

Abstract
The plant pathogen Agrobacterium tumefaciens responds to three main signals at the plant-bacterium interface: phenolics, such as acetosyringone (AS), monosaccharides, and acidic pH (∼5.5). These signals are transduced via the chromosomally encoded sugar binding protein ChvE and the Ti plasmid-encoded VirA/VirG two-component regulatory system, resulting in the transcriptional activation of the Ti plasmid virulence genes. Here, we present genetic and physical evidence that the periplasmic domain of VirA dimerizes independently of other parts of the protein, and we examine the effects of several engineered mutations in the periplasmic and transmembrane regions of VirA on vir-inducing capacity as indicated by AS sensitivity and maximal level of vir-inducing activity at saturating AS levels. The data indicate that helix-breaking mutations throughout the periplasmic domain of VirA or mutations that reposition the second transmembrane domain (TM2) of VirA relieve the periplasmic domain's repressive effects on the maximal activity of this kinase in response to phenolics, effects normally relieved only when ChvE, sugars, and low pH are also present. Such relief, however, does not sensitize VirA to low concentrations of phenolics, the other major effect of the ChvE-sugar and low pH signals. We further demonstrate that amino acid residues in a small Trg-like motif in the periplasmic domain of VirA are crucial for transmission of the ChvE-sugar signal to the cytoplasmic domain. These experiments provide evidence that small perturbations in the periplasmic domain of VirA can uncouple sugar-mediated changes in AS sensitivity from the sugar-mediated effects on maximal activity.
Agrobacterium tumefaciens is a Gram-negative pathogen that is the etiological agent of crown gall tumor disease in a wide variety of plant species. Proteins encoded by the virulence (vir) genes of the tumor-inducing (Ti) plasmid are critical for tumorigenesis that occurs as a result of the transfer of oncogenic DNA into the plant cell (80, 81). This process is very demanding in energetic terms, and the bacterium has evolved complex and precise regulation of the vir genes that are transcriptionally silent unless they are exposed to plants or plant exudates (8). Several classes of plant-derived molecules serve as signals for the induction of the vir genes. Phenol derivatives, such as acetosyringone (AS), are the primary inducers, absolutely required for induction. Though they are incapable of vir induction on their own, aldose sugars and low pH (5.2 to 5.8) are secondary inducers, affecting the sensitivity of the bacteria to phenols as well as the maximum level of induction by those molecules (46, 56).
At the crux of this signal-responsive virulence pathway is the VirA/VirG two-component system, in which VirA is the histidine kinase and VirG is its cognate response regulator (65). Signal transduction by VirA/G follows a common theme found in many prokaryotes comprised of a two-component sensing system that consists of a membrane-bound histidine kinase and a cytoplasmically localized response regulator (69). These systems respond to environmental signals and control a variety of bacterial processes, including virulence and chemotaxis, among others (45). VirA is a homodimeric, 92-kDa membrane-bound protein that has 7 domains: a short 18-amino-acid N-terminal cytoplasmic domain, 2 membrane-spanning domains (TM1 and TM2) that flank a 220-amino-acid periplasmic sensor domain (P), and a 550-amino-acid cytoplasmic C-terminal domain that can be subdivided into the linker, kinase, and receiver domains (L, K, and R, respectively; see Fig. 1A) (15, 47, 48). The kinase domain of VirA carries the conserved phosphoacceptor histidine residue, H474, that is critical for both autophosphorylation and phosphotransfer (38, 40). VirG is the response regulator, and its VirA-dependent phosphorylation at a conserved aspartate residue is necessary for transcriptional activation (39).
FIG. 1.

(A) Schematic depiction of VirA domain organization in a VirA monomer and known functions of each domain. Two transmembrane domains, TM1 and TM2 (amino acids highlighted in yellow in panel B), delineate the periplasmic domain. The cytoplasmic domain contains the sites of phenol perception (linker) and the kinase domain that has a histidine residue essential for phosphotransfer at position 474. Note that the receiver domain, though assigned an inhibitory role by Chang and Winans (15), has recently been found to have a positive regulatory effect on VirA kinase activity (72). (B) Predicted secondary structure of the N-terminal region of wild-type VirA. Secondary structure predictions were made from the amino acid sequence by using SABLE (1, 2, 68) and rendered using Polyview (60, 61). Note the predominantly α-helical nature of this region and the sites of different four-amino-acid insertions used in this study. Residues in green signify a small motif identical to a periplasmic binding protein interaction motif found in the E. coli Trg chemoreceptor. Amino acids N226, Q229, and I236 were the subjects of a site-directed mutagenesis study reported here to evaluate the importance of this motif in signal integration by VirA. A previously reported E255-to-L amino acid mutation that causes a loss of AS sensitivity (5) and was used in this study is also depicted (residue in blue). Also shown here are the positions where four helix-breaking amino acids were inserted into various predicted α-helices (shown by arrows pointing down), and the two amino acids, R259 and L276 (depicted in red), that were replaced by a serine and arginine, respectively, in this study. The R259S/L276R double mutation results in a TM2 domain that ends at position 276 instead of 278, according to prediction programs.
One of the distinguishing features of the VirA histidine kinase is that plant-released stimuli are perceived by both the periplasmic and the cytoplasmic domains. Genetic evidence indicates that phenolic signals are required for vir gene expression and are recognized by the linker domain of VirA (15, 67). In contrast, the periplasmic domain is necessary for sugar and pH signaling (13, 26, 64), though neither of these conditions is absolutely required for induction. The chromosomally encoded periplasmic sugar binding protein ChvE is also required for the detection and transduction of these signals and is thought to bind to the periplasmic region of VirA (30, 64). Point mutations and small deletions in the periplasmic region result in a defective sugar response. For example, the VirAE255L mutant (with an E-to-L amino acid change at position 255) and the mutant with a 16-amino-acid deletion from amino acids (aa) 242 to 257 are sugar insensitive, but interestingly, these retain the requirement for an acidic pH (5). Other alterations in the periplasmic domain, for example, a large deletion from aa 63 to 240 or some of the mutations described in this report, cause a phenotype corresponding to insensitivity to the monosaccharide stimulus but exhibition of greater maximal activity than that of the wild type at saturating AS concentrations in the absence of sugars as well as a loss of pH sensitivity (47).
Analogous to the VirA-ChvE system, the periplasmic domains of the Escherichia coli chemoreceptors Tar and Trg bind the maltose binding protein (MBP) and the glucose/galactose binding protein (GBP), respectively, in the presence of their sugar ligands (34, 49). This information is transduced through transmembrane domains to the cytoplasmic domains of these receptors, ultimately resulting in activation of the cytoplasmic histidine kinase CheA (34). Like VirA, Tar and Trg have two transmembrane domains and a large periplasmic domain and exist as a dimer in the inner membrane (4, 53). Though Tar and Trg lack extensive sequence homology, a common feature of their periplasmic domains is a preponderance of α-helices (11, 27). Similarly, the periplasmic domain of VirA (VirAperi) does not share extensive sequence homology with Tar and Trg. It does, however, share a sequence motif with Trg that lies within the proposed binding site for the periplasmic ribose binding protein (RBP) through which Trg responds to ribose (13, 74). Moreover, as we describe below, VirAperi, like the periplasmic domain of Tar (77), is almost exclusively α-helical as predicted by secondary structure analysis tools (10, 41).
The studies reported here utilize genetic and physical approaches to demonstrate that the periplasmic domain of VirA dimerizes and, as predicted by modeling programs, has an extensive α-helical character. The role of these predicted α-helical regions in dimerization and in the biological activity of VirA was tested via engineering 10 different helix-breaking four-amino-acid insertions across the periplasmic domain. The results indicate that all helix-breaking insertions had similar effects on vir gene expression, but only one such insertion affected dimerization. Interestingly, mutations predicted to relocate the second transmembrane domain of VirA such that amino acids normally associated with the cytoplasmic face of that domain are positioned further into the cytoplasm confer the same phenotype as the helix-breaking insertions. We also demonstrate here that the short Trg-like motif found in the periplasmic domain of VirA is crucial for sugar signaling, specifically resulting in different forms of sugar-insensitive phenotypes which are suppressed to different extents by a newly isolated, constitutively active form of ChvE (35).
MATERIALS AND METHODS
Bacterial strains and plasmids.
Bacterial and Saccharomyces cerevisiae (yeast) strains and plasmids used in this study are listed in Table S1 in the supplemental material. All cloning was performed using standard protocols, and all enzymes were used as recommended by manufacturers.
Culture media, growth conditions, and chemicals.
Escherichia coli strains used for cloning were grown in Luria-Bertani medium (LB) containing the appropriate antibiotics at 37°C. A. tumefaciens strains were grown at 25°C in AB minimal medium (22), AB induction medium (71), or LB containing the appropriate concentrations of antibiotics and acetosyringone (if used). The following antibiotics for E. coli were used (concentrations in μg/ml, in liquid medium/solid medium): spectinomycin (50/100), ampicillin (50/125), kanamycin (40/75), chloramphenicol (10/30), and gentamicin (12.5/25). The following antibiotics for A. tumefaciens were used (concentrations in μg/ml, in liquid medium/solid medium): spectinomycin (50/100), carbenicillin (30/100), and gentamicin (400/400). Acetosyringone or 3′,5′-dimethoxy-4′-hydroxyacetophenone (AS), isopropyl-β-d-thiogalactopyranoside (IPTG), and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), 2-[N-morpholino]ethanesulfonic acid (MES), and bis-Tris were obtained from Sigma Chemicals. Restriction enzymes were obtained from New England BioLabs, Pfu polymerase was obtained from Stratagene, and Taq polymerase was obtained from Perkin Elmer. Invitrogen Inc. supplied primers for cloning and sequencing. For AS stock, 100 mM stocks were made fresh in dimethyl sulfoxide (DMSO) and added to induction medium as necessary, at the appropriate concentration. For A. tumefaciens, X-Gal was used at a concentration of 50 μg/ml in AB minimal medium plates. For E. coli, X-Gal was used at 50 μg/ml in conjunction with IPTG (5 μg/ml). Plasmids were introduced into E. coli by electroporation at 200 Ω and 2.5 V by using a Bio-Rad electroporator. Cells were recovered in LB for 1 h at 37°C and plated on selection media. Plasmids were electroporated into competent A. tumefaciens strains at 400 Ω and 2.5 V by using a Bio-Rad electroporator as described by Nair et al. in 2003 (54). Basic protocols as described by Golemis et al. were used with regard to growth and use of Saccharomyces cerevisiae strains for yeast 2-hybrid analysis (33).
Construction of mutant strains and plasmids.
Standard methods were used for plasmid isolation, restriction analysis, agarose gel electrophoresis, DNA ligation, and PCR amplification of DNA. Ten insertion mutants were originally placed in a 660-bp fragment encoding the periplasmic region of VirA in the vector pJG4-5. Insertions are denoted by the amino acid position after which the insertion was made. For example, i64 is an insertion after amino acid position 64. VirAi64 defines the protein product of this mutation, and virAi64 defines the gene encoding this protein. Because the original virA coding region from pTiA6 is carried on a large 4.6-kb fragment flanked by KpnI sites and has very few unique restriction sites (described previously in reference 42), the 2.6-kb virA open reading frame (ORF) was amplified using primers with KpnI sites and placed in the vector pACYC184K in which the EcoRV site had been replaced with a KpnI site for use as a cloning template (plasmid pBMD0). A PvuI-AatII-digested virAperi fragment from the pMJ series of plasmids (see Table 1) was used to replace the wild-type virAperi in pBMD0, resulting in the pGN30 plasmid series. Plasmid pGN20 has the original 4.6-kb fragment with the wild-type virA gene cloned at the KpnI site of pBBR1MCS5. pGN20 was used as a template for finally placing virAperi mutant sequences into the full-length 4.6-kb virA gene. A 1.3-kb fragment amplified from the pGN30 plasmid series was digested with XmnI and BstEII prior to ligation into pGN20 digested with the same enzymes. To enable expression in A. tumefaciens, the KpnI fragments from the pGN20 series were moved into the low-copy-number vector pAW50 or the plasmid pRG109. In the case of i248, we used pAW19 as the template, since this mutation lies outside the PvuI-AatII sites on the virA sequence.
TABLE 1.
Trypsin-digested peptides of VirAperi monomer and dimer bands identified by LC-MS-MSa
| Band | Peptideb | Xcorr | DeltaCn |
|---|---|---|---|
| Dimer | R.QSHIVSESNAAQLLR.Q | 4.450 | 0.6 |
| R.SINADSASLQR.D | 4.040 | 0.8 | |
| R.LQDSLASFTR.A | 3.907 | 0.8 | |
| K.NLEDLKQLFR.Q | 3.763 | 0.7 | |
| R.QLEVSLNSADAAVAAFGAQNVR.L | 3.355 | 0.8 | |
| K.ASTDQTLEKPTELASMMLQFLR.Q | 3.213 | 0.6 | |
| R.QPSPAISFEISLELER.L | 2.730 | 0.8 | |
| Monomer | R.QLEVSLNSADAAVAAFGAQNVR.L | 6.882 | 0.9 |
| R.SINADSASLQR.D | 4.345 | 0.8 | |
| R.QSHIVSESNAAQLLR.Q | 4.259 | 0.7 | |
| K.ASTDQTLEKPTELASMMLQFLR.Q | 4.013 | 0.6 | |
| K.NLEDLKQLFR.Q | 3.752 | 0.7 | |
| R.EGPIILSLLPQVK.D | 3.730 | 0.8 | |
| R.LQDSLASFTR.A | 3.405 | 0.9 | |
| R.AHTGTVANYRPIISR.L | 2.612 | 0.7 | |
| R.GLDEAPVR.I | 2.105 | 0.8 |
The two VirAperi bands were excised from a Coomassie blue G-250-stained gel and subjected to mass spectrometry analysis as described in Materials and Methods. Xcorrrepresents the cross-correlation score, which is an indicator of the correlation between expected and observed spectra of the fragment in question. DeltaCn is the normalized correlation score. Xcorr values of >2.5 and deltaCn values of >0.1 are indicative of a strong correlation between theoretical and observed data (76).
Periods indicate trypsin cleavage sites; mass spectrum-identified fragments are between periods.
The site-specific mutations corresponding to virA(N226Y), virA(I236F), and virA(Q229K) were created by using overlap extension PCR with oligonucleotides that carried these mutations and external primers that amplified a 1.7-kb fragment of virA (see Table S2 in the supplemental material). The resulting PCR product was digested with XmnI and BstEII, and the 1.3-kb digestion product was ligated with pGN20 to yield 4.6-kb virA fragments that had these mutations. The 4.6-kb virA was moved into the plasmid pRG109 for expression in A. tumefaciens, resulting in plasmids pGN71, pGN72, and pGN74. All the Agrobacterium expression plasmids were sequenced at the predicted mutation site. The chvE(T187P) allele was isolated in a mutagenesis screen for chvE mutants by screening for blue colonies on pH 5.5 AB induction medium plates with 10 mM glycerol as the sole carbon source (35).
pAW139 [virA(L276R)] and pAW140 [virA(R259S)] were constructed using sequential PCR. The initial rounds of PCR used primer 1705m with either L276Rc or R259Sc and primer 2850c with either primer L276Rm or R259Rm. pAW154 [virA(R259S,L276R)] was constructed using the same method but with pAW139 as the template in PCRs using 1705m/R259Sc and 2850c/R259Sm in the initial PCRs. The secondary rounds of PCRs used the products of the initial PCRs with both 1705m and 2850c to create fragments extending from positions 1705 to 2850 in the 4.6-kb virA sequence which carried either or both the R259S and the L276R mutations. These fragments were digested with NheI and BstEII and used to replace the wild-type NheI-BstEII fragment of pAW19. Following confirmation of the mutant virA sequences, the KpnI fragments of pAW139, pAW140, and pAW154 were cloned into pAW50 to make pAW142, pAW143, and pAW167, respectively.
Yeast two-hybrid β-galactosidase interaction analysis on solid media.
Interaction analysis was performed on solid media as described previously (24). Briefly, pJK202 carrying the lexA-virAperi fusion (pMJ13) and pMJ0, pMJ10, pMJ11, and pMJ12 (see the supplemental material for plasmid descriptions) were introduced into the Saccharomyces cerevisiae strain EGY48 carrying pSH18-34 and grown on leucine-free solid media containing the chromogenic substrate X-Gal. A positive interaction is indicated by blue. The control experiment had the pJK202 plasmid expressing the A. tumefaciens P21 protein (42).
Yeast two-hybrid liquid β-galactosidase assays.
Saccharomyces cerevisiae EGY48 containing the plasmid pSH18-34, a reporter plasmid that carries a Gal1-lexAop-lacZ gene cluster, pMJ13 (see the supplemental material) carrying the wild-type VirA (VirAwt) periplasmic domain, and the empty prey plasmid pJG4-5 was used as a negative control for this assay. The positive control was EGY48 containing pMJ13 with a wild-type periplasmic domain and pMJ0. The experimental subjects were EGY48 containing pMJ13 with the wild-type periplasmic domain and pMJ1 through pMJ9 expressing VirAperi mutants. Using the method described by Miller in 1972 (50), strains were assayed for β-galactosidase activity. Assays were performed in quintuplicate, and results are shown in Miller units.
Nicotiana tabacum virulence assays.
Greenhouse grown Nicotiana tabacum L. cv. Havana 425 plants were used for all leaf transformation protocols, by using procedures previously described (5). Briefly, overnight cultures of A. tumefaciens strains normalized to an optical density at 600 nm (OD600) of ∼0.5 were cocultivated with Nicotiana tabacum L. cv. Havana 425 leaf explants for 48 h on hormone-free MS medium (29) containing the indicated concentrations of AS, followed by transfer to hormone-free MS medium containing 200 μg vancomycin per ml and 200 μg ticarcillin-clavulanate (Timentin) per ml to inhibit bacterial growth. Tumors were scored after 21 days. At least 16 leaf explants per strain were used at each AS concentration.
vir gene induction assays.
pSW209Ω (see Table S1 in the supplemental material) is a reporter plasmid that carries a PvirB::lacZ fusion (PvirB from pTiA6). pRG109 constructs carry PN25virG along with a PvirB::lacZ fusion. Strains were assayed for β-galactosidase by the method of Miller (50) after a 20-h induction at 28°C in AB induction medium (70) buffered at pH 5.5 with 20 mM MES or at pH 7.0 with 75 mM bis-Tris, containing 10 mM l-arabinose, glucose, or glycerol at different AS concentrations. Note that arabinose and glucose have the same effect on vir gene induction (13). The addition of a carboxy-terminal FLAG epitope to wild-type VirA has no discernible effect on its activity (data not shown). Assays were done in triplicate, and results are expressed in Miller units. Fifty percent effective doses (ED50) were obtained by curve fitting normalized dose-response data at different AS concentrations under different conditions to the equation 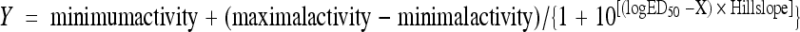 , where X = log [AS]. Y is given as the percent response. Maximal activity was constrained to 100, minimal activity to 0, and the Hill slope to 1.
, where X = log [AS]. Y is given as the percent response. Maximal activity was constrained to 100, minimal activity to 0, and the Hill slope to 1.
Expression of VirAperi.
The cloning, expression, and purification of the periplasmic domain of VirA (aa 39 to 259), utilizing the intein-based Impact-Twin system (New England Biolab, Ipswich, MA), were performed according to the manufacturer's instructions. Briefly, the periplasmic region of VirA was amplified by PCR from pTiA6, which is carried by A. tumefaciens strain A348 (see Table S1 in the supplemental material), using primers Peri-f/Peri-r (see Table S2 in the supplemental material). The C-terminal fusion construct was obtained by cloning the PCR product into pTWIN1 vector at the NdeI and SapI restriction sites. The C-terminal intein tag allows for affinity purification on a chitin column. Primers C244Af/C244Ar were used in overlap extension PCRs for replacing cysteine 244 with an alanine (TGT→GCT). The sequences encoding the periplasmic domains of the two insertion mutants (VirAi169 and VirAi248) were amplified from pGN46 and pGN49a, respectively, using peri-f/peri-r as primers. Constructs were transformed into E. coli BL21(DE3). For protein purification, a single colony was inoculated into 3 ml LB containing 50 μg/ml of carbenicillin and grown at 37°C overnight. Following this, the overnight culture was diluted 100-fold into fresh LB containing 50 μg/ml of carbenicillin and incubated at 37°C until an OD600 of 0.5 to 0.7 was obtained (∼2 h and 15 min). IPTG was added to a final concentration of 0.2 mM, and the culture was grown at 15°C overnight. Cells were then collected by centrifugation at 5,000 × g for 10 min and resuspended in buffer B2 (20 mM HEPES, pH 7.0, 500 mM NaCl, 1 mM EDTA) to an OD600 of ∼20. The cells were then sonicated on ice and clarified cell extracts obtained by centrifugation at 19,000 × g for 30 min. Cell extracts were then applied to a chitin column (∼2 ml) equilibrated in buffer B2. The column was washed with 15 column volumes of buffer B2 to remove unbound proteins. To induce on-column cleavage of periplasmic domain of VirAperi from the intein tag, the column was flushed with three column volumes of buffer B3 (20 mM HEPES, pH 8.5, 500 mM NaCl, 1 mM EDTA, 40 mM dithiothreitol [DTT]). The cleavage reaction was allowed to proceed overnight at 4°C. The target protein was then eluted with buffer B3.
Sample preparation and mass spectrometry analysis.
Gel pieces were excised from a Coomassie blue G-250-stained gel, washed with double-distilled water by vortexing, destained with 50% methyl cyanide (CH3CN) for 15 min, and washed repeatedly for 15 min at room temperature with 100 mM ammonium carbonate-methyl cyanide [(NH3)2CO3-CH3CN] (50/50, vol/vol) until completely destained. They were then shrunk for another 5 min by adding 100% CH3CN, dried in a SpeedVac, and then suspended in a solution containing 6 ng/μl trypsin (Promega Inc., Madison, WI) in 50 mM ammonium bicarbonate (NH4HCO3) and incubated at 37°C overnight. After incubation, 30 μl of a solution containing 1% formic acid (HCOOH) and 2% CH3CN together was added to each digested sample and vortexed for 30 min at room temperature to complete extraction of digested peptides. After a brief centrifugation cycle, the supernatant was collected and the sample was dried in a SpeedVac. Following this, samples were subject to liquid chromatography-tandem mass spectrometry (LC-MS-MS) analysis. Autosampling and chromatography were performed essentially as described previously (55).
Mass spectra were measured with an LCQ Deca XP Plus ion trap mass spectrometer (Thermo Finnigan). Mass spectrometry scans as well as high-performance liquid chromatography (HPLC) solvent gradients were controlled by the XCalibur software (Thermo Finnigan). Experimentally collected tandem spectra were searched against the pTiA6 proteome database using SEQUEST software by comparison with the theoretical spectra of all possible peptide fragments from the SEQUEST database (43, 76).
Far-UV CD spectroscopy.
The circular dichroism (CD) spectra were obtained with 7.5 μM wild-type VirAperi in 50 mM NaHPO4 buffer (pH 7.6) and 50 mM NaCl or 7.5 μM VirAi248 in the same buffer. Measurements were taken in 1-nm increments from 260 to 200 nm in a 0.1-cm-path-length cuvette with a bandwidth of 1 nm on a Jasco J-810 spectropolarimeter. The spectrum was the average of three scans at 20°C.
Analytical ultracentrifugation.
The sedimentation equilibrium experiment and data analysis were performed as previously described (78). This experiment was conducted at 25°C using a Beckman XL1 analytical ultracentrifuge with absorbance optics. A 1-mg/ml volume of purified VirAperi in 10 mM Tris-HCl, pH 8.0, 50 mM NaCl, and 5% glycerol was loaded, and equilibrium data were collected at 20,000, 25,000, 30,000, and 35,000 rpm. Data obtained were globally fitted by nonlinear least-squares curve by IGOR Pro (Wavemetrics).
RESULTS
The dimerization of the VirA periplasmic domain.
The periplasmic domain of VirA (VirAperi) lies between amino acids 40 and 259. Secondary structure prediction programs indicate that VirAperi has at least 10 major helical regions located throughout this domain, beginning with amino acid 40 (Fig. 1) (10, 41). This is similar to the α-helical content of the periplasmic domains of Tar and Trg, each of which interacts with periplasmic sugar binding proteins similar to ChvE (11, 34). To develop a better understanding of VirAperi, we utilized genetic and biochemical tools to test the hypothesis that this domain, in isolation from the transmembrane and cytoplasmic domains, can participate in dimer formation and to determine which, if any, of the predicted α-helical regions may be required for dimerization. Our first approach utilized the yeast two-hybrid system (28). To this end, the periplasmic domain of VirA (aa 40 to 259) was cloned into a bait vector (see Materials and Methods) and either the wild type or mutant versions of this domain were cloned into the prey vector and tested for interaction. The results (Fig. 2) indicate that the wild-type periplasmic domains do interact, as indicated by the very high levels of β-galactosidase activity. Not surprisingly, the negative control, the empty vector, and a version of this domain with a very large deletion from amino acids 63 to 240 (47) were not able to interact with the wild-type domain. Two other versions of the periplasmic domain known to affect sugar signaling first described by Banta et al. (5), Glu-254,Glu-255→Gln-254,Leu-255 (EE-QL) and that with a deletion of amino acids 242 to 257 (Fig. 2, fourth and fifth rows), were both capable of interacting with the wild-type domain, indicating that these residues are not essential for interaction.
FIG. 2.
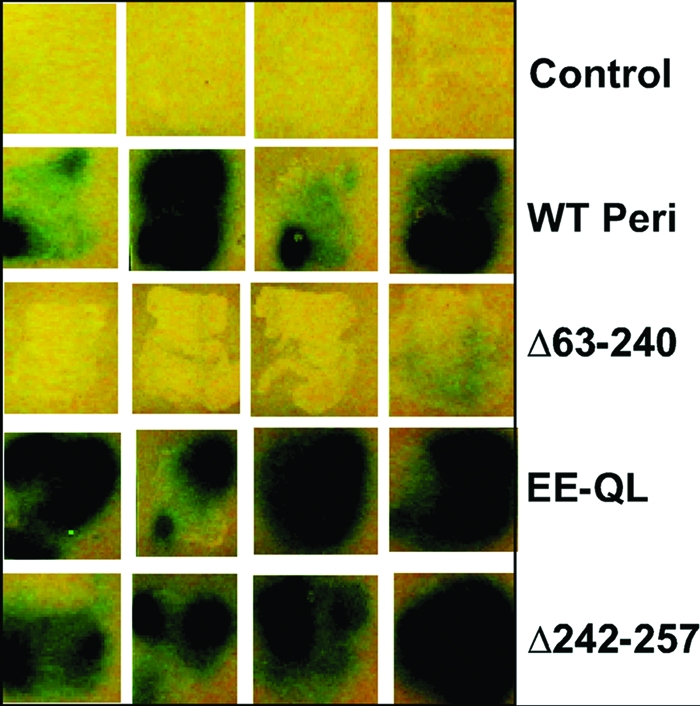
Yeast two-hybrid analysis of VirAperi interactions. Saccharomyces cerevisiae EGY48 containing the plasmid pSH18-34, a reporter plasmid that carries a Gal1-lexAop-lacZ gene cluster, with a wild-type periplasmic domain (pMJ13) and the prey plasmid pJG4-5 expressing the P21 protein, was used as a negative control for this assay. The positive control was EGY48 containing pJK202 with a wild-type periplasmic domain and pMJ0 expressing the wild-type periplasmic domain of VirA (WT Peri). The experimental subjects were EGY48 containing pJK202 with a wild-type periplasmic domain and prey plasmids expressing VirAperi mutants as shown. Shown here are four independent streaks of each strain on leucine-free solid media containing the chromogenic substrate X-Gal (5-bromo-4-chloro-3-indolyl-d-galactopyranoside). Blue is indicative of a positive interaction. See the text for a discussion of the results. EE-QL, Glu-254,Glu-255→Gln-254,Leu-255 mutant.
These results are consistent with a model in which the VirA periplasmic domain is capable of dimerizing independently of the transmembrane and cytoplasmic domains. To further test this, the periplasmic domain was purified as described in Materials and Methods. Briefly, a PCR product encoding amino acids 39 to 259 was cloned into the pTWIN1 vector (NEB), creating an in-frame fusion with intein and the chitin binding protein (CBP), yielding plasmid pXL10. Soluble proteins from E. coli BL21(DE3) induced with IPTG were passed over a chitin column and eluted with a DTT-containing buffer which cleaves the periplasmic domain from the VirAperi-intein-CBP fusion protein, yielding a >90% pure preparation of VirAperi (Fig. 3A). Interestingly, when this eluate was dialyzed against buffers without DTT, there appeared on the denaturing gel a band (Fig. 3A, lane 3) that ran at about twice the size of the monomer (47 versus 23 kDa). This apparent dimerization could be partially reversed via the reintroduction of DTT and high NaCl (500 mM) (data not shown). To test the hypothesis that the 47-kDa band is in fact a dimer of VirAperi, both the 23-kDa and 47-kDa species were purified from the gel and subjected to analysis via LC-MS-MS. These results demonstrate that the only peptides found at significant levels from either band are those from VirAperi (Table 1). Given that the higher-molecular-mass species runs at the size of a VirAperi dimer and includes only VirAperi peptides, we conclude that it is, in fact, a dimer of this domain. Analytical ultracentrifugation was used to investigate the dimerization of isolated VirAperi (Fig. 3B). The data best fit a monomer-dimer model with a Kd (dissociation constant) of 39.8 μM, providing additional evidence that the periplasmic domain can dimerize by itself without requiring either the transmembrane domain or the cytoplasmic domain as a dimerization tether.
FIG. 3.

VirAperi isolation, dimerization, and helical abundance. (A) SDS-PAGE (10%) analysis of the wild-type VirAperi and the insertion mutant (i248). Lane 1, protein standards with molecular sizes labeled in kDa; lanes 2 and 3, wild-type VirAperi before and after dialysis against buffer A (20 mM HEPES, pH 8.5, 50 mM NaCl), respectively; lanes 4 and 5, VirAperi with the i248 mutation before and after dialysis against buffer A, respectively. The elution buffer for purification is 20 mM HEPES, pH 8.5, 500 mM NaCl, 40 mM DTT, and 1 mM EDTA. (B) Analytical ultracentrifugation analysis of VirAperi. The dots in the lower panel represent measured absorbances at 280 nm taken at 20,000, 25,000, 30,000, and 35,000 rpm. The best-fit curves (continuous lines) for the equilibrium model in a monomer-dimer equilibrium with a Kd of 39.8 μM are superimposed. AutoCorr, autocorrelation; R, radius. (C) Far-UV CD spectroscopy of wild-type (wt) VirAperi and VirAperi with i248. The spectra of both wt VirAperi and VirAperi with i248 were obtained at a concentration of 7.5 μM in 50 mM NaHPO4 buffer (pH 7.6) and 50 mM NaCl. Measurements were taken in 1-nm increments from 260 to 200 nm in a 0.1-cm-path-length cuvette with a bandwidth of 1 nm on a Jasco J810 spectropolarimeter. The spectrum was the average of three scans at 20°C. res, residue.
Because of the DTT sensitivity of the putative VirAperi dimer, we postulated that it ran as a dimer in the denaturing gel system as a result of a disulfide bond that could form via the sole cysteine residue (C244) in the periplasmic domain. This cysteine was converted to alanine via site-directed mutagenesis, and the resultant VirAperi-C244A was purified and tested as described above. Surprisingly, VirAperi-C244A exhibited the same phenotype as the wild-type VirAperi—it was able to dimerize after dialysis, and the dimer continued to be DTT sensitive (data not shown). We conclude that C244 and a disulfide bond between the C244 residues of participating monomers are not required for the in vitro dimerization of VirAperi.
The CD spectrum of purified VirAperi (Fig. 3C) is consistent with the prediction that the periplasmic domain of VirA contains abundant α-helices (Fig. 1). CD spectrum analysis programs predicted the α-helical content to be approximately 87% (59). However, further structural analysis would be required to definitively confirm these results. The ratio of molar ellipticity at 220 to that at 208 nm was found to be 0.99 for VirAperi, which might indicate the presence of a two-stranded α-helical structure (51). We next examined the role such helices may play in the interaction between VirAperi monomers by engineering helix-disrupting four-amino-acid insertions (amino acid sequence GSPW) in predicted helices at regular intervals across VirAperi by overlap extension PCR. These were then cloned into the yeast two-hybrid prey vector and tested for interaction with wild-type VirAperi as described above. The results of a quantitative β-galactosidase assay indicate that only one insertion (i169) significantly affected interaction (Fig. 4). VirAperi domains with insertions i169 and i248 were also purified as described above and tested for in vitro dimerization. VirAi169 was not soluble and could not be tested via this assay. Consistent with the yeast two-hybrid analysis, VirAi248 was soluble and exhibited the same capacity to form a dimer as did the wild type (Fig. 3A, lanes 4 and 5). Moreover, it has a CD spectrum similar to that of the wild type (Fig. 3C) except with less α-helical content, indicating that the insertion did not cause major structural changes to the protein or affect its ability to dimerize. We conclude that the insertions, with the exception of i169, allow the periplasmic domain to participate in the dimer formation.
FIG. 4.

β-Galactosidase activities of VirAperi insertion mutants. The graph depicts results of a representative yeast two-hybrid assay to characterize the interactions of the VirA periplasmic domain with VirAperi mutations. Liquid assays for β-galactosidase activity of yeast strains carrying a wild-type VirAperi bait domain and various insertion mutants of the periplasmic domain were performed to study interaction as described in the Materials and Methods. Activity is expressed in Miller units. See the text for a description of the results. This assay was performed in triplicate, and similar results were observed in at least two independent experiments. neg, negative control.
Biological characterization of VirAperi insertion mutants.
To gain more insight into the contribution of the highly structured periplasmic domain to VirA activity, the helix-breaking insertion mutations described above were made in full-length VirA, and the mutants were cloned into the low-copy-number plasmid pAW50 (73; see also Materials and Methods and Table S1 in the supplemental material). Plasmids carrying mutant alleles were introduced into A. tumefaciens strain A348-3 (see the supplemental material), which does not carry a functional copy of the virA allele, as was pSW209Ω, a PvirB::lacZ reporter plasmid. These strains were tested under a variety of induction conditions, different AS concentrations, inducing sugars, and pHs. Unexpectedly, despite being distributed across the entire periplasmic domain, all of these mutations had very similar effects on vir gene induction. In contrast to wild-type VirA, which shows high activity at low concentrations of AS in the presence of sugar, none of these mutants was able to induce vir gene expression at 10 mM arabinose and an AS concentration of 1 μM (Fig. 5A). At 100 μM AS in the presence of arabinose, the mutants are active, though at levels approximately 2- to 3-fold lower than those of the wild type. Strikingly, in the absence of the inducing sugar (Fig. 5B), each of the insertion mutants induced higher levels of vir gene expression than wild-type VirA at both 10 and 100 μM AS. Finally, all of the insertion mutants supported high levels of activity at both pH 5.5 and pH 7.0 both in the presence and absence of sugar (Fig. 5C), contrasting with wild-type VirA, which requires low pH to support vir gene induction. Taken together, these results indicate that the insertion mutations across the domain (i) relieve the apparent repression of VirA activity by the periplasmic domain when sugar is absent and (ii) disrupt the capacity for this domain to respond to the ChvE/sugar signaling mechanism that increases the sensitivity of the system to AS.
FIG. 5.

vir induction profiles of insertion mutants (see the text for a detailed discussion of the results). A348-3 strains carrying the PvirB::lacZ reporter fusion on pSW209Ω and virA mutant alleles on pAW50 were grown at 25°C in pH 5.5 induction medium with 10 mM l-arabinose (a) or glycerol (b), at low and high concentrations of the phenolic inducer acetosyringone (AS), and in pH 7.0 medium with 10 mM arabinose or glycerol with 200 μM AS (c). β-Galactosidase activity was determined after 20 h. Results are reported in Miller units. Samples were assayed in triplicate, and results are plotted as means with standard errors. Similar results were observed in three independent assays.
Detailed analysis of the AS sensitivity of the VirAperi proteins, particularly in relation to pH, is complicated by the fact that virG expression is induced by low pH (17). To eliminate the pH-dependent effect on virG expression, the virA-containing plasmids pGN60 and pGN66 (both pRG109 derivatives, described in Materials and Methods and Table S1 of the supplemental material) were moved into strain A136 (no Ti plasmid). The plasmid pRG109 carries virG under the expression of the constitutive, pH-independent PN25 promoter (30) as well as a PvirB-lacZ reporter construct. Analysis of strains carrying either the wild-type virA or virAi169 revealed that (i) VirAi169 is ∼30-fold less sensitive to AS than wild-type VirA is when tested in the presence of an inducing sugar, (ii) the sensitivity of VirAi169 to AS is unaffected by sugar, and (iii) in the absence of inducing sugars, the maximal vir gene induction level with VirAi169 is nearly as high as that supported by wild-type VirA in the presence of sugar (Fig. 6A). At pH 7.0, regardless of inducing sugar supplement, VirAi169 supported significantly higher levels of vir gene expression than did wild-type VirA (Fig. 6B). Virtually identical results were obtained in tests of VirAi248 (data not shown). Because the virAi169 and virAi248 alleles result in at least one phenotype that ordinarily requires ChvE (high maximal activity), we tested them for this activity in the absence of ChvE by moving the alleles into strain DL8 (lacking chvE) (30), also carrying pRG109 as described above. The results were identical to those described above for the chvE+ strain, A136 (data not shown), indicating that ChvE is not required for the high-maximal-activity phenotype observed in the absence of inducing sugar.
FIG. 6.

Comparative AS dose-response determinations for wild-type VirA and VirAi169 and the effect of AS sensitivity on tumor initiation. A136 (wild-type chvE) strains carrying wild-type virA and virAi169 on pRG109 (pGN60 and pGN66; see the supplemental material for a description) were grown at 25°C in induction medium with 10 mM l-arabinose and glycerol at either pH 5.5 (a) or pH 7.0 (b). AS concentrations from 0 to 300 μM were used. β-Galactosidase activity was determined after 20 h, and the results are expressed in Miller units. The top right corners of panels a and b depict ED50 values (the concentrations of the phenolic inducers that elicit 50% of the maximal response; calculated as described in Materials and Methods) in μM AS. Samples were assayed in triplicate and plotted as means with standard errors. Three independent experiments were performed and yielded similar results. (c) Tumorigenesis assay. A348-3 strains carrying plasmids expressing wild-type VirA, VirAi169, or VirAi248 were evaluated for the ability to form tumors by cocultivation with tobacco leaf explants at different external AS concentrations on hormone-free MS medium. This graph represents the average number of tumors per leaf explant for each strain at cell concentrations of 0.5 OD600. The error bars on the graph represent standard errors (n = 14). Two independent virulence assays were performed before conclusions were drawn.
To study the relationship between the tumorigenic capacities of the insertion mutants and their abilities to sense AS, virA and two representative mutants, virAi169 and virAi248, cloned into the low-copy-number vector pAW50, were electroporated into A348-3, a virA deletion strain. These strains were cocultivated with tobacco leaf explants at different concentrations of AS in the cocultivation medium, and tumors were scored after 12 to 14 days (see Materials and Methods). The concentrations of AS ranged from 0 μM to 500 μM. Figure 6C shows the average numbers of tumors for each strain at different concentrations of AS. When no exogenous AS is provided in the cocultivation medium, the inciting strain has to rely on phenolics present at the wound site. Only the wild-type strain produced tumors under those conditions. VirAi169 and VirAi248 required higher concentrations of AS to stimulate any tumor formation. Thus, the virulence profile for the mutants was a reflection of their loss of sensitivity to AS in the presence of arabinose.
Repositioning of the TM2-spanning helix.
A working model for the physical response of periplasmic sensor domains of both chemoreceptors and histidine kinases comprises the ligand inducing a shift of the second transmembrane helix toward the cytoplasm (“piston model”), resulting in an alteration in cytoplasmic activity of the protein (19, 27, 36). Support for this model has come in several forms, including engineering of the TM2-spanning helix so that it would be shifted as if a piston-like motion had moved the helix toward the cytoplasm. Such mutations (for example, the W209R mutation in the TM2 domain of TAR or others in the E. coli nitrate sensors NarQ and NarX) have been shown to result in a ligand-independent “locked” configuration with a phenotype of the ligand-bound wild-type receptor (21). We therefore examined the effect that certain mutations (specifically, R259 and L276; Fig. 1) at the flanks of the TM2-spanning region would have on the predicted position of that helix. Both TM-pred and E-Z potential (37, 63) predict that the transmembrane domain of TM2 of the double mutant VirAR259S,L276R is shifted dramatically compared to the wild type (ig. 1), with two residues (277 and 278) previously associated with the inner face of the membrane now predicted to be in the cytoplasm, whereas each individual mutation was not predicted to cause such a shift. The “outside” border of the predicted TM2 transmembrane domain of VirAR259S was not altered according to prediction programs, though the inside border showed a one-amino-acid change, with the valine 277 residue predicted to be the last residue of this domain. This shift was confirmed by using vir gene induction assays. For example, VirAR259S had only a small effect on vir gene expression, being sugar sensitive though having much lower sensitivity to AS than VirAwt had (Fig. 7A). The other single TM2 mutant, VirAL276R, was inactive at all AS concentrations tested. It is possible that the mutant 276R residue did not allow for appropriate insertion into the membrane and thus yielded an inactive protein. In contrast to the single mutants, VirAR259S,L276R had a profound effect on the vir gene expression profile: this form of VirA supported high maximal activity, but low AS sensitivity, in the absence of sugar and was unresponsive to the effects of inducing sugar (Fig. 7B). This phenotype is the same as that of the insertion mutants described above—in fact, VirAR259S,L276R also supports vir gene induction at pH 7 (data not shown), as do the insertion mutants.
FIG. 7.

TM2 mutants. A348-3 strains carrying the PvirB::lacZ reporter fusion on pSW209Ω and wild-type virA or the TM2 R259S and L276R single mutants or the R259S/L276R double mutant on the plasmid pAW50 were grown at 25°C in pH 5.5 induction medium with 10 mM glucose (a) or glycerol (b), at increasing concentrations of the phenolic inducer acetosyringone (AS). β-Galactosidase activity was determined after 20 h. Results are reported in Miller units. Samples were assayed in triplicate, and results are plotted as means with standard deviations. Three independent assays were performed.
Analysis of the Trg-like region within the periplasmic domain of VirA.
The Trg chemoreceptor of E. coli interacts with the periplasmic galactose and ribose binding proteins (GBP and RBP, respectively) to induce chemotaxis toward these sugars (see the introduction). ChvE shows ∼30% sequence similarity to these two periplasmic binding proteins (3). While the VirA periplasmic domain is not closely related by sequence to the Trg periplasmic domain, it shares a short region of identity with a Trg domain that is proposed to be the ligand interaction region of Trg (Fig. 8) (13, 74). This region lies between amino acids 226 and 239 of VirA or, more pertinently, between the E210 and E255 residues, which are the sites of mutations that have previously been shown to result in loss of ChvE-mediated sugar sensitivity. To test whether site-specific mutations at residues of VirA that are identical to those of the proposed ligand interaction domain of Trg (Fig. 8) result in similar effects in the VirA-ChvE interaction, we generated the virA(N226Y), virA(Q229K), virA(Q229P), and virA(I236F) alleles. These were chosen because the analogous mutations in Trg had three distinct and interesting signaling properties that would be interesting to replicate in the VirA-ChvE system. For example, TrgN76Y, analogous to VirAN226Y, displayed a “mimicked-occupancy” phenotype, i.e., the chemoreceptor acted as if sugar-bound RBP was bound to it, whereas TrgI86F was insensitive to the RBP signal. The Trg residue Q at position 79 was interesting because a conversion to a lysine (Q79K) resulted in a mimicked-occupancy phenotype, whereas a change to proline rendered the protein inactive (74). Each of the virA allele mutants was tested in the absence of ChvE or in the presence of wild-type ChvE or ChvET187P. The latter corresponds to a hyperactive allele of chvE that mimics the sugar-bound form of ChvE, that is, inducing sugars are not required to cause high sensitivity to phenols and maximal vir-inducing activity by wild-type VirA (35).
FIG. 8.

Sequence alignment of a portion of the VirA periplasmic domain and the ligand recognition domain of Trg, showing the presence of an identical 4-XXXXX-4 amino acid motif with two regions of identity (depicted in gray and underlined). This motif in Trg is thought to be the ligand interaction site for ribose binding protein and has been the site of extensive study (7, 58, 74). VirA amino acids that were the mutagenized for this study are boxed, with substitutions at these sites shown beneath them.
The mutant virA alleles were cloned into the pRG109 plasmid described above and transformed into the DL8 strain (ΔchvE, no Ti plasmid) carrying ChvE or ChvET187P in the pBBR1-5 vector (described in Materials and Methods). Each strain was then tested at various AS concentrations in the presence of glucose, yielding estimates of maximal vir gene expression at saturating concentrations of AS (maximal activity) and AS sensitivity, presented as the ED50 (concentration of AS at half the maximal activity) (Table 2; see Fig. S1 in the supplemental material). As expected, the AS sensitivity and maximal activity supported by wild-type VirA are both dramatically increased when ChvE is present, and interestingly, ChvET187P had a modest effect on maximal activity but increased AS sensitivity even further (∼10-fold) compared to wild-type ChvE. Also as reported previously (5), VirAE255L tested in the presence of ChvE exhibited a marked decrease in AS sensitivity (nearly the same as wild-type VirA tested in the absence of ChvE), but its maximal activity was not affected. ChvET187P was capable of increasing the AS sensitivity exhibited by the strain with VirAE255L to near-wild-type levels but had no effect on maximal activity. As reported above, in the absence of ChvE (and/or sugar), VirAi169 supports a level of maximal activity significantly above that of wild-type VirA but a low sensitivity to AS. Neither ChvE nor ChvET187P affected either maximal activity or AS sensitivity. Analysis of the point mutations in the Trg-like domain demonstrates that the corresponding residues are indeed critical for ChvE-mediated sugar signaling. In terms of AS sensitivity, VirAN226Y and VirAI236F supported phenotypes similar to that supported by VirAE255L: when tested in the presence of ChvE, their AS sensitivity was low compared to that of wild-type VirA, and ChvET187P dramatically increased the AS sensitivity. However, the maximal activity supported by VirAN226Y and VirAI236F in the presence of ChvE was significantly lower than that supported by VirAE255L. ChvET187P was able, though, to significantly enhance the maximal-activity phenotype supported by either VirAN226Y or VirAI236F.
TABLE 2.
Effect of ChvE on AS sensitivity and maximal activity of VirA as a function of β-galactosidase activity
| Protein | No ChvE |
Wild-type ChvE |
ChvET187P |
|||
|---|---|---|---|---|---|---|
| ED50 (μM)a | Maximal activityb | ED50 (μM)a | Maximal activityb | ED50 (μM)a | Maximal activityb | |
| VirAwt | 33 | 660 ± 92 | 2 | 5,717 ± 548 | 0.3 | 7,928 ± 188 |
| VirAE255L | 44 | 950 ± 37 | 19 | 6,066 ± 38 | 4 | 5,916 ± 351 |
| VirAN226Y | 42 | 550 ± 33 | 15 | 2,340 ± 232 | 1 | 7,052 ± 112 |
| VirAI236F | 75 | 655 ± 212 | 43 | 1,531 ± 125 | 7 | 4,213 ± 595 |
| VirAQ229K | 108 | 2,076 ± 362 | 121 | 1,807 ± 73 | 78 | 4,541 ± 254 |
| VirAi169 | 26 | 2,838 ± 219 | 19 | 2,342 ± 15 | 23 | 2,190 ± 133 |
Expressed as ED50, or the micromolar concentration of the phenolic inducer that elicits 50% of the maximal response (calculated as described in Materials and Methods).
β-Galactosidase activities ± standard deviations are expressed in Miller units and represented here as averages (and standard deviations) of at least three experiments.
The glutamine residue at position 229 was converted to either a proline or a lysine. VirAQ229P was inactive under all conditions tested (data not shown), as was the Q79P form of Trg. VirAQ229K exhibited a novel set of phenotypes (Table 2) compared to the other forms with mutations in the periplasmic domain that affect sugar signaling. As with VirAN226Y and VirAI236F, VirAQ229K was much less sensitive to AS than wild-type VirA when they were tested in the presence of wild-type ChvE and inducing sugar. In contrast to those supported by VirAN226Y and VirAI236F, the AS sensitivity supported by VirAQ229K was unaffected by ChvET187P. The maximal activity supported by VirAQ229K tested in the absence of ChvE was significantly higher (∼3-fold) than that supported by wild-type VirA tested under the same conditions, analogous to the mimicked-occupancy phenotype of TrgQ79K. While the maximal activity supported by VirAQ229K was not increased in the presence of wild-type ChvE, it showed a dramatic increase when tested in the presence of ChvET187P. Thus, VirAQ229K cannot respond to ChvET187P in terms of AS sensitivity but is still capable of responding in terms of maximal activity.
DISCUSSION
The periplasmic domain of VirA is hypothesized to interact with sugar-bound ChvE, possibly in a pH-dependent fashion (30, 64). This results in the transmission of information to the cytoplasmic domain of VirA, increasing sensitivity to phenolics, and leading to high levels of vir gene expression at saturating levels of phenolics. Our results are consistent with the model in which the periplasmic domain is a repressive element in the context of maximal activity, whereas it serves as a positive element in relation to phenolic sensitivity. Moreover, these phenotypes can be genetically uncoupled, strongly suggesting that distinct transducing mechanisms must exist.
Dimerization of the VirA periplasmic domain.
The periplasmic domain of VirA has a high proportion of α-helices, as predicted via secondary structure prediction programs (Fig. 1) and subsequently confirmed by the CD spectrum of purified VirAperi (Fig. 3C). An important feature of the sensor domains of most histidine kinases is that their existence as dimers is integral to their ability to transmit signals within the cell (19). The presence of biologically relevant dimers has been demonstrated both biophysically and genetically for the Tar chemoreceptor and histidine kinases such as PhoQ, DcuS, NarX, and TorS (20, 32, 77, 79). The evidence for VirA dimers comes by way of genetic analysis using intersubunit complementation (9, 66, 73) and by chemical cross-linking analysis indicating that VirA exists as a dimer even in the absence of the inducing ligands (57). While these earlier studies affirm that VirA exists as a dimer and that dimerization is critical to VirA function, our investigations demonstrate periplasmic domain dimerization in the absence of tethering via transmembrane and cytoplasmic domains. A yeast two-hybrid analysis revealed that the wild-type VirAperi domain interacted strongly with itself but not with a form carrying a large deletion (from aa 63 to 240 [VirAΔ63-240]). In an effort to identify sites within the periplasmic domain that are necessary for monomer-monomer interaction, helix-breaking insertions were engineered in all predicted α-helices across the entire domain. Interestingly, with the sole exception of that corresponding to i169, the insertion mutants were able to interact readily with the wild-type periplasmic domain in the yeast two-hybrid system (Fig. 4).
In vitro studies demonstrated that the periplasmic domain has significant α-helical character and can form a dimer in solution. Interestingly, this dimer appears extremely stable even when subjected to SDS-PAGE. Analytical ultracentrifugation of the soluble, isolated periplasmic domain indicates that the dimer forms with a Kd of 39.8 μM, in line with the Kd observed for the isolated periplasmic domain of the trimethylamine N-oxide (TMAO)-responsive histidine kinase TorS (52). The dissociation constant is indicative of a strong and biologically relevant association. Moreover, as noted by Moore and Hendrickson, the actual Kds of such domains when tethered to the membrane via their transmembrane domains are likely to be significantly lower (52). The observation that the soluble VirA periplasmic domain can migrate as a dimer under denaturing conditions also indicates a very strong interaction. There have been some reports of histidine kinases migrating as dimers when treated with SDS, one of the earliest examples being the Arabidopsis thaliana ethylene-responsive ETR1 (16). Surprisingly, replacement of the one cysteine residue in the VirA periplasmic domain did not alter its capacity to dimerize (data not shown), suggesting that a very stable nondisulfide interaction between the VirAperi monomers must occur. Such stable dimerization has also been noted for the BM2 integral membrane protein of influenza B virus and other histidine kinases, like NarX, whose structures have been resolved (12, 20). SDS-stable coiled-coil domains have been reported for other proteins, for example, the Epstein-Barr virus transcription factor, Zta (62). Programs to predict coiled coils in proteins, such as Marcoils and PCOILS, show that there is a high probability that VirAperi forms coiled coils even though it lacks the presence of heptad motifs associated with coiled-coil formation (see Fig. S2 in the supplemental material) (25, 43, 44). Earlier studies have shown that the yeast Sir4 dimerization motif, also a coiled coil, does not share a consensus sequence with coiled coils (18). Thus, one possibility is that the SDS stability of VirAperi could be the result of a strong coiled-coil interaction.
Consistent with the yeast two-hybrid analysis, characterization of isolated VirAi248 indicated that it also continued to be capable of dimerization in solution. The VirAi169 protein expressed in E. coli was insoluble, indicating that the i169 mutation had a more deleterious effect on the structure of the periplasmic domain. However, because the full-length protein with the i169 mutation is capable of successfully inducing vir gene expression, we assume that it is forming a homodimer by virtue of its cytoplasmic and TM domains, a phenomenon that has been observed for E. coli EnvZ (62). Our analysis indicates that periplasmic domain dimerization is quite strong and likely to result from interactions at more than one helical domain within the protein. This is consistent with results for other histidine kinase periplasmic domains that have multiple sites of interaction (20, 52).
The VirA periplasmic domain is both a repressive and activating element.
The role of the periplasmic domain as a negative regulator of histidine kinase activity has been observed before in the form of mimicked-occupancy or “locked-on” phenotypes seen in mutant versions of other systems, for example, the CpxAR system in E. coli, the nitrate sensors NarQ and NarX, the chemoreceptor Trg, and most recently, the PhoQ system in Salmonella enterica serovar Typhimurium (14, 21, 23, 74). The insertion mutants described in this study can be viewed, in one context, as being in the locked-on state: they relieve the repressive effects of the periplasmic domain on maximal activity in the absence of inducing sugars or at neutral pH (Fig. 5 and 6). However, these mutants cannot respond to ChvE and sugars that normally increase AS sensitivity of wild-type VirA. Taken together, the available data support a model in which the ChvE-VirA periplasmic domain interaction is required for two layers of mechanistic control: (i) release of negative regulation by the periplasmic domain that affects maximal activity under suboptimal inducing conditions, for example, in the absence of inducing sugars or at neutral pH, and (ii) stimulation of positive regulation on the cytoplasmic domain in the presence of inducing sugars and low pH, leading to increased sensitivity to AS.
Current models for the control of cytoplasmic activity of structurally related histidine kinases and chemoreceptors suggest that the periplasmic domain responds to ligands by the displacement of the α-helix spanning TM2 by one or two amino acids toward the cytoplasm, with the rigidity of this helix causing perturbation of a cytoplasmic “signal conversion” domain resulting in effects on kinase activity (19, 34, 36). As a first step to determine whether either or both of the two phenotypes controlled by the VirA periplasmic domain (maximal activity and phenolic sensitivity) are related to such a piston-like movement, we performed site-specific mutations that were predicted, via several different predictive programs (37, 63), to result in a VirA protein (VirAR259S,L276R) in which the TM2-spanning α-helix is shifted by 1 or 2 amino acids toward the cytoplasm. The results reported here showed that this mutant form of VirA exhibited a high maximal vir-inducing activity compared to that of wild-type VirA when tested in the absence of inducing sugars (Fig. 7) but could not respond to inducing sugars and had extremely low sensitivity to phenolics. These results, essentially the same as those found in the analysis of the insertion mutants, are consistent with a model in which the periplasmic domain of VirA is in a highly constrained, repressive conformation. While the ChvE/sugar signaling mechanism is capable of not only releasing the repression but also activating an increase in phenolic sensitivity, the insertion mutations and the TM2 mutations appear to release the repressive effect, possibly by allowing (causing) an uncontrolled displacement of the TM2-spanning α-helix toward the cytoplasm. However, this type of release appears to completely eliminate the capacity for VirA to respond to the activating properties of ChvE. One possibility is that an uncontrolled displacement of the TM2 α-helix by the mutations described disrupts the capacity for some signal conversion module in the cytoplasmic domain to achieve an activated status in terms of phenolic sensitivity. One candidate for a regulatory scheme has been proposed by Gao and Lynn, who suggest that the rotation of α-helices just cytoplasmic to TM2 are critical in controlling VirA activity (31) (see also last paragraph in the text).
In the biological context, the abilities of VirAi169 and another insertion mutant, VirAi248, to form tumors on tobacco leaf explants correlate directly with their sensitivity to AS in the presence of inducing sugars (Fig. 6C). Interestingly, VirA carrying a large periplasmic deletion (VirAΔ63-240), exhibiting a vir induction phenotype similar to that of the insertion mutants, was found to confer an intermediate level of tumorigenesis when the external AS concentration was varied in quantitative tumor assays (5). Importantly, the results of Banta et al. and the concurrent tumorigenic and in vitro dose-response assays of the VirA mutants studied here prove that the ability to form tumors is a function of sensitivity of VirA to plant-derived phenolics like AS and not maximal activity. This suggests that at least these plant tissues have limiting amounts of phenolics available and demonstrate the critical role of sugars in the virulence process.
Critical role of the Trg-like region.
The results presented here demonstrate that the small region in VirA that has strong similarities in sequence to a region in the periplasmic domain of the E. coli methyl-accepting chemoreceptor Trg has a critical role in sugar signaling (13). Genetic and biochemical analyses suggest that this small motif in Trg represents the putative ligand binding protein interaction (74, 75). Amino acid substitutions in this region of Trg (e.g., N76Y and Q79K) can result in a mimicked ligand (sugar periplasmic binding protein [PBP]) occupancy phenotype. An earlier study examined a mutation at an amino acid residue corresponding to Q79 in Trg (74). In this case, VirAQ229L showed an ∼3-fold increase in activity over the wild-type level in the absence of inducing sugars, suggesting a mimicked-occupancy phenotype, but the corresponding strain was still capable of high AS sensitivity and maximal activity in the presence of sugars, resulting in a conclusion that the region was not critical in the sugar signal transduction process (66). In contrast, our examination of mutations in VirA that correspond to the Trg mutants described by Yaghmai and Hazelbauer (74, 75) shows that this region is crucial in sugar signal transduction and provides further evidence of the degree to which two key sugar-related phenotypes, maximal sensitivity and maximal activity, can be uncoupled. Strains carrying VirAN226Y, VirAI236F, or VirAQ229K exhibit phenotypes distinctly different from those of the previously studied VirAE255L (Table 2; see Fig. S1 in the supplemental material). For VirAN226Y, VirAI236F, and VirAQ229K, both AS sensitivity and maximal vir-inducing activity at saturating AS concentrations were, when tested in the presence of inducing sugars and wild-type ChvE, significantly lower than those for wild-type VirA, whereas in the case of VirAE255L, only AS sensitivity was affected. One interpretation of these data is that the mutations in the Trg-like region affect the interaction of ChvE with VirA, whereas the E255L mutation affects only signal transduction related to AS sensitivity.
The mutations in the Trg-like region do not eliminate interaction with ChvE, however, because VirAN226Y and VirAI236F still respond to both wild-type ChvE and the hyperactive ChvET187P by progressive increases in both sensitivity and maximal activity, though their responses are still weaker than those of the wild type tested in an identical manner (Table 2). These could be interpreted as deficiencies in the interaction of ChvE and VirA. In contrast, VirAQ229K appears to represent a distinctly different case. In the absence of ChvE, VirAQ229K supports higher maximal activity than that supported by wild-type VirA (Table 2). Because VirAQ229K responds in a manner that is seen with wild-type VirA only in the presence of inducing sugar and wild-type ChvE, this could be considered mimicked occupancy as in the case of the corresponding trg allele (encoding TrgQ79K). Interestingly, the presence of ChvET187P results in even higher maximal activity, indicating that the VirAQ229K protein can still interact productively with this version of ChvE. An interpretation of the mimicked-occupancy phenotype is that the substituted amino acid residues result in conformational changes that are similar to temporary effects caused by ligand binding (6, 74). However, although strains expressing VirAQ229K exhibit a mimicked-occupancy phenotype in relation to maximal vir-inducing activity, the effect on their AS sensitivity is just the opposite: such a strain exhibits very low AS sensitivity, and this is unaffected by either wild-type ChvE or ChvET187P when tested in the presence of inducing sugar. Of all the residues for which site-specific substitutions were made, glutamine at 229 seems to be pivotal for postsignaling effects of the interaction of VirA with ChvE, because mutations at this residue result in a more severe sugar-sensing effect.
The question raised by these results is how the observed genetic uncoupling of the VirA/ChvE-sugar-mediated phenotypes (maximal activity and phenolic sensitivity) can be understood in a biochemical context. That is, does ChvE mediate physical responses by the periplasmic domain of VirA that are transduced independently of one another? The repressive activity of the periplasmic domain could be the result of its constraining the protein into a configuration of high phosphatase activity (9). However, phosphatase activity has not been demonstrated in isolated VirA, and other potential repressive activities may be at play. According to the model of Gao and Lynn, net VirA kinase activity is a sum of piston and rotational movements of the periplasmic, TM, and linker domains (31). In their model, a piston movement set in motion by the ChvE-sugar-pH signals lowers the energy barrier for phenol perception by the linker domain, thus increasing sensitivity to phenols. Our data support a refinement of this model, in which the piston in and of itself can lead to an increase in maximal vir-inducing activity, whereas some controlled version of this movement (or possibly a more significant movement than is achieved by the insertion mutations or the TM2 mutations studied here) is required to achieve the proposed lower energy barrier for phenol perception. The ChvE-mediated displacement of the TM2-spanning helix would be, in this model, precisely controlled so as to appropriately modulate the movement allowing increased phenolic sensitivity, as well as maximum activity. We suggest that the residues in the Trg-like region of the VirA periplasmic domain may be integral in this control.
Supplementary Material
Acknowledgments
We thank William Degrado and Paul Billings in the Department of Biochemistry and Biophysics at the University of Pennsylvania for assistance with biophysical experiments (analytical ultracentrifugation and far-UV circular dichroism spectroscopy) and many useful discussions. We are grateful to Fevzi Daldal and Nur Selamoglu in the Department of Biology at the University of Pennsylvania for assistance with the mass spectroscopy. We also thank David G. Lynn and Yi-Han Lin in the Department of Chemistry at Emory University and Kevin S. McIver in the Department of Cell Biology and Molecular Genetics at the University of Maryland, College Park, MD, for critical reading of the manuscript.
A portion of this work was part of G.R.N.'s doctoral work at the University of Pennsylvania. These studies were funded by the NIH (GM47369), the NSF (0818613), and a University Research Foundation grant to A.N.B.
Footnotes
Published ahead of print on 7 January 2011.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Adamczak, R., A. Porollo, and J. Meller. 2004. Accurate prediction of solvent accessibility using neural networks-based regression. Proteins 56:753-767. [DOI] [PubMed] [Google Scholar]
- 2.Adamczak, R., A. Porollo, and J. Meller. 2005. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins 59:467-475. [DOI] [PubMed] [Google Scholar]
- 3.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 4.Amin, D. N., and G. L. Hazelbauer. 2010. The chemoreceptor dimer is the unit of conformational coupling and transmembrane signaling. J. Bacteriol. 192:1193-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banta, L. M., R. D. Joerger, V. R. Howitz, A. M. Campbell, and A. N. Binns. 1994. Glu-255 outside the predicted ChvE binding site in VirA is crucial for sugar enhancement of acetosyringone perception by Agrobacterium tumefaciens. J. Bacteriol. 176:3242-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beel, B. D., and G. L. Hazelbauer. 2001. Signalling substitutions in the periplasmic domain of chemoreceptor Trg induce or reduce helical sliding in the transmembrane domain. Mol. Microbiol. 40:824-834. [DOI] [PubMed] [Google Scholar]
- 7.Beel, B. D., and G. L. Hazelbauer. 2001. Substitutions in the periplasmic domain of low-abundance chemoreceptor trg that induce or reduce transmembrane signaling: kinase activation and context effects. J. Bacteriol. 183:671-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brencic, A., and S. C. Winans. 2005. Detection of and response to signals involved in host-microbe interactions by plant-associated bacteria. Microbiol. Mol. Biol. Rev. 69:155-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brencic, A., Q. Xia, and S. C. Winans. 2004. VirA of Agrobacterium tumefaciens is an intradimer transphosphorylase and can actively block vir gene expression in the absence of phenolic signals. Mol. Microbiol. 52:1349-1362. [DOI] [PubMed] [Google Scholar]
- 10.Bryson, K., et al. 2005. Protein structure prediction servers at University College London. Nucleic Acids Res. 33:W36-W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burrows, G. G., M. E. Newcomer, and G. L. Hazelbauer. 1989. Purification of receptor protein Trg by exploiting a property common to chemotactic transducers of Escherichia coli. J. Biol. Chem. 264:17309-17315. [PubMed] [Google Scholar]
- 12.Buschmann, H., L. Sanchez-Pulido, M. A. Andrade-Navarro, and C. W. Lloyd. 2007. Homologues of Arabidopsis microtubule-associated AIR9 in trypanosomatid parasites: hints on evolution and function. Plant Signal Behav. 2:296-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cangelosi, G. A., R. G. Ankenbauer, and E. W. Nester. 1990. Sugars induce the Agrobacterium virulence genes through a periplasmic binding protein and a transmembrane signal protein. Proc. Natl. Acad. Sci. U. S. A. 87:6708-6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavicchioli, R., R. C. Chiang, L. V. Kalman, and R. P. Gunsalus. 1996. Role of the periplasmic domain of the Escherichia coli NarX sensor-transmitter protein in nitrate-dependent signal transduction and gene regulation. Mol. Microbiol. 21:901-911. [DOI] [PubMed] [Google Scholar]
- 15.Chang, C.-H., and S. C. Winans. 1992. Functional roles assigned to the periplasmic, linker, and receiver domains of the Agrobacterium tumefaciens virA protein. J. Bacteriol. 174:7033-7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang, C., S. F. Kwok, A. B. Bleecker, and E. M. Meyerowitz. 1993. Arabidopsis ethylene-response gene etr1: similarity of products to two-component regulators. 262:539-544. [DOI] [PubMed] [Google Scholar]
- 17.Chang, C. H., and S. C. Winans. 1996. Resection and mutagenesis of the acid pH-inducible P2 promoter of the Agrobacterium tumefaciens virG gene. J. Bacteriol. 178:4717-4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang, J. F., et al. 2003. Structure of the coiled-coil dimerization motif of Sir4 and its interaction with Sir3. Structure 11:637-649. [DOI] [PubMed] [Google Scholar]
- 19.Cheung, J., and W. A. Hendrickson. 2010. Sensor domains of two-component regulatory systems. Curr. Opin. Microbiol. 13:116-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung, J., and W. A. Hendrickson. 2009. Structural analysis of ligand stimulation of the histidine kinase NarX. Structure 17:190-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiang, R. C., R. Cavicchioli, and R. P. Gunsalus. 1997. ‘Locked-on’ and ‘locked-off’ signal transduction mutations in the periplasmic domain of the Escherichia coli NarQ and NarX sensors affect nitrate- and nitrite-dependent regulation by NarL and NarP. Mol. Microbiol. 24:1049-1060. [DOI] [PubMed] [Google Scholar]
- 22.Chilton, M.-D., et al. 1974. Is there foreign DNA in crown gall tumor DNA?, p. 297-311. In R. Markham, D. R. Davies, D. Hopwood, and R. W. Horne (ed.), Modification of the information content of plant cells. Elsevier, New York, NY.
- 23.Cho, U. S., et al. 2006. Metal bridges between the PhoQ sensor domain and the membrane regulate transmembrane signaling. J. Mol. Biol. 356:1193-1206. [DOI] [PubMed] [Google Scholar]
- 24.Das, A., L. B. Anderson, and Y.-H. Xie. 1997. Delineation of the interaction domains of Agrobacterium tumefaciens VirB7 and VirB9 by the yeast two-hybrid assay. 179:3404-3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delorenzi, M., and T. Speed. 2002. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18:617-625. [DOI] [PubMed] [Google Scholar]
- 26.Doty, S. L., M. C. Yu, J. I. Lundin, J. D. Heath, and E. W. Nester. 1996. Mutational analysis of the input domain of the VirA protein of Agrobacterium tumefaciens. J. Bacteriol. 178:961-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falke, J. J., and G. L. Hazelbauer. 2001. Transmembrane signaling in bacterial chemoreceptors. Trends Biochem. Sci. 26:257-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fields, S., and O. Song. 1989. A novel genetic system to detect protein-protein interactions. 340:245-246. [DOI] [PubMed] [Google Scholar]
- 29.Gamborg, O. L., T. Murashige, T. A. Thorpe, and I. K. Vasil. 1976. Plant tissue culture media. In Vitro 12:473-478. [DOI] [PubMed] [Google Scholar]
- 30.Gao, R., and D. G. Lynn. 2005. Environmental pH sensing: resolving the VirA/VirG two-component system inputs for Agrobacterium pathogenesis. J. Bacteriol. 187:2182-2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao, R., and D. G. Lynn. 2007. Integration of rotation and piston motions in coiled-coil signal transduction. J. Bacteriol. 189:6048-6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg, S. D., C. S. Soto, C. D. Waldburger, and W. F. Degrado. 2008. Determination of the physiological dimer interface of the PhoQ sensor domain. J. Mol. Biol. 379:656-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golemis, E., J. Gyuris, and R. Brent. 1999. Interaction trap/two-hybrid system to identify interacting proteins, p. 20.21.21-20.21.40. In F. M. Ausubel et al. (ed.), Current protocols in molecular biology. John Wiley and Sons, Inc., Hoboken, NJ.
- 34.Hazelbauer, G. L., and W. C. Lai. 2010. Bacterial chemoreceptors: providing enhanced features to two-component signaling. Curr. Opin. Microbiol. 13:124-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He, F., et al. 2009. Molecular basis of ChvE function in sugar binding, sugar utilization, and virulence in Agrobacterium tumefaciens. J. Bacteriol. 191:5802-5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hendrickson, W. A. 2005. Transduction of biochemical signals across cell membranes. Q. Rev. Biophys. 38:321-330. [DOI] [PubMed] [Google Scholar]
- 37.Hofmann, K., and W. Stoffel. 1993. TMBASE—a database of membrane spanning protein segments. Biol. Chem. Hoppe-Seyler 374:166. [Google Scholar]
- 38.Huang, Y., P. Morel, B. Powell, and C. Kado. 1990. VirA, a coregulator of Ti-specified virulence genes, is phosphorylated in vitro. J. Bacteriol. 172:1142-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin, S., R. K. Prusti, T. Roitsch, R. G. Ankenbauer, and E. W. Nester. 1990. Phosphorylation of the virG protein of Agrobacterium tumefaciens by the autophosphorylated virA protein: essential role in biological activity of virG. J. Bacteriol. 172:4945-4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin, S., T. Roitsch, R. G. Ankenbauer, M. P. Gordon, and E. W. Nester. 1990. The VirA protein of Agrobacterium tumefaciens is autophosphorylated and is essential for vir gene regulation. J. Bacteriol. 172:525-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones, D. T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195-202. [DOI] [PubMed] [Google Scholar]
- 42.Lee, K., et al. 1992. Mechanisms of activation of Agrobacterium virulence genes: identification of phenol-binding proteins. Proc. Natl. Acad. Sci. U. S. A. 89:8666-8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lupas, A., M. Van Dyke, and J. Stock. 1991. Predicting coiled coils from protein sequences. Science 252:1162-1164. [DOI] [PubMed] [Google Scholar]
- 44.Lupas, A. N., and M. Gruber. 2005. The structure of alpha-helical coiled coils. Adv. Protein Chem. 70:37-78. [DOI] [PubMed] [Google Scholar]
- 45.Mascher, T., J. D. Helmann, and G. Unden. 2006. Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol. Mol. Biol. Rev. 70:910-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCullen, C. A., and A. N. Binns. 2006. Agrobacterium tumefaciens and plant cell interactions and activities required for interkingdom macromolecular transfer. Annu. Rev. Cell Dev. Biol. 22:101-127. [DOI] [PubMed] [Google Scholar]
- 47.Melchers, L. S., et al. 1989. Membrane topology and functional analysis of the sensory protein VirA of Agrobacterium tumefaciens. EMBO J. 8:1919-1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melchers, L. S., et al. 1987. Molecular characterization of the virulence gene virA of the Agrobacterium tumefaciens octopine Ti plasmid. Plant Mol. Biol. 9:635-645. [DOI] [PubMed] [Google Scholar]
- 49.Miller, A. F., and J. J. Falke. 2004. Chemotaxis receptors and signaling. Adv. Protein Chem. 68:393-444. [DOI] [PubMed] [Google Scholar]
- 50.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- 51.Monera, O. D., N. E. Zhou, C. M. Kay, and R. S. Hodges. 1993. Comparison of antiparallel and parallel two-stranded alpha-helical coiled-coils. Design, synthesis, and characterization. J. Biol. Chem. 268:19218-19227. [PubMed] [Google Scholar]
- 52.Moore, J. O., and W. A. Hendrickson. 2009. Structural analysis of sensor domains from the TMAO-responsive histidine kinase receptor TorS. Structure 17:1195-1204. [DOI] [PubMed] [Google Scholar]
- 53.Mowbray, S. L. 1999. Bacterial chemoreceptors: recent progress in structure and function. Mol. Cells 9:115-118. [PubMed] [Google Scholar]
- 54.Nair, G. R., Z. Liu, and A. N. Binns. 2003. Reexamining the role of the accessory plasmid pAtC58 in the virulence of Agrobacterium tumefaciens strain C58. Plant Physiol. 133:989-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naumann, B., E. J. Stauber, A. Busch, F. Sommer, and M. Hippler. 2005. N-terminal processing of Lhca3 is a key step in remodeling of the photosystem I-light-harvesting complex under iron deficiency in Chlamydomonas reinhardtii. J. Biol. Chem. 280:20431-20441. [DOI] [PubMed] [Google Scholar]
- 56.Palmer, A. G., R. Gao, J. Maresh, W. K. Erbil, and D. G. Lynn. 2004. Chemical biology of multi-host/pathogen interactions: chemical perception and metabolic complementation. Annu. Rev. Phytopathol. 42:439-464. [DOI] [PubMed] [Google Scholar]
- 57.Pan, S. Q., T. Charles, S. Jin, Z. Wu, and E. W. Nester. 1993. Preformed dimeric state of the sensor protein VirA is involved in plant-Agrobacterium signal transduction. 90:9939-9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peach, M. L., G. L. Hazelbauer, and T. P. Lybrand. 2002. Modeling the transmembrane domain of bacterial chemoreceptors. Protein Sci. 11:912-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perez-Iratxeta, C., and M. A. Andrade-Navarro. 2008. K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct. Biol. 8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Porollo, A., and J. Meller. 2007. Versatile annotation and publication quality visualization of protein complexes using POLYVIEW-3D. BMC Bioinformatics 8:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Porollo, A. A., R. Adamczak, and J. Meller. 2004. POLYVIEW: a flexible visualization tool for structural and functional annotations of proteins. Bioinformatics 20:2460-2462. [DOI] [PubMed] [Google Scholar]
- 62.Schelcher, C., et al. 2007. Atypical bZIP domain of viral transcription factor contributes to stability of dimer formation and transcriptional function. J. Virol. 81:7149-7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Senes, A., et al. 2007. E (z), a depth-dependent potential for assessing the energies of insertion of amino acid side-chains into membranes: derivation and applications to determining the orientation of transmembrane and interfacial helices. J. Mol. Biol. 366:436-448. [DOI] [PubMed] [Google Scholar]
- 64.Shimoda, N., A. Toyoda-Yamamoto, S. Shinsuke, and Y. Machica. 1993. Genetic evidence for an interaction between the VirA sensor protein and the ChvE sugar-binding protein of Agrobacterium. J. Biol. Chem. 268:26552-26558. [PubMed] [Google Scholar]
- 65.Stachel, S. E., and P. C. Zambryski. 1986. virA and virG control the plant-induced activation of the T-DNA transfer process of A. tumefaciens. Cell 46:325-333. [DOI] [PubMed] [Google Scholar]
- 66.Toyoda-Yamamoto, A., N. Shimoda, and Y. Machida. 2000. Genetic analysis of the signal-sensing region of the histidine protein kinase VirA of Agrobacterium tumefaciens. Mol. Gen. Genet. 263:939-947. [DOI] [PubMed] [Google Scholar]
- 67.Turk, S. C. H. J., R. P. van Lange, T. J. G. Regensbuirg-Tuink, and P. J. J. Hooykaas. 1994. Localization of the VirA domain involved in acetosyringone-mediated vir gene induction in Agrobacterium tumefaciens. Plant Mol. Biol. 1994. 25:899-907. [DOI] [PubMed] [Google Scholar]
- 68.Wagner, M., R. Adamczak, A. Porollo, and J. Meller. 2005. Linear regression models for solvent accessibility prediction in proteins. J. Comput. Biol. 12:355-369. [DOI] [PubMed] [Google Scholar]
- 69.West, A. H., and A. M. Stock. 2001. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem. Sci. 26:369-376. [DOI] [PubMed] [Google Scholar]
- 70.Winans, S. C., R. A. Kerstetter, and E. W. Nester. 1988. Transcriptional regulation of the virA and virG genes of Agrobacterium tumefaciens. J. Bacteriol. 170:4047-4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Winans, S. C., R. A. Kerstetter, J. E. Ward, and E. W. Nester. 1989. A protein required for transcriptional regulation of Agrobacterium virulence genes spans the cytoplasmic membrane. J. Bacteriol. 171:1616-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wise, A. A., et al. 2010. The receiver domain of hybrid histidine kinase VirA: an enhancing factor for vir gene expression in Agrobacterium tumefaciens. J. Bacteriol. 192:1534-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wise, A. A., L. Voinov, and A. N. Binns. 2005. Intersubunit complementation of sugar signal transduction in VirA heterodimers and posttranslational regulation of VirA activity in Agrobacterium tumefaciens. J. Bacteriol. 187:213-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yaghmai, R., and G. L. Hazelbauer. 1992. Ligand occupancy mimicked by single residue substitutions in a receptor: transmembrane signaling induced by mutation. Proc. Natl. Acad. Sci. U. S. A. 89:7890-7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yaghmai, R., and G. L. Hazelbauer. 1993. Strategies for differential sensory responses mediated through the same transmembrane receptor. EMBO J. 12:1897-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yates, J. R., III, J. K. Eng, A. L. McCormack, and D. Schieltz. 1995. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67:1426-1436. [DOI] [PubMed] [Google Scholar]
- 77.Yeh, J. I., et al. 1996. High-resolution structures of the ligand binding domain of the wild-type bacterial aspartate receptor. J. Mol. Biol. 262:186-201. [DOI] [PubMed] [Google Scholar]
- 78.Zhang, Y., D. W. Kulp, J. D. Lear, and W. F. DeGrado. 2009. Experimental and computational evaluation of forces directing the association of transmembrane helices. J. Am. Chem. Soc. 131:11341-11343. [DOI] [PubMed] [Google Scholar]
- 79.Zhang, Z., and W. A. Hendrickson. 2010. Structural characterization of the predominant family of histidine kinase sensor domains. J. Mol. Biol. 400:335-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu, J., et al. 2000. The bases of crown gall tumorigenesis. J. Bacteriol. 182:3885-3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zupan, J., T. R. Muth, O. Draper, and P. Zambryski. 2000. The transfer of DNA from Agrobacterium tumefaciens into plants: a feast of fundamental insights. Plant J. 23:11-28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.