

Abstract
The biosyntheses of the [NiFe]-hydrogenase and urease enzymes in Helicobacter pylori require several accessory proteins for proper construction of the nickel-containing metallocenters. The hydrogenase accessory proteins HypA and HypB, a GTPase, have been implicated in the nickel delivery steps of both enzymes. In this study, the metal-binding properties of H. pylori HypB were characterized, and the effects of metal binding on the biochemical behavior of the protein were examined. The protein can bind stoichiometric amounts of Zn(II) or Ni(II), each with nanomolar affinity. Mutation of Cys106 and His107, which are located between two major GTPase motifs, results in undetectable Ni(II) binding, and the Zn(II) affinity is weakened by 2 orders of magnitude. These two residues are also required for the metal-dependent dimerization observed in the presence of Ni(II) but not Zn(II). The addition of metals to the protein has distinct impacts on GTPase activity, with zinc significantly reducing GTP hydrolysis to below detectable levels and nickel only slightly altering the kcat and Km of the reaction. The regulation of HypB activities by metal binding may contribute to the maturation of the nickel-containing enzymes.
Helicobacter pylori is a Gram-negative, microaerophilic bacterium that infects nearly half of the world's population (11). It is responsible for chronic gastric inflammation, peptic ulcers, and atrophic gastritis, the precursor lesion to gastric cancer (14, 25). Successful colonization of the human gastric mucosa by H. pylori requires the activity of two nickel-containing enzymes: urease and hydrogenase (15, 54). Urease contains a dinuclear nickel cluster in the active site and can catalyze the hydrolysis of urea to produce ammonia and bicarbonate (50, 51). The activity of urease is believed to prevent dramatic drops in the cytosolic pH of the bacterium, thus allowing it to withstand acid shock upon initial colonization of the gastric tract (59) and thrive upon continuous exposure to milder acidic conditions. The [NiFe]-hydrogenase enzyme provides a means for H. pylori to utilize hydrogen gas, a by-product of carbohydrate fermentation by other bacteria, as an energy source (54, 67).
The metallocenters of urease and [NiFe]-hydrogenase are complex and require the participation of multiple dedicated accessory proteins for bioassembly. To this end, H. pylori contain two operons encoding the UreIEFGH and HypABCDEF accessory proteins that aid in the maturation of the urease and hydrogenase enzymes, respectively (66). Furthermore, gene deletion mutants in H. pylori revealed that interruption of either hypA or hypB not only abrogated hydrogenase activity but also disrupted urease activity (55). Upon nickel supplementation of the growth media both hydrogenase and urease activity can be partially restored in the mutant strains, suggesting a role for HypA and HypB in the nickel delivery steps of both biosynthetic pathways (55).
Significant progress has been made in understanding the overall maturation process of the [NiFe]-hydrogenase enzyme (4, 40). After the iron is incorporated, along with its carbon monoxide and cyanide ligands, into the large subunit of the hydrogenase precursor protein, HypA and HypB are believed to cooperate to insert the nickel ion (37, 43), aided by SlyD in some organisms (30). The exact mechanism of this nickel delivery step is still not known, although it is clear that GTP hydrolysis by HypB is required for full hydrogenase maturation (45, 47, 53). In addition, HypB forms a complex with HypA in vitro (2, 48). The latter protein contains what appears to be an intrinsic structural zinc site, as well as a low-affinity nickel site (Kd ≈ 60 μM) in a separate domain that includes the N terminus of the protein (2, 32, 48, 68, 69). In vitro studies demonstrated that several HypB proteins also bind metal at different types of sites, depending on the homolog, with the variability occurring at the N terminus (30). A few HypB proteins have sequences rich in histidine residues that contribute to the binding of multiple nickel ions and are thought to function in nickel storage (18, 53, 57). Furthermore, Escherichia coli HypB (EcHypB) binds stoichiometric nickel with subpicomolar affinity to a three-cysteine motif at the N terminus of the protein (8, 36). Although this latter nickel site is essential for hydrogenase production in E. coli (13), it is not found in all HypB homologs. Finally, either nickel or zinc can bind with micromolar affinity to an essential site localized to the GTPase domain (G-domain) of EcHypB (13, 19, 36), and a crystal structure of HypB from Methanocaldococcus jannaschii revealed a corresponding dinuclear zinc site bridging two monomers of a homodimer (19). Due to the diverse metal-binding abilities of the HypB homologs, it is possible that the mechanisms of nickel delivery to the hydrogenase enzyme may vary between organisms. However, how the metal-binding activities of the HypB proteins affect other biochemical properties and contribute to metallocenter assembly is not known.
H. pylori HypB (HpHypB) is of significant interest because of several unusual features. HpHypB lacks both the N-terminal high-affinity metal-binding site and the histidine stretch found in many other HypB homologs. Furthermore, previous studies suggested that HpHypB is incapable of binding nickel (48). This finding was surprising as several of the residues that comprise the metal-binding site in the G-domain of EcHypB are conserved in HpHypB (see Fig. S1 in the supplemental material) (36). Finally, as mentioned above, HpHypB, along with HypA, also participates in urease maturation in addition to the UreDEFG accessory proteins, indicating that it has a key role in the nickel homeostasis of this pathogenic organism.
In order to learn more about the mechanism of action of this critical nickel homeostasis protein, we examined the metal-binding properties of HpHypB. Our results indicate that if the protein is fully reduced it can bind one equivalent of nickel or zinc. The affinity of the protein for these metals was determined, and the role of two absolutely conserved Cys and His residues as metal ligands was established. Next, we investigated whether metal binding has any influence on the other attributes of HypB, including GTP hydrolysis and secondary and quaternary structures, and the results demonstrate that nickel and zinc are indeed allosteric effectors, each modulating HpHypB in a distinctive fashion. This report constitutes the first detailed biochemical analysis of the effects of metal binding on the oligomeric state and enzymatic activity of a HypB protein, revealing that the activities of the protein are all intimately connected, which may be important for the function of HypB in the biosynthetic pathways of the nickel-containing urease and [NiFe]-hydrogenase enzymes.
MATERIALS AND METHODS
Materials.
Restriction endonucleases and T4 DNA ligase were obtained from New England Biolabs. Primers (see Table S1 in the supplemental material) were purchased from Sigma Genosys. All chromatography media were from GE Healthcare. Kanamycin, tris(2-carboxyethyl)phosphine (TCEP), and IPTG (isopropyl-β-d-thiogalactopyranoside) were purchased from BioShop (Toronto, Ontario, Canada). Nickel chloride salt (as a minimum, 99.9% pure) was purchased from Sigma, and the concentrations of stock solutions in water were verified by inductively coupled plasma-atomic emission spectroscopy. Other metal stocks were atomic absorption standard solutions. All other reagents were analytical or molecular biology grade from Sigma. Electronic absorption measurements were conducted on an Agilent 8453 spectrophotometer with a 1-cm-path-length cuvette. The buffers for all metal assays were treated with Chelex-100 (Bio-Rad) to minimize trace metal contamination. All solutions were prepared with Milli-Q water, 18.2 MΩ-cm resistance (Millipore).
HpHypB expression vector and mutants.
The coding sequence of HpHypB was amplified from genomic H. pylori DNA (strain 26695) by using primers designed with restriction sites for NdeI (HpHypB forward) and XhoI (HpHypB reverse) (see Table S1 in the supplemental material). The digested PCR product was ligated with T4 DNA ligase into the pET24b vector (Novagen) digested with NdeI and XhoI and dephosphorylated with calf intestinal phosphatase (New England Biolabs). The C106A, H107A HpHypB mutant was created from the HpHypB-pET24b construct by QuikChange PCR mutagenesis (Stratagene) with Pfu Turbo polymerase by using the primers listed in Table S1 in the supplemental material. The template strand was subsequently digested with DpnI. For production of large amounts of the parent HpHypB-pET24b and mutant plasmids, the plasmids were transformed into XL-2 Blue E. coli competent cells (Stratagene) and isolated by using the Fermentas GeneJET plasmid miniprep kit. All plasmids were sequenced (ACGT, Toronto, Ontario, Canada) in the forward and reverse directions by using the T7 promoter and terminator primers.
Protein expression and purification.
For expression of wild-type (WT) and mutant HpHypB, the plasmids were transformed into BL21 Star (DE3) E. coli cells (Invitrogen). Overnight cultures were grown, and 25 ml was used to inoculate 1.5 liters of LB medium supplemented with 50 μg of kanamycin/ml. The cells were grown aerobically at 37°C until the A600 reached 0.6, at which point they were induced with 0.25 mM IPTG. For some purifications, 1.5 mM NiSO4 was added to the medium prior to IPTG, and nickel-loaded protein was purified, but all subsequent steps were the same for both apo- or holo-proteins. After shaking at 37°C for an additional 5 h, the cells were harvested by centrifugation and resuspended in 40 ml of 20 mM Tris (pH 7.5) and 100 mM NaCl supplemented with 4 mM TCEP and two Complete, Mini, EDTA-free protease inhibitor cocktail tablets (Roche Applied Science). For a single protein purification preparation, a total of 6 liters of cell culture was used. All subsequent steps were performed at 4°C or on ice. The resuspended cells were sonicated and centrifuged at 25,000 × g for 40 min. The supernatant was passed through a 0.45-μm-pore-size syringe filter and then loaded onto a DEAE Sepharose anion-exchange column (GE Healthcare) equilibrated with buffer A (20 mM Tris [pH 7.5], 1 mM TCEP). Fractions from a NaCl gradient were screened by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 12.5% gels. WT and mutant HpHypB eluted at approximately 50 mM NaCl. Fractions containing the protein of interest were pooled and dialyzed against buffer A for at least 3 h. The sample was then loaded onto a Hi-TrapQ anion-exchange column (GE Healthcare) equilibrated with buffer A. Once again, fractions from a NaCl gradient were screened by SDS-PAGE, and fractions containing the protein of interest (eluting at approximately 50 mM NaCl) were pooled. After concentration of the pooled fractions to 1 ml using Amicon Ultra 3K MWCO centrifuge concentrators (Millipore), the sample was loaded onto a Superdex 200 gel filtration column (GE Healthcare) equilibrated with 25 mM HEPES (pH 7.6), 200 mM NaCl, and 1 mM TCEP. Fractions containing the protein of interest were pooled and concentrated such that the final concentration was in the range of 250 to 500 μM. The protein concentrations were calculated by using the extinction coefficient of 7,450 M−1 cm−1 for both WT and mutant HpHypB at 280 nm in 4 M guanidinium HCl (GuHCl) (21). A sample of each protein was sent for electrospray ionization mass spectrometry (ESI-MS; Department of Chemistry, University of Toronto), and the determined molecular masses of the WT and the double mutant were determined to be 27,178.0 and 27,080.0 Da, respectively. These values are 132 Da lower than the calculated molecular masses of 27,310.4 and 27,212.2 Da, suggesting that the N-terminal methionine residue is removed. All proteins were >90% pure, as estimated by Coomassie blue-stained SDS-PAGE and by using the public domain ImageJ program (U.S. National Institutes of Health [http://rsb.info.nih.gov/ij/]).
HPLC metal analysis.
A high-pressure liquid chromatography (HPLC)-based method for the detection and identification of metal ions in solution was previously developed by our laboratory (1). For HPLC analysis, at least 50 μg of protein was dried by centrifugation under vacuum, reconstituted with metal-free concentrated HCl (SeaStar Chemicals), followed by incubation overnight at 95°C for protein hydrolysis. The sample was once again dried to remove HCl and reconstituted in 80 μl of Milli-Q water. This sample was injected onto a Dionex IonPak CS5A column equilibrated with 7 mM pyridine-2,6-dicarboxylic acid, 66 mM KOH, 5.6 mM K2SO4, and 74 mM HCOOH and attached to a metal-free Dionex BioLC HPLC system. The metals were detected at 500 nm after post-column mixing with 4-(2-pyridylazo)-resorcinol (PAR).
Preparation of proteins.
Reduced, apo-protein was produced by incubating the protein with 10 mM EDTA and 20 mM TCEP in a Coy anaerobic glove box at 4°C for 48 h. The TCEP and EDTA were removed in the glove box by exhaustive dialysis into protein buffer (25 mM HEPES, 100 mM NaCl [pH 7.6]). The absence of any bound metal to the protein was confirmed by HPLC metal analysis (1). The free thiol content of the proteins was measured via reaction of the protein with DTNB [5,5′-dithiobis(2-nitrobenzoic acid)] in the presence of 6 M GuHCl and 1 mM EDTA. β-Mercaptoethanol was used as a standard, and the absorbance of the 5-mercapto-2-nitrobenzoic acid product was measured at 412 nm. Protein samples were >90% reduced after treatment with TCEP.
Metal binding and stoichiometry.
Individual samples containing 20 μM apo-HpHypB in protein buffer and 0 to 120 μM NiCl2 were prepared in the glove box and incubated overnight at 4°C. The electronic absorption spectrum was monitored between 250 and 500 nm and corrected by background subtraction at 600 nm. Metal stoichiometry experiments were conducted by incubating 120 μM apo-HpHypB with either 360 μM nickel or zinc overnight at 4°C in the glove box. Excess metal was removed by passing the protein through a PD-10 gel filtration column (GE Healthcare) equilibrated with protein buffer in the glove box. The protein concentration was subsequently determined (see above). The metal content was determined via a PAR assay (26) in which the protein was denatured with 4 M GuHCl, and 50 μM PAR was added to the sample. The absorbance at 500 nm, corresponding to the formation of a 2:1 PAR-Me2+ complex, was monitored and compared to a standard curve prepared with 50 μM PAR in 4 M GuHCl and known metal concentrations. To confirm the results of the PAR assay and to identify the metal present, HPLC metal analysis was also conducted on these samples (1).
HpHypB metal affinity.
The nickel titration suggested that HpHypB binds nickel quantitatively under the conditions described above, indicating that the metal affinity would be difficult to determine without a competitor. As such, the competitor Mag-fura-2 was selected due to its reported mid-nM Kd for nickel and zinc (22). Stocks of Mag-fura-2 (Invitrogen) were prepared in Milli-Q water and quantified by using the reported extinction coefficient of 22,000 M−1 cm−1 at 369 nm (22). To determine the metal-binding affinity of Mag-fura-2 with Ni(II) and Zn(II) under our experimental conditions, fluorescence spectroscopy was used. For determination of the Ni(II) and Zn(II) Kd values, 0 to 6 μM metal was titrated into a sample of 5 nM Mag-fura-2 in protein buffer and allowed to equilibrate at room temperature for 5 min between metal additions. Mag-fura-2 was excited at 366 nm, and the decreasing fluorescence upon metal addition was monitored at 500 nm. All fluorescence experiments were conducted on a JY HORIBA Fluorolog-3 spectrofluorimeter.
Competition experiments were prepared by incubating 10 μM WT or mutant HpHypB, together with 10 μM Mag-fura-2 and various amounts of Ni(II) or Zn(II). The samples were incubated overnight at 4°C in the glove box. The absorbance of Mag-fura-2 at 366 nm was monitored, and the data were analyzed by using DYNAFIT (33) using a custom DYNAFIT script that describes the competition between the protein and Mag-fura-2 for the metal.
To determine the affinity of HpHypB for Zn(II), a competition between Zn(II) and Ni(II) was conducted. A solution containing 10 μM HpHypB was incubated with 50 μM Ni(II) overnight at 4°C in the glove box. Various concentrations of Zn(II) were then titrated into the sample, and the decrease of the 350-nm LMCT peak was monitored. The data were analyzed with DynaFit using a custom script describing a direct competition for the protein by Ni(II) and Zn(II).
Analytical gel filtration chromatography.
Samples containing 140 μM HpHypB were incubated with the desired metal or GDP at various concentrations (see Tables S2 and S3 in the supplemental material) overnight at 4°C in the glove box. For samples containing GTP, the protein was incubated with GTP for at least 2 h at 4°C in the glove box prior to injection onto the gel filtration column. All samples contained 25 mM HEPES, 100 mM NaCl, and 5 mM MgCl2 (pH 7.6). Apo-protein and metal-containing samples were loaded onto a Superdex 200 10/300 analytical gel filtration column (GE Healthcare) equilibrated with filtered and chelexed 25 mM HEPES, 200 mM NaCl, and 5 mM MgCl2 (pH 7.6). The column was calibrated with thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.4 kDa) from Bio-Rad. No difference in the elution profile of these standard proteins was observed when 140 μM Ni(II) or Zn(II) was added prior to gel filtration chromatography. Molecular masses were determined by plotting the log molecular masses of the standards versus the partition coefficients (Kav), where Kav = (Ve − Vo)/(Vt − Vo), Ve represents the elution volume, Vo is the void volume, and Vt is the total column volume. The eluted peaks were collected and subjected to inductively coupled plasma-mass spectrometry (ICP-MS; Department of Chemistry, University of Toronto) to ensure the presence of the added metal.
CD spectroscopy.
WT and mutant HpHypB samples were prepared for circular dichroism (CD) spectroscopy by diluting the protein in Milli-Q water to a final concentration of ∼10 μM in the glove box. For metal titrations, either Zn(II) or Ni(II) was added to the diluted samples and allowed to equilibrate overnight at 4°C in the glove box. Protein samples with GDP or GTP included 1 mM Mg(II) and were also incubated overnight at 4°C in the glove box. All samples were analyzed on a Jasco J-170 spectropolarimeter with a capped 1-mm path-length cuvette in order to minimize exposure to the air. Spectra were collected at 1 nm intervals over a spectral range of 200 to 260 nm with a scan speed of 20 nm min−1 at room temperature. The final spectra obtained are averages of six scans and corrected by subtracting the background buffer signal. The observed ellipticity was converted into mean residue ellipticity ([θ]mre) in degrees cm2 dmol−1 by using the following equation (31):
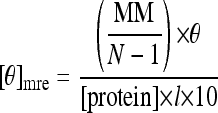 |
where MM is the molecular mass of the protein in Da, N is the number of amino acids, θ is the observed ellipticity in degrees, [protein] is the concentration of protein in g/ml, and l is the path-length.
GTPase assay.
GTPase activity was determined by a Malachite Green assay for free phosphate adapted from Lanzetta et al. (34). A series of 160-μl samples containing 2 μM HpHypB (in 25 mM HEPES, 100 mM NaCl [pH 7.6]), 5 mM Mg(II), and various GTP concentrations between 12.5 and 700 μM were incubated at 37°C in the glove box for 2.5 h. Controls containing only buffer, 5 mM Mg(II), and the corresponding GTP concentrations were prepared alongside the protein samples and received the same treatment. After incubation, the samples were quickly plated on a 96-well plate, and 40 μl of the phosphate detection reagent (2.6 mM Malachite Green, 1.5% ammonium molybdate, and 0.2% Tween 20) was added to each sample. The samples were then gently mixed by shaking the plate for 3 min, after which sodium citrate was added to a final concentration of 3.5%. The plate was then mixed again by shaking, and the color was allowed to develop for 30 min before the absorbance was measured at 630 nm with an EL808 Ultra microplate reader (Bio-Tek Instruments). The amount of phosphate released was determined by using a standard curve from a phosphate standard (Molecular Probes). The data were analyzed by fitting to the Michaelis-Menten equation by using OriginPro 7.5. Samples containing nickel or zinc were incubated with the metal overnight prior to the assay (prepared as a stock of 40 μM protein with either 80 μM Zn(II) or 200 μM Ni(II)). These stocks were then diluted to a final protein concentration of 2 μM for the assay in a buffer that contained either 10 μM Zn(II) or 20 μM Ni(II).
HpHypB structural modeling.
A homology model of HpHypB was generated by using MODELLER 9v7 (58) based on the general protocol as outlined in Eswar et al. (17). Template searches using the HpHypB sequence as the inquiry yielded the M. jannaschii HypB (MjHypB) X-ray crystal structure (PDB 2HF9) as the only hit. A global alignment in MODELLER was conducted to align the two structures, and this alignment was used to generate 100 models. All of these sequences featured a large unstructured loop at the N terminus. Due to this loop, all models yielded poor pseudo-energy profiles compared to the template, and PROSAII analysis (62) indicated that none of these models were acceptable. In order to avoid this loop region, the amino acids in the HpHypB sequence that align with the unresolved first 10 amino acids of MjHypB, corresponding to residues 1 to 23 of HpHypB, were deleted. This sequence was then used for the alignment and model generation as described above. The resulting models yielded better pseudo-energy profiles and the PROSAII analysis indicated far better model quality. The best 25 models based on their DOPE (49) score were further analyzed and submitted for PROCHECK (35), ERRAT (10), and VERIFY3D (7, 41) analysis on the University of California at Los Angeles Structural Analysis and Verification Server (http://nihserver.mbi.ucla.edu/SAVES/). Based on these analyses, the best model was selected. All images were created by using UCSF Chimera (16).
RESULTS
HpHypB metal binding.
Due to the proposed role of HpHypB in the bioassembly pathways of the nickel-containing urease and hydrogenase enzymes, determining whether the protein could bind nickel was the initial focus of the present study. After treatment with TCEP and incubation with stoichiometric NiCl2, an intense charge-transfer band centered at 350 nm was apparent (Fig. 1 A). This absorption can be attributed to a Cys—S− → Ni(II) ligand-to-metal charge transfer (LMCT) (27, 39) and suggests a contribution of at least one Cys residue as a metal ligand in HpHypB. The difference spectrum, obtained by subtracting the signal of apo-HpHypB from that of HpHypB loaded with 1 equivalent of Ni(II), revealed a second peak at 293 nm (Fig. 1A inset), which is also attributable to a Cys—S− → Ni(II) LMCT (27). Upon titration of 20 μM apo-HpHypB with increasing amounts of nickel, a linear increase was observed and saturation of the signal occurred upon the addition of 1 equivalent of nickel, suggesting a 1:1 Ni(II):HpHypB stoichiometry (Fig. 1B). The linear portion of the titration yielded an extinction coefficient of (4.1 ± 0.3) × 103 M−1 cm−1 at 350 nm. In order to confirm the metal-binding stoichiometry, 120 μM apo-HpHypB was incubated with 360 μM Ni(II), followed by gel filtration chromatography to remove excess metal. Subsequent metal analysis revealed that 1 equivalent of nickel remained bound to the protein (Table 1) . In contrast, when unreduced HpHypB was incubated with stoichiometric amounts of NiCl2, little change was evident in the electronic absorption spectrum (Fig. 1, inset), and the amount of nickel bound to protein was <20% of the protein concentration upon gel filtration and analysis with PAR. This lack of nickel binding may be due to cysteine oxidation, which was confirmed by analysis with DTNB (data not shown).
FIG. 1.

Nickel binding to HpHypB. (A) Upon the addition of 1 equivalent of NiCl2 to apo-HpHypB (dotted line), an overall increase in absorbance is noted between 250 and 450 nm (solid line), with the formation of a new peak centered at 350 nm. A difference spectrum (inset, solid line) was generated by subtracting the signal of apo-HpHypB from that of HpHypB loaded with 1 equivalent of Ni(II). Metal analysis after gel filtration chromatography confirmed stoichiometric nickel binding (Table 1). When HpHypB is oxidized, the peaks in the difference spectrum upon the addition of nickel are significantly less intense (inset, dashed line), suggesting that the protein's ability to bind Ni(II) is diminished. (B) Upon titration of 20 μM apo-HpHypB with NiCl2, a linear increase in the absorbance at 350 nm is observed until approximately 1 equivalent of metal has been added (circles). The linear region of the titration curve yields an extinction coefficient of (4.1 ± 0.3) × 103 M−1 cm−1. A similar titration with oxidized HpHypB does not yield an increase in the signal at 350 nm (triangles).
TABLE 1.
Stoichiometry of metal binding to WT and mutant HpHypBa
| Protein | Metal added | Metal boundb |
|---|---|---|
| WT HpHypB | Ni(II) | 1.0 ± 0.3 |
| Zn(II) | 1.0 ± 0.1 | |
| C106A, H107A HpHypB | Ni(II) | ND |
| Zn(II) | 1.2 ± 0.3 |
Apo-HpHypB (120 μM) was incubated with 360 μM either Ni(II) or Zn(II) overnight at 4°C in an anaerobic glove box. Excess metal was removed by passing the proteins over a PD-10 gel filtration column, and metal was detected either via a PAR assay or by HPLC metal analysis.
The data listed are average values and standard deviations of the number of metal ions bound per protein monomer from three independent experiments. ND, no metal detected.
To measure the affinity of WT HpHypB for nickel, a competition for nickel between HpHypB and the fluorimetric metal chelator Mag-fura-2 was conducted. Mag-fura-2 was originally developed as a fluorescent dye for measuring free magnesium in the cytosol (56), but several studies have also used it for other divalent metals (12, 22, 61). Upon metal binding to Mag-fura-2, a blue-shift of the 366 nm absorbance to 325 nm is observed (Fig. 2 A). The affinity of Mag-fura-2 for Ni(II) has been previously reported (12, 22, 61), but under different conditions than those used in these competitions. Thus, the Mag-fura-2 nickel Kd was determined via fluorescence under our conditions and was found to be 150 ± 10 nM, a value in close agreement to that determined by Golynskiy et al. (22). Upon titration of equivalent amounts of HpHypB and Mag-fura-2 with nickel, a decrease in the 366-nm signal of apo-Mag-fura-2 was observed, and an affinity of HpHypB for nickel (KdNi(II)) of 150 ± 20 nM was calculated (Fig. 2B).
FIG. 2.

Affinity of HpHypB for Ni(II). (A) In order to measure the strength of the HpHypB-Ni(II) interaction, a competition between HpHypB and the metal chelator Mag-fura-2 for Ni(II) was established. The spectrum of apo-Mag-fura-2 features a peak at 366 nm that blue-shifts to 325 nm upon metal binding. The spectra shown depict the change in absorbance of 10 μM Mag-fura-2 upon the addition of up to 50 μM Ni(II) in the presence of 10 μM HpHypB. (B) By monitoring the decrease of the Mag-fura-2 366 nm signal upon metal binding and fitting these data versus the amount of Ni(II) added, the HpHypB KdNi(II) was determined to be 150 ± 20 nM. The closed circles are the average of three replicate datasets; the error bars represent the standard deviation, and the black line is the best-fit curve as determined by a custom DynaFit script.
Given the clear nickel-binding ability of HpHypB and the evidence for zinc binding to other HypB homologs (19, 36), zinc binding was also investigated. Incubation of the protein with excess zinc, followed by gel filtration chromatography and metal analysis revealed stoichiometric zinc bound (Table 1). When a titration of 10 μM protein was conducted with zinc in the presence of Mag-fura-2 an initial plateau region was noted in which the 366 nm signal of apo-Mag-fura-2 did not change until after 10 μM zinc was added, suggesting that the protein was outcompeting Mag-fura-2 for the available zinc (data not shown). In order to determine the zinc affinity, the ability of zinc to compete with nickel binding to HpHypB was exploited. In this competition, HpHypB was loaded with excess nickel and zinc was titrated into the holo-protein. As zinc bound to the protein, a decrease in the 350-nm signal of the Ni(II)-protein complex was monitored (Fig. 3 A). This experiment yielded a KdZn(II) of 1.2 ± 0.3 nM, which is 2 orders of magnitude tighter than the Kd for nickel.
FIG. 3.

Affinity of WT and C106A, H107A HpHypB for Zn(II). (A) A solution containing 10 μM HpHypB was incubated with 50 μM Ni(II) overnight at 4°C in an anaerobic glove box prior to the addition of increasing amounts of Zn(II). The decrease in the absorbance at 350 nm was monitored and used to calculate the fractional saturation of the protein with nickel. The data from several experiments were analyzed using DynaFit to determine a KdZn(II) of 1.2 ± 0.3 nM. The data shown are from a representative data set, and the line is the line of best fit as determined by DynaFit. (B) The affinity of the C106A, H107A HpHypB mutant (10 μM) for Zn(II) was determined by using a competition with Mag-fura-2 (10 μM). The absorbance of apo-Mag-Fura-2 was monitored at 366 nm upon titration with zinc and used to calculate a Kd Zn(II) for the double mutant of 110 ± 40 nM. The closed circles represent a representative raw titration data set, and the line is the best-fit curve as determined by a custom DynaFit script.
Finally, the CD spectrum of WT HpHypB suggests that the protein contains significant α-helical content as indicated by the presence of two minima at approximately 208 and 222 nm (31) (data not shown). The overall secondary structure of the protein does not appear to change upon the addition of Ni(II), Zn(II), or nucleotide (data not shown).
Identification of metal-binding residues.
Residues C106 and H107 are conserved in all HypB homologs (see Fig. S1 in the supplemental material). In EcHypB, these residues correspond to C166 and H167 and upon mutation to alanine, the nickel-binding ability of the protein is diminished (36). To determine whether these residues participate in metal binding to HpHypB, both C106 and H107 were mutated to alanines. The CD spectrum of the double mutant was identical to that of the WT protein, demonstrating that these mutations do not affect the secondary structure of the protein (data not shown). Mutating C106 and H107 to alanine in HpHypB resulted in a loss of detectable nickel binding following gel filtration and metal analysis (Table 1), and a change in the electronic absorption spectrum was not observed upon titration with nickel (data not shown). In contrast, stoichiometric zinc binding was maintained (Table 1). However, zinc binding by the mutant protein was sufficiently weakened such that the affinity of the C106A, H107A HpHypB mutant for Zn(II) could be determined via competition between the protein and Mag-fura-2. Similar to the affinity for nickel, the affinity of Mag-fura-2 for zinc was first determined by using fluorescence to be 101 ± 6 nM. This affinity is slightly weaker than some previously published values (22) but close to that determined by de Seny et al., who used a buffer composition similar to that of the present study but at a lower pH (12). A clear competition was observed between Mag-fura-2 and mutant HpHypB (Fig. 3B), yielding a KdZn(II) of 110 ± 40 nM. The disruption of nickel binding and weakened zinc binding upon mutation of C106 and H107, coupled to the ability of zinc to compete with nickel, suggests that the coordination spheres of the two metals share some ligands but are not identical.
HpHypB quaternary structure.
Analytical gel filtration chromatography was used to investigate the oligomeric state of apo-HpHypB, as well as how it is affected by metal and/or nucleotide. In the apo form, HpHypB eluted as a monomer from the column (Fig. 4). A previous study reported that HpHypB exists as a homodimer in solution (48), and this discrepancy may be due to different experimental conditions such as protein oxidation. Upon the addition of half an equivalent of nickel, a significant portion of the protein dimerizes, with all of the protein existing as a dimer upon the addition of 1 equivalent of nickel (Fig. 4). Further addition of nickel did not change the dimeric state (data not shown). When the conserved residues C106 and H107 were mutated to alanine, the protein lost the ability to dimerize upon the addition of nickel (see Table S2 in the supplemental material), indicating that these residues are necessary for metal-induced dimerization of the protein. Zinc, on the other hand, did not affect the oligomeric state of the protein (Fig. 4). Even with 420 μM Zn(II) in the mobile phase, the protein still eluted as a monomer (data not shown). The presence of metal in all holo-samples following elution from the column was confirmed by ICP-MS. When the protein was incubated with equimolar amounts of zinc and nickel, little dimerization was observed (see Table S2 in the supplemental material). ICP-MS analysis indicated that zinc was bound to the protein instead of nickel, supporting the finding that HpHypB has a higher affinity for zinc than nickel and that it preferentially binds zinc over nickel.
FIG. 4.

Effect of metal on the quaternary structure of HpHypB. In the absence of metal, HpHypB (140 μM) elutes from a gel filtration column at a volume corresponding to a monomer (solid line). Upon the addition of half an equivalent of nickel (dashed line), a significant proportion of the protein dimerizes, with all protein eluting as dimer upon the addition of a single equivalent of metal (dash-dot line). The addition of 1 equivalent of zinc did not affect the protein's oligomeric state (dotted line). The chromatographic traces shown are representative experiments using a Superdex 200 10/300 analytical column with 25 mM HEPES (pH 7.6), 200 mM NaCl, and 5 mM MgCl2 as the mobile phase. The eluate was monitored at 280 nm, and the nickel-bound protein absorbs much more strongly at this wavelength than apo-protein (Fig. 1). The ticks at the top of the graph denote the elution volumes of the protein standards. From left to right, the identity of the standards and their respective molecular masses are as follows: γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.4 kDa).
The calculated molecular mass of the apo-HpHypB species from the gel filtration chromatography experiments (28.1 ± 2.6 kDa) is in close agreement with the predicted molecular mass (27.2 kDa). However, the monomeric species in the presence of metal elutes as a slightly smaller protein (23.0 to 25.8 kDa), suggesting the formation of a more compact protein structure upon binding of metal. Furthermore, the observed molecular mass of the nickel-induced dimeric species is smaller than expected (MMobs = 50.9 ± 1.9 kDa and MMpred = 54.2 kDa, respectively), suggesting that the metal-bound dimer is also more compact in its overall shape.
If the protein was incubated with nucleotides and applied to a gel filtration column, little change in the oligomeric state of the protein was observed (see Table S3 in the supplemental material). However, when 400 μM GDP or GTP was included in the mobile phase, some nucleotide-dependent dimerization of HpHypB was detected (see Table S3 in the supplemental material). The requirement of additional nucleotides in the mobile phase suggests that the interaction between the protein and nucleotide is weak enough for dissociation to occur during the time scale of the gel filtration experiment. GTP hydrolysis by HpHypB is slow enough (see below) such that over the course of the gel filtration experiment significant hydrolysis of the GTP to GDP should not be an issue. Finally, the addition of zinc to the protein inhibited both GTP- and GDP-dependent dimerization (see Table S3 in the supplemental material).
GTPase activity.
As previously reported (48), HpHypB catalyzes the hydrolysis of GTP (Fig. 5 and Table 2). The experiments shown here reveal that the enzymatic activity of HpHypB is quite low, with a kcat of only (6 ± 1) × 10−4 s−1 and a Km of (5 ± 1) × 10−5 M (kcat/Km of 12 ± 3 M−1 s−1). To determine whether metal binding modulates the GTPase kinetics, the protein was incubated with extra nickel or zinc. In the presence of Ni(II), the kcat increases severalfold to (1.6 ± 0.1) × 10−3 s−1 and the Km of the protein increases to (1.6 ± 0.5) × 10−4 M, suggesting a slightly weakened affinity for GTP compared to that of apo-HpHypB. Overall, the addition of Ni(II) to the protein does not affect the kcat/Km, since it remains at 11 ± 3 M−1 s−1. In contrast, the addition of Zn(II) inhibits the enzymatic activity, and no activity was detectable in the presence of 10 μM Zn(II) (Fig. 5). Mutating the C106 and H107 residues to alanine did not dramatically affect GTPase activity, and the double mutant had a Km = (4.0 ± 0.2) × 10−5 M, a kcat = (1.0 ± 0.1) × 10−4 s−1, and a kcat/Km = 2.4 ± 0.3 M−1 s−1 (data not shown).
FIG. 5.

GTPase activity of HpHypB. Apo-HpHypB (2 μM, squares) was incubated with increasing concentrations of GTP at 37°C for 2 h in an anaerobic glove box prior to detection of released inorganic phosphate by using a colorimetric assay. Fitting the data from several experiments to the Michaelis-Menten equation yields a kcat of (6 ± 1) × 10−4 s−1, a Km of (5 ± 1) × 10−5 M, and a kcat/Km of 12 ± 3 M−1 s−1. The presence of 20 μM nickel (circles) results in an increased kcat of (1.6 ± 0.1) × 10−3 s−1 and Km of (1.6 ± 0.5) × 10−4 M. Thus, overall the kcat/Km does not change and remains at 11 ± 3 M−1 s−1. The addition of 10 μM zinc inhibits GTPase activity (triangles). All datasets are the average of at least three independent replicates, and error bars represent the standard deviations of the replicates.
TABLE 2.
Kinetics of GTP hydrolysis by HpHypBa
| Buffer | Avg ± SDb |
||
|---|---|---|---|
| kcat (s−1) | Km (M) | kcat/Km (M−1 s−1) | |
| Apo WT HpHypB | (6 ± 1) × 10−4 | (5 ± 1) × 10−5 | 12 ± 3 |
| + 20 μM Ni(II) | (1.6 ± 0.1) × 10−3 | (1.6 ± 0.5) × 10−4 | 11 ± 3 |
| + 10 μM Zn(II) | NH | NH | NH |
All GTPase assays were conducted with 2 μM HpHypB in protein buffer supplemented with 5 mM MgCl2. Samples containing metal were preincubated with either zinc or nickel overnight at 4°C in an anaerobic glove box. The amount of released phosphate was detected using a modified Malachite Green assay.
The data listed are average values from three independent experiments. NH, no measurable hydrolysis.
HpHypB structural modeling.
The crystal structure of MjHypB (19) provided an excellent template upon which a structural model of HpHypB could be constructed. The sequences of HpHypB and MjHypB are 30% identical and 51% similar, sufficient to create a reasonable structural model of HpHypB (42). In order to verify the validity of our final homology model, several tests were conducted. ERRAT analyzes the nonbonded interactions between different atoms and scores the overall structure based on a comparison to highly refined structures (10). The final model had an ERRAT score of 71.09, indicating an acceptable model (score > 50). Furthermore, PROCHECK, an analysis of the overall stereochemistry, indicated no major deviations from ideal protein stereochemistry, with 100% of the residues falling within acceptable regions of the Ramachandran plot. VERIFY 3D analyzes the compatibility of a model with its own amino acid sequence (7, 41). For the HpHypB homology model, 82.27% of the residues have a score of greater than 0.2, indicating a good quality model. The calculated Cα RMSD between the homology model and the template was 0.285 Å, demonstrating close homology between the model and template.
The constructed model of HpHypB is a globular protein with a seven-strand parallel β-sheet core surrounded by α-helices, a structure characteristic of P-loop NTPases of the SIMIBI class (Fig. 6) (38). This classification is in accordance with the large P-loop NTPase phylogenetic study conducted by Leipe et al. in which the authors found that the HypB proteins are evolutionarily related to the UreG, COG0523 (also known as CobW), and ArgK proteins, which together comprise the G3E family of GTPases (38). This family is defined by a glutamate residue in the Walker B motif (hhhhEXXG, where h is any hydrophobic amino acid; also referred to as the G3 motif), which substitutes for the conserved Mg(II)-binding aspartate residue (hence the name G3E) (38). An alignment of the HpHypB and MjHypB sequences (see Fig. S1 in the supplemental material) reveals a conservation of all of the major GTPase motifs, as well as the CH motif located between the Walker A and B motifs. In the homology model, these two residues are located on the surface of the protein (Fig. 6) adjacent to an additional conserved cysteine, C142. The surface-exposed position of these residues is ideal for mediating nickel-dependent dimerization. Furthermore, an overlay of the protein model with that of the crystal structure of MjHypB demonstrates that the major nucleotide motifs are located in similar positions, as would be expected (data not shown).
FIG. 6.

Homology model of HpHypB. A homology model of HpHypB was created by using Modeler 9v7 with the MjHypB crystal structure as a template (PDB: 2HF9). The resulting model is a typical P-loop GTPase with a central seven-stranded parallel β-sheet core surrounded by α-helices. The proposed metal-binding residues mutated in the present study, C106 and H107, are located on the surface of the protein adjacent to another absolutely conserved C142, also believed to be involved in the metal binding (inset). The surface-exposed nature of these residues supports their role in the nickel-induced dimerization of the protein. The only other likely metal-binding residue within the area of C106 and H107 is E109, which is also depicted.
DISCUSSION
HpHypB contributes to the maturation of both urease and hydrogenase in H. pylori (55). Initial characterization of the protein suggested that it does not bind Ni(II) (48); however, the experiments reported here demonstrate that HpHypB can bind both nickel and zinc when fully reduced. The protein is clearly sensitive to oxygen, but given that hydrogenase enzymes involved in utilizing H2 as an energy source are typically associated with anaerobic metabolism (9), this oxygen sensitivity is likely not an issue for HpHypB in vivo. It is possible that the disruption of metal binding by oxidation is a mechanism to regulate the function of HpHypB and hydrogenase and/or urease production in H. pylori, as has been suggested for other types of systems (for examples, see references 20, 23, 46, and 72), but there is no evidence yet to support this hypothesis.
Although HpHypB lacks both the N-terminal high-affinity metal-binding sequence and a histidine-rich sequence, it is still capable of binding stoichiometric metal. The fact that zinc can compete with nickel suggests that the metal ions have some common ligands. It is also possible that binding of zinc at a separate location of HpHypB has an allosteric effect on the conformation of the protein that inhibits nickel binding, but the lack of any detectable change in the CD spectrum upon binding of either metal does not support a dramatic change in the protein conformation. Furthermore, mutating the conserved C106/H107 amino acids completely abrogates detectable nickel binding and weakens the affinity for zinc by 2 orders of magnitude, indicating that both residues can coordinate either nickel or zinc. However, the distinct effects of each type of metal ion on the properties of the protein indicate that the sites are not superimposable. Upon closer examination of the homology model of HpHypB, very few residues located near the CH motif appear to be capable of binding metals. One possibility is C142, located adjacent to the CH motif (Fig. 6). Preliminary experiments with a C142A mutant suggested that nickel binding is significantly weakened when this residue is removed, but extensive experimentation was hindered due to metal-induced protein aggregation of the mutant protein (data not shown). This cysteine residue is conserved in all HypB homologs, suggesting a critical role in the protein (see Fig. S1 in the supplemental material). A second possible metal ligand is E109, located near H107 (Fig. 6). This residue is also well-conserved in HypB homologs, either as a glutamic acid or an aspartic acid (see Fig. S1 in the supplemental material).
The conserved CH motif serves to bind metal in the MjHypB crystal structure, which revealed a bimetallic site at the interface between two monomers (19). The presence of a similar nonsymmetrical site in the isolated G-domain of EcHypB loaded with zinc was suggested by X-ray absorption spectroscopy (13). However, despite the evidence for a metal-binding site at a dimeric interface, no prior biochemical study has investigated the effect of metal on the oligomeric structure of HypB. In the case of HpHypB, metal-induced dimerization of HpHypB is not observed in the presence of zinc, and in fact zinc blocks dimerization stimulated by nucleotide, indicating that zinc binding at a dimer interface is not conserved. Dimerization is only induced by nickel, further supporting the differences between the metal coordination properties of each metal.
The disparate effects of nickel versus zinc on the quaternary structure of HpHypB may explain the metal-dependent modulation of the GTPase activity observed in the present study. The structure of MjHypB revealed nucleotide-binding sites composed of residues from both monomers, suggesting that the dimer is the form required for GTP hydrolysis (19). Thus, a metal that enhanced dimerization, such as nickel in the case of HpHypB, would either activate the GTPase or have no effect, whereas metal binding that blocked dimerization would inhibit hydrolysis, as observed upon addition of zinc. However, the situation might not be so straightforward because the quaternary structure required for active HpHypB has not yet been established, and work with full-length EcHypB indicates that the protein is active as a monomer (F. Cai and D. B. Zamble, unpublished data). In support of an alternative explanation, the invariant C142, located at an appropriate position to coordinate a metal ion bound to C106/H107 on the monomeric protein (Fig. 6), is within the highly conserved Switch II motif (see Fig. S1 in the supplemental material) that connects GTP hydrolysis to conformational changes in the protein (63). Given this location of C142, it is feasible that metal binding to HpHypB could influence GTP hydrolysis in a manner that depends on if and/or how C142 coordinates the metal but that is independent of quaternary structure.
It has been shown that several isolated HypB proteins are poor GTPases, and HpHypB is no exception (18, 44, 57). This low level of hydrolysis is typical of GTPases that are examined in vitro, where they lack additional protein partners and cofactors that may stimulate GTPase activity in vivo (65). For example, mammalian p21ras, one of the best-characterized GTPases involved in control of critical regulatory pathways such as cell proliferation and differentiation (6), has low GTPase activity in vitro, with a kcat of 3.4 × 10−4 s−1 (52), a number comparable to that of HpHypB. Given the metal-binding activity of the protein, it was reasonable to predict that metal could activate GTP hydrolysis. However, nickel binding has a minimal effect and zinc regulates catalysis in a negative manner. Thus, it is likely that the GTPase activity of HpHypB is activated in the context of a functional maturation pathway through the interaction with another protein or cofactor. Thus far, HpHypB has been shown to also interact with HypA in vitro (48), as well as SlyD and UreG in vivo (64), but it may also be a part of larger, multicomponent complexes that cooperate for hydrogenase and urease assembly. Studies conducted by Mehta et al. demonstrated that in vitro, addition of equimolar amounts of HypA fail to alter the GTPase activity of HpHypB (48). Further work is needed to determine whether any of these additional proteins activates GTPase turnover of HpHypB.
The GTPase activity of HpHypB is critical for hydrogenase and urease activity in H. pylori (47). Upon mutation of K59, a critical residue involved in nucleotide binding, the GTPase activity of the protein is nearly abolished in vitro (48), and hydrogenase and urease activity is diminished in vivo (47). Supplementation of the media with nickel partially restored hydrogenase activity and fully restored urease activity, supporting the role of HpHypB in the nickel-insertion steps of these two maturation pathways (47). The participation of metal-binding NTPases in the biosynthetic pathways of complex metallocenters is an emerging trend (30), although whether a common theme of metal-regulated NTP hydrolysis exists is not yet clear. For example, metal-dependent dimerization was observed for CooC (28, 29), the ATPase involved in carbon monoxide dehydrogenase biosynthesis, but ATP hydrolysis was not dramatically affected by the addition of nickel. Furthermore, the GTPase dedicated to urease maturation, UreG, binds nickel or zinc with micromolar affinities at a site that likely includes the CxH motif found at a similar location with respect to the GTPase motifs as C106/H107 of HpHypB (5, 70, 71). However, the relative affinities reported for nickel versus zinc, and whether the metals affect quaternary structure, vary depending on the UreG homolog. Finally, comparative genomic analysis of the COG0523 proteins, another part of the G3E family of P-loop GTPases that also includes HypB and UreG, lead to the prediction that several subgroups have roles in metal metabolism with some of them linked to zinc homeostasis (24), so it is likely that metal binding is a common property of the G3E GTPases that contributes to pathways including, but not limited to, metallocenter assembly.
HpHypB lacks the high-affinity metal-binding sequence found at the N terminus of EcHypB, a site postulated to be dispensable in various organisms and only required under certain growth conditions (36). On the other hand, the affinities of the metal site in HpHypB are at least several orders of magnitude tighter than the corresponding site in the G domain of EcHypB, perhaps indicating adaptation of the respective metal-binding sites to the specific conditions of each organism. Along the same lines, H. pylori expresses several other proteins that bind multiple nickel ions and appear to be involved in the maturation pathway of hydrogenase and/or urease, including the GroES homolog HspA, two small polypeptides called Hpn and Hpn-like, and the peptidyl-prolyl isomerase SlyD (40, 43). These additional proteins have regions rich in various combinations of metal-binding amino acids and can bind multiple nickel ions, which may compensate for the lack of a His-rich region in either HpHypB or the H. pylori UreE protein (30). Furthermore, the reduced nickel-binding capacity of H. pylori UreE has been suggested to be a plausible reason for the supplemental involvement of HpHypB in urease bioassembly (60). In support of this hypothesis, H. pylori UreE with a synthetically attached His-rich sequence can rescue the urease activity in H. pylori hypA and hypB deletion mutants (3).
The results presented here demonstrate that HpHypB binds stoichiometric nickel or zinc and that metal modulates the GTPase activity and dimerization state, revealing for the first time a connection between metal binding and other activities of a HypB protein. However, which metal is physiologically relevant or whether both metals have a role is not yet clear. In the context of EcHypB, Leach et al. speculated that a metal site in the G domain could be responsible for detecting nickel delivery to the hydrogenase precursor protein and accelerating GTP hydrolysis (36), thus activating the next stage in the process such as disengagement with the other factors of the nickel-insertion complex. However, nickel binding does not dramatically enhance the GTP hydrolysis activity of HpHypB, so other consequences of the metal-protein interactions must be considered. Alternatively, perhaps control of the formation of the HpHypB dimer is a regulatory mechanism that modulates interactions with other proteins. In the holo-homodimer state, nickel binding to HpHypB could act as an “off” switch, thereby preventing any further interactions with partner proteins such as HypA. However, further research to elucidate the effect of metal on the formation of the HypA-HpHypB heterodimer relative to that of the HpHypB homodimer is required. Another nonexclusive method of control could also be achieved in the zinc-bound state because the activity of the protein is significantly inhibited upon the addition of zinc. In the zinc-bound state, the enzyme's GTPase activity is turned off but can be restored by the removal or replacement of the zinc by nickel. Perhaps other factors involved in hydrogenase bioassembly, such as SlyD, are required for this zinc removal and only when HpHypB is required for urease or hydrogenase maturation is the GTPase activity turned on in this manner. Whether this metal-dependent control of GTPase activity is relevant in vivo is unclear, but it does highlight an interesting mechanism through which the enzyme's activity may be regulated. The distinct effects of nickel versus zinc may reflect regulation of the protein by metal availability in the H. pylori cytoplasm, which could change depending on the needs of the cell and signal an increase or decrease in nickel enzyme requirements. Further elucidation of this metal-dependent GTPase activity is required to understand what role it plays in the nickel insertion process of hydrogenase and urease bioassembly.
Supplementary Material
Acknowledgments
We thank Harini Kaluarachchi, Kim Chan Chung, Jessica Flood, and Yanjie Li for a critical reading of the manuscript and members of the Zamble lab for helpful discussions.
This study was supported in part by funding from the Canadian Institutes of Health Research and the Canada Research Chair program.
Footnotes
Published ahead of print on 14 January 2011.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Atanassova, A., R. Lam, and D. B. Zamble. 2004. A high-performance liquid chromatography method for determining transition metal content in proteins. Anal. Biochem. 335:103-111. [DOI] [PubMed] [Google Scholar]
- 2.Atanassova, A., and D. B. Zamble. 2005. Escherichia coli HypA is a zinc metalloprotein with a weak affinity for nickel. J. Bacteriol. 187:4689-4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benoit, S., and R. J. Maier. 2003. Dependence of Helicobacter pylori urease activity on the nickel-sequestering ability of the UreE accessory protein. J. Bacteriol. 185:4787-4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Böck, A., P. W. King, M. Blokesch, and M. C. Posewitz. 2006. Maturation of hydrogenases. Adv. Microbiol. Physiol. 51:1-71. [DOI] [PubMed] [Google Scholar]
- 5.Boer, J. L., S. Quiroz-Valenzuela, K. L. Anderson, and R. P. Hausinger. 2010. Mutagenesis of Klebsiella aerogenes UreG to probe nickel binding and interactions with other Urease-related proteins. Biochemistry 49:5859-5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourne, H. R., D. A. Sanders, and F. McCormick. 1990. The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348:125-132. [DOI] [PubMed] [Google Scholar]
- 7.Bowie, J. U., R. Luthy, and D. Eisenberg. 1991. A method to identify protein sequences that fold into a known three-dimensional structure. Science 253:164-170. [DOI] [PubMed] [Google Scholar]
- 8.Chan Chung, K. C., et al. 2008. A high-affinity metal-binding peptide from Escherichia coli HypB. J. Am. Chem. Soc. 130:14056-14057. [DOI] [PubMed] [Google Scholar]
- 9.Ciurli, S., and S. Mangani. 2001. Nickel-containing enzymes, p. 669-707. In I. Bertini, A. Sigel, and H. Sigel (ed.), Handbook on metalloproteins. Marcel Dekker, New York, NY.
- 10.Colovos, C., and T. O. Yeates. 1993. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 2:1511-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Covacci, A., J. L. Telford, G. Del Giudice, J. Parsonnet, and R. Rappuoli. 1999. Helicobacter pylori virulence and genetic geography. Science 284:1328-1333. [DOI] [PubMed] [Google Scholar]
- 12.de Seny, D., et al. 2001. Metal ion binding and coordination geometry for wild type and mutants of metallo-beta-lactamase from Bacillus cereus 569/H/9 (BcII) J. Biol. Chem. 276:45065-45078. [DOI] [PubMed] [Google Scholar]
- 13.Dias, A. V., et al. 2008. Structural and biological analysis of the metal sites of Escherichia coli hydrogenase accessory protein HypB. Biochemistry 47:11981-11991. [DOI] [PubMed] [Google Scholar]
- 14.Dunn, B. E., H. Cohen, and M. J. Blaser. 1997. Helicobacter pylori. Clin. Microbiol. Rev. 10:720-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eaton, K. A., and S. Krakowka. 1994. Effect of gastric pH on urease-dependent colonization of gnotobiotic piglets by Helicobacter pylori. Infect. Immun. 62:3604-3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eric, F. P., et al. 2004. UCSF Chimera: a visualization system for exploratory research and analysis. J. Comp. Chem. 25:1605-1612. [DOI] [PubMed] [Google Scholar]
- 17.Eswar, N., D. Eramian, B. Webb, M.-Y. Shen, and A. Sali. 2008. Protein structure modeling with MODELLER, p. 145-159. In B. Kobe, M. Guss, and T. Huber (ed.), Structural proteomics: high-throughput methods, vol. 426. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 18.Fu, C., J. W. Olson, and R. J. Maier. 1995. HypB protein of Bradyrhizobium japonicum is a metal-binding GTPase capable of binding 18 divalent nickel ions per dimer. Proc. Natl. Acad. Sci. U. S. A. 92:2333-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gasper, R., A. Scrima, and A. Wittinghofer. 2006. Structural insights into HypB, a GTP-binding protein that regulates metal binding. J. Biol. Chem. 281:27492-27502. [DOI] [PubMed] [Google Scholar]
- 20.Giles, N. M., et al. 2003. Metal and redox modulation of cysteine protein function. Chem. Biol. 10:677-693. [DOI] [PubMed] [Google Scholar]
- 21.Gill, S. C., and P. H. von Hippel. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182:319-326. [DOI] [PubMed] [Google Scholar]
- 22.Golynskiy, M. V., W. A. Gunderson, M. P. Hendrich, and S. M. Cohen. 2006. Metal-binding studies and EPR spectroscopy of the manganese transport regulator MntR. Biochemistry 45:15359-15372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graf, P. C. F., and U. Jakob. 2002. Redox-regulated molecular chaperones. Cell Mol. Life Sci. 59:1624-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haas, C., et al. 2009. A subset of the diverse COG0523 family of putative metal chaperones is linked to zinc homeostasis in all kingdoms of life. BMC Genomics 10:470-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herrera, V., and J. Parsonnet. 2009. Helicobacter pylori and gastric adenocarcinoma. Clin. Microbiol. Infect. 15:971-976. [DOI] [PubMed] [Google Scholar]
- 26.Hunt, J. B., S. H. Neece, and A. Ginsburg. 1985. The use of 4-(2-pyridylazo)resorcinol in studies of zinc release from Escherichia coli aspartate transcarbamoylase. Anal. Biochem. 146:150-157. [DOI] [PubMed] [Google Scholar]
- 27.Jenkins, R. M., M. L. Singleton, E. Almaraz, J. H. Reibenspies, and M. Y. Darensbourg. 2009. Imidazole-containing (N3S)-NiII complexes relating to nickel-containing biomolecules. Inorg. Chem. 48:7280-7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeoung, J.-H., T. Giese, M. Grunwald, and H. Dobbek. 2009. CooC1 from Carboxydothermus hydrogenoformans is a nickel-binding ATPase. Biochemistry 48:11505-11513. [DOI] [PubMed] [Google Scholar]
- 29.Jeoung, J.-H., T. Giese, M. Grünwald, and H. Dobbek. 2009. Crystal structure of the ATP-dependent maturation factor of Ni,Fe-containing carbon monoxide dehydrogenases. J. Mol. Biol. 396:1165-1179. [DOI] [PubMed] [Google Scholar]
- 30.Kaluarachchi, H., K. C. C. Chung, and D. B. Zamble. 2010. Microbial nickel proteins. Nat. Prod. Rep. 27:681-694. [DOI] [PubMed] [Google Scholar]
- 31.Kelly, S. M., T. J. Jess, and N. C. Price. 2005. How to study proteins by circular dichroism. Biochim. Biophys. Acta 1751:119-139. [DOI] [PubMed] [Google Scholar]
- 32.Kennedy, D. C., R. W. Herbst, J. S. Iwig, P. T. Chivers, and M. J. Maroney. 2007. A dynamic Zn site in Helicobacter pylori HypA: a potential mechanism for metal-specific protein activity. J. Am. Chem. Soc. 129:16-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuzmic, P. 1996. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237:260-273. [DOI] [PubMed] [Google Scholar]
- 34.Lanzetta, P. A., L. J. Alvarez, P. S. Reinach, and O. A. Candia. 1979. An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 100:95-97. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283-291. [Google Scholar]
- 36.Leach, M. R., S. Sandal, H. Sun, and D. B. Zamble. 2005. Metal-binding activity of the Escherichia coli hydrogenase maturation factor HypB. Biochemistry 44:12229-12238. [DOI] [PubMed] [Google Scholar]
- 37.Leach, M. R., and D. B. Zamble. 2007. Metallocenter assembly of the hydrogenase enzymes. Curr. Opin. Chem. Biol. 11:159-165. [DOI] [PubMed] [Google Scholar]
- 38.Leipe, D. D., Y. I. Wolf, E. V. Koonin, and L. Aravind. 2002. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 317:41-72. [DOI] [PubMed] [Google Scholar]
- 39.Lever, A. B. P. 1984. Inorganic electronic spectroscopy, 2nd ed., vol. 33. Elsevier Science B.V., Amsterdam, Netherlands.
- 40.Li, Y., and D. B. Zamble. 2009. Nickel homeostasis and nickel regulation: an overview. Chem. Rev. 109:4617-4643. [DOI] [PubMed] [Google Scholar]
- 41.Luthy, R., J. U. Bowie, and D. Eisenberg. 1992. Assessment of protein models with three-dimensional profiles. Nature 356:83-85. [DOI] [PubMed] [Google Scholar]
- 42.Madhusudhan, M. S., et al. 2005. Comparative protein structure modeling, p. 831-860. In J. M. Walker (ed.), The proteomics protocols handbook. Humana Press, Totowa, NJ.
- 43.Maier, R. J., S. Benoit, and S. Seshadri. 2007. Nickel-binding and accessory proteins facilitating Ni-enzyme maturation in Helicobacter pylori. Biometals 20:655-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maier, T., A. Jacobi, M. Sauter, and A. Böck. 1993. The product of the hypB gene, which is required for nickel incorporation into hydrogenases, is a novel guanine nucleotide-binding protein. J. Bacteriol. 175:630-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maier, T., F. Lottspeich, and A. Bock. 1995. GTP hydrolysis by HypB is essential for nickel insertion into hydrogenases of Escherichia coli. Eur. J. Biochem. 230:133-138. [PubMed] [Google Scholar]
- 46.Maret, W. 2009. Fluorescent probes for the structure and function of metallothionein. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 877:3378-3383. [DOI] [PubMed] [Google Scholar]
- 47.Mehta, N., S. Benoit, and R. J. Maier. 2003. Roles of conserved nucleotide-binding domains in accessory proteins, HypB and UreG, in the maturation of nickel-enzymes required for efficient Helicobacter pylori colonization. Microb. Pathog. 35:229-234. [DOI] [PubMed] [Google Scholar]
- 48.Mehta, N., J. W. Olson, and R. J. Maier. 2003. Characterization of Helicobacter pylori nickel metabolism accessory proteins needed for maturation of both urease and hydrogenase. J. Bacteriol. 185:726-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Min-yi, S., and S. Andrej. 2006. Statistical potential for assessment and prediction of protein structures. Protein Sci. 15:2507-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mobley, H. L., M. D. Island, and R. P. Hausinger. 1995. Molecular biology of microbial ureases. Microbiol. Mol. Biol. Rev. 59:451-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mobley, H. L. T. 2001. Urease, p. 179-191. In S. L. Hazell, G. L. Mendz, and H. L. T. Mobley (ed.), Helicobacter pylori: physiology and genetics. ASM Press, Washington, DC. [PubMed]
- 52.Neal, S. E., J. F. Eccleston, A. Hall, and M. R. Webb. 1988. Kinetic analysis of the hydrolysis of GTP by p21N-ras. The basal GTPase mechanism. J. Biol. Chem. 263:19718-19722. [PubMed] [Google Scholar]
- 53.Olson, J. W., and R. J. Maier. 2000. Dual roles of Bradyrhizobium japonicum nickelin protein in nickel storage and GTP-dependent Ni mobilization. J. Bacteriol. 182:1702-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olson, J. W., and R. J. Maier. 2002. Molecular hydrogen as an energy source for Helicobacter pylori. Science 298:1788-1790. [DOI] [PubMed] [Google Scholar]
- 55.Olson, J. W., N. S. Mehta, and R. J. Maier. 2001. Requirement of nickel metabolism proteins HypA and HypB for full activity of both hydrogenase and urease in Helicobacter pylori. Mol. Microbiol. 39:176-182. [DOI] [PubMed] [Google Scholar]
- 56.Raju, B., E. Murphy, L. A. Levy, R. D. Hall, and R. E. London. 1989. A fluorescent indicator for measuring cytosolic free magnesium. Am. J. Physiol. 256:C540-C548. [DOI] [PubMed] [Google Scholar]
- 57.Rey, L., J. Imperial, J. M. Palacios, and T. Ruiz-Argueso. 1994. Purification of Rhizobium leguminosarum HypB, a nickel-binding protein required for hydrogenase synthesis. J. Bacteriol. 176:6066-6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sali, A., and T. L. Blundell. 1993. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 234:779-815. [DOI] [PubMed] [Google Scholar]
- 59.Scott, D. R., et al. 1998. The role of internal urease in acid resistance of Helicobacter pylori. Gastroenterol. 114:58-70. [DOI] [PubMed] [Google Scholar]
- 60.Shi, R., et al. 2010. Crystal structures of apo and metal-bound forms of the UreE protein from Helicobacter pylori: role of multiple metal binding sites. Biochemistry 49:7080-7088. [DOI] [PubMed] [Google Scholar]
- 61.Simons, T. J. B. 1993. Measurement of free Zn2+ ion concentration with the fluorescent probe mag-fura-2 (furaptra). J. Biochem. Biophys. Methods 27:25-37. [DOI] [PubMed] [Google Scholar]
- 62.Sippl, M. J. 1993. Recognition of errors in three-dimensional structures of proteins. Proteins 17:355-362. [DOI] [PubMed] [Google Scholar]
- 63.Sprang, S. R. 1997. G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem. 66:639-678. [DOI] [PubMed] [Google Scholar]
- 64.Stingl, K., et al. 2008. In vivo interactome of Helicobacter pylori urease revealed by tandem affinity purification. Mol. Cell Proteom. 7:2429-2441. [DOI] [PubMed] [Google Scholar]
- 65.Takai, Y., T. Sasaki, and T. Matozaki. 2001. Small GTP-binding proteins. Physiol. Rev. 81:153-208. [DOI] [PubMed] [Google Scholar]
- 66.Tomb, J.-F., et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539-547. [DOI] [PubMed] [Google Scholar]
- 67.Vignais, P. M., and B. Billoud. 2007. Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107:4206-4272. [DOI] [PubMed] [Google Scholar]
- 68.Watanabe, S., et al. 2009. Crystal structure of HypA, a nickel-binding metallochaperone for [NiFe] hydrogenase maturation. J. Mol. Biol. 394:448-459. [DOI] [PubMed] [Google Scholar]
- 69.Xia, W., H. Li, K.-H. Sze, and H. Sun. 2009. Structure of a nickel chaperone, HypA, from Helicobacter pylori reveals two distinct metal binding sites. J. Am. Chem. Soc. 131:10031-10040. [DOI] [PubMed] [Google Scholar]
- 70.Zambelli, B., et al. 2005. UreG, a chaperone in the Urease assembly process, is an intrinsically unstructured GTPase that specifically binds Zn2+. J. Biol. Chem. 280:4684-4695. [DOI] [PubMed] [Google Scholar]
- 71.Zambelli, B., P. Turano, F. Musiani, P. Neyroz, and S. Ciurli. 2009. Zn(II)-linked dimerization of UreG from Helicobacter pylori, a chaperone involved in nickel trafficking and urease activation. Proteins 74:222-239. [DOI] [PubMed] [Google Scholar]
- 72.Zumbrennen, K. B., M. L. Wallander, S. J. Romney, and E. A. Leibold. 2009. Cysteine oxidation regulates the RNA-binding activity of iron regulatory protein 2. Mol. Cell. Biol. 29:2219-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.