

Abstract
The soluble tungsten, iron-sulfur enzyme acetylene hydratase (AH) from mesophilic Pelobacter acetylenicus is a member of the dimethyl sulfoxide (DMSO) reductase family. It stands out from its class as it catalyzes a nonredox reaction, the addition of H2O to acetylene (H—C☰C—H) to form acetaldehyde (CH3CHO). Caught in its active W(IV) state, the high-resolution three-dimensional structure of AH offers an excellent starting point to tackle its unique chemistry and to identify catalytic amino acid residues within the active site cavity: Asp13 close to W(IV) coordinated to two molybdopterin-guanosine-dinucleotide ligands, Lys48 which couples the [4Fe-4S] cluster to the W site, and Ile142 as part of a hydrophobic ring at the end of the substrate access channel designed to accommodate the substrate acetylene. A protocol was developed to express AH in Escherichia coli and to produce active-site variants which were characterized with regard to activity and occupancy of the tungsten and iron-sulfur centers. By this means, fusion of the N-terminal chaperone binding site of the E. coli nitrate reductase NarG to the AH gene improved the yield and activity of AH and its variants significantly. Results from site-directed mutagenesis of three key residues, Asp13, Lys48, and Ile142, document their important role in catalysis of this unusual tungsten enzyme.
Molybdenum and tungsten are the only transition metals of the second (Mo) and third (W) row of the periodic table of elements with known biological functions (7). In their biologically active form, both metals are bound to the cofactor molybdopterin (Moco), which is present in all molybdenum and tungsten enzymes with the exception of nitrogenase, where molybdenum is coordinated to a large iron-sulfur cluster, MoFe7S9 (9). Virtually all organisms including plants and mammals use either molybdenum or tungsten proteins in important metabolic pathways (35). Microorganisms carry a wide variety of molybdenum enzymes, such as nitrate reductase (NAR), formate dehydrogenase (FDH), dimethyl sulfoxide reductase (DMSOR), or trimethylamine N-oxide reductase (TMAOR) (7). These enzymes are involved in either oxygen atom transfer reactions or in reductive hydroxylations. By this means, the metal shuttles between the oxidation states +IV and +VI (16). Notably, the tungsten, iron-sulfur enzyme acetylene hydratase ([AH] EC 4.2.1.112), isolated from the soluble fraction of the mesophilic anaerobe Pelobacter acetylenicus, is an exception (26). It catalyzes the hydration of acetylene to acetaldehyde via an enol intermediate as an initial step for the fermentation of acetylene by P. acetylenicus, clearly a nonredox reaction (equation 1):
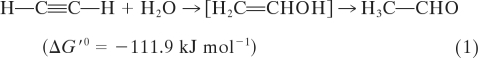 |
Except for nitrogenase, which reduces acetylene to ethylene (H2C=CH2), AH is the only enzyme known to accept acetylene as a substrate. However, acetylene is well known to act as an inhibitor for numerous metal-dependent enzymes (10). AH is a member of the DMSOR family and carries one [4Fe-4S] cluster and two molybdopterin-guanosine-dinucleotide (referred to as P- and Q-MGD) ligands that coordinate the tungsten atom (Fig. 1) (18). The enzyme is sensitive toward dioxygen, and its [4Fe-4S] cluster is converted to a truncated [3Fe-4S] cluster upon exposure to air, as shown by electron paramagnetic resonance (EPR) (18). In AH prepared under the exclusion of dioxygen, the EPR signal of the [3Fe-4S] cluster was absent, and reaction with sodium dithionite led to a rhombic EPR signal (gz of 2.048, gy of 1.939, and gx of 1.920) originating from a [4Fe-4S]+ cluster. Upon oxidation with hexacyanoferrate(III), a new signal appeared (gx of 2.007, gy of 2.019, and gz of 2.048; average g value [gav] of 2.022), which was assigned to a W(V) center (18).
FIG. 1.

Acetylene hydratase from P. acetylenicus. (Left) Overall structure, with the [4Fe-4S] cluster and W(MGD)2 buried inside the protein; the peptide backbone is shown in dark blue at the N-terminal end, and continues as light blue, cyan, green, and yellow to orange at the C-terminal end. (Right) [4Fe-4S] cluster and W(MGD)2 center. C is shown in gray, N in blue, O in red, P in orange, S in yellow, Fe in brown, and W in cyan (Protein Data Bank [PDB] code 2E7Z).
For catalytic activity, AH requires a strong reductant, such as sodium dithionite or titanium(III) citrate (18). Recently, the X-ray structure of AH in the reduced state could be solved at 1.26-Å resolution (28) which gave a first view of its active site: W(IV) is coordinated by four sulfur atoms delivered by the two dithiolene ligands (MGD), one cysteinyl sulfur (Cys141), and one oxygen ligand at a distance of 2.04 Å (Fig. 1). Mechanistically, the nature of this oxygen ligand is critical. The observed W-O distance of 2.04 Å is right between the values expected for a hydroxide ligand (1.9 to 2.1 Å) and a coordinated water (2.0 to 2.3 Å), thus not allowing an unequivocal assignment of the sixth ligand of the WS5O core. Two different reaction mechanisms have been proposed: (i) nucleophilic attack of the hydroxide group and (ii) electrophilic attack of a polarized water molecule, on the C,C triple bond of acetylene (28). As a consequence of theoretical calculations, and in agreement with the observed bond distances, the active W(IV) state should favor a water ligand and therefore an electrophilic addition mechanism (28). Acetylene can access the tungsten ion through a well-defined channel close to the N-terminal domain that harbors the [4Fe-4S] cluster. One residue, Asp13, interacts with the oxygen ligand bound to the W ion, forming a short hydrogen bond of 2.4 Å. Above the W ion and the coordinated H2O molecule, the substrate channel ends in a ring of six hydrophobic residues. These residues build a cavity with dimensions perfect for accommodating acetylene. Experiments to bind the substrate acetylene, ethylene, the inhibitor propargyl alcohol (H—C☰C—CH2OH), and dinitrogen or carbon monoxide have failed thus far to produce a complex in the crystal. However, computer docking of one acetylene molecule at this position led to a reasonable fit, positioning the two carbon atoms of the substrate exactly above the H2O molecule coordinated to tungsten (28).
To gain further information about the reaction mechanism of AH, we initiated a study by site-directed mutagenesis and exchanged several amino acids which have been suggested to be important for catalysis at the active site cavity. To achieve this goal, we had to develop a suitable procedure for the heterologous expression of AH in Escherichia coli. Notably, E. coli uses a chaperone system for the insertion of Moco into its enzymes (6). These chaperones of the TorD superfamily act in two ways. First, they bind at the N-terminal signal sequence, similar to the sequence of the TAT export system, thereby delaying the folding of the newly synthesized molybdenum enzyme until Moco has been properly inserted (25). Second, they actively facilitate the incorporation of the molybdopterin cofactor by binding to a second, yet unknown site (13). To improve the assembly of the metal sites as well as to increase the enzymatic activity of the recombinant AH, the N-terminal chaperone binding site of the nitrate reductase, NarG, from E. coli was fused to the AH gene in the expression vector.
MATERIALS AND METHODS
Bacterial strains, plasmids, and cultivation.
Pelobacter acetylenicus strain WoAcy1 (DSMZ 3246) was grown in freshwater mineral medium at 30°C, as described previously (24). E. coli JM109 (Stratagene) was used for plasmid proliferation, and E. coli Rosetta (DE3) (Novagen) was used for expression of AH; the pET24a(+) vector was from Novagen. Two-milliliter cultures of E. coli JM109 cells were grown aerobically in DYT medium (16 g/liter tryptone, 10 g/liter yeast extract, and 5 g/liter NaCl), supplemented with 50 mg/ml kanamycin sulfate. Expression of AH was carried out using 1-liter cultures of E. coli Rosetta (DE3) in anaerobic mineral medium (100 mM KPi, 10 mM NH4Cl, 2 mM MgCl2, and 0.5 g/liter protein hydrolysate), supplemented with 1 ml/liter SL10 (33), 10 mM Na2WO4, 1 mM Na2S, 1 ml/liter seven-vitamin solution (33), 15 mg/ml kanamycin sulfate, and 17 mg/ml chloramphenicol. Glycerol (0.5%) was used as a carbon source, and 50 mM Na-fumarate was used as an electron acceptor.
Cloning of the AH gene.
The AH gene was amplified from genomic DNA of P. acetylenicus strain WoAcy1 and ligated into the NheI/XhoI restriction sites of the pET24a(+) vector by Trenzyme GmbH (Konstanz, Germany). The resulting vector was called pET24_AH.
Addition of NarG chaperone binding site.
The 108-bp N-terminal chaperone binding site of the E. coli nitrate reductase NarG was amplified by Trenzyme GmbH and ligated into the NdeI/NheI restrictions sites of the pET24a(+) vector already containing the AH gene. The resulting vector was called pET24_NarG-AH.
Site-directed mutagenesis.
Exchange of single amino acid residues was done by PCR. The mismatch primers are listed in Table S1 in the supplemental material. The vectors pET24_AH and pET24_NarG-AH were used as templates. High-fidelity PCR enzyme mix was obtained from Fermentas; deoxynucleoside triphosphate (dNTP) bundles were from Jena Bioscience. The PCR was performed in a Master cycler gradient thermocycler (Eppendorf). DNA polymerase (0.05 U/μl), 0.2 mM dNTPs, 10× high-fidelity PCR buffer (Fermentas), 2 mM MgCl2, 1 μM primer I, 1 μM primer II, and 0.2 ng/μl template were used in the PCR. After a test restriction with NheI/XhoI, PCR products with the correct restriction pattern were amplified in E. coli JM109. The plasmids were then isolated using a GeneElute Plasmid miniprep kit (Sigma) and sequenced at GATC (Konstanz, Germany). Plasmids with the correct amino acid exchange were then transformed into E. coli Rosetta (DE3) for expression, using the method of Inoue et al. (12).
Expression of AH in E. coli.
The first experiments were carried out with E. coli BL21(DE3), E. coli BL21(DE3) pLys, and E. coli Rosetta (DE3) under aerobic conditions, but only insoluble protein was obtained (32). Soluble AH could be obtained by heterologous expression in E. coli Rosetta (DE3) using the medium described above; cells carrying the pET24_AH or the pET24_NarG-AH vector grew anaerobically at 37°C to an optical density at 600 nm (OD600) of 1.0 within 2 days (1-liter batch cultures). The cultures were cooled to 25°C, and expression of AH was induced by addition of 100 μM IPTG. For the expression of AH with the chaperone binding site of E. coli NarG, 100 μM NaNO3 was added to the culture 1 h prior the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to induce the formation of the chaperones. After induction with 100 μM IPTG and 24 h of expression at 25°C, typically 1.5 ± 0.5 g of wet cells/liter was harvested.
Enzyme purification.
AH from P. acetylenicus was purified under the exclusion of dioxygen, as described previously (28). Heterologously expressed AH was also purified under the exclusion of dioxygen; cells were disrupted by passage (three times) through a French press at 110 MPa. The soluble and insoluble fractions were separated by ultracentrifugation (100,000 × g). The soluble fraction was subjugated to two steps of ammonium sulfate precipitation (2.0 and 3.2 M). The pellet of the second step was dissolved in 50 mM Tris, pH 8.0, and loaded on a Co2+-charged Chelating Sepharose Fast Flow column (Amersham). Bound protein was eluted by applying a pH (8.0 to 7.5) gradient, followed by an imidazole (0 to 500 mM) gradient. AH-containing fractions were identified by SDS-PAGE, pooled, concentrated by ultra-centrifugal filter devices (30-kDa cutoff; Millipore), and loaded on a Superdex 200 gel filtration column (Amersham). Fractions containing pure AH were analyzed by SDS-PAGE, pooled, and concentrated to a final concentration of 10 mg/ml protein.
SDS-PAGE.
Electrophoresis was performed according to Laemmli (17), using 12% gels; proteins were fixed with 12% trichloroacetic acid (1 h) and stained overnight with 0.1% Coomassie brilliant blue G250, 10% (NH4)2SO4, 20% methanol, and 3% H3PO4 (21).
Protein concentration.
Protein was determined with the bicinchoninic acid method, with bovine serum albumin as a standard (30).
Molybdopterin cofactor.
Molybdopterin was determined fluorometrically (excitation wavelength [λex], 375 nm; emission wavelength [λem], 445 nm) on a Perkin Elmer LS50 luminescence spectrometer (15). Fifty microliters of AH in 50 mM Tris, pH 7.5 (1 to 2 mg/ml), was added to 200 μl of 55 mM KMnO4 in 0.1 M NaOH. The samples were boiled at 100°C for 20 min to oxidize molybdopterin to the fluorescent form A. Excess KMnO4 was precipitated by addition of 700 μl of ethanol (EtOH; 99%). After centrifugation, the fluorescence of the supernatant was measured; commercially available pterin-6-carboxylic acid (Fluka) served as a reference.
Metal analysis.
The metals of AH and variants were analyzed by inductively coupled plasma mass spectroscopy (ICP-MS) at the Spurenanalytisches Laboratorium Dr. Baumann (Maxhütte-Haidhof, Germany). Iron, molybdenum, and tungsten were determined in samples from different cultivations and purifications (200-μl samples; 2.5 mg/ml protein).
CD spectroscopy.
The secondary structure elements of AH from P. acetylenicus and heterologously expressed AH were compared by circular dichroism (CD) spectroscopy. Spectra were recorded on a J-810 spectropolarimeter (Jasco) in cuvettes of 1.0-mm and 0.1-mm path lengths. Samples were prepared in 10 mM Tris, pH 7.5, with 0.4 mg of protein/ml. The secondary structure elements were calculated for the range 195 to 260 nm, with the program CD Spectra Deconvolution, version 2.1 (3).
Electron paramagnetic resonance spectroscopy.
X-band EPR spectra were recorded with Suprasil quartz tubes (outer diameter of EPR tube [øout], 4 mm; sample volume, 250 μl) on a Bruker Elexsys 500 instrument equipped with an ER 049X microwave bridge, a 4122 SHQE cavity (perpendicular mode, 9.38 GHz microwave frequency, 100 kHz modulation frequency, and modulation amplitude of 0.1 to 1 mT), and an Oxford ESR 900 helium cryostat connected to an ITC 503 temperature controller (Oxford Instruments). Spectra were evaluated with the Bruker software and simulated with the program WEPR (20).
Enzyme activity.
The activity of AH and variants was determined in a coupled assay, with alcohol dehydrogenase (ADH) from Saccharomyces cerevisiae (18, 24). The assay is based on the formation of acetaldehyde (AH) and the subsequent NADH-dependent reduction of acetaldehyde to ethanol (ADH). Briefly, 960 μl of the reducing buffer [50 mM Tris, pH 7.5, 1.5 mM Ti(III)-citrate] was mixed with 20 μl of 10 mM NADH, 10 μl of 2,000 U/ml ADH, and 10 μl of AH (10 mg/ml) in quartz cuvettes under an N2/H2 (94%/6%, vol/vol) atmosphere, and cuvettes were sealed with rubber stoppers. The mixture was incubated for 30 min at 30°C; thereafter, the reaction was started by the addition of 2 ml of acetylene. Oxidation of NADH was measured photometrically at 365 nm, and the activity was calculated using a ɛ365(NADH) of 3.4 mM−1 cm−1 (36). AH and variants were also tested with ethylene as a substrate, but no activity was found.
Crystallization of AH and variants.
Crystallization experiments were done under exclusion of dioxygen following the protocol developed by Seiffert et al. (28). Briefly, crystallization screens of heterologously expressed AH were performed applying both the sitting- and the hanging-drop vapor diffusion methods. Small crystals began to grow in both cases over a period of 3 to 4 weeks from a solution of E. coli NarG-AH (6.5 to 10 mg/ml) in 5 mM HEPES-NaOH, pH 7.5, containing 7.5 mM Na2S2O4 as a reductant. For diffraction experiments, crystals were transferred to a cryoprotectant solution containing all substances of the reservoir solution and 20% (vol/vol) 2-methyl-2,4-pentanediol (MPD). After incubation with the cryoprotectant solution, crystals were flash frozen in liquid nitrogen. So far, no suitable X-ray diffraction data could be collected from theses crystals, most likely due to their size and irregular shape.
RESULTS
Several procedures were explored to express AH in E. coli under aerobic and anaerobic conditions and to produce active-site variants. The purified proteins have been characterized with regard to the molybdopterin content, the occupancy of the metal sites, and their specific activity, as compiled in Table 1. Activities are reported as μmol of C2H2 converted per minute and mg of protein; in addition, activity values have been normalized to the tungsten content (given in parentheses).
TABLE 1.
Specific and relative activity of AH and variantsa
| Enzyme | Expression profile (mol/mol of AH) |
MGD/W ratio | Specific activity (μmol of C2H2/min/mg of protein) | Relative activity (μmol of C2H2/min/nmol of W) | ||
|---|---|---|---|---|---|---|
| W | Fe | MGD | ||||
| P. acetylenicus AHb | 0.37 ± 0.04 | 3.69 ± 0.04 | 0.94 ± 0.04 | 2.54 | 14.2 ± 0.9 | 38.4 |
| E. coli AH | 0.06 ± 0.02 | 1.22 ± 0.26 | 0.17 ± 0.08 | 2.83 | 2.6 ± 0.8 | 43.3 |
| E. coli AH D13A | 0.09 ± 0.02 | 1.17 ± 0.29 | 0.21 ± 0.03 | 2.33 | 0.2 ± 0.1 | 2.2 |
| E. coli AH D13E | 0.05 ± 0.01 | 1.11 ± 0.30 | 0.20 ± 0.10 | 4.00 | 2.5 ± 0.3 | 50.0 |
| E. coli NarG-AH | 0.14 ± 0.06 | 3.17 ± 0.49 | 0.31 ± 0.09 | 2.21 | 9.7 ± 1.9 | 69.3 |
| E. coli NarG-AH K48A | 0.15 ± 0.01 | 3.56 ± 0.31 | ND | ND | 7.2 ± 0.3 | 48.0 |
| E. coli NarG-AH I142A | 0.18 ± 0.02 | 3.20 ± 0.22 | 0.29 ± 0.10 | 1.61 | 2.2 ± 0.2 | 12.2 |
Measurements were performed in triplicate. ND, not done.
The first preparation of AH from P. acetylenicus (as isolated in the presence of air) had 4.8 mol of Fe and 0.4 mol of W/mol of AH. The Km for acetylene was 14 μM; Vmax was 69 μmol min−1 mg of protein−1; optimum temperature was 50°C; the pH optimum was 6.0 to 6.5 (24).
Purification of heterologously expressed AH.
The purification of AH heterologously expressed in E. coli typically yielded 3.25 ± 0.7 mg of pure AH/g of wet cells. Compared to a yield of ≈0.8 ± 0.1 mg of AH/g of wet cells purified from P. acetylenicus, a 4-fold overexpression was achieved in E. coli. E. coli AH had a specific activity of 2.6 ± 0.8 (43.3) μmol of C2H2 min−1 mg−1, which is lower than the value of 14.2 ± 0.9 (38.4) μmol of C2H2 min−1 mg−1 found for P. acetylenicus AH. However, when normalized to the tungsten content, the activity values were similar (38.4 versus 43.3 μmol of C2H2 min−1 mg−1) (Table 1).
Purification of the heterologously expressed AH with the NarG chaperone binding site.
Addition of the N-terminal chaperone binding site of the E. coli nitrate reductase NarG increased the yield of AH to ≈4.2 ± 0.3 mg/g of wet cells. Notably, the specific activity of the recombinant enzyme also increased significantly to 9.7 ± 1.9 (69.3) μmol of C2H2 min−1 mg−1 (Table 1).
The homogeneity of the individual preparations was checked by SDS-PAGE (Fig. 2), and protein folding was controlled by CD spectroscopy as discussed below. None of the protein samples purified under the exclusion of dioxygen showed any EPR signal; incubation with sodium dithionite led to the appearance of the rhombic EPR signal described earlier (gz of 2.048, gy of 1.939, and gx of 1.920) originating from the iron-sulfur cluster in the [4Fe-4S]+ redox state (data not shown) (18, 24, 27).
FIG. 2.

SDS-PAGE (12%) of acetylene hydratase and variants. Lane 1, molecular mass markers; lane 2, P. acetylenicus AH (2 μg); lane 3, E. coli AH (2 μg); lane 4, E. coli AH D13A variant (3 μg); lane 5, E. coli AH D13E variant (2 μg); lane 6, E. coli NarG-AH (1 μg); lane 7, E. coli NarG-AH I142A variant (1 μg); lane 8, E. coli NarG-AH K48A variant (3 μg).
The N-terminal chaperone binding site of E. coli TMAO reductase, TorA, was also cloned in frame into the AH expression vector in front of the AH gene, as described for the NarG sequence, producing TorA-AH (32). The protein could be purified to homogeneity and was active; however, the yield of TorA-AH was significantly lower than that of NarG-AH. Consequently, investigation of this expression system was not pursued (32).
Molybdopterin cofactor in heterologously expressed AH.
The enzyme isolated from P. acetylenicus carries two MGD ligands/mol of enzyme, as documented in the crystal structure (Fig. 1) (28). Fluorimetric analysis of standard preparations of wild-type AH used in this work and for the crystallization of the enzyme usually gave 0.94 ± 0.04 mol of MGD per mol of enzyme. The highest content amounted to 1.3 ± 0.1 mol of MGD; the theoretical value of two MGD ligands per mol of enzyme could not be achieved for AH as isolated (18). Recombinant AH from E. coli contained 0.17 ± 0.08 mol of MGD; fusion of the NarG chaperone binding site increased the content of MGD to 0.31 ± 0.09 mol per mol of enzyme, corresponding to 33% of the value found in standard preparations of wild-type AH (Table 1).
Metals in heterologously expressed AH and variants.
The content of metals in AH from P. acetylenicus and protein heterologously expressed in E. coli was determined by ICP-MS. Typically, standard preparations of wild-type AH contained 3.69 ± 0.04 mol of Fe and 0.37 ± 0.03 mol of W/mol of AH (ratio of MGD/W, 2.54). The metal content of recombinant AH from E. coli was lower, 1.22 ± 0.26 mol of Fe and 0.06 ± 0.02 mol of W/mol of AH. However, after attachment of the N-terminal chaperone site of NarG, the content of iron increased to 3.17 ± 0.49 mol of Fe/mol of AH, representing 86% of the value determined for AH purified from P. acetylenicus. In line with this result, the content of tungsten increased to 0.14 ± 0.06 mol of W/mol of AH (ratio of MGD/W of 2.14) (Table 1). Molybdenum was absent in all samples.
Control of protein folding.
One measure for the quality of heterologously expressed AH and variants is the proper folding of the protein, which can be checked by CD spectroscopy (Fig. 3). With regard to the secondary structure elements, heterologously expressed AH proteins exhibited a slightly lower content of antiparallel β-sheets than the native P. acetylenicus enzyme. On the other hand, the recombinant proteins had a slightly higher content of α-helices. The only region with antiparallel β-sheets within the structure of AH is the N-terminal domain I, which harbors the [4Fe-4S] cluster (Fig. 1) (28). Most likely, the minor changes in the secondary structure result from conformational changes induced by the lower occupancy of the metal sites. In the NarG-AH fusion proteins, antiparallel β-sheets were even less abundant, according to CD spectroscopy. Here, 30 amino acids were attached to the N-terminal end of the protein. The secondary structure of this tail and its influence on the conformation of domain I are currently not known. However, the increase in iron content indicates that the structure of the [4Fe-4S] cluster has not been disturbed by the addition of the chaperone binding site, which is also supported by the EPR properties described above.
FIG. 3.

Representative CD spectra of acetylene hydratase and derived secondary structure elements. The spectra were recorded as described in Materials and Methods; the spectra of the variants did not differ from the spectra of the corresponding proteins, E. coli AH and E. coli NarG-AH; the secondary structure elements were calculated with the program CD Spectra Deconvolution, version 2.1.
Site-directed mutagenesis.
Three amino acid residues at the active site of AH were successfully exchanged by site-directed mutagenesis to investigate their functional role in the reaction mechanism of AH. Residue Asp13, which forms a hydrogen bond to the oxygen ligand of the tungsten atom (28), was replaced with glutamate (D13E) and alanine (D13A). Lys48, which has been shown to play a critical role in electron transfer between the [4Fe-4S] cluster and the Mo(MGD)2 center in enzymes of the DMSOR family (5), was replaced by alanine (K48A) (Fig. 4 A). Finally, Ile142, a constituent of the hydrophobic ring between the tungsten active site and the substrate channel (Fig. 4B) (28), was successfully replaced with alanine (I142A), whereas attempts to exchange a second residue of the hydrophobic ring, Trp472, remained unsuccessful.
FIG. 4.

(A) Active-site structure of acetylene hydratase from P. acetylenicus indicating the positions of residues Asp13 and Lys48; W is shown in cyan, and H2O is in red. The radii of the spheres correspond to the covalence radii of the atoms according to http://www.periodensystem.info/. (B) Active-site structure of acetylene hydratase from P. acetylenicus indicating the position of residue Ile142; W is shown in cyan, and H2O is in red. The surface of the hydrophobic ring formed by three isoleucine and three tryptophan residues, with one acetylene molecule (in gray) placed into the pocket, is shown in the top figure. In the bottom figure, distances between acetylene and catalytic residues Ile142, Asp13, and the water ligand are shown. The radii of the spheres correspond to the covalence radii of the atoms according to http://www.periodensystem.info/.
AH activity of variants.
In both reaction mechanisms based on the crystal structure of AH (28), Asp13 plays a critical role, either by donating a second proton after the nucleophilic attack of the hydroxide group on the acetylene C,C triple bond or by activation of the coordinated water molecule to perform an electrophilic attack (28). In the coupled reaction assay, the activity of the D13A variant was reduced to close to zero, while the D13E variant exhibited nearly the identical activity as the wild-type enzyme, documenting the important role of the carboxylic group of Asp13 in AH catalysis (Table 1).
Lys48 is located between the [4Fe-4S] cluster and the Q-MGD ligand (Fig. 4A). As the reaction of AH does not involve a net electron transfer, it was not too surprising that the K48A variant had practically the same activity as reported for the wild-type enzyme (Table 1).
Ile142 is part of a hydrophobic ring of six bulky amino acid residues (Fig. 4B) (28), forming a small cavity for binding the substrate at the end of the channel. Acetylene placed in this cavity would be positioned directly above the oxygen ligand, in close proximity to both the tungsten atom and Asp13. The activity of the I142A variant amounted to 2.2 ± 0.2 (12.2) μmol of C2H2 min−1 mg−1 (Table 1). The marked loss of activity upon exchanging Ile142 for alanine supports the idea that the cavity formed by the hydrophobic ring is the substrate binding site of AH.
Ethylene, in addition to acetylene, was also tested as a substrate for the heterologously expressed AH and variants, and as in the case of wild-type AH ethylene, was not accepted as a substrate.
DISCUSSION
The strictly anaerobic bacterium P. acetylenicus can grow with acetylene as a single carbon and energy source. The first step in the fermenting pathway is the transformation of acetylene to acetaldehyde, which is consecutively converted to acetate and ethanol. It appears that P. acetylenicus conserves only the free energy available in the acetate kinase reaction and not the amount of free energy available from hydration of acetylene (equation 1). Earlier, it was speculated that the conversion of acetylene to acetaldehyde might represent a bifunction of an unspecific hydratase enzyme which mainly acts in the natural environment in detoxification of acetylenic compounds, nitriles, or cyanides (24).
Catalysis of AH is rather peculiar in the sense that two complex metal sites and a strong reductant are required for the addition of one molecule of water to the C☰C bond. Notably, there do exist iron-sulfur proteins that catalyze hydration reactions, with aconitase being among the first discovered examples (2).
Heterologous expression of AH.
Acetylene hydratase has been found exclusively in the soluble fraction of P. acetylenicus (24). Following the procedures originally described for the heterologous expression of Rhodobacter sphaeroides DMSO reductase (8), the first attempts were carried out with three strains of E. coli under different experimental conditions including the variation of the copper concentration (19). By this means, only insoluble protein was obtained (32). Upon anaerobic cultivation of E. coli Rosetta (DE3), with glycerol as a carbon source and sodium fumarate as an electron acceptor, soluble E. coli AH could be isolated and purified to homogeneity. The protein was active but exhibited a low content of molybdopterin, iron, and tungsten (Table 1). Notably, the wild-type enzyme purified from P. acetylenicus (as isolated under the exclusion of dioxygen) was also always low in molybdopterin and tungsten (1.3 mol of Moco and 0.5 mol of W per mol of AH), whereas the iron content usually reached the theoretical value of 4 mol of Fe per mol of AH (18, 24). In crystalline AH, however, the sites of the two MGD ligands were fully occupied, in contrast to the occupancy of the tungsten site, which remained low (≈40%). Thus, at a resolution of 1.10 to 1.26 Å, the MGD sites with and without tungsten could be clearly differentiated (28; also G. Seiffert, P. M. H. Kroneck, and O. Einsle, submitted for publication).
Extensive studies on the maturation of Moco-containing enzymes had revealed a family of chaperones that facilitated the incorporation of the cofactor during protein biosynthesis and prevented the export of periplasmic enzymes before its proper insertion (11, 22, 23, 25). Complementation studies indicated that these chaperones were highly specific for their partner and could not complement the absence of another chaperone (11). Amino acid sequence alignments showed that such an N-terminal chaperone binding site was missing in the P. acetylenicus AH gene. In order to improve the insertion of cofactors and metals, the first 108 bp of the NarG gene or the first 117 bp of the TorA gene were successfully fused to the AH gene. This operation not only increased the yield of protein in the case of the NarG-AH enzyme but also helped to increase the content of molybdopterin, tungsten, and iron accompanied by a significant increase in activity (Table 1). Earlier, a protocol had been published for the heterologous expression of R. sphaeroides DMSO reductase. In contrast to AH, this enzyme carried an N-terminal chaperone binding site which had been removed prior to the expression in E. coli. In addition, the binding site TorA had been fused to the DMSOR gene which, in contrast to the experiments with TorA-AH, led to a significant decrease in activity (8).
At this point, the different steps leading to the maturation of the soluble enzyme AH in both P. acetylenicus and E. coli are not understood and will require further investigations. Obviously, fusion of the AH gene with the N-terminal chaperone binding site of the E. coli nitrate reductase NarG improved the quality of the protein. This finding suggests that the main function of the chaperone is to keep the protein unfolded for a longer time period and therefore extend the time frame for cofactor assembly and metal insertion during biosynthesis of AH. To get more information about this issue, experiments with radioactive metal isotopes are planned (29).
Substrate specificity.
As the hydration of acetylene depends on tungsten and as most tungsten enzymes described to date have been purified from strictly anaerobic, thermophilic, or extremely thermophilic bacteria, one might speculate that metabolism of acetylene represents an early form of life (7). However, P. acetylenicus is a mesophilic organism, and the temperature optimum of AH has been determined to be 55°C. Furthermore, a molybdenum-dependent active form of AH could be obtained from P. acetylenicus cultivated on molybdate (2 μM) in the presence of nanomolar concentrations of tungstate (18), as reported for DMSOR from Rhodobacter capsulatus (31). Attempts to replace tungsten with vanadium, however, have thus far failed (1).
AH is highly specific toward its substrate acetylene as no other substrates have been found to date. Our search included ethylene and derivatives of acetylene (propargyl alcohol and acetylene mono- and dicarboxylic acid), cyanide, nitriles, and isonitriles. In summary, a possible physiological function of AH beyond the conversion of acetylene to acetaldehyde cannot be defined at present.
Active-site access, active-site architecture, and reaction mechanism.
In the structures of proteins of the DMSO reductase family available to date, access to the active center is provided through a funnel-like entrance whose position is conserved in enzymes such as DMSO and TMAO reductases, as well as in formate and nitrate reductases. In AH, however, this entire region has been completely rearranged. The substrate acetylene must approach the tungsten site from a different angle through a funnel close to the N-terminal domain that harbors the [4Fe-4S] cluster (28). Above the tungsten ion, the substrate funnel ends in a ring of six hydrophobic residues (three Ile and three Trp) which form the substrate cavity. Through shape complementarity, the residues of the hydrophobic ring are a key determinant for the enzyme's substrate specificity (Fig. 4B). Numerous attempts to pressurize crystals of AH with acetylene, ethylene, carbon monoxide, nitric oxide, or dinitrogen, as well as soaking of crystals of AH with different compounds including the inhibitor propargyl alcohol, have failed thus far to produce a crystalline substrate complex of AH. With xenon gas, one Xe atom could be trapped in the funnel but not, however, in bonding distance to the tungsten center (27, 32).
The formation of acetaldehyde is accomplished by activation of a water molecule bound to a W(IV) ion interacting with residue Asp13 and the [4Fe-4S] cluster, one of whose ligands, Cys12, is an immediate neighbor of Asp13. Usually, the [4Fe-4S] cluster is involved in electron transfer in enzymes of the DMSOR family, as Lys48 is considered to be essential for electron transfer from the iron-sulfur cluster to the Q-MGD ligand (5). In AH, however, the active site is found at a different side of the tungsten ion, closer to the [4Fe-4S] cluster. Furthermore, in the structure of AH, the conserved water molecule is missing (27), which has been assumed to be a crucial component of the electron transfer pathway (reference 14 and Fig. 5 therein). Thus, the tungsten center remains in the W(IV) state during catalysis, and electron transfer does not occur. Instead, a significant increase in pKa for Asp13 is caused by the desolvation of this residue, and the [4Fe-4S] cluster appears to push electrons toward Asp13 and thus helps to increase its proton affinity (28). The increased specific activity of AH under reducing conditions is in part explained by this finding, as the shift in pKa and the degree of activation of the water ligand will be stronger in a cluster in the reduced [4Fe-4S]+ state (28). Recent redox titrations of the iron-sulfur cluster in P. acetylenicus AH gave a midpoint potential E0′ of −410 mV ± 20 mV (Nernst coefficient n = 1), and the enzyme activity depended on the potential of the solution, with 50% of maximum activity at −340 ± 20 mV (n = 2). AH (as isolated) reacted with [Fe(CN)6]3− to give a W(V) EPR signal (gav of 2.022). The oxidative reaction proceeded very sluggishly even in the presence of mediating redox dyes; attempts to determine midpoint potentials for the tungsten center by EPR-monitored titrations remained unsuccessful (18, 27).
In accordance with these results, the biomimetic complex [Et4N]2[WIVO(mnt)2] (where mnt is 1,2-dicyanoethylenedithiolate) had been reported to catalyze the same reaction as AH, while the corresponding oxidized W(VI) complex was inactive but could be reactivated by addition of strong reductants (34).
Replacing residue Asp13 of AH with either alanine or glutamate gives strong support for the crucial catalytic role of this residue, as predicted from the crystal structure and theoretical calculations (28). Assuming that Asp13 will be protonated in the catalytically active W(IV) state, its replacement by alanine will not have a marked influence on the redox potential of the W(IV) center according to these calculations.
Second, as catalysis does not include an electron transfer between the two metal centers, replacing residue Lys48 with alanine did not affect the acetylene hydratase activity of this variant as expected (Table 1). Cys46 (3.27 Å) which ligates the [4Fe-4S] cluster, and Q-MGD (2.94 Å) are the closest neighbors of Lys48 (Fig. 4A). Replacement of the positively charged Lys48 residue with the neutral alanine is expected to influence the reduction potential of the [4Fe-4S] cluster; however, its value will still be quite negative (4).
Third, in view of the strongly reduced activity of the I142A variant, a more detailed picture of the mode of substrate binding can be drawn (Fig. 4B). The hydrophobic cavity formed by Ile142 and five other amino acid residues is perfectly suited to position H—C☰C—H above the W-OH2 moiety. On the other hand, the more polar molecules ethenol (CH2=CHOH) or acetaldehyde (CH3CHO) will readily leave the hydrophobic region of the active site, facilitating the removal of the product of the enzymatic reaction. Notably, in the structure of AH two molecules of the cryoprotectant 2-methyl-2,4-pentanediol (MPD) were present in the funnel, one of them relatively close to the active site (27).
Supplementary Material
Acknowledgments
We thank Oliver Einsle (Universität Freiburg) for helpful discussions.
Financial support by the Deutsche Forschungsgemeinschaft (Kr 451/40-1 and -42-1) is gratefully acknowledged.
Footnotes
Published ahead of print on 30 December 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Abt, D. J. 2001. Tungsten-acetylene hydratase from Pelobacter acetylenicus and molybdenum-transhydraxylase from Pelobacter acidigallici: two novel molybdopterin and iron-sulfur containing enzymes. Ph.D. dissertation. University of Konstanz, Konstanz, Germany.
- 2.Beinert, H., M. C. Kennedy, and C. D. Stout. 1996. Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev. 96:2335-2374. [DOI] [PubMed] [Google Scholar]
- 3.Böhm, G. 1997. CDNN: CD spectra deconvolution software, version 2.1. University of Halle-Wittenberg, Halle, Germany.
- 4.Dey, A., et al. 2007. Solvent tuning of electrochemical potentials in the active sites of HiPIP versus ferredoxin. Science 318:1464-1468. [DOI] [PubMed] [Google Scholar]
- 5.Dobbek, H., and R. Huber. 2002. The molybdenum and tungsten cofactors: a crystallographic view. Met. Ions Biol. Syst. 39:227-263. [PubMed] [Google Scholar]
- 6.Genest, O., et al. 2008. Dedicated metallochaperone connects apoenzyme and molybdenum cofactor biosynthesis components. J. Biol. Chem. 283:21433-21440. [DOI] [PubMed] [Google Scholar]
- 7.Hille, R. 2002. Molybdenum and tungsten in biology. Trends Biochem. Sci. 27:360-367. [DOI] [PubMed] [Google Scholar]
- 8.Hilton, J. C., C. A. Temple, and K. V. Rajagopalan. 1999. Re-design of Rhodobacter sphaeroides dimethyl sulfoxide reductase. Enhancement of adenosine N1-oxide reductase activity. J. Biol. Chem. 274:8428-8436. [DOI] [PubMed] [Google Scholar]
- 9.Hine, F. J., A. J. Taylor, and C. D. Garner. 2010. Dithiolene complexes and the nature of molybdopterin. Coord. Chem. Rev. 254:1570-1579. [Google Scholar]
- 10.Hyman, M. R., and D. J. Arp. 1988. Acetylene inhibition of metalloenzymes. Anal. Biochem. 173:207-220. [DOI] [PubMed] [Google Scholar]
- 11.Ilbert, M., V. Mejean, and C. Iobbi-Nivol. 2004. Functional and structural analysis of members of the TorD family, a large chaperone family dedicated to molybdoproteins. Microbiology 150:935-943. [DOI] [PubMed] [Google Scholar]
- 12.Inoue, H., H. Nojima, and H. Okayama. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23-28. [DOI] [PubMed] [Google Scholar]
- 13.Jack, R. L., et al. 2004. Coordinating assembly and export of complex bacterial proteins. EMBO J. 23:3962-3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jepson, B. J., et al. 2007. Spectropotentiometric and structural analysis of the periplasmic nitrate reductase from Escherichia coli. J. Biol. Chem. 282:6425-6437. [DOI] [PubMed] [Google Scholar]
- 15.Johnson, J. L., and K. V. Rajagopalan. 1982. Structural and metabolic relationship between the molybdenum cofactor and urothione. Proc. Natl. Acad. Sci. U. S. A. 79:6856-6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kisker, C., H. Schindelin, and D. C. Rees. 1997. Molybdenum-cofactor-containing enzymes: structure and mechanism. Annu. Rev. Biochem. 66:233-267. [DOI] [PubMed] [Google Scholar]
- 17.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 18.Meckenstock, R. U., R. Krieger, S. Ensign, P. M. Kroneck, and B. Schink. 1999. Acetylene hydratase of Pelobacter acetylenicus. Molecular and spectroscopic properties of the tungsten iron-sulfur enzyme. Eur. J. Biochem. 264:176-182. [DOI] [PubMed] [Google Scholar]
- 19.Morrison, M. S., P. A. Cobine, and E. L. Hegg. 2007. Probing the role of copper in the biosynthesis of the molybdenum cofactor in Escherichia coli and Rhodobacter sphaeroides. J. Biol. Inorg Chem. 12:1129-1139. [DOI] [PubMed] [Google Scholar]
- 20.Neese, F. 1995. The program EPR. Quantum Chem. Program Exch. Bull. 15:5. [Google Scholar]
- 21.Neuhoff, V., N. Arold, D. Taube, and W. Ehrhardt. 1988. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9:255-262. [DOI] [PubMed] [Google Scholar]
- 22.Palmer, T., et al. 1996. Involvement of the narJ and mob gene products in distinct steps in the biosynthesis of the molybdoenzyme nitrate reductase in Escherichia coli. Mol. Microbiol. 20:875-884. [DOI] [PubMed] [Google Scholar]
- 23.Pommier, J., V. Mejean, G. Giordano, and C. Iobbi-Nivol. 1998. TorD, a cytoplasmic chaperone that interacts with the unfolded trimethylamine N-oxide reductase enzyme (TorA) in Escherichia coli. J. Biol. Chem. 273:16615-16620. [DOI] [PubMed] [Google Scholar]
- 24.Rosner, B. M., and B. Schink. 1995. Purification and characterization of acetylene hydratase of Pelobacter acetylenicus, a tungsten iron-sulfur protein. J. Bacteriol. 177:5767-5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sargent, F. 2007. Constructing the wonders of the bacterial world: biosynthesis of complex enzymes. Microbiology 153:633-651. [DOI] [PubMed] [Google Scholar]
- 26.Schink, B. 1985. Fermentation of acetylene by an obligate anaerobe, Pelobacter acetylenicus sp. nov. Arch. Microbiol. 142:295-301. [Google Scholar]
- 27.Seiffert, G. 2007. Structural and functional studies on two molybdopterine and iron-sulfur containing enzymes: transhydroxylase from Pelobacter acidigallici and aceytlene hydratase from Pelobacter acetylenicus. Ph.D. dissertation. University of Konstanz, Konstanz, Germany.
- 28.Seiffert, G. B., et al. 2007. Structure of the non-redox-active tungsten/[4Fe:4S] enzyme acetylene hydratase. Proc. Natl. Acad. Sci. U. S. A. 104:3073-3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sevcenco, A. M., et al. 2010. Molybdenum incorporation in tungsten aldehyde oxidoreductase enzymes from Pyrococcus furiosus. J. Bacteriol. 192:4143-4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith, P. K., et al. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150:76-85. [DOI] [PubMed] [Google Scholar]
- 31.Stewart, L. J., et al. 2000. Dimethylsulfoxide reductase: an enzyme capable of catalysis with either molybdenum or tungsten at the active site. J. Mol. Biol. 299:593-600. [DOI] [PubMed] [Google Scholar]
- 32.tenBrink, F. 2010. Acetylene hydratase from Pelobacter acetylenicus: functional studies on a gas-processing tungsten, iron-sulfur enzyme by site directed mutagenesis and crystallography. Ph.D. dissertation. University of Konstanz, Konstanz, Germany.
- 33.Widdel, F., and N. Pfennig. 1981. Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids. I. Isolation of new sulfate-reducing bacteria enriched with acetate from saline environments. Description of Desulfobacter postgatei gen. nov., sp. nov. Arch. Microbiol. 129:395-400. [DOI] [PubMed] [Google Scholar]
- 34.Yadav, J., K. S. Das, and S. Sarkar. 1997. A functional mimic of the new class of tungstoenzyme, acetylene hydratase. J. Am. Chem. Soc. 119:4315-4316. [Google Scholar]
- 35.Zhang, Y., and V. N. Gladyshev. 2008. Molybdoproteomes and evolution of molybdenum utilization. J. Mol. Biol. 379:881-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziegenhorn, J., M. Senn, and T. Bucher. 1976. Molar absorptivities of beta-NADH and beta-NADPH. Clin. Chem. 22:151-160. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.