

Abstract
Despite the essential function of lipopolysaccharides (LPS) in Gram-negative bacteria, it is largely unknown how the exact amount of this molecule in the outer membrane is controlled. The first committed step in LPS biosynthesis is catalyzed by the LpxC enzyme. In Escherichia coli, the cellular concentration of LpxC is adjusted by the only essential protease in this organism, the membrane-anchored metalloprotease FtsH. Turnover of E. coli LpxC requires a length- and sequence-specific C-terminal degradation signal. LpxC proteins from Salmonella, Yersinia, and Vibrio species carry similar C-terminal ends and, like the E. coli enzyme, were degraded by FtsH. Although LpxC proteins are highly conserved in Gram-negative bacteria, there are striking differences in their C termini. The Aquifex aeolicus enzyme, which is devoid of the C-terminal extension, was stable in E. coli, whereas LpxC from the alphaproteobacteria Agrobacterium tumefaciens and Rhodobacter capsulatus was degraded by the Lon protease. Proteolysis of the A. tumefaciens protein required the C-terminal end of LpxC. High stability of Pseudomonas aeruginosa LpxC in E. coli and P. aeruginosa suggested that Pseudomonas uses a proteolysis-independent strategy to control its LPS content. The differences in LpxC turnover along with previously reported differences in susceptibility against antimicrobial compounds have important implications for the potential of LpxC as a drug target.
Gram-negative cells are surrounded by an asymmetric outer membrane containing mostly lipopolysaccharides (LPS) in its outer leaflet. The LPS layer serves as a permeability barrier protecting the cell from harmful compounds, including antibiotics (46). Hence, a proper equilibrium between LPS and phospholipid molecules in the outer membrane is crucial for viability of most Gram-negative bacteria. LPS also plays an important role in symbiotic and pathogenic plant-microbe interactions as well as in mammalian infections (45, 49). The biosynthesis of LPS has gained a lot of attention (i) because lipid A, the hydrophobic anchor of LPS, is an endotoxin that causes severe sepsis upon Gram-negative infections and (ii) because LPS biosynthesis is both unique and essential for Gram-negative bacteria and therefore an attractive target for the design of novel antibiotics and vaccines (54, 55).
Both too much and too little LPS are detrimental in Escherichia coli. To avoid toxic accumulation of LPS, the membrane-bound and essential AAA protease (ATPases associated with various cellular activities) FtsH degrades two enzymes of the LPS biosynthesis pathway: LpxC and KdtA (35, 47). LpxC catalyzes the first committed step in biosynthesis of lipid A (64). Therefore, both accumulation and lack of LpxC are lethal for E. coli (15, 47, 59), making this enzyme the target of choice for drug design (8, 10, 36, 38, 48, 50). KdtA attaches the KDO sugar core moieties to lipid A, forming a minimal lipid A structure which is thought to allow growth of Gram-negative microorganisms (22).
The molecular mechanism of LpxC degradation is not yet fully understood (44). Degradation by the FtsH protease requires a length- and sequence-specific C-terminal degradation signal containing an LAXXXXXAVLA motif consisting of six nonpolar amino acids within the last 11 residues (15). Like the SsrA tag (9, 21, 28), this tail serves as a general degradation signal. Additional, not yet defined internal regions of LpxC are required to direct the enzyme exclusively to FtsH (16).
The exact composition of LPS is known to vary between different Gram-negative bacteria and can be modulated in response to changing environmental conditions, e.g., cold shock in E. coli (7, 54). Despite this variability, the first steps in lipid A biosynthesis are highly conserved and are referred to as the constitutive part of the LPS biosynthesis pathway. Interestingly, the C termini of LpxC proteins differ significantly between species (Fig. 1) although the overall sequence of LpxC enzymes is highly conserved (Table 1). Whether FtsH is essential in Gram-negative bacteria other than E. coli and its close relatives is not known. The finding that FtsH is not needed for viability in the alphaproteobacterium Caulobacter crescentus (14) suggests that the requirement of the protease in control of LPS biosynthesis is not entirely conserved.
FIG. 1.

Comparison of the C-terminal sequences of LpxC from selected Gram-negative bacteria. Sequences identical to LpxCEc are underlined, and residues conforming to the C-terminal degradation signal of LpxCEc are shown in bold. The length of the LpxC proteins is given as the number of amino acids.
TABLE 1.
Sequence identity of LpxC, FtsH, and Lon from selected Gram-negative bacteria compared to E. coli
| Organism | Sequence identity to the corresponding E. coli protein (%)a |
||
|---|---|---|---|
| LpxC | FtsH | Lon | |
| S. enterica | 98 | 97 | 99 |
| Y. pseudotuberculosis | 92 | 91 | 91 |
| V. cholerae | 73 | 77 | 82 |
| P. aeruginosa | 57 | 67 | 69 |
| A. tumefaciens | 39 | 58 | 62 |
| R. capsulatus | 39 | 50 | 58 |
| A. aeolicus | 30 | 46 | 48 |
Identities were measured using Clustal W2 (39).
Our present study was motivated by three interesting observations. (i) The degradation tag of E. coli LpxC (LpxCEc) is entirely missing in the Aquifex aeolicus protein (Fig. 1) whose three-dimensional structure has been solved (3, 19, 62). (ii) The C-terminal ends of alphaproteobacterial LpxC proteins differ substantially from the E. coli sequence (Fig. 1). (iii) Several differences in the susceptibility toward chemical LpxC inhibitors have been described between the E. coli, Pseudomonas aeruginosa, A. aeolicus, and Rhizobium leguminosarum enzymes (3, 4, 30, 32, 41, 42, 48). These findings raised the possibility that the mechanisms controlling LPS biosynthesis differ in Gram-negative bacteria. Therefore, we set out to analyze the stability of LpxC enzymes from representative Gram-negative species, including various human or plant pathogens, a photosynthetic bacterium, and A. aeolicus as the most distant relative of E. coli (Fig. 1). We provide evidence that FtsH-dependent proteolysis of LpxC is a widespread but not entirely conserved mechanism.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains used in this study are listed in Table 2. E. coli W3110, BL21(DE3), RH166, and Δlon strains and Salmonella enterica cells were cultivated in liquid LB medium or on LB agar plates at 37°C. Main cultures were grown at 30°C. Cultivation of E. coli ΔftsH and AR5088 strains was routinely performed at 30°C. P. aeruginosa cells were cultivated on Pseudomonas isolation agar plates (PIA; Difco) or in liquid LB medium at 37°C. When needed, antibiotics were used in the following concentrations: 100 μg ml−1 ampicillin (Amp), 50 μg ml−1 kanamycin (Kan), and 200 μg ml−1 chloramphenicol (Cm) for E. coli and S. enterica; 500 μg ml−1 carbenicillin (replaces Amp), 25 μg ml−1 Cm, and 200 μg ml−1 gentamicin (Gm) for P. aeruginosa.
TABLE 2.
Bacterial strains, genomic DNA, and plasmids used in this study
| Strain, genomic DNA source, or plasmid | Relevant characteristic(s)a | Reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | λ− φ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | 56 |
| S17I-λpir | recA pro thi hsdR−M+ RP4-2 Tc::Mu-Km::Tn7-λpir | 12 |
| W3110 | F− IN(rrnD-rrnE)1 | 1 |
| ΔftsH strain | W3110 zad220::Tn10 sfhC21 ΔftsH3::kan | 60 |
| RH166 | MC4100 Δara Δleu lac mutant | 6 |
| Δlon strain | RH166 Δlon::Tn10 | 5 |
| BL21(DE3) | F−ompT hsdSB(rB− mB−)gal dcm (DE3) | 57 |
| AR5088 | BL21(DE3) zad220::Tn10 sfhC21 ΔftsH3::kan | 33 |
| S. enterica serovar Typhimurium M556 | SL1344 sseD::aphT | 26 |
| P. aeruginosa PAO1 | Wild type | 13 |
| Origins of genomic DNA | ||
| E. coli K12 | Wild type | 1 |
| S. enterica serovar Typhimurium M556 | SL1344 sseD::aphT | 26 |
| P. aeruginosa PAO1 | Wild type | 13 |
| A. aolicus VF5 | Wild type | 11 |
| R. capsulatus B10S | Wild type | 37 |
| A. tumefaciens C58 | Wild type | 61 |
| V. cholerae O1 serovar El Tor strain N16961 | Wild type | 27 |
| Y. pseudotuberculosis YPIII | Inv+ (pIB1) | 18 |
| Plasmids | ||
| pBAD24 | Ampr; PBADaraC | 25 |
| pBO110 | pBAD24 derivative coding for LpxCEc | 15 |
| pBO197 | pBAD24 derivative coding for N-terminal His6 tag fusions | H. Baumann |
| pET19b | Ampr; PT7; codes for N-terminal His10 tag fusions | Novagen |
| pASK-IBA5(+) | Ampr; P/OtettetR; codes for N-terminal Strep tag fusions | IBA GmbH |
| pBO1721 | pASK-IBA5(+) derivative; the coding region for the Strep tag was replaced by a His6 sequence | S. Hagen |
| pHERD20T | Ampr; PBADaraC; E. coli-P. aeruginosa shuttle vector | 52 |
| pBO1702 | pET19b derivative coding for His10-LpxCEc | This study |
| pBO2382 | pASK-IBA5(+) derivative coding for His10-LpxCEc | This study |
| pBO2381 | pASK-IBA5(+) derivative coding for His10-LpxCAa | This study |
| pBO1718 | pASK-IBA5(+) derivative coding for His6-LpxCRc | This study |
| pBO1144 | pBAD24 derivative coding for His6-LpxCAt | This study |
| pBO1187 | pBAD24 derivative coding for His6-LpxCAtΔC13 | This study |
| pBO1177 | pBAD24 derivative coding for His6-LpxCPa | This study |
| pBO1146 | pBAD24 derivative coding for His6-LpxCVc | This study |
| pBO1173 | pET19b derivative coding for His10-LpxCYp | This study |
| pBO1142 | pBAD24 derivative coding for LpxCSe | This study |
| pBO1172 | pBAD24 derivative coding for LpxCSeΔC5 | This study |
| pBO1745 | pHERD20T derivative coding for His6-LpxCPa | This study |
Ampr, ampicillin resistance. For truncated proteins, the number of C-terminally deleted residues is indicated (ΔC).
Construction of LpxC expression plasmids.
Plasmids used in this study are listed in Table 2, and oligonucleotides and restriction sites used for construction are shown in Table S1 in the supplemental material. All recombinant DNA techniques were performed using standard protocols (56) with E. coli DH5α as a cloning host.
Heterologous expression of LpxC in E. coli.
E. coli cells transformed with the corresponding plasmids coding for full-length LpxC or LpxC variants from A. aeolicus (LpxCAa), Rhodobacter capsulatus (LpxCRc), Agrobacterium tumefaciens (LpxCAt), P. aeruginosa (LpxCPa), Vibrio cholerae (LpxCVc), Yersinia pseudotuberculosis (LpxCYp), or S. enterica (LpxCSe) were inoculated to an optical density at 580 nm (OD580) of 0.05 in liquid LB medium. Cultures were grown in a water bath shaker (180 rpm) at 30°C to an OD580 of 0.5. To gain equal and soluble amounts of LpxC, inducer concentrations and plasmids used were as follows: 0.2 to 0.25 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) with pBO1173 for the PT7 expression system, 200 ng ml−1 anhydrotetracycline (AHT) with pBO1718 and pBO2381 for the Ptet expression system, and 0.05% arabinose with pBO1142, 0.05 to 0.1% arabinose with pBO1172, 0.05 to 0.5% arabinose with pBO1177, 0.1% arabinose with pBO1146, 0.1 to 0.5% arabinose with pBO1144, and 0.3% arabinose with pBO1187 for the PBAD expression system.
LpxC activity assays.
Activity of LpxC from different Gram-negative bacteria in E. coli cells was determined by phenotypic analysis in liquid LB medium or on MacConkey agar plates and by measurement of 3-deoxy-D-manno-oct-2-ulopyranosonic acid (KDO) amounts. Growth of LpxC-expressing cells in LB medium (30°C) was monitored with three different concentrations of arabinose as inducer (0, 0.1, and 0.5%) by measuring the OD580. As a comparison, growth of cells with the corresponding vector control or an lpxCEc expression plasmid was analyzed likewise. For phenotypic analysis on solid medium, 2 μl of serial 1:10 dilutions of cultures adjusted to an OD580 of 0.5 were spotted on MacConkey agar plates with 0 and 0.1 mM IPTG or with 0 and 100 ng ml−1 AHT, respectively. Growth was monitored after incubation at 30°C for 1 to 2 days. KDO amounts in the membranes of LpxC-expressing cells were analyzed using a KDO assay as described previously (16, 34).
Heterologous in vivo degradation of LpxC in E. coli.
Stability of plasmid-encoded LpxC proteins in E. coli was measured using in vivo degradation experiments. Cells carrying the corresponding plasmids were grown at 30°C in a water bath shaker (180 rpm) in liquid LB medium until an OD580 of 0.5 was reached. Depending on the vector, expression was induced by addition of arabinose, IPTG, or AHT at the concentrations described above. After 30 min, translation was blocked by addition of Cm. Samples were taken at different time points and were frozen in liquid nitrogen.
In vivo degradation experiments in S. enterica or P. aeruginosa.
Cells of S. enterica were electroporated with pBO1142 (LpxCSe) or pBO1172 (for LpxCSe with a deletion of 5 amino acids [aa] in the C terminus [LpxCSeΔC5]). Degradation experiments were performed as described for E. coli with addition of 0.05% arabinose for 10 min to induce LpxC expression. For protein expression in P. aeruginosa, the shuttle vector pBO1745 (LpxCPa) was transferred by conjugational transfer using E. coli S17I-λpir as a donor. Growth of E. coli on PIA plates was inhibited by addition of Cm. LpxC expression was induced by addition of 1% arabinose for 30 min. Translation was blocked by addition of Gm.
Protein preparation and LpxC detection.
To gain protein extracts, cell pellets were resuspended in TE buffer (10 mM Tris-HCl, pH 8, 1 mM EDTA) according to their optical densities (100 μl for an OD580 of 1). After addition of protein sample buffer (final concentration: 2% [wt/vol] SDS, 0.1% [wt/vol] bromophenol blue, 10% glycerol, 50 mM Tris-HCl, pH 6.8), cells were heated (at 100°C for 10 min) and centrifuged (for 1 min at 16,000 × g). The protein extract was subjected to SDS gel electrophoresis and Western transfer following standard protocols (56). LpxC proteins were detected either with a polyclonal LpxC antibody (LpxCEc and LpxCSe [15]) combined with a secondary goat anti-rabbit horseradish peroxidase (HRP) conjugate (Bio-Rad) or with a His5-HRP conjugate (Qiagen) (all other LpxC proteins). Chemiluminescence signals were visualized with an ECL chemiluminescence detection system (Amersham) and a ChemiImager Ready (Alpha Innotec). Half-lives of LpxC were calculated with the AlphaEaseFC software (version 4.0.0; Alpha Innotec).
RESULTS
LpxC proteins from various Gram-negative bacteria can be expressed as active enzymes in E. coli.
As a basis for further analysis, it was important to establish whether the LpxC enzymes from the model organisms used were active enzymes in E. coli as a host. To this end, the lpxC genes from S. enterica, Y. pseudotuberculosis, V. cholerae, P. aeruginosa, A. tumefaciens, R. capsulatus, and A. aeolicus were cloned and expressed in E. coli to moderate levels using either the arabinose-inducible pBAD system, the IPTG-inducible pET system, or the AHT-inducible pASK-IBA system, depending on which system worked best (Table 2). Except for LpxC from S. enterica (LpxCSe), which can be detected by the antiserum raised against E. coli LpxC, all other LpxC variants carried an N-terminal hexa- or decahistidine tag to facilitate protein detection. All LpxC enzymes were expressed as soluble proteins (see Fig. S1 in the supplemental material).
Three different assays were used to monitor the activity and subsequent phenotypic consequences of heterologously expressed LpxC proteins in E. coli. Figure 2 illustrates representative results for three different phenotypic assays. Figures S2 to S5 in the supplemental material summarize the data for all three activity assays for all LpxC variants used in this study. Figures S2 to S5 additionally include the microscopic analysis of cells expressing the LpxC variants showing that overexpression of active LpxC leads to the formation of elongated cells (16). In the first two assays shown in Fig. 2, we took advantage of the fact that overproduction of active LpxC is toxic in E. coli due to the formation of abnormal membrane stacks in the periplasm (47, 59). The effect of LpxC from S. enterica and V. cholerae (LpxCVc) on growth in liquid LB medium is depicted in Fig. 2A. As shown previously (15), induction of plasmid-encoded E. coli LpxC (LpxCEc) expression in E. coli caused severe growth defects. Nontreated cultures or cells harboring the empty vector grew normally. The observed growth effects are not due to overexpression of plasmid-encoded proteins but are related to LpxC activity because production of an inactive LpxC variant (LpxCEc with an N-terminal deletion of 2 amino acids [LpxCEcΔN2]) does not impair growth (see Fig. S1 in the supplemental material) (15, 16). Protein expression was validated by Western transfer and immunodetection (see Fig. S1). Expression of LpxCSe and LpxCVc was toxic in E. coli, suggesting that both proteins were expressed as active enzymes. Alternatively, the toxic effect of LpxC overproduction can be monitored on MacConkey agar plates due to increased sensitivity against bile salts (15). The toxic effect of LpxC expression can also be monitored on LB agar plates, yet the growth defect is not as evident as on MacConkey plates, as shown in Fig. S2 to S5. Serial dilutions of E. coli cultures expressing LpxC from Y. pseudotuberculosis (LpxCYp), A. aeolicus (LpxCAa), or R. capsulatus (LpxCRc) were spotted on MacConkey agar plates (Fig. 2B). Cultures carrying the corresponding empty vector or a plasmid coding for LpxCEc were used as a reference. Growth of all cultures was comparable on plates without inducer. While the vector control was unaffected by addition of 1 mM IPTG or 100 ng ml−1 AHT, growth of E. coli was compromised upon expression of each of the four heterologous LpxC proteins.
FIG. 2.

Functional expression of LpxC variants from Gram-negative bacteria in E. coli. (A) Expression of LpxC from S. enterica (LpxCSe; pBO1142) and V. cholerae (LpxCVc; pBO1146) impaired growth of E. coli W3110 cells as effectively as the expression of E. coli LpxC (LpxCEc; pBO110). Growth of cells containing the vector control (pBO197) is given as a reference. Cultures carrying the corresponding plasmids were grown in LB medium at 30°C supplemented with 0 (▪), 0.1 (⧫), or 0.5% arabinose (▴) for protein induction. (B) E. coli BL21 or W3110 cells showed increased sensitivity against bile salts in MacConkey agar when LpxC from Y. pseudotuberculosis (LpxCYp; pBO1173) or A. aeolicus (LpxCAa; pBO2381) and R. capsulatus (LpxCRc; pBO1718) was expressed. Serial dilutions of cultures (OD580 of 0.5) containing the corresponding plasmids were spotted on MacConkey agar plates with 0 and 0.1 mM IPTG or 0 and 100 ng ml−1 AHT. Cells harboring the plasmids pET19b or pBO1721 were used as vector controls, and pBO1702 or pBO2382 was used as a positive control. (C) Induction of active LpxC from A. tumefaciens (LpxCAt; pBO1144) and P. aeruginosa (LpxCPa; pBO1177) with 0 (light gray), 0.1 (gray), and 0.5% arabinose (black) resulted in accumulating KDO amounts per cell compared to the vector control (pBO197), as has been shown for LpxCEc (pBO110) (16). t, time.
In the third assay, we measured the amounts of KDO since production of active LpxC is known to result in KDO accumulation (16). Expression of LpxC from A. tumefaciens (LpxCAt) and P. aeruginosa (LpxCPa) led to increased amounts of KDO in E. coli membranes (Fig. 2C). While addition of 0.1 and 0.5% of arabinose did not significantly alter KDO amounts in the presence of the empty vector, expression of LpxCAt and LpxCPa increased KDO amounts almost as efficiently as expression of LpxCEc. This was true for all other LpxC variants tested in this study (see Fig. S2 to S5 in the supplemental material). Finally, the toxic effect of LpxC was documented by the filamentous growth of E. coli strains expressing recombinant lpxC genes (see Fig. S2 to S5). Using four different approaches, all LpxC proteins used in this study were shown to be active when expressed in E. coli.
Some, but not all, LpxC proteins are subject to FtsH-mediated degradation in E. coli.
To gain insights into the susceptibility of the chosen LpxC proteins toward FtsH, their half-lives were determined in E. coli wild-type (WT) and ΔftsH strains as described in Materials and Methods. LpxC proteins from S. enterica, Y. pseudotuberculosis, and V. cholerae were degraded in an FtsH-dependent manner with half-lives comparable to the half-life of E. coli LpxC (8 to 12 min) (Fig. 3) (15). LpxCPa appeared stable within the documented time period of 60 min (Fig. 3) but turned out to be degraded slowly, with a half-life of 78 ± 9.9 min when stability was monitored for longer time periods (data not shown).
FIG. 3.

Degradation experiments of LpxC proteins from Gram-negative bacteria in E. coli. Stability of plasmid-encoded LpxC from different bacteria was measured in E. coli W3110 or BL21 (WT) and the corresponding ΔftsH strain depending on the expression system. LpxC was encoded on pBO1142 (LpxCSe), pBO1173 (LpxCYp), pBO1146 (LpxCVc), pBO1177 (LpxCPa), pBO1718 (LpxCRc), pBO2381 (LpxCAa), and pBO1144 (LpxCAt). Half-lives (T1/2) of LpxC variants were analyzed using in vivo degradation experiments and Western blot analysis. Chloramphenicol (Cm) was used to block translation. The sample taken before induction of LpxC expression is indicated by a minus sign. Standard deviations were calculated from at least three independent experiments. The asterisk indicates that LpxCPa turned out to be a poor protease substrate with a half-life of 78 ± 9.9 min when degradation experiments were performed for 120 min.
LpxCAa was stable both in the E. coli wild-type and ΔftsH strains, indicating that this protein is not degraded by FtsH or any other protease present in E. coli. LpxCRc and LpxCAt were degraded with half-lives of about 68 min and 20 min, respectively. The turnover was independent of the FtsH protease because these LpxC proteins were not stabilized in the absence of FtsH (Fig. 3). To determine which protease is responsible for proteolysis of LpxCAt and LpxCRc in E. coli, half-lives were determined in various protease-deficient strains. Degradation of LpxCAt and LpxCRc was not affected by loss of either HslUV or ClpP (data not shown). However, the absence of Lon stabilized both LpxCAt and LpxCRc, indicating that these proteins are substrates of the E. coli Lon protease (Fig. 4).
FIG. 4.

LpxC proteins from A. tumefaciens and R. capsulatus are Lon substrates in E. coli. Stabilities of LpxCAt and LpxCRc were measured in the E. coli Δlon strain and the corresponding parental strain (RH166; WT). The half-lives were analyzed using in vivo degradation experiments and Western blot analysis. Chloramphenicol (Cm) was used to block translation. The sample taken before induction of LpxC expression is indicated by a minus sign. Standard deviations were calculated from three independent experiments.
The C terminus of LpxC serves as a degradation signal for FtsH and Lon substrates.
The C-terminal residues of LpxCEc have been characterized as the pivotal signal needed for degradation in E. coli (15, 16). LpxC proteins from S. enterica, Y. pseudotuberculosis, and V. cholerae share significant sequence similarities in their C terminus and a comparable overall length with LpxCEc(Fig. 1 and Table 1). To analyze whether the C terminus of LpxC also mediates degradation in other organisms, several LpxC variants were constructed. Both solubility and activity of these proteins were verified (see Fig. S1 in the supplemental material). The stability of a C-terminally truncated LpxC from S. enterica (LpxCSeΔC5) was measured in E. coli. The C terminus was found to be essential for degradation, as LpxCSeΔC5 was completely stable (Fig. 5). LpxCAt was used as example of a Lon substrate. Although its C terminus does not resemble that of LpxCEc, a C-terminal truncation of 13 amino acids (LpxCAtΔC13), making it the same length as the E. coli enzyme (305 aa), stabilized the protein (Fig. 5).
FIG. 5.

The LpxC C terminus is conserved as a degradation signal. Truncation of 5 or 13 amino acids from the C terminus from LpxCSe (pBO1172) or LpxCAt (pBO1187), respectively, led to stabilization of these proteins in E. coli. Half-lives of LpxC variants were analyzed using in vivo degradation experiments and Western blot analysis. Chloramphenicol (Cm) was used to block translation. The sample taken before induction of LpxC expression is indicated by a minus sign. Standard deviations were calculated from at least three independent experiments.
Proteolysis of LpxCSe and LpxCPa in E. coli reflects the situation in the homologous background.
To verify that the stability of LpxC from different Gram-negative bacteria in E. coli as a heterologous host agrees with the homologous systems, degradation experiments with LpxCSe and LpxCPa were performed in S. enterica and P. aeruginosa, respectively. Activity and solubility of LpxCSe and LpxCSeΔC5 in the homologous system were determined (see Fig. S6 and S7 in the supplemental material). In full agreement with the results obtained in E. coli, LpxCSe was turned over with a half-life of ∼10 min. Likewise, the C terminus of LpxCSe was also crucial for degradation in S. enterica as LpxCSeΔC5 was stabilized (Fig. 6).
FIG. 6.

LpxCSe is a protease substrate in S. enterica, and the C terminus is crucial for degradation. LpxCSe or LpxCSeΔC5 encoded on pBO1142 or pBO1172, respectively, was expressed in S. enterica cells. Chloramphenicol (Cm) was used to block translation. Half-lives were analyzed using in vivo degradation experiments and Western blot analysis. The sample taken before induction of LpxC expression is indicated by a minus sign. Standard deviations were calculated from at least three independent experiments.
Comparable to its stability in E. coli, LpxCPa was degraded only slowly, with a half-life of 93 min in P. aeruginosa (Fig. 7 A). LpxCPa was confirmed to be an active enzyme in E. coli because overproduction was toxic for the cells and because LpxCPa overproduction led to KDO accumulation in the membranes (Fig. 2; see also Fig. S3 in the supplemental material). Overproduction of LpxCPa in the homologous background was not toxic as growth of P. aeruginosa was not impaired when increasing amounts of inducer were added (Fig. 7B and C). LpxCPa overexpression in P. aeruginosa did not elevate KDO amounts and had no effect on cell morphology (see Fig S8). This suggests a different mode of LPS biosynthesis control in this organism.
FIG. 7.
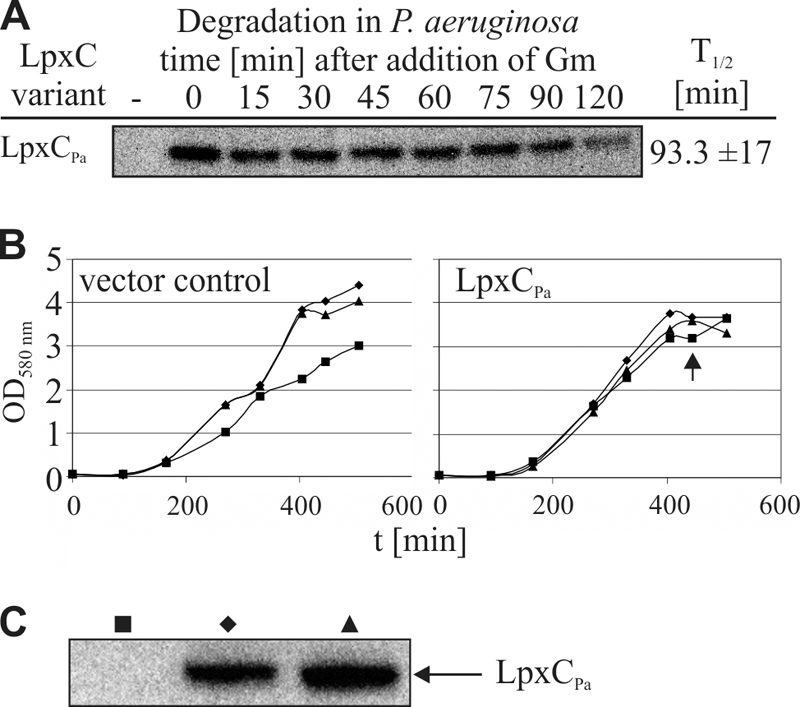
LpxCPa is a poor protease substrate in P. aeruginosa, and overexpression of this enzyme is not toxic in this organism. (A) Stability of LpxCPa was measured in P. aeruginosa using gentamicin (Gm) to block translation. The sample taken before induction of LpxC expression is indicated by a minus sign. (B) Overexpression of LpxCPa in P. aeruginosa is not toxic. Cultures carrying pBO1745 were grown in LB medium at 30°C supplemented with 0 (▪), 0.5 (⧫), or 1% arabinose (▴) for LpxCPa protein induction. Growth of cells containing the vector control (pHERD20T) is given as a reference. Samples of P. aeruginosa cells harboring pBO1745 were taken after 445 min (indicated by the arrow), and LpxCPa production was verified using Western blot analysis (C).
DISCUSSION
FtsH-dependent proteolysis of LpxC is conserved in enterobacteria.
FtsH is the only essential AAA protease in E. coli. The indispensable activity of FtsH is the turnover of LpxC, the key enzyme of lipid A biosynthesis. LpxC belongs to the constitutive enzymes of the lipid A pathway, shows no sequence similarity to other deacetylases or amidases, and is encoded by a highly conserved single-copy gene (55). To fully exploit its potential as a target for the design of novel antibiotics, detailed understanding of the regulation of this protein in Gram-negative bacteria is required.
The high conservation of LpxC proteins from organisms used in this study is reflected by the activity of these enzymes in E. coli, as shown by four different in vivo phenotypic analyses (Fig. 2; see also Fig. S2 to S5 in the supplemental material). In vivo degradation experiments with E. coli as a host provided the first evidence that turnover of LpxC by FtsH is a common mechanism in enterobacteria (Fig. 3). The E. coli ΔftsH strain is viable due to a suppressor mutation in the fabZ gene. This mutation improves the outbalanced ratio of LPS to phospholipids when LpxC degradation is abolished (43, 47). No such mutant is available in S. enterica, and specific inhibitors of FtsH in vivo are not established. Yet we confirmed that LpxCSe is degraded in S. enterica in an ATP-dependent manner as addition of arsenate stabilized the protein (see Fig. S9). The high conservation of both LpxC and FtsH (Table 1) and the comparable turnover rates of LpxCSe in Salmonella and in E. coli (Fig. 6) strongly suggest that FtsH-dependent proteolysis is a conserved strategy to ensure balanced biosynthesis of LPS in these species.
LpxC proteins from alphaproteobacteria are degraded by the Lon protease.
It has been reported previously that during evolution, degradation of certain proteins has switched to a different protease. Polypeptides trapped in stalled ribosomes are labeled with the SsrA tag and are commonly degraded by the ClpXP machinery (21, 63). The genes for clpXP in Mycoplasma were lost during genome reduction, and both the SsrA tag and the Lon protease have coevolved to allow a switch to this substrate-protease pair (23).
Here, we provide evidence that turnover of LpxC in alphaproteobacteria like A. tumefaciens and R. capsulatus is coupled to proteolysis as in enterobacteria but has switched from FtsH to the Lon protease. Given that FtsH is not responsible for degradation of LpxCAt, it might not be an essential protease in this organism, as has been reported for another alphaproteobacterium, namely, C. crescentus (14). To the best of our knowledge, construction of an ftsH knockout strain in A. tumefaciens has not yet been attempted. On the other hand, an A. tumefaciens lon mutant is available (58) although Lon is expected to be essential due to its potential role in LpxC degradation in this organism. It is important to note, however, that the mutant displayed severe defects and heterogeneity in cell morphology. This might indicate the accumulation of suppressor mutations, which ensured viability of these cells, as has been demonstrated for the E. coli ΔftsH strain (43). Interestingly, absence of Lon impairs virulence of A. tumefaciens. This effect was ascribed to the slower growth of the protease mutant (58). Another possibility raised by our study is an imbalance in LPS production due to the absence of the Lon protease. It is known that LPS is important for plant-microbe interactions (45).
The extended C terminus of LpxC is a degradation signal for various proteases.
The exceptionally short A. aeolicus LpxC protein does not carry a C-terminal extension and was resistant to proteolysis in E. coli, lending weight to the importance of the C terminus for turnover of LpxC enzymes. LpxC from A. aeolicus is the evolutionarily oldest LpxC variant analyzed in this study. The Gram-negative membranes of this hyperthermophilic organism contain variants in lipid A that might confer thermal tolerance (51, 55). Although unknown, it is likely that unbalanced amounts of lipid A are toxic in Aquifex. Novel regulatory mechanisms might be responsible for control of LPS biosynthesis in this organism. Since LpxC, FtsH, and Lon share only limited identity with the corresponding E. coli proteins (Table 1), it cannot be excluded that in Aquifex these proteins have evolved to establish proteolytic control of LPS biosynthesis by a mechanism independent of a C-terminal degradation signal.
Degradation of E. coli LpxC depends on a sequence- and length-specific, nonpolar C-terminal degradation signal (15, 16). Consistent with this, the five C-terminal amino acids of LpxCEc have recently been shown by nuclear magnetic resonance (NMR) analysis to be highly dynamic and disordered (2). Enterobacterial LpxC enzymes have C termini of similar lengths and sequences (Fig. 1). Individual point mutations in the C terminus of E. coli LpxC still permitted FtsH-dependent degradation (15, 16). In agreement with these reports are variations in the C termini of enterobacterial LpxC proteins which did not interfere with proteolysis. Compared to the E. coli LpxC sequence, LpxCSe and LpxCVc sequences carry single amino acid substitutions (T302 and M302, respectively, instead of A302). Nonpolar residues are exchanged in LpxCVc(V295 instead of L295), and LpxCYp is extended by a Y residue.
The C termini of alphaproteobacterial LpxC proteins are even longer than the enterobacterial ones and share no significant sequence homology with them. Interestingly, shortening of the C terminus of LpxCAt(LpxCAtΔC13) and LpxCSe (LpxCSeΔC5) led to drastic stabilization of these proteins (Fig. 5) without affecting the activity of the enzymes (see Fig. S2 in the supplemental material). This suggests that C-terminally elongated LpxC proteins are subject to proteolytic control independent of the executing protease. Since Lon is known to recognize aromatic residues in both native and nonnative substrates (20, 24), it is tempting to speculate that the C-terminal residues W316 and F313 in LpxCAt are crucial for Lon degradation.
Control of LPS biosynthesis in P. aeruginosa is independent of proteolysis.
LpxCPa was a poor protease substrate both in E. coli and P. aeruginosa (Fig. 3 and 5). If FtsH is not involved in LpxC control, the protease should not be essential in P. aeruginosa. In support of this assumption, a P. aeruginosa transposon insertion mutant for ftsH (strain 18778) is viable (31). Transposon mutants for all additional AAA proteases known so far (HslV, Lon, ClpP, ClpP2, and ClpP3) also have been reported (31, 40, 53), strongly suggesting that LPS biosynthesis is not controlled by proteolysis.
The presence of LPS is critically important for P. aeruginosa because LpxC activity is essential for growth (42). Notably, LpxCPa accumulation was toxic for E. coli but not for P. aeruginosa (Fig. 2 and 7). This indicates that lipid A biosynthesis by LpxC in P. aeruginosa is kept in balance by a yet unknown mechanism not available in E. coli. The E. coli deacetylase does not seem to be subject to this control mechanism since overexpression of LpxCEc in P. aeruginosa resulted in sick cells with a strong tendency to aggregate (see Fig. S8C in the supplemental material).
The divergent control strategies acting on LpxC from E. coli and P. aeruginosa suggest significant structural differences between both proteins despite a sequence identity of 57% and a similarity of 82%. In fact, several differences have already been reported. Many β-strands in LpxCEc are predicted to be longer than those in LpxCPa (2). Clearly indicative of conformational deviations are the different responses to synthetic inhibitors. Except for CHIR-090 (41), most LpxC inhibitors active against LpxCEc did not effectively inhibit LpxCPa (10, 36, 42, 48). Most likely, the structure, catalytic activity, and regulation of LpxC proteins are dictated not only by their sequence but also by intracellular molecules, such as proteins (42) or the metals Zn and Fe (17, 29). Revealing the complexity of LpxC regulation will be a challenging task of future research. Comparative analyses of LpxC proteins from P. aeruginosa and E. coli will largely depend on insights from the not yet determined three-dimensional structure of the E. coli LpxC enzyme.
Supplementary Material
Acknowledgments
This work was supported by a grant of the German Research Foundation (DFG; SFB 642, ATP- and GTP-dependent membrane processes) to F.N. and by a scholarship of the Konrad Adenauer Foundation to S.L.
We thank Frank Führer for initial help with this project, Alexandra Müller and Kai Westphal for critical reading of the manuscript, Holger Baumann and Stefanie Hagen for construction of plasmids that we used for cloning, Yvonne von der Gathen and Niels Pfennigwerth for experimental assistance, and Thomas Happe for providing the ChemiImager. We are grateful to Nicole Frankenberg-Dinkel, Bernd Masepohl, Regine Hengge, Dagmar Willkomm, Petra Dersch, and Joachim Reidl for the generous gift of strains or genomic DNA.
Footnotes
Published ahead of print on 30 December 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Bachmann, B. J. 1972. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol. Rev. 36:525-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barb, A. W., L. Jiang, C. R. Raetz, and P. Zhou. 2010. Assignment of 1H, 13C and 15N backbone resonances of Escherichia coli LpxC bound to L-161,240. Biomol. NMR Assign. 4:37-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barb, A. W., L. Jiang, C. R. Raetz, and P. Zhou. 2007. Structure of the deacetylase LpxC bound to the antibiotic CHIR-090: time-dependent inhibition and specificity in ligand binding. Proc. Natl. Acad. Sci. U. S. A. 104:18433-18438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barb, A. W., et al. 2009. Uridine-based inhibitors as new leads for antibiotics targeting Escherichia coli LpxC. Biochemistry 48:3068-3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barembruch, C., and R. Hengge. 2007. Cellular levels and activity of the flagellar sigma factor FliA of Escherichia coli are controlled by FlgM-modulated proteolysis. Mol. Microbiol. 65:76-89. [DOI] [PubMed] [Google Scholar]
- 6.Becker, G., and R. Hengge-Aronis. 2001. What makes an Escherichia coli promoter σS dependent? Role of the −13/−14 nucleotide promoter positions and region 2.5 of σS. Mol. Microbiol. 39:1153-1165. [DOI] [PubMed] [Google Scholar]
- 7.Carty, S. M., K. R. Sreekumar, and C. R. Raetz. 1999. Effect of cold shock on lipid A biosynthesis in Escherichia coli. Induction at 12 °C of an acyltransferase specific for palmitoleoyl-acyl carrier protein. J. Biol. Chem. 274:9677-9685. [DOI] [PubMed] [Google Scholar]
- 8.Chen, M. H., et al. 1999. Carbohydroxamido-oxazolidines: antibacterial agents that target lipid A biosynthesis. Bioorg. Med. Chem. Lett. 9:313-318. [DOI] [PubMed] [Google Scholar]
- 9.Choy, J. S., L. L. Aung, and A. W. Karzai. 2007. Lon protease degrades transfer-messenger RNA-tagged proteins. J. Bacteriol. 189:6564-6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clements, J. M., et al. 2002. Antibacterial activities and characterization of novel inhibitors of LpxC. Antimicrob. Agents Chemother. 46:1793-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deckert, G., et al. 1998. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature 392:353-358. [DOI] [PubMed] [Google Scholar]
- 12.de Lorenzo, V., and K. N. Timmis. 1994. Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 235:386-405. [DOI] [PubMed] [Google Scholar]
- 13.Dunn, N. W., and B. W. Holloway. 1971. Pleiotrophy of p-fluorophenylalanine-resistant and antibiotic hypersensitive mutants of Pseudomonas aeruginosa. Genet. Res. 18:185-197. [DOI] [PubMed] [Google Scholar]
- 14.Fischer, B., G. Rummel, P. Aldridge, and U. Jenal. 2002. The FtsH protease is involved in development, stress response and heat shock control in Caulobacter crescentus. Mol. Microbiol. 44:461-478. [DOI] [PubMed] [Google Scholar]
- 15.Führer, F., S. Langklotz, and F. Narberhaus. 2006. The C-terminal end of LpxC is required for degradation by the FtsH protease. Mol. Microbiol. 59:1025-1036. [DOI] [PubMed] [Google Scholar]
- 16.Führer, F., et al. 2007. Sequence and length recognition of the C-terminal turnover element of LpxC, a soluble substrate of the membrane-bound FtsH protease. J. Mol. Biol. 372:485-496. [DOI] [PubMed] [Google Scholar]
- 17.Gattis, S. G., M. Hernick, and C. A. Fierke. 2010. Active site metal ion in UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) switches between Fe(II) and Zn(II) depending on cellular conditions. J. Biol. Chem. 285:33788-33796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gemski, P., J. R. Lazere, T. Casey, and J. A. Wohlhieter. 1980. Presence of a virulence-associated plasmid in Yersinia pseudotuberculosis. Infect. Immun. 28:1044-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gennadios, H. A., D. A. Whittington, X. Li, C. A. Fierke, and D. W. Christianson. 2006. Mechanistic inferences from the binding of ligands to LpxC, a metal-dependent deacetylase. Biochemistry 45:7940-7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez, M., E. G. Frank, A. S. Levine, and R. Woodgate. 1998. Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: in vitro degradation and identification of residues required for proteolysis. Genes Dev. 12:3889-3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottesman, S., E. Roche, Y. Zhou, and R. T. Sauer. 1998. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 12:1338-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gronow, S., and H. Brade. 2001. Lipopolysaccharide biosynthesis: which steps do bacteria need to survive? J. Endotoxin Res. 7:3-23. [PubMed] [Google Scholar]
- 23.Gur, E., and R. T. Sauer. 2008. Evolution of the ssrA degradation tag in Mycoplasma: specificity switch to a different protease. Proc. Natl. Acad. Sci. U. S. A. 105:16113-16118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gur, E., and R. T. Sauer. 2008. Recognition of misfolded proteins by Lon, a AAA+ protease. Genes Dev. 22:2267-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hapfelmeier, S., et al. 2004. Role of the Salmonella pathogenicity island 1 effector proteins SipA, SopB, SopE, and SopE2 in Salmonella enterica subspecies 1 serovar Typhimurium colitis in streptomycin-pretreated mice. Infect. Immun. 72:795-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heidelberg, J. F., et al. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herman, C., D. Thévenet, P. Bouloc, G. C. Walker, and R. D'Ari. 1998. Degradation of carboxy-terminal-tagged cytoplasmic proteins by the Escherichia coli protease HflB (FtsH). Genes Dev. 12:1348-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hernick, M., S. G. Gattis, J. E. Penner-Hahn, and C. A. Fierke. 2010. Activation of Escherichia coli UDP-3-O-[(R)-3-hydroxymyristoyl]-N-acetylglucosamine deacetylase by Fe2+ yields a more efficient enzyme with altered ligand affinity. Biochemistry 49:2246-2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackman, J. E., et al. 2000. Antibacterial agents that target lipid A biosynthesis in gram-negative bacteria. Inhibition of diverse UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylases by substrate analogs containing zinc binding motifs. J. Biol. Chem. 275:11002-11009. [DOI] [PubMed] [Google Scholar]
- 31.Jacobs, M. A., et al. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 100:14339-14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadam, R. U., A. V. Shivange, and N. Roy. 2007. Escherichia coli versus Pseudomonas aeruginosa deacetylase LpxC inhibitors selectivity: surface and cavity-depth-based analysis. J. Chem. Infect. Model. 47:1215-1224. [DOI] [PubMed] [Google Scholar]
- 33.Karata, K., T. Inagawa, A. J. Wilkinson, T. Tatsuta, and T. Ogura. 1999. Dissecting the role of a conserved motif (the second region of homology) in the AAA family of ATPases. Site-directed mutagenesis of the ATP-dependent protease FtsH. J. Biol. Chem. 274:26225-26232. [DOI] [PubMed] [Google Scholar]
- 34.Karkhanis, Y. D., J. Y. Zeltner, J. J. Jackson, and D. J. Carlo. 1978. A new and improved microassay to determine 2-keto-3-deoxyoctonate in lipopolysaccharide of Gram-negative bacteria. Anal. Biochem. 85:595-601. [DOI] [PubMed] [Google Scholar]
- 35.Katz, C., and E. Z. Ron. 2008. Dual role of FtsH in regulating lipopolysaccharide biosynthesis in Escherichia coli. J. Bacteriol. 190:7117-7122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kline, T., et al. 2002. Potent, novel in vitro inhibitors of the Pseudomonas aeruginosa deacetylase LpxC. J. Med. Chem. 45:3112-3129. [DOI] [PubMed] [Google Scholar]
- 37.Klipp, W., B. Masepohl, and A. Pühler. 1988. Identification and mapping of nitrogen fixation genes of Rhodobacter capsulatus: duplication of a nifA-nifB region. J. Bacteriol. 170:693-699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langsdorf, E. F., et al. 2010. Screening for antibacterial inhibitors of the UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) using a high-throughput mass spectrometry assay. J. Biomol. Screen. 15:52-61. [DOI] [PubMed] [Google Scholar]
- 39.Larkin, M. A., et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947-2948. [DOI] [PubMed] [Google Scholar]
- 40.Lewenza, S., et al. 2005. Construction of a mini-Tn5-luxCDABE mutant library in Pseudomonas aeruginosa PAO1: a tool for identifying differentially regulated genes. Genome Res. 15:583-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McClerren, A. L., et al. 2005. A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciprofloxacin. Biochemistry 44:16574-16583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mdluli, K. E., et al. 2006. Molecular validation of LpxC as an antibacterial drug target in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 50:2178-2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohan, S., T. Kelly, S. Eveland, C. Raetz, and M. Anderson. 1994. An Escherichia coli gene (fabZ) encoding (3R)-hydroxymyristoyl acyl carrier protein dehydrase. J. Biol. Chem. 269:32896-32903. [PubMed] [Google Scholar]
- 44.Narberhaus, F., M. Obrist, F. Führer, and S. Langklotz. 2009. Degradation of cytoplasmic substrates by FtsH, a membrane-anchored protease with many talents. Res. Microbiol. 160:652-659. [DOI] [PubMed] [Google Scholar]
- 45.Newman, M. A., J. M. Dow, A. Molinaro, and M. Parrilli. 2007. Priming, induction and modulation of plant defence responses by bacterial lipopolysaccharides. J. Endotoxin Res. 13:69-84. [DOI] [PubMed] [Google Scholar]
- 46.Nikaido, H., and M. Vaara. 1985. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 49:1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ogura, T., et al. 1999. Balanced biosynthesis of major membrane components through regulated degradation of the committed enzyme of lipid A biosynthesis by the AAA protease FtsH (HflB) in Escherichia coli. Mol. Microbiol. 31:833-844. [DOI] [PubMed] [Google Scholar]
- 48.Onishi, H. R., et al. 1996. Antibacterial agents that inhibit lipid A biosynthesis. Science 274:980-982. [DOI] [PubMed] [Google Scholar]
- 49.Opal, S. M. 2007. The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int. J. Med. Microbiol. 297:365-377. [DOI] [PubMed] [Google Scholar]
- 50.Pirrung, M. C., et al. 2002. Inhibition of the antibacterial target UDP-(3-O-acyl)-N-acetylglucosamine deacetylase (LpxC): isoxazoline zinc amidase inhibitors bearing diverse metal binding groups. J. Med. Chem. 45:4359-4370. [DOI] [PubMed] [Google Scholar]
- 51.Plötz, B. M., B. Lindner, K. O. Stetter, and O. Holst. 2000. Characterization of a novel lipid A containing d-galacturonic acid that replaces phosphate residues. The structure of the lipid A of the lipopolysaccharide from the hyperthermophilic bacterium Aquifex pyrophilus. J. Biol. Chem. 275:11222-11228. [DOI] [PubMed] [Google Scholar]
- 52.Qiu, D., F. H. Damron, T. Mima, H. P. Schweizer, and H. D. Yu. 2008. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl. Environ. Microbiol. 74:7422-7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qiu, D., V. M. Eisinger, N. E. Head, G. B. Pier, and H. D. Yu. 2008. ClpXP proteases positively regulate alginate overexpression and mucoid conversion in Pseudomonas aeruginosa. Microbiology 154:2119-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raetz, C. R., C. M. Reynolds, M. S. Trent, and R. E. Bishop. 2007. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 76:295-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raetz, C. R., and C. Whitfield. 2002. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71:635-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 57.Studier, F. W., A. H. Rosenberg, J. J. Dunn, and J. W. Dubendorff. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185:60-89. [DOI] [PubMed] [Google Scholar]
- 58.Su, S., B. B. Stephens, G. Alexandre, and S. K. Farrand. 2006. Lon protease of the α-proteobacterium Agrobacterium tumefaciens is required for normal growth, cellular morphology and full virulence. Microbiology 152:1197-1207. [DOI] [PubMed] [Google Scholar]
- 59.Sullivan, N. F., and D. Donachie. 1984. Transcriptional organization within an Escherichia coli cell division gene cluster: direction of transcription of the cell separation gene envA. J. Bacteriol. 160:724-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tatsuta, T., et al. 1998. Heat shock regulation in the ftsH null mutant of Escherichia coli: dissection of stability and activity control mechanisms of σ32 in vivo. Mol. Microbiol. 30:583-593. [DOI] [PubMed] [Google Scholar]
- 61.Van Larebeke, N., et al. 1974. Large plasmid in Agrobacterium tumefaciens essential for crown gall-inducing ability. Nature 252:169-170. [DOI] [PubMed] [Google Scholar]
- 62.Whittington, D. A., K. M. Rusche, H. Shin, C. A. Fierke, and D. W. Christianson. 2003. Crystal structure of LpxC, a zinc-dependent deacetylase essential for endotoxin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 100:8146-8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wiegert, T., and W. Schumann. 2001. SsrA-mediated tagging in Bacillus subtilis. J. Bacteriol. 183:3885-3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Young, K., et al. 1995. The envA permeability/cell division gene of Escherichia coli encodes the second enzyme of lipid A biosynthesis. J. Biol. Chem. 270:30384-30391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.