

Abstract
Bacteria regulate the frequency and timing of DNA replication initiation by controlling the activity of the replication initiator protein DnaA. SirA is a recently discovered regulator of DnaA in Bacillus subtilis whose synthesis is turned on at the start of sporulation. Here, we demonstrate that SirA contacts DnaA at a patch of 3 residues located on the surface of domain I of the replication initiator protein, corresponding to the binding site used by two unrelated regulators of DnaA found in other bacteria. We show that the interaction of SirA with domain I inhibits the ability of DnaA to bind to the origin of replication. DnaA mutants containing amino acid substitutions of the 3 residues are functional in replication initiation but are immune to inhibition by SirA.
Fundamental to an organism's survival is the ability to create one complete and accurate copy of its genome for every cell division. Underreplication leaves some cells without any genetic information, and overreplication wastes resources and could lead to aneuploidy. For these reasons, controlling the frequency of DNA replication initiation is critical to the cell cycle. Bacteria accomplish this by regulating the availability and activity of the replication initiator protein DnaA. An AAA+ ATPase, DnaA binds cooperatively to the origin of replication when in the ATP-bound state and distorts and melts the double helix. This then allows recruitment and loading of the replicative helicase onto the single-stranded DNA.
The DnaA protein consists of four domains (23). The most N-terminal of these, domain I, dimerizes with other domain I monomers (1) and interacts with other proteins that can modulate DnaA function (13). Domain II is unstructured and flexible and is poorly conserved. Domain III contains the AAA+ ATPase motif, multimerizes when bound to ATP, and forms contacts with the single-stranded DNA of the open complex (24). The most C-terminal domain, domain IV, binds double-stranded DNA via a helix-turn-helix DNA binding motif, recruiting DnaA to the consensus DnaA boxes at the origin of replication (5).
The mechanisms of DnaA function and the regulators of its activity are well studied in Escherichia coli. In this and other Gram-negative bacteria, the activity of DnaA is controlled by several mechanisms. These include regulation of the nucleotide binding state of DnaA (6, 11, 12, 28), regulation of the availability of the origin region (sequestration) to interact with DnaA (21, 26), binding to high-affinity DnaA-binding sites (cis-acting elements) that reduce the levels of DnaA that are free to interact with origin DNA (15, 16), and regulation of DnaA's affinity for DNA by interaction with other proteins (10, 13, 14). In Gram-positive bacteria, however, there are no homologs of the proteins or cis-acting elements responsible for these well-understood regulatory mechanisms in E. coli. In the Gram-positive model organism Bacillus subtilis, only two regulators of DnaA function have been identified, YabA (3, 8, 22) and Soj (17, 18), and the mechanisms by which these proteins control DnaA are not yet known.
In response to nutrient starvation, cells of B. subtilis undergo a regulated and complex developmental program to create a robust and dormant spore. Recently, we discovered a gene, sirA, that is induced at the start of sporulation and whose product is an inhibitor of DNA replication (25). Cells lacking sirA have more chromosomes than wild-type cells when challenged to sporulate under conditions that promote replication initiation. When synthesis of SirA is artificially induced during growth, replication is impaired, resulting in cells that lack a chromosome (25). Wagner et al. have shown that SirA acts through DnaA (29). Cells that initiate replication through a plasmid origin, oriN, that uses an initiator protein other than DnaA are unaffected by sirA expression. Additionally, SirA and DnaA interact in a yeast two-hybrid assay (29), suggesting that SirA affects DnaA function directly.
To investigate further the interaction between SirA and DnaA and the mechanism by which SirA regulates replication initiation, we selected for mutations in dnaA that rendered initiation of replication immune to SirA. We discovered four such mutations, which altered three codons for amino acids located in domain I of DnaA. Using a yeast two-hybrid assay, we demonstrate that these residues are required for the interaction of DnaA with SirA and that domain I alone is sufficient for interaction with SirA. Finally, taking advantage of one of the mutant proteins, we demonstrate that at the start of sporulation SirA reduces the amount of wild-type DnaA bound at the origin of replication and does so in a manner that depends on its interaction with DnaA.
MATERIALS AND METHODS
Strains, plasmids, and primers.
Strains were derived from B. subtilis PY79. All strains, plasmids, and primers used in this work are described in Table 1.
TABLE 1.
Strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Description or sequence (reference or source) |
|---|---|
| Bacillus strains | |
| LR158 | thrC::Phyperspank-sirA (25) |
| LR166 | cat inserted 406 bp upstream from dnaA translational start by LFHa with primers LRL96, LRL170, LRL171, and LRL100 (9) |
| LR227 | amyE::Phyperspank-kinA from MF2250 (lab strain), cat upstream of dnaA (LR166) |
| LR228 | amyE::Phyperspank-kinA, cat upstream of dnaA(A50V) |
| LR229 | amyE::Phyperspank-kinA, cat upstream of dnaA, sirA::tet from LR74 (25) |
| LR230 | amyE::Phyperspank-kinA, cat upstream of dnaA(A50V), sirA::tet |
| S. cerevisiae strains | |
| LR192 | Y2HGold with pGBKT7 (Clontech) |
| LR193 | Y187 with pGADT7 (Clontech) |
| LR197 | Y187 with pLR55 |
| LR198 | Y2HGold with pLR48 |
| LR199 | Y2HGold with pLR49 |
| LR200 | Y2HGold with pLR50 |
| LR201 | Y2HGold with pLR51 |
| LR202 | Y2HGold with pLR52 |
| LR203 | Y2HGold with pLR53 |
| LR204 | Y2HGold with pLR54 |
| LR205 | Y2HGold with pLR57 |
| LR206 | Y2HGold with pLR58 |
| Plasmids | |
| pLR48 | pGBKT7 (Clontech) with B. subtilis dnaA cloned into NdeI/BamHI with primers LRL195 and LRL196 |
| pLR49 | pGBKT7 with B. subtilis dnaA domain I cloned into NdeI/BamHI with primers LRL195 and LRL198 |
| pLR50 | pGBKT7 with B. subtilis dnaA domains I and II cloned into NdeI/BamHI with primers LRL195 and LRL197 |
| pLR51 | pLR48 with A50V point mutation made using primers LRL205 and LRL206 |
| pLR52 | pLR48 with N47D point mutation made using primers LRL207 and LRL208 |
| pLR53 | pLR48 with F49Y point mutation made using primers LRL209 and LRL210 |
| pLR54 | pLR48 with A50T point mutation made using primers LRL211 and LRL212 |
| pLR55 | pGADT7 (Clontech) with sirA cloned into EcoRI/BamHI with primers LRL193 and LRL194 |
| pLR57 | pGBKT7 with E. coli dnaA cloned into NdeI/BamHI with primers LRL224 and LRL225 |
| pLR58 | pLR57 with V47A point mutation made using primers LRL230 and LRL231 |
| Primers | |
| LRL83 | 5′-ATGGTACCACTCCCTGTAAAGGATACTC-3′ |
| LRL96 | 5′-CATTTCCGGCTGTAAAGCCTGCA-3′ |
| LRL99 | 5′-CTAAGCCGACGCCCCCATAGAT-3′ |
| LRL100 | 5′-CTTAAAATTGCAATTCTCGTTTCTAG-3′ |
| LRL101 | 5′-ATTTTTTCATGCGCATGAATAACGGT-3′ |
| LRL170 | 5′-CAATTCGCCCTATAGTGAGTCGTGATATCCACATGCGTGATTTTAGG-3′ |
| LRL171 | 5′-CCAGCTTTTGTTCCCTTTAGTGAGTTTTTCGGCTTTTTTTAGTATCCACAG-3′ |
| LRL174 | 5′-GATGCACGATCAATGGCCTGAAG-3′ |
| LRL193 | 5′-GCCGAATTCATGGAACGTCACTACTATACG-3′ |
| LRL194 | 5′-ATGAGGGATCCGGTTTTAGACAAAATTTCTTTC-3′ |
| LRL195 | 5′-CGGCATATGGAAAATATATTAGACCTGTGG-3′ |
| LRL196 | 5′-AGACTGGATCCTATTTAAGCTGTTCTTTAATTTC-3′ |
| LRL197 | 5′-ATGCAGGATCCTTTATTTTGAGGAAAATCAGATGTATCTTC-3′ |
| LRL198 | 5′-AGCAGGATCCTTTAATGACAAACTTAATGCTCAATTCTTC-3′ |
| LRL205 | 5′-CACGGCTCCCAATGAATTTGTCAGAGACTGG-3′ |
| LRL206 | 5′-CCAGTCTCTGACAAATTCATTGGGAGCCGTG-3′ |
| LRL207 | 5′-CACGGCTCCCGATGAATTTGCCAGAGACTGG-3′ |
| LRL208 | 5′-CCAGTCTCTGGCAAATTCATCGGGAGCCGTG-3′ |
| LRL209 | 5′-CACGGCTCCCAATGAATATGCCAGAGACTGG-3′ |
| LRL210 | 5′-CCAGTCTCTGGCATATTCATTGGGAGCCGTG-3′ |
| LRL211 | 5′-CACGGCTCCCAATGAATTTACCAGAGACTGG-3′ |
| LRL212 | 5′-CCAGTCTCTGGTAAATTCATTGGGAGCCGTG-3′ |
| LRL224 | 5′-CGGCATATGGTGTCACTTTCGCTTTGGCAGC-3′ |
| LRL225 | 5′-AGACTGGATCCGTTTACGATGACAATGTTCTGATTAA-3′ |
| LRL230 | 5′-CCATATTGCATGGTATGTAGCTATGGATCAATTATGGTACATTGGAGATC-3′ |
| LRL231 | 5′-GATCTCCAATGTACCATAATTGATCCATAGCTACATACCATGCAATATGG-3′ |
| WKS139 | 5′-GGAGGACGTGATCATACGA-3′ |
| WKS140 | 5′-TAGGGCCTGTGGATTTGTG-3′ |
| WKS145 | 5′-CGAGCAAGGTGTCGCTTA-3′ |
| WKS146 | 5′-GCAGGCGGTCATCATGTA-3′ |
LFH, long flanking homology.
Mutagenic PCR.
PCR was performed with AcuStart PCR supermix (Quanta Biosciences) on chromosomal DNA from strain LR166 with primers LRL96 and LRL174. Reaction mixtures (400 μl) were run in 12-μl aliquots to increase the diversity of mutations and then pooled. Reaction mixtures were cycled at 94°C for 2 min, followed by 10 cycles of 94°C for 1 min, 65°C − 1°C per cycle for 1 min, and 72°C for 5 min. Finally, 25 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 5 min were run. Pooled PCR products were transformed directly into strain LR158 as described previously (30).
Identifying mutations in dnaA.
Chromosomal DNA was isolated from colonies able to grow on plates containing LB medium with chloramphenicol and IPTG (isopropyl-β-d-thiogalactopyranoside). PCRs were run on these templates using primers LRL171 and LRL83. The reactions were set up with 1 μl of template DNA, 100 pM of each primer, 20 μl of 10× Platinum Pfx polymerase buffer (Invitrogen), 1 μl of 25 mM deoxynucleoside triphosphates, 2 μl of 50 mM MgSO4, and 1 μl of platinum Pfx polymerase (Invitrogen) in a 100-μl volume, and mixtures were thermocycled under standard conditions. PCR products were purified with a PCR purification kit (Qiagen) and sequenced with primers LRL171, LRL99, LRL100, LRL101, and LRL83.
Yeast two-hybrid assays.
Yeast two-hybrid experiments were performed using a Matchmaker Gold yeast two-hybrid system (Clontech). sirA was cloned into the EcoRI/BamHI sites of the prey plasmid pGADT7, and dnaA, the two truncated dnaA genes, and E. coli dnaA were cloned into the NdeI/BamHI sites of the bait plasmid pGBKT7, as described in Table 1. To make the dnaA point mutations, pLR48 and pLR57 were mutated with a QuikChange site-directed mutagenesis kit (Stratagene) as described in Table 1. The prey plasmids (pLR55 and empty pGADT7) were transformed into Saccharomyces cerevisiae strain Y187 as per the manufacturer's directions, and transformants were selected on SD-Leu plates. The bait plasmids (pLR48 to pLR54, pLR57, pLR58, and empty pGBKT7) were transformed into yeast strain Y2HGold, and transformants were selected on SD-Trp plates. Prey and bait strains were mated by being streaked together on yeast extract-peptone-dextrose (YPD) agar and then replica plated onto an minimal synthetic defined (SD)-Trp-Leu plate to select for both bait and prey plasmids. To assay for prey-bait interactions, the mated strains were restreaked onto SD-Trp-Leu-His-Ade plates, grown at 30°C for 3 days, and then photographed.
Chromatin immunoprecipitation.
Cultures were grown in 25% LB to an optical density at 600 nm (OD600) of 0.25. kinA expression was induced with 100 mM IPTG. After 1 h, 50-ml cultures were cross-linked in 1% formaldehyde for 20 min and then quenched with 5 ml of 2.5 M glycine. Cell pellets were washed once in cold phosphate-buffered saline (PBS) and then frozen at −80°C. Pellets were resuspended in 3 ml of 10 mM Tris, pH 8, 20% sucrose, 50 mM NaCl, 10 mM EDTA, 1 mg/ml lysozyme, and 1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF) and then incubated at 37°C for 30 min. Three milliliters of 2× immunoprecipitation (IP) buffer (100 mM Tris, pH 7, 300 mM NaCl, 2% Triton X-100) was added, and the mixture was sonicated for 1 min at 15% power output with a microtip. Cell debris was pelleted and 40 μl of the lysate set aside in 40 μl TE (10 mM Tris, pH 8, 0.1 mM EDTA) and 9 μl of 10% sodium dodecyl sulfate (SDS) as the total DNA sample. One-milliliter aliquots of the lysate were used for each chromatin immunoprecipitation (ChIP) pulldown. Two microliters polyclonal rabbit antiserum for DnaA, which behaves similarly to the previously described chicken antibodies against DnaA (27), was added, and the samples were incubated at 4°C with gentle agitation overnight. Thirty microliters 50% protein A-Sepharose slurry was added, and the samples were incubated for an additional hour at room temperature. The lysate was removed, and the beads were washed six times with 1 ml of 1× IP buffer, followed by a 1-ml wash in TE. The DNA-protein complexes were eluted from the beads by incubation with 250 μl TE with 0.67% SDS at 65°C for 10 min. Immunoprecipitate and total samples were incubated overnight at 65°C to reverse cross-linking. Proteinase K (20 mg/ml; Qiagen) was added to 250 μl of TE, pH 7.6, and the reaction mixtures were incubated at 37°C for 2 h. LiCl was added to 400 mM, and the DNA was purified with phenol chloroform extraction. Glycogen (20 μg) was added, and the DNA was precipitated in 75% ethanol at −80°C for 20 min. DNA pellets were resuspended in 75 μl TE.
Quantitative PCR.
Quantitative PCRs (qPCRs) (20-μl mixtures) were set up with LightCycler 480 SYBR green Master mix (Roche) and 2 μl immunoprecipitated DNA without dilution or total DNA diluted 1:500. To amplify the origin, primers WKS139 and WKS140 were used. To amplify yhaX, a site not bound by DnaA (2), primers WKS145 and WKS146 were used. The reactions were run on a LightCycler 480 II instrument (Roche). Signals were analyzed using LightCycler 480 SW 1.5 software (Roche), according to the manufacturer (Advanced RelQuant; second derivative of Max, using Median Cp for calculation). Reactions were run in duplicate, and the average crossing point (Cp) value was used. Standard curves of total DNA samples from strain LR227 were used to find the concentration of the template DNA in arbitrary units. To find the fold enrichment, the concentration of the origin in each IP sample normalized to the concentration of the origin in the total for that sample was divided by the concentration of yhaX in that sample normalized to the concentration of yhaX in the total.
RESULTS
Isolation of DnaA mutants that are resistant to the growth defect caused by engineered expression of sirA.
We sought to isolate mutants of DnaA that were immune to the action of SirA. To achieve this, we took advantage of the severe growth defect caused by the addition of inducer to cells engineered to produce SirA during growth in response to IPTG. dnaA was mutagenized using error-prone PCR. As a template, we used chromosomal DNA from a strain in which a gene conferring resistance to chloramphenicol had been inserted just upstream of the origin of replication (LR166). Primers LRL96 and LRL174 resulted in a PCR product that contained the antibiotic resistance gene, the origin of replication, dnaA, and flanking regions on either side for homologous recombination (Fig. 1).
FIG. 1.

Replication origin region of the B. subtilis chromosome. Shown is the site of insertion of the gene for resistance to chloramphenicol (cat) in strain LR166. The promoter for dnaA is marked by an arrow. The essential elements of the origin of replication are shown as white boxes. Primers LRL96 and LRL174 were used to amplify this region, which also includes the genes rpnA and rpmH and portions of the genes spoIIIJ and dnaN.
This PCR product was transformed into strain LR158, which was engineered to express sirA in response to IPTG. The cells were allowed to grow on LB plates containing chloramphenicol and IPTG at 22°C for 3 days. Transformants that were chloramphenicol resistant and able to grow in the presence of IPTG were colony purified and subjected to further analysis as follows. The origin of replication, including dnaA, was amplified from each transformant and subjected to sequence analysis. Four different mutants were isolated, and all contained single nucleotide substitutions in the coding sequence of dnaA (Table 2). The resulting amino acid substitutions, N47D, F49Y, A50V, and A50T, were clustered in the N-terminal region of DnaA domain I. The same mutation causing the N47D substitution was isolated independently from two PCRs.
TABLE 2.
Mutations in the isolated DnaA mutants
| Nucleotide position | Nucleotide substitution | Amino acid substitution |
|---|---|---|
| 139 | A to G | N47D |
| 146 | T to A | F49Y |
| 148 | G to A | A50T |
| 149 | C to T | A50V |
Next, we analyzed the locations of the amino acid substitutions in the structure of domain I, as inferred from the nuclear magnetic resonance (NMR) structure of Escherichia coli DnaA domain I (1). As shown in Fig. 2, all of the residues are clustered in a patch on the surface of the protein.
FIG. 2.
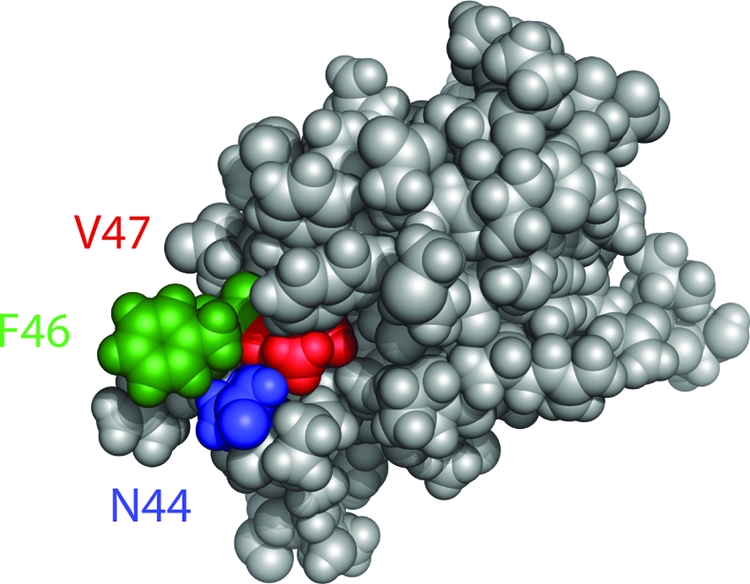
SirA contacts a patch of residues on the surface of domain I of DnaA. The structure of E. coli DnaA domain I is shown from Abe et al. (1). N44 (B. subtilis N47) is shown in blue, V47 (B. subtilis A50) is shown in red, and F46 (B. subtilis F49) is shown in green.
Strains separately containing each of the four dnaA point mutations showed no obvious growth defect and had a doubling time in liquid culture indistinguishable from that of strains containing a wild-type allele of dnaA, with the possible exception of the strain containing the F49Y substitution, whose doubling time appeared to be slightly slower than that of the wild type (data not shown).
The DnaA mutants are defective in interacting with SirA.
Wild-type DnaA interacts with SirA as judged by a yeast two-hybrid assay (29). We sought to determine if our DnaA mutants were defective in interacting with SirA. We used SirA as the prey, fusing the coding sequence in yeast to that for the activation domain of the GAL4 transcription factor. Wild-type DnaA and the four point mutants were used as bait, with their coding sequences fused to that for the DNA-binding domain of the GAL4 transcription factor. As expected, wild-type DnaA was able to interact with SirA, activating expression of HIS3 and ADE2 as judged by the ability of auxotrophic yeast cells to grow on medium lacking histidine and adenine (Table 3 and Fig. 3 A). In contrast, none of the four yeast strains in which mutant DnaA proteins had been used as bait were able to grow on the selective media (Table 3 and Fig. 3C). These results suggest that the amino acid substitutions in DnaA that allowed for growth in the presence of SirA were defective in the interaction between DnaA and SirA and likely form part of the site where DnaA contacts SirA.
TABLE 3.
Summary of yeast two-hybrid assay resultsa
| GAL BDb fusion | Interaction with GAL ADc-SirA |
|---|---|
| B. subtilis | |
| DnaA | Yes |
| DnaA residues 1-109 | Yes |
| DnaA residues 1-80 | Yes |
| DnaA(N47D) | No |
| DnaA(F49Y) | No |
| DnaA(A50T) | No |
| DnaA(A50V) | No |
| E. coli | |
| DnaA | No |
| DnaA(V47A) | Yes |
Results from Fig. 3 and data not shown.
GAL BD, DNA-binding domain of the GAL4 transcription factor.
GAL AD, activation domain of the GAL4 transcription factor.
FIG. 3.

A yeast two-hybrid assay identifies residues required for the binding of SirA to DnaA. Shown are streaks of yeast cells in which SirA was used as prey and the following forms of DnaA were used as bait: wild-type B. subtilis DnaA, B. subtilis DnaA residues 1 to 80, B. subtilis DnaA(A50V), and E. coli DnaA(V47A). For each plate, streaked on the left as a control is yeast harboring the experimental DnaA vector and the empty prey vector, streaked in the center is yeast harboring the experimental DnaA bait vector and the SirA prey vector, and streaked on the right as a control is yeast harboring the empty bait vector and the SirA prey vector.
Domain I of DnaA is sufficient for interaction with SirA.
Because the amino acid substitutions in DnaA were clustered near each other in domain I of the protein, we wondered whether domain I was sufficient for interaction with SirA. If this is the case, then SirA should be able to interact with a truncated version of DnaA that contains only domain I. We tested two truncations in our yeast two-hybrid assay, one that retained domain II, the flexible linker between the N-terminal domain and the rest of the protein, and a more severe truncation that retained only domain I. In both cases, SirA was able to interact with the truncated protein (Table 3 and Fig. 3B), supporting the idea that domain I contains the entire binding site for SirA.
DnaA A50 is coconserved with SirA.
Of the three residues that underwent substitution in the DnaA mutants, two (N47 and F49) are widely conserved. Residue 50, however, which is an alanine in B. subtilis, is often a valine or a threonine in other species. In fact, as shown in Fig. 4, an alanine at this position correlates with the presence of a SirA homolog. In species without SirA, valine or threonine is found at this position. The coconservation of A50 and SirA is consistent with the view that DnaA A50 is important for SirA function.
FIG. 4.

Alanine 50 of DnaA is conserved among DnaA homologs in species that contain a SirA homolog. Shown is a portion of the amino acid sequence of DnaA domain I for the indicated species (L. monocytogenes, Listeria monocytogenes; S. aureus, Staphylococcus aureus; C. difficile, Clostridium difficile; C. botulinum, Clostridium botulinum). Residues at which substitutions conferred immunity to SirA are marked with asterisks. B. subtilis, Geobacillus kaustophilus, and Bacillus anthracis possess SirA homologs. Sequences were aligned with ClustalW.
A single amino acid substitution enables E. coli DnaA to interact with SirA.
As just indicated, species that lack a SirA homolog contain valine or threonine at the residue corresponding to A50. In Escherichia coli, the corresponding residue (residue 47) is a valine. Thus, E. coli DnaA is naturally equivalent to our B. subtilis A50V mutant. Indeed, as shown in Table 3, we could detect little or no interaction between E. coli DnaA and B. subtilis SirA in the yeast two-hybrid assay. We therefore wondered whether we could convert E. coli DnaA into a SirA-interacting protein simply by switching the valine at residue 47 (corresponding to residue 50 in B. subtilis) to an alanine. Strikingly, simply switching V47 to an alanine in E. coli DnaA was sufficient to confer on the heterologous protein the ability to interact with SirA (Table 3 and Fig. 3D). This finding reinforces the view that A50 in B. subtilis DnaA forms part of the DnaA-SirA binding site. It would be interesting in future work to engineer an E. coli strain that produces the V47A substitution and ask whether engineered synthesis of SirA blocks DNA replication in the heterologous host.
SirA inhibits the binding of wild-type but not mutant DnaA to the origin of replication.
Evidence based on the use of a DnaA-green fluorescent protein (GFP) fusion indicated that SirA interferes with the association of DnaA with the origin region of the chromosome (29). We wondered whether we could observe this effect more directly and at higher resolution by use of chromatin immunoprecipitation (ChIP) followed by quantitative PCR (qPCR) and, if so, whether DnaA harboring the A50V substitution would be refractory to the effect of SirA. To measure DnaA binding to the origin, we performed ChIPs using anti-DnaA antibodies followed by qPCR and measured the amount of origin DNA that had been cross-linked to DnaA compared to that for a control locus unrelated to DnaA.
To carry out the experiment, we artificially induced sporulation using cells that were engineered to produce KinA in response to an inducer. KinA is one of several histidine kinases that phosphorylate the master regulator for sporulation Spo0A via a phosphorelay. Overexpression of KinA efficiently triggers sporulation even in rich medium in which cells are growing rapidly and would otherwise sporulate poorly (7). We had previously shown that, under these conditions, cells lacking sirA overreplicate their chromosomes (25). Indeed, as shown in Fig. 5 A, about 3-fold more DnaA was observed at the origin of replication in cells of a strain lacking sirA (LR229) than in cells of the wild type (LR227). In a separate experiment, Western blotting showed that expression of sirA had little or no effect on the total amount of DnaA protein present in the cell (data not shown). Thus, the results from ChIP analysis support the idea that SirA interferes with the binding of DnaA to the origin or removes it from the origin once bound.
FIG. 5.

SirA reduces the amount of wild-type but not mutant DnaA bound at the origin of replication. Cells were induced to sporulate in rich medium. The graphs show the results of ChIP experiments, expressed as fold enrichment of the origin of replication over that for an unrelated locus in the pulldown of DnaA from cell lysates of strains with or without sirA. (A) Wild-type DnaA (strains LR227 and LR229). (B) DnaA(A50V) (strains LR228 and LR230). Error bars represent the standard errors from six replicates.
Next, we performed the same experiment with strains that produced DnaA(A50V) instead of wild-type DnaA (LR228 and LR230). In this case, the presence or absence of sirA had no measurable effect on the amount of the mutant protein observed at the origin of replication (Fig. 5B).
Interestingly, and independently of the presence or absence of sirA, the A50V substitution mutation seemed to alter the amount of protein at the origin. We observed twice as much DnaA(A50V) at the origin of replication (20-fold enrichment) (Fig. 5B) as we did with wild-type DnaA (10-fold enrichment) (Fig. 5A). Evidently, the A50V substitution increased the affinity of DnaA for origin DNA or, conceivably, its affinity for our antibody.
Taken together, these data support a model in which SirA binds to DnaA via a cluster of residues located in domain I. Moreover, our findings together with those of Wagner et al. (29) indicate that the effect of SirA is to antagonize the ability of DnaA to associate with the origin and hence to initiate replication. Thus, the induction of SirA synthesis at the start of sporulation helps to ensure that further rounds of DNA replication are suppressed so that the sporangium will contain no more than two chromosomes.
DISCUSSION
The principal contribution of the present investigation is the demonstration that SirA interacts with DnaA by contacting a small patch of amino acids that sit at the surface of domain I of the initiator protein and that this interaction inhibits the binding of DnaA to the origin of replication or acts to remove DnaA that is already bound at the origin. For comparison, YabA, a negative regulator of DnaA function, is thought to interact with domain III of the protein (3), but it is not known how this interaction interferes with DnaA function. Soj, which is also a negative regulator, is known to interact with DnaA, but it is not known which part of the protein it contacts (18).
Domain I is also the contact site for two regulators of DnaA in Gram-negative bacteria that promote replication initiation. These are DiaA from E. coli (13) and HobA from Helicobacter pylori (20). DiaA interacts with DnaA in a way that increases the initiator's affinity for DnaA boxes, resulting in more DnaA binding to the origin and faster initiation (14). HobA, which is essential in H. pylori, dimerizes DnaA domain I monomers (20). H. pylori DnaA lacks the W6 residue that is essential for dimerization of this domain in E. coli (1), and it is thought that HobA is required to replace this function.
Interestingly, DiaA and HobA interact with DnaA at residues corresponding to those we have identified as the binding site for SirA (13, 20). This is the case even though these three proteins alter DnaA function in different ways. Both E. coli and B. subtilis DnaA have the W6 residue for which HobA compensates in H. pylori. Like DiaA, SirA influences DnaA's interaction with the origin of replication, but DiaA promotes this interaction and SirA antagonizes it. Thus, the residues on the surface of domain I seem to have evolved as a common interaction site that different species have exploited for diverse purposes.
HobA and DiaA have been called “structural homologs,” because they share no primary sequence homology but have the same three-dimensional shape (19). SirA does not share sequence similarity with either of these proteins, but it is similar to them in size (18 kDa for SirA, versus 21 kDa for both HobA and DiaA) and binds to the same site on DnaA. Conceivably, SirA is a homolog of HobA and DiaA whose relatedness has been preserved at the structural level. Initial attempts to model the structure of SirA by protein threading (4) through HobA and DiaA were unsuccessful. If SirA proves to be structurally related, then all three proteins may have a common evolutionary origin even though their functions have diverged.
Acknowledgments
We thank Danaya Pakotiprapha and David Jeruzalmi for assistance with structural modeling.
This work was supported by NIH grant GM18568 to R.L., NIH postdoctoral fellowship GM093408 to H.M., and NIH grant GM41934 to A.D.G.
Footnotes
Published ahead of print on 14 January 2011.
REFERENCES
- 1.Abe, Y., et al. 2007. Structure and function of DnaA N-terminal domains: specific sites and mechanisms in inter-DnaA interaction and in DnaB helicase loading on oriC. J. Biol. Chem. 282:17816-17827. [DOI] [PubMed] [Google Scholar]
- 2.Breier, A. M., and A. D. Grossman. 2009. Dynamic association of the replication initiator and transcription factor DnaA with the Bacillus subtilis chromosome during replication stress. J. Bacteriol. 191:486-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho, E., N. Ogasawara, and S. Ishikawa. 2008. The functional analysis of YabA, which interacts with DnaA and regulates initiation of chromosome replication in Bacillus subtilis. Genes Genet. Syst. 83:111-125. [DOI] [PubMed] [Google Scholar]
- 4.Eswar, N., et al. 2006. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics 5:5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujikawa, N., et al. 2003. Structural basis of replication origin recognition by the DnaA protein. Nucleic Acids Res. 31:2077-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujimitsu, K., T. Senriuchi, and T. Katayama. 2009. Specific genomic sequences of E. coli promote replicational initiation by directly reactivating ADP-DnaA. Genes Dev. 23:1221-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujita, M., and R. Losick. 2005. Evidence that entry into sporulation in Bacillus subtilis is governed by a gradual increase in the level and activity of the master regulator Spo0A. Genes Dev. 19:2236-2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goranov, A. I., A. M. Breier, H. Merrikh, and A. D. Grossman. 2009. YabA of Bacillus subtilis controls DnaA-mediated replication initiation but not the transcriptional response to replication stress. Mol. Microbiol. 74:454-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guerout-Fleury, A. M., K. Shazand, N. Frandsen, and P. Stragier. 1995. Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167:335-336. [DOI] [PubMed] [Google Scholar]
- 10.Ishida, T., et al. 2004. DiaA, a novel DnaA-binding protein, ensures the timely initiation of Escherichia coli chromosome replication. J. Biol. Chem. 279:45546-45555. [DOI] [PubMed] [Google Scholar]
- 11.Katayama, T., T. Kubota, K. Kurokawa, E. Crooke, and K. Sekimizu. 1998. The initiator function of DnaA protein is negatively regulated by the sliding clamp of the E. coli chromosomal replicase. Cell 94:61-71. [DOI] [PubMed] [Google Scholar]
- 12.Kato, J., and T. Katayama. 2001. Hda, a novel DnaA-related protein, regulates the replication cycle in Escherichia coli. EMBO J. 20:4253-4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keyamura, K., Y. Abe, M. Higashi, T. Ueda, and T. Katayama. 2009. DiaA dynamics are coupled with changes in initial origin complexes leading to helicase loading. J. Biol. Chem. 284:25038-25050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keyamura, K., et al. 2007. The interaction of DiaA and DnaA regulates the replication cycle in E. coli by directly promoting ATP DnaA-specific initiation complexes. Genes Dev. 21:2083-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitagawa, R., H. Mitsuki, T. Okazaki, and T. Ogawa. 1996. A novel DnaA protein-binding site at 94.7 min on the Escherichia coli chromosome. Mol. Microbiol. 19:1137-1147. [DOI] [PubMed] [Google Scholar]
- 16.Kitagawa, R., T. Ozaki, S. Moriya, and T. Ogawa. 1998. Negative control of replication initiation by a novel chromosomal locus exhibiting exceptional affinity for Escherichia coli DnaA protein. Genes Dev. 12:3032-3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee, P. S., and A. D. Grossman. 2006. The chromosome partitioning proteins Soj (ParA) and Spo0J (ParB) contribute to accurate chromosome partitioning, separation of replicated sister origins, and regulation of replication initiation in Bacillus subtilis. Mol. Microbiol. 60:853-869. [DOI] [PubMed] [Google Scholar]
- 18.Murray, H., and J. Errington. 2008. Dynamic control of the DNA replication initiation protein DnaA by Soj/ParA. Cell 135:74-84. [DOI] [PubMed] [Google Scholar]
- 19.Natrajan, G., D. R. Hall, A. C. Thompson, I. Gutsche, and L. Terradot. 2007. Structural similarity between the DnaA-binding proteins HobA (HP1230) from Helicobacter pylori and DiaA from Escherichia coli. Mol. Microbiol. 65:995-1005. [DOI] [PubMed] [Google Scholar]
- 20.Natrajan, G., M. F. Noirot-Gros, A. Zawilak-Pawlik, U. Kapp, and L. Terradot. 2009. The structure of a DnaA/HobA complex from Helicobacter pylori provides insight into regulation of DNA replication in bacteria. Proc. Natl. Acad. Sci. U. S. A. 106:21115-21120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nievera, C., J. J. Torgue, J. E. Grimwade, and A. C. Leonard. 2006. SeqA blocking of DnaA-oriC interactions ensures staged assembly of the E. coli pre-RC. Mol. Cell 24:581-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noirot-Gros, M. F., et al. 2006. Functional dissection of YabA, a negative regulator of DNA replication initiation in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 103:2368-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozaki, S., and T. Katayama. 2009. DnaA structure, function, and dynamics in the initiation at the chromosomal origin. Plasmid 62:71-82. [DOI] [PubMed] [Google Scholar]
- 24.Ozaki, S., et al. 2008. A common mechanism for the ATP-DnaA-dependent formation of open complexes at the replication origin. J. Biol. Chem. 283:8351-8362. [DOI] [PubMed] [Google Scholar]
- 25.Rahn-Lee, L., B. Gorbatyuk, O. Skovgaard, and R. Losick. 2009. The conserved sporulation protein YneE inhibits DNA replication in Bacillus subtilis. J. Bacteriol. 191:3736-3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slater, S., et al. 1995. E. coli SeqA protein binds oriC in two different methyl-modulated reactions appropriate to its roles in DNA replication initiation and origin sequestration. Cell 82:927-936. [DOI] [PubMed] [Google Scholar]
- 27.Smits, W. K., A. I. Goranov, and A. D. Grossman. 2010. Ordered association of helicase loader proteins with the Bacillus subtilis origin of replication in vivo. Mol. Microbiol. 75:452-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su'etsugu, M., T. R. Shimuta, T. Ishida, H. Kawakami, and T. Katayama. 2005. Protein associations in DnaA-ATP hydrolysis mediated by the Hda-replicase clamp complex. J. Biol. Chem. 280:6528-6536. [DOI] [PubMed] [Google Scholar]
- 29.Wagner, J. K., K. A. Marquis, and D. Z. Rudner. 2009. SirA enforces diploidy by inhibiting the replication initiator DnaA during spore formation in Bacillus subtilis. Mol. Microbiol. 73:963-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson, G. A., and K. F. Bott. 1968. Nutritional factors influencing the development of competence in the Bacillus subtilis transformation system. J. Bacteriol. 95:1439-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]