11,000 × g, and purified by ethidium bromide/CsCl gradient centrifugation as described previously (27).
Cloning of the genes encoding DMS monooxygenase.
N-terminal sequence data and peptide sequences obtained by interpretation of MS/MS spectra were used to design PCR primers taking into account codon usage of H. sulfonivorans, estimated from partial sequencing of the gene encoding the large subunit of methanol dehydrogenase (mxaF). Primer dmoA6F (5′-GTYCTNAACGCNTTCGACATG-3′) was based on the underlined part of the N-terminal sequence (MKKRIVLNAFDMTXV). Primers dmoARI (5′-GCCTGRAAGATNACNGGNGT-3′) and dmoARL (5′-GCCTGRAAVAGNACNGGNGT-3′) were based on the underlined part of an internal peptide sequence (TPV[I/L]FQAGASGR, where the fourth position could be a leucine or isoleucine residue). The primer combinations dmoA6F/dmoARI and dmoA6F/dmoARL were used to amplify gene fragments of the DMS monooxygenase subunit gene from 50 ng H. sulfonivorans DNA in a PCR consisting of an initial denaturation for 5 min at 95°C, followed by 35 cycles of 95°C for 1 min, 50°C for 1 min, and 72°C for 1 min, followed by a final extension step at 72°C for 10 min using a PCR setup which was otherwise as described previously (31). Genomic DNA was digested with restriction endonucleases suitable for subsequent cloning of restriction fragments into pUC18. Digested DNA was electrophoresed and transferred onto HybondN+ membrane (Amersham, Little Chalfont, United Kingdom) for Southern analysis essentially as described previously (31). A 25-ng sample of the PCR product obtained with primer combination dmoA6F/dmoARI was purified, and the probe was labeled using [α-32P]dCTP. Probe labeling and Southern hybridization were carried out as described previously (31). An EcoRI fragment of approximately 6.5 kb and a KpnI fragment of approximately 10 kb were identified as targets for cloning into pUC18 as described previously (31). Transformants containing inserts from size-fractionated EcoRI and KpnI digests were picked into 96-well plates containing 150 μl LB broth (100 μg ampicillin/ml) per well and incubated overnight at 37°C. Clones with dmo-containing inserts were identified by PCR as described above except that the reaction volume was 20 μl and transformants were directly used as templates. PCR-positive transformants for both EcoRI and KpnI were identified (plasmid pE2 and pK5, respectively) and used for plasmid preparation using the Qiagen maxiprep kit according to the manufacturer's recommendations. Plasmids pE2 and pK5 (10-μg portions) were digested with EcoRI and KpnI, respectively, to release the insert, which was gel purified (Qiagen gel extraction kit) and digested with Bsp143I for sequencing of random insert fragments. Bsp143I restriction fragments of 1 to 1.5 kb were gel purified, ligated into pUC18 digested with BamHI, and electroporated into TOP10 cells (Invitrogen, Paisley, United Kingdom). Transformants were picked into multiwell plates as described above, and inserts were amplified using M13F and M13R primers, purified using a Qiagen PCR purification kit, and sequenced using standard M13 primers using the ABI BigDye dye terminator ready reaction kit. Further sequencing was carried out using custom-designed primers. Open reading frames (ORFs) were identified using the ORF-Finder tool on the NCBI website (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) and the Artemis sequence annotation program (29). Translated ORF sequences were analyzed using BLASTP (17) to assign putative function.
Nucleotide accession number.
The nucleic acid sequence data reported in this study have been deposited in the GenBank database under accession number GQ980036.
RESULTS
Growth of H. sulfonivorans on DMSO2 and SDS-PAGE analysis of total cell extracts.
Similarly to the previous work by Borodina et al. (7), SDS-PAGE analysis of DMSO2-grown Hyphomicrobium sulfonivorans showed a 53-kDa peptide which was not detected after growth on methanol. This band was transferred to a PVDF membrane by electroblotting, and the N-terminal sequence was determined to be MKKRIVLNAFDMTXV, where the position indicated by X was not identified by HPLC analysis. A search of the GenBank database using the BLASTP algorithm (2) indicated that the peptide was related to flavin-dependent monooxygenases of the NtaA/SnaA/DszA family, which includes nitrilotriacetate monooxygenase, pristinamycin IIA synthetase, dibenzothiophene sulfone monooxygenase, and other two-component monooxygenases from the bacterial luciferase family. FMNH2-dependent monooxygenases are typically coupled to NADH-dependent FMN oxidoreductases. It was therefore assumed that a second subunit (“DmoB”) would be required for optimal DMS monooxygenase activity. Previous work (7) showed the presence of a 19-kDa polypeptide which was found to be coexpressed with DmoA during growth on DMS, DMSO, or DMSO2. This suggested that DMS monooxygenase might also be a two-component enzyme.
Assay of DMS monooxygenase with addition of FMN to the assay.
DMS monooxygenase activity in cell extracts was assayed in the presence or absence of 3 μM FMN. The specific activity of DMS monooxygenase in the absence of FMN was 6.8 ± 0.4 nmol NADH oxidized min−1 mg protein−1 (n = 9). When FMN was added to the assay mix, the specific activity increased to 83.4 ± 1.9 nmol NADH oxidized min−1 mg protein−1 (n = 9). This approximately 12-fold increase in specific activity supports the hypothesis that DMS monooxygenase is FMNH2 dependent.
Purification of DMS monooxygenase.
Fractions from gel filtration experiments using Superdex S200 that contained polypeptides of approximately 53 kDa and 19 kDa (both the presumed subunits of DMS monooxygenase) contained maximal DMS monooxygenase activity. Following a second round of gel filtration using Sephadex G75, the active fractions were found to contain predominantly the 53-kDa and 19-kDa peptides, in addition to polypeptides of approximately 66 kDa and 8 kDa, which were identified by tandem mass spectrometry as the methanol dehydrogenase subunits MxaF and MxaI, respectively (result not shown). An additional purification step using hydrophobic interaction chromatography with phenyl-Sepharose led to isolation of the pure enzyme (Fig. 2 and Table 1).
FIG. 2.

Silver-stained NuPAGE gel showing the purification of dimethylsulfide monooxygenase from H. sulfonivorans grown under DMSO2 limitation. Lanes: MW, molecular size markers; 1, soluble protein fraction; 2, concentrated pooled active fractions from Superdex 200 PG column; 3, pooled active fractions from Sephadex G75 column; 4, concentrated purified protein following hydrophobic interaction chromatography with phenyl-Sepharose.
TABLE 1.
Purification of DMS monooxygenase from Hyphomicrobium sulfonivorans cells obtained from a DMSO2-limited chemostat
| Purification step | Protein (mg) | Activity (nmol NADH oxidized/min) | Sp act (nmol NADH oxidized/min/mg protein) | Purification (fold) | % Recovery |
|---|---|---|---|---|---|
| Crude CFE | 24,000 | 2,688,000 | 112 | 0 | 100.0 |
| 150,000 × g | 19,040 | 2,341,920 | 123 | 1.1 | 87.1 |
| Superdex 200 PG | 9,870 | 2,645,160 | 268 | 2.4 | 98.4 |
| Amicon Ultra-15 | 9,705 | 2,620,350 | 270 | 2.4 | 97.5 |
| Sephadex G75 | 3,420 | 2,855,700 | 835 | 7.5 | 106.2 |
| Amicon Ultra-15 | 3,340 | 2,772,200 | 830 | 7.4 | 103.0 |
| Phenyl-Sepharose | 560 | 644,000 | 1,150 | 10.3 | 24.0 |
| Amicon Ultra-15 | 495 | 638,550 | 1,290 | 11.5 | 23.8 |
| HiTrap desalting | 450 | 565,200 | 1,256 | 11.2 | 20.0 |
Analytical gel filtration.
Separate analyses of DmoA and DmoB revealed the masses of the subunits to be 53 kDa and 19 kDa, respectively. The holoenzyme was found to be approximately 73 kDa, indicating an αβ structure.
Enzyme kinetics.
Kinetic parameters were determined using the Lineweaver-Burk, Eadie-Hofstee, and Hanes-Woolf plots. The resulting values for Km were 16.5, 16.5, and 18.2 μM, respectively, giving an average of 17.2 (± 0.48). The Vmax values determined by the same plots were all 1.25 μmol NADH min−1 mg protein−1, corresponding to a turnover number (kcat) of 5.45 s−1.
Metal dependence.
CFE preparations previously depleted of cations (14) had virtually no residual monooxygenase activity (1% of the activity of an untreated control). Restoration of activity was tested after incubation of depleted CFE on ice for 30 min with each of the following (at 1 mM): Ca2+, Co2+, Hg2+, Cd2+, and Zn2+ (as their chlorides) and Mg2+, Cu2+, Fe2+, and Mn2+ (as their sulfates). No significant restoration of activity was achieved by incubation with Ca2+ and Co2+ (both 0%), Cu2+ (1% ± 0.0%), Hg2+ (2% ± 0.8%), Mn2+ (2% ± 0.4%), Cd2+ (2% ± 0.2%), and Zn2+ (3% ± 0.0%). None of the divalent cations, added alone or in combination, fully restored activity, possibly due to inefficient reassociation of the enzyme with the correct cations; however, significant restoration of activity was seen with the addition of Mg2+ (10% ± 1.2%), Fe2+ (18% ± 0.1%), or Mg2+ and Fe2+ together (63% ± 3%).
Substrate range.
No activity was recorded with alkanes, alcohols, alkyl thiols, alkyl sulfoxides, alkyl sulfones, sodium alkanesulfonates, sodium nitrilotriacetate, dimethyl disulfide, or dibenzothiophene sulfone. Activities with symmetric alkyl sulfides and asymmetric methyl alkyl sulfides as percentages of the Vmax for DMS are given in Fig. 3. The enzyme had some activity with symmetric alkyl sulfides (R-S-R) up to dipentylsulfide (2% activity), but increasing the alkyl residue to C5 reduced the activity compared to that with DMS. With asymmetric methyl-alkyl sulfides (Me-S-R), activity was higher than with the equivalent symmetric compounds, and some activity was still discernible with methylhexylsulfide. No activity was recorded with diphenylsulfide or methylphenylsulfide. These data indicate that DmoAB is a DMS monooxygenase with some limited activity with the higher dialkylsulfides. No activity was observed with the substrates of enzymes in the same class (i.e., aldehydes, alkanesulfonates, dibenzothiophene sulfone etc.), indicating that DMS monooxygenase is a distinct enzyme in terms of its catalytic specificity.
FIG. 3.

Relative specific activities of DMS monooxygenase with symmetric alkyl sulfides and asymmetric methyl alkyl sulfides as percentages of Vmax obtained with DMS as a substrate. Error bars indicate standard errors of the means (n = 7). Abbreviations: DMS, dimethylsulfide; DES, diethylsulfide; DPrS, dipropylsulfide; DBS, dibutylsulfide; DPeS, dipentylsulfide; DHS, dihexylsulfide; EMS, ethylmethylsulfide; PrMS, propylmethylsulfide; BMS, butylmethylsulfide; PeMS, pentylmethylsulfide; HMS, hexylmethylsulfide.
Enzyme cofactors.
Activities with alternative cofactor combinations were determined in the presence of nicotinamides (1 mM) and flavins (3 μM). DMS monooxygenase activity was highest with the NADH-FMN combination. Only 4% of that activity (percentage of Vmax for DMS with the NADH-FMN combination) was achieved with the NADPH-FMN combination. Addition of NADH and FAD to purified DMS monooxygenase resulted in 64% and NADH-lumiflavin addition in 17% activity. Little or no activity (5% or less) was observed with FAD, lumiflavin, riboflavin, or lumichrome in the presence of NADPH. In isolation, DmoB oxidized NADH in the presence of FMN but not with the other flavins. DmoB could also reduce FMN in the presence of NADPH. These data indicate that DmoB is an FMN-specific oxidoreductase with low specificity for its reductant, accepting reducing equivalents from both NADH and NADPH.
Inhibitors.
DMS monooxygenase specific activities in the presence of various potential inhibitory compounds are given as percentages of control activity (Fig. 4). No inhibition was observed with MTBE, chloroform, or amitrole (previously shown to inhibit DMS oxidation in bacteria [36]). EVS, which is known to inhibit the oxidation of organic sulfides in various systems, being a suicide inhibitor of methyltransferases (5, 37), had no effect on DMS oxidation by DmoAB. Coumarin did not inhibit the enzyme, though umbelliferone (7-hydroxycoumarin) was the strongest inhibitor tested in this study, potentially due to competitive binding to the NADH-binding sites of the enzyme (BRENDA database [33]). ANS gave the second-highest level of inhibition of all of the inhibitors screened, presumably due to binding to hydrophobic regions of the active site. Metal-chelating agents (bathocuproinedisulfonate, EDTA, EGTA) all showed significant inhibition of the enzyme. Owing to the stimulation of enzyme activity observed in the presence of Fe2+ and Mg2+ in combination, we conclude that such compounds inhibit the enzyme by removing the Fe2+ and Mg2+ necessary for activity. Cd2+, Hg2+, and Pb2+ (1 mM) inhibited activity by 85%, 94%, and 98%, respectively, and KCN (1 mM) by about 50%. The products of the oxidation of DMS, formaldehyde and methanethiol, only slightly inhibited the monooxygenase (22% and 21% inhibition, respectively). Formaldehyde is known to form methylene cross-links within protein molecules (19), and methanethiol is known to denature proteins and to chelate metals (16), so both compounds would be expected to inhibit most enzymes; however, the stability of DmoAB in the presence of formaldehyde and methanethiol would be beneficial to H. sulfonivorans.
FIG. 4.

Relative specific activities (1 mM DMS) of the purified DMS monooxygenase exposed to various inhibitors for 30 min on ice prior to the assay. Error bars give standard errors of the means (n = 7).
Gene cloning.
PCR amplification using primers designed on N-terminal and internal peptide sequences of the 53-kDa polypeptide resulted in single PCR products; sequencing confirmed that the amplified partial gene encoded a large subunit of an enzyme of the NtaA family. Southern hybridization of genomic DNA of H. sulfonivorans resulted in single hybridization signals, indicating that the gene was present as a single copy (result not shown). EcoRI and KpnI fragments of approximately 6.5 and 10 kb were cloned into pUC18, sequenced, and assembled into a 14.1-kb contig (Fig. 5). The genes identified and the functions of the predicted protein sequences are summarized in Table 2. The gene dmoA encoding the enzyme subunit DmoA was identified by mass spectrometry analysis of excised purified protein. Several peptides of tryptic digests matched the predicted amino acid sequences (see Fig. S1 in the supplemental material). Furthermore, the N-terminal sequence derived from the dmoA gene matched the experimentally determined N terminus, whereas the X position that did not contain data in the N-terminal sequence was predicted as a cysteine residue. In addition, the contig contained a gene (orf176) predicted to encode a flavin reductase of 19 kDa which was located two genes upstream of dmoA. A single-peptide match was obtained by mass spectrometry analysis of DmoB against orf176, suggesting it may encode the small subunit DmoB. The contig also contained genes that may be associated with the metabolism of organic and inorganic sulfur compounds, namely, an alkanesulfonate monooxygenase family protein (orf380), and genes encoding a sulfite oxidase (SoxC) family protein (orf454) and a cytochrome c superfamily protein (orf182). There were several genes potentially involved in the synthesis of flavin cofactors (compare Table 2) and a tetR-like transcriptional regulator and genes encoding a monoamine oxidase domain protein (orf149) and a major facilitator protein (orf409).
FIG. 5.

Region of the genome of Hyphomicrobium sulfonivorans containing dmoA encoding the large subunit of DMS monooxygenase. The regions cloned as inserts of plasmids pE2 and pK5 are indicated by lines.
TABLE 2.
Predicted genes colocated in the dmo gene cluster and inferred properties of proteins encoded
| Gene | Positiona | Length of gene (bp) | Length of polypeptide/molecular mass (kDa)b | Inferred function | Blastp e-value against conserved domain |
|---|---|---|---|---|---|
| orf136 | 1-408 (partial) | NAc | NA | Putative flavin reductase | 8.42e-39 |
| orf380 | 472-1614 | 1,143 | 380/43.1 | Alkanesulfonate monooxygenase family protein | 5.18e-66d |
| orf376 | 1730-2860 | 1,131 | 376/41.3 | Putative bifunctional 3,4-dihydroxy-2-butanone-4-phosphate synthase/GTP cyclohydrolase II-like protein | 9.71e-116 |
| orf176 | 2887-3417 | 531 | 176/19.1 | Putative flavin reductase | 1.07e-38 |
| orf313 | 3473-4414 | 942 | 313/34.0 | Putative riboflavin biosynthesis protein RibF (bifunctional riboflavin kinase, FMN adenylyltransferase) | 2.83e-65 |
| dmoA | 4824-6266 | 1443 | 480/53.1 | Dimethylsulfide monooxygenase large subunit DmoA | NA |
| orf100 | 6509-6811 | 303 | 100/10.7 | Conserved hypothetical protein | NA |
| orf454 | 6884-8248 | 1,365 | 454/50.5 | Sulfite oxidase (SoxC) family protein | 1.72e-135 |
| orf182 | 8271-8819 | 549 | 182/20.5 | Cytochrome c superfamily protein | 7.30e-08 |
| ribA | 8837-9457 | 621 | 206/22.4 | GTP cyclohydrolase II | 3.53e-58d |
| tetR/acrR | 9693-10556c | 864 | 287/10.6 | Putative TetR/AcrR family transcriptional regulator | 2.77e-11 |
| orf149 | 10780-11229 | 450 | 149/15.9 | (R)-specific enoyl-coenzyme A hydratase | 5.77e-30d |
| orf409 | 11279-12508 | 1,230 | 409/43.3 | Putative major facilitator superfamily protein | 1.31e-03 |
| orf202 | 12514-13122 | 609 | 202/21.3 | Putative riboflavin synthase alpha subunit | 7.50e-68 |
| orf170 | 13179-13691 | 513 | 170/18.3 | Putative 6,7-dimethyl-8-ribityllumazine synthase | 6.55e-32 |
Position in gene cluster. c, gene is located on complementary strand.
In the absence of biochemical evidence, molecular mass is based on translation of predicted gene sequences.
NA, not applicable.
Specific hit against CD domain entry.
Comparative sequence analysis of DmoA.
Derived amino acid sequences of the DmoA subunit were aligned with other FMNH2-dependent monooxygenases, namely, pristinamycin IIA synthase (SnaA) from Streptomyces pristinaespiralis, EDTA monooxygenase (EmoA) from Mesorhizobium sp. BNC1 (previously bacterium BNC1), nitrilotriacetate monooxygenase (NtaA) from Aminobacter aminovorans (previously Chelatobacter heintzii), dibenzothiophene sulfone monooxygenase (DszA) from Rhodococcus sp. IGTS8, and alkanesulfonate monooxygenase from Pseudomonas putida (SsuD). A maximum likelihood tree based on the aligned sequences (Fig. 6) showed DmoA to branch separately from the other monooxygenases.
FIG. 6.

Phylogenetic analysis of FMNH2-dependent monooxygenase subunits with similarity to DmoA. Sequences were aligned with Clustal W (23), and the phylogeny was derived using the maximum likelihood algorithm PROTML using ARB (25). The alignment on which the phylogenetic analysis was based is provided as Fig. S2 in the supplemental material.
DISCUSSION
An enzyme capable of oxidizing DMS to methanethiol and formaldehyde at the expense of NADH was purified from H. sulfonivorans. The enzyme has been reported as being unstable (H. Op den Camp, personal communication), which may previously have hindered its purification. The enzyme acts in a manner similar to that of the monooxygenases which oxidize dibenzothiophene sulfone to 2-(2-hydroxyphenyl) benzenesulfinate and alkanesulfonates to aldehydes and sulfite. The enzyme is an FMNH2-dependent DMS monooxygenase, which couples with an NADH-dependent FMN oxidoreductase (see Fig. S3 in the supplemental material) with the overall reaction:
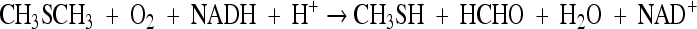 |
Despite the relatively low turnover number of the enzyme, calculations using the specific activity measured in cell extracts indicated that the enzyme should account for at least 70% of substrate turnover. Assuming a steady-state population of 280 mg (dry weight) bacteria per liter (from Y at D = 0.03 h−1 of 7 g dry weight/mol [6]) containing approximately 182 mg cell protein which consumes 1.74 mmol DMSO2 per hour (40 mM DMSO2 in inflowing medium at a dilution rate of 0.03), the potential specific activity of the DMS monooxygenase is 112 nmol min−1 mg−1 × 60 min × 182 mg = 1.22 mmol DMSO2 consumed h−1 liter−1 of culture, or 70% of the observed consumption rate. The highest specific activity (Table 1) was 1,290 nmol min−1 mg−1 enzyme protein, at a purification of 11.5-fold. This indicated that the enzyme comprised 100/11.5 = 8.7% of the cell protein, hence compensating for the low turnover number with a high abundance of the enzyme. Therefore, the steady-state 182-mg cell protein contained 15.8 mg enzyme protein, allowing 15.8 mg × 60 min × 1,290 nmol min−1 mg−1 enzyme protein = 1.22 mmol DMSO2 consumed h−1 liter−1 of culture, as calculated for the CFE.
The FMNH2-dependent monooxygenases are poorly understood in comparison to the FMN oxidoreductases with which they are associated (10, 15, 20, 22, 28, 35). Initial N-terminal sequencing data obtained from the 53-kDa peptide induced during growth on DMS in H. sulfonivorans suggested that the peptide was from a two-component FMNH2-dependent monooxygenase. We avoided anion-exchange chromatography during the purification, as this might lead to loss of enzyme activity. Purification showed that the enzyme comprised two subunits. Highest enzyme activities were recorded with FMN as added cofactor, indicating it to be an FMNH2-dependent monooxygenase. Specific activities of DMS monooxygenase reported for a range of bacteria growing on DMS, DMSO, and DMSO2 are comparatively low (7), possibly due to the assay conditions used which included NADH as a reductant. We showed that with FMN in the assay, higher activities of the enzyme can be measured. The findings reported here will facilitate analysis of the mechanisms of DMS degradation in other bacteria, by providing improved enzyme assay conditions and providing molecular masses of potentially diagnostic enzyme subunits that can be visualized by SDS-PAGE. Indeed, similar 53-kDa polypeptides have been detected during growth on DMS in Arthrobacter methylotrophus and A. sulfonivorans (7), and 53-kDa and 19-kDa polypeptides were seen in Methylobacterium podarium strain FM1 (3) grown on dimethylsulfone, the metabolism of which involves DMS as an intermediate; notably, these polypeptides were absent from M. podarium grown on methylamine (J. Vohra and A. P. Wood, unpublished data). These observations suggest that similar DMS monooxygenases may be present in a wider range of DMS-degrading bacteria.
DMS monooxygenase had a narrow substrate range. It was specific for alkyl sulfides, having highest activities with DMS but no activity with thiols, suggesting that DMS is the true substrate of the enzyme. The lack of activity with substrates of related enzymes such as nitrilotriacetate, dibenzothiophene sulfone, and alkanesulfonate monooxygenases showed that DMS monooxygenase is a catalytically distinct FMNH2-dependent monooxygenase.
No inhibition was observed with MTBE, chloroform, or amitrole (previously shown to inhibit DMS oxidation in bacteria [36]). Chloroform has previously been suggested as being an inhibitor of DMS metabolism by methyltranferases (21), but this remains untested with a purified DMS methyltransferase and has, to our knowledge, been based only on the observation that DMS consumption rates of environmental samples and of an anaerobic DMS-degrading Thiobacillus isolate (strain ASN-1 [36]) were significantly reduced by the addition of chloroform. Amitrole was reported to inhibit the growth on DMS by Thiobacillus thioparus T5 (36) and Rhodovulum sulfidophilum SH1 (26). In T. thioparus the inhibitory effect of amitrole was ascribed to inhibition of catalase, which would be required for degrading hydrogen peroxide produced by the oxidation of the intermediate methanethiol. The failure of chloroform and amitrole to inhibit DMS monooxygenase is therefore conceivable. MTBE was suggested to inhibit DMS monooxygenase based on inhibition of oxygen-dependent DMS oxidation in whole-cell suspensions of T. thioparus T5 (36). The observation that MTBE did not inhibit the purified DMS monooxygenase from H. sulfonivorans might therefore suggest that there may be more than one DMS monooxygenase family, one of which could be sensitive to MTBE and at least one other, represented by the enzyme here, that is insensitive to MTBE. Alternatively, it is possible that inhibition of DMS oxidation by MTBE in whole-cell suspensions of T. thioparus was due to a different physiological effect of MTBE. Inhibition of DMS monooxygenase by metal-chelating agents and in metal depletion experiments suggested that DMS monooxygenase requires Mg2+ and Fe2+ for activity. Work by Adoki (1) on the effect on DMS oxidation of addition of divalent cations to cell suspensions of Thiobacillus thioparus Tk-m suggested that Ca2+, Mg2+, and Mn2+ stimulated DMS oxidation in that strain, in contrast to the Fe2+ and Mg2+ dependence of the DMS monooxygenase of H. sulfonivorans. While Fe2+ addition was not tested by Adoki and data from cell-free extracts are not directly comparable to those obtained with cell suspensions, these observations might point to differences in the properties of DMS monooxygenase of the two strains.
Cloning of the gene encoding the large subunit of DMS monooxygenase allowed some insight into the evolutionary relationship of this enzyme to other monooxygenases, notably other organosulfur compound-degrading enzymes. Comparative analysis of the predicted amino acid sequences of DmoA clearly separates DMS monooxygenase from other related enzymes. The colocation of genes encoding putative flavin reductases and other enzymes potentially involved in inorganic and organic sulfur metabolism, including homologues of the alkanesulfonate monooxygenase, suggests that these might be coordinately expressed from a common promoter. However, preliminary bioinformatic analyses using a promoter prediction tool (18) suggest the potential presence of a promoter region between the gene encoding a putative riboflavin biosynthesis protein (RibF) and the gene encoding the large subunit of the DMS monooxygenase. If that was the case, then neither of the two genes encoding flavin reductases (orf136 and orf176) that might be candidates for encoding the small subunit DmoB may be present in the same operon. Only a single match against a peptide from the protein encoded by orf176 was obtained by mass spectrometry, which is insufficient for linking this gene to DmoB. The identity of the gene encoding the NADH-dependent FMN reductase therefore cannot be established at present. Several genes encoding homologues of enzymes of the riboflavin synthesis pathway were closely associated with the gene encoding DMS monooxygenase: the cluster encoded putative homologous enzymes for all the functions required to synthesize FMN from GTP and ribulose-5-phosphate, except for the bifunctional enzyme responsible for deamination and reduction of 2,5-diamino-5-amino-6-ribitylamino-2,4(1H,3H)-pyrimidinone-5′-phosphate. Colocation of riboflavin synthesis genes with oxidoreductases containing flavin cofactors is not unusual, e.g., in the lux operons of Photobacterium phosporeum (24).
Overall, the results indicate that the DMS monooxygenase of H. sulfonivorans represents a new type of FMNH2-dependent monooxygenase, biochemically and functionally distinct from other FMNH2-dependent monooxygenases. The identification of the gene encoding the large subunit of DMS monooxygenase will be beneficial to future analysis of the distribution and diversity of DMS monooxygenase in a range of DMS-degrading isolates and in environmental samples.
Supplementary Material
Acknowledgments
This work was supported by grants from the Natural Environment Research Council (NERC) of the United Kingdom. R.B. was supported through a NERC Ph.D. studentship; H.S. was supported by a NERC Advanced Fellowship (NE/E01333/1).
N. Patel, E. Blatherwick, M. Edgeworth, and S. E. Slade from the Biological Mass Spectrometry and Proteomics Facility, Department of Biological Sciences, University of Warwick, are thanked for performing liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) analyses. G. Chapman is thanked for technical support, and N. Myronova is acknowledged for many stimulating discussions on protein purification strategies.
Footnotes
Published ahead of print on 7 January 2011.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Adoki, A. 2007. Influence of divalent metal ions on degradation of dimethylsulphide by intact cells of Thiobacillus thioparus Tk-m. Afr. J. Biotechnol. 6:1343-1347. [Google Scholar]
- 2.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 3.Anesti, V., et al. 2004. Molecular detection and isolation of facultatively methylotrophic bacteria, including Methylobacterium podarium sp. nov., from the human foot microflora. Environ. Microbiol. 6:820-830. [DOI] [PubMed] [Google Scholar]
- 4.Bentley, R., and T. G. Chasteen. 2004. Environmental VOSCs—formation and degradation of dimethyl sulfide, methanethiol and related materials. Chemosphere 55:291-317. [DOI] [PubMed] [Google Scholar]
- 5.Boden, R., D. P. Kelly, J. C. Murrell, and H. Schäfer. 2010. Oxidation of dimethylsulfide to tetrathionate by Methylophaga thiooxidans sp. nov.: a new link in the sulfur cycle. Environ. Microbiol. 12:2688-2699. [DOI] [PubMed] [Google Scholar]
- 6.Borodina, E., D. P. Kelly, F. A. Rainey, N. L. Ward-Rainey, and A. P. Wood. 2000. Dimethylsulfone as a growth substrate for novel methylotrophic species of Hyphomicrobium and Arthrobacter. Arch. Microbiol. 173:425-437. [DOI] [PubMed] [Google Scholar]
- 7.Borodina, E., et al. 2002. Enzymes of dimethylsulfone metabolism and the phylogenetic characterization of the facultative methylotrophs Arthrobacter sulfonivorans sp. nov., Arthrobacter methylotrophus sp. nov., and Hyphomicrobium sulfonivorans sp. nov. Arch. Microbiol. 177:173-183. [DOI] [PubMed] [Google Scholar]
- 8.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 9.Charlson, R. J., J. E. Lovelock, M. O. Andreae, and S. G. Warren. 1987. Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature 326:655-661. [Google Scholar]
- 10.Chiu, H.-J., E. Johnson, I. Schröder, and D. C. Rees. 2001. Crystal structures of a novel ferric reductase from the hyperthermophilic archaeon Archaeoglobus fulgidus and its complex with NADP+. Structure 9:311-319. [DOI] [PubMed] [Google Scholar]
- 11.Chongcharoen, R., T. J. Smith, K. P. Flint, and H. Dalton. 2005. Adaptation and acclimatization to formaldehyde in methylotrophs capable of high-concentration formaldehyde detoxification. Microbiology 151:2615-2622. [DOI] [PubMed] [Google Scholar]
- 12.De Bont, J. A. M., J. P. van Dijken, and W. Harder. 1981. Dimethyl sulphoxide and dimethyl sulphide as a carbon, sulphur and energy source for growth of Hyphomicrobium S. J. Gen. Microbiol. 127:315-323. [Google Scholar]
- 13.Dickinson, R. G., and N. W. Jacobson. 1970. A new sensitive and specific test for the detection of aldehydes: formation of 6-mercapto-3-substituted-s-triazole [4,3-b]-s-tetrazines. Chem. Commun. (Camb.) 24:1719-1721. [Google Scholar]
- 14.Duewel, H. S., and R. W. Woodard. 2000. A metal bridge between two enzyme families. J. Biol. Chem. 275:22824-22831. [DOI] [PubMed] [Google Scholar]
- 15.Filisetti, L., N. Fontecave, and V. Nivière. 2003. Mechanism and substrate specificity of the flavin reductase ActVB from Streptomyces coelicolor. J. Biol. Chem. 278:296-303. [DOI] [PubMed] [Google Scholar]
- 16.Finkelstein, A., and N. J. Benevenga. 1986. The effect of methanethiol and methionine toxicity on the activities of cytochrome c oxidase and enzymes involved in protection from peroxidative damage. J. Nutr. 116:204-215. [DOI] [PubMed] [Google Scholar]
- 17.Gish, W., and D. J. States. 1993. Identification of protein coding regions by database similarity search. Nat. Genet. 3:266-272. [DOI] [PubMed] [Google Scholar]
- 18.Gordon, L., A. Y. Chervonenkis, A. J. Gammerman, I. A. Shahmuradov, and V. V. Solovyev. 2003. Sequence alignment kernel for recognition of promoter regions. Bioinformatics 19:1964-1971. [DOI] [PubMed] [Google Scholar]
- 19.Hopwood, D. 1969. Fixatives and fixation: a review. Histochem. J. 1:323-360. [DOI] [PubMed] [Google Scholar]
- 20.Kendrew, S. G., S. E. Harding, D. A. Hopwood, and E. N. G. Marsh. 1995. Identification of a flavin:NADH oxidoreductase involved in the biosyntheis of actinorhodin. Purification and characterisation of the recombinant enzyme. J. Biol. Chem. 270:17339-17343. [DOI] [PubMed] [Google Scholar]
- 21.Kiene, R. P., and T. S. Bates. 1990. Biological removal of dimethyl sulfide from sea-water. Nature 345:702-705. [Google Scholar]
- 22.Kirchner, U., A. H. Westphal, R. Müller, and W. J. H. van Berkel. 2003. Phenol hydroxylase from Bacillus thermoglucosidasius A7, a two-protein component monooxygenase with a dual role for FAD. J. Biol. Chem. 278:47545-47553. [DOI] [PubMed] [Google Scholar]
- 23.Larkin, M. A., et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947-2948. [DOI] [PubMed] [Google Scholar]
- 24.Lee, C. Y., D. J. Okane, and E. A. Meighen. 1994. Riboflavin synthesis genes are linked with the lux operon of Photobacterium phosphoreum. J. Bacteriol. 176:2100-2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ludwig, W., et al. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDevitt, C. A., P. Hugenholtz, G. R. Hanson, and A. G. McEwan. 2002. Molecular analysis of dimethyl sulphide dehydrogenase from Rhodovulum sulfidophilum: its place in the dimethyl sulphoxide reductase family of microbial molybdopterin-containing enzymes. Mol. Microbiol. 44:1575-1587. [DOI] [PubMed] [Google Scholar]
- 27.Oakley, C. J., and J. C. Murrell. 1988. nifH genes in obligate methane oxidising bacteria. FEMS Microbiol. Lett. 49:53-57. [Google Scholar]
- 28.Parry, R. J., and W. Li. 1997. An NADPH:FAD oxidoreductase from the valanimycin producer, Streptomyces viridifaciens. Cloning, analysis, and overexpression. J. Biol. Chem. 272:23303-23311. [DOI] [PubMed] [Google Scholar]
- 29.Rutherford, K., et al. 2000. Artemis: sequence visualisation and annotation. Bioinformatics 16:944-945. [DOI] [PubMed] [Google Scholar]
- 30.Schäfer, H. 2007. Isolation of Methylophaga spp. from marine dimethylsulfide-degrading enrichment cultures and identification of polypeptides induced during growth on dimethylsulfide. Appl. Environ. Microbiol. 73:2580-2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schäfer, H., I. R. McDonald, P. D. Nightingale, and J. C. Murrell. 2005. Evidence for the presence of a CmuA methyltransferase pathway in novel marine methyl halide-oxidising bacteria. Environ. Microbiol. 7:839-852. [DOI] [PubMed] [Google Scholar]
- 32.Schäfer, H., N. Myronova, and R. Boden. 2010. Microbial degradation of dimethylsulphide and related C1-sulphur compounds: organisms and pathways controlling fluxes of sulphur in the biosphere. J. Exp. Bot. 61:315-334. [DOI] [PubMed] [Google Scholar]
- 33.Schomburg, I., A. Chang, and D. Schomburg. 2002. BRENDA, enzyme data and metabolic function. Nucleic Acids Res. 30:47-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith, N. A., and D. P. Kelly. 1988. Isolation and physiological characterization of autotrophic sulfur bacteria oxidizing dimethyl disulfide as sole source of energy. J. Gen. Microbiol. 134:1407-1417. [Google Scholar]
- 35.Valton, J., L. Filisetti, N. Fontecave, and V. Nivière. 2004. A two-component flavin-dependent monooxygenase involved in actinorhodin biosynthesis in Streptomyces coelicolor. J. Biol. Chem. 279:44362-44369. [DOI] [PubMed] [Google Scholar]
- 36.Visscher, P. T., and B. F. Taylor. 1993. A new mechanism for the aerobic catabolism of dimethyl sulfide. Appl. Environ. Microbiol. 59:3784-3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warner, D. R., and J. L. Hoffman. 1996. Suicide inactivation of thioether S-methyltransferase by ethyl vinyl sulfide. Biochemistry 35:4480-4484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.