

Abstract
The diarrheal potential of a Bacillus cereus strain is essentially dictated by the amount of secreted nonhemolytic enterotoxin (Nhe). Expression of genes encoding Nhe is regulated by several factors, including the metabolic state of the cells. To identify metabolic sensors that could promote communication between central metabolism and nhe expression, we compared four strains of the B. cereus group in terms of metabolic and nhe expression capacities. We performed growth performance measurements, metabolite analysis, and mRNA measurements of strains F4430/73, F4810/72, F837/76, and PA cultured under anoxic and fully oxic conditions. The results showed that expression levels of nhe and ldhA, which encodes lactate dehydrogenase A (LdhA), were correlated in both aerobically and anaerobically grown cells. We examined the role of LdhA in the F4430/73 strain by constructing an ldhA mutant. The ldhA mutation was more deleterious to anaerobically grown cells than to aerobically grown cells, causing growth limitation and strong deregulation of key fermentative genes. More importantly, the ldhA mutation downregulated enterotoxin gene expression under both anaerobiosis and aerobiosis, with a more pronounced effect under anaerobiosis. Therefore, LdhA was found to exert a major control on both fermentative growth and enterotoxin expression, and it is concluded that there is a direct link between fermentative metabolism and virulence in B. cereus. The data presented also provide evidence that LdhA-dependent regulation of enterotoxin gene expression is oxygen independent. This study is the first report to describe a role of a fermentative enzyme in virulence in B. cereus.
Bacillus cereus strains express various putative virulence genes, including genes encoding hemolysins, various enterotoxins, phospholipases, metalloproteases, collagenases, and other proteins (7, 38). The contribution of these putative virulence factors to the pathogenicity of B. cereus strains has not been fully elucidated. However, the amount of secreted nonhemolytic enterotoxin (Nhe) appeared to essentially dictate B. cereus-associated cytotoxic activity (29). Nhe is a three-component enterotoxin encoded by the PlcR-regulated nheABC operon (nhe) (1, 16, 26). In in vitro culture, nhe transcription is responsive to a number of cues linked to carbohydrate catabolism, including carbon source concentration, carbon source type, extracellular oxidoreduction potential, and growth phase (10, 28, 31, 42), which suggests that there are mechanisms to promote communication between carbohydrate catabolism and the nhe regulatory network. Carbohydrates such as glucose are catabolized primarily through the glycolytic pathway and to a lesser extent through the pentose phosphate pathway in growing B. cereus cells (42). Glycolysis produces two molecules of pyruvate per molecule of glucose consumed and in the process reduces two molecules of NAD+ to NADH. The catabolic fate of pyruvate is determined by growth conditions, specifically oxygen availability. During fermentative anaerobic growth, pyruvate or acetyl coenzyme A (acetyl-CoA) derived from pyruvate serves as an electron acceptor for the maintenance of redox balance and as the substrate for generating ATP and acetate.
The enzymes responsible for redox balance in anoxic B. cereus cells are lactate dehydrogenase (Ldh), malate dehydrogenase (Mdh), succinate dehydrogenase (Sdh), pyruvate formate lyase (Pfl), aldehyde dehydrogenase (AdhE), and alcohol dehydrogenase (AdhA). The production of acetate from acetyl-CoA is dependent on the phosphotransacetylase-acetate kinase (Pta-Ack) pathway (see Fig. 1A). The main fermentative by-products of B. cereus are a mixture of organic acids, such as lactate, succinate, acetate and formate, and ethanol. Lactate is the major product of fermentation and normally accounts for more than 65% of the total produced from glucose by B. cereus F4430/73 culture. Acetate and formate are produced in similar amounts (each accounting for 15% of total production), while ethanol and succinate are minor fermentation products (less than 3% and 2% of total by-products produced from glucose, respectively [11, 34]). Producing this mixture of by-products in the appropriate ratio helps maintain redox balance under anoxic conditions while also maximizing ATP yield per glucose molecule to support high growth rates and cell yields. During aerobic growth, the pyruvate dehydrogenase complex (Pdh) connects the glycolytic reactions to the tricarboxylic acid cycle (TCA cycle) by catalyzing the production of acetyl-CoA from pyruvate. The NADH generated during the complete oxidation of glucose is reoxidized to NAD+ by O2 through the aerobic respiratory chain with accompanying ATP production. Acetate excretion can occur aerobically when carbon flux into the cells exceeds TCA cycle capacity (11, 34). In B. cereus, the expression of most of the genes encoding metabolic enzymes is modulated by the two-component ResDE system (11), the one-component Fnr system (43), and the catabolite control protein CcpA (39). These three systems also contribute to virulence. ResDE and Fnr may act as components of transcriptional regulatory complexes (13). Understanding how these regulatory complexes detect metabolic cues is an important step toward understanding how gene regulation contributes to the pathogenic potential of a B. cereus strain.
FIG. 1.

Metabolic responses of B. cereus strains F4430/73, F4810/72, F837/76, and PA. (A) Pathways for the excretion of partially oxidized metabolites. The reactions are represented by the names of the corresponding genes, as follows: ldhA, ldhB, and ldhC, l-lactate dehydrogenase; pfl, pyruvate formate lyase; pdh, pyruvate dehydrogenase; pta, phosphotransacetylase; ack, acetate kinase; adhE, acetaldehyde dehydrogenase; adhA, alcohol dehydrogenase; mdh, malate dehydrogenase; sdh, succinate dehydrogenase; citB, aconitase hydratase; citZ, citrate synthase. Reactions that normally function during aerobiosis are represented by gray arrows, and reactions that normally function during anaerobiosis are represented by black arrows. CIT, citrate; ICT, isocitrate; AKG, α-ketoglutarate; SUC, succinate; FUM, fumarate; MAL, malate; OAA, oxaloacetate; TCA, tricarboxylic acid cycle. (B) Extracellular metabolite analyses. (C) Specific Nhe production. Error bars indicate the standard errors of the means.
In the present study, we first compared the catabolic capacities of four B. cereus strains (F4430/73, F4810/72, F837/76, and PA) grown under anoxic and oxic conditions in relation to their capacities for expressing nhe. This led us to study the role of lactate dehydrogenase A (LdhA) as a fermentative enzyme contributing to nhe expression regulation. We showed that disruption of ldhA affected the fermentative capacity of anaerobically grown F4430/73 cells and the nhe expression capacities of both anaerobically and aerobically grown F4430/73 cells. We demonstrated that ldhA plays a role in the transcription of several toxin or putative toxin genes under both aerobic and anaerobic growth conditions. These effects were not observed with other B. cereus l-lactate dehydrogenase genes (ldhB and ldhC). We discuss the possible role of LdhA in regulation of enterotoxin gene expression.
MATERIALS AND METHODS
Bacterial strains, mutant construction, and growth conditions.
The four B. cereus strains used in this study were the food-poisoning strains F4430/73 (37), F4810/72 (AH187) (12), and F837/76 (DSM4222) (20) and the food-borne strain PA (20). ldhA and pfo deletion mutants were constructed as follows. Two SalI-BglII DNA fragments of 1,309 bp and 1,011 bp encompassing the ldhA and pfo open reading frames (ORFs), respectively, were amplified by PCR using chromosomal DNA as the template and the primer pairs ldhAmutF-ldhAmutR and pfomutF-pfomutR, respectively (see Table S1 in the supplemental material). The amplified DNA fragments were digested with the appropriate enzymes and inserted between the corresponding pMAD sites (4), producing pMADldhA and pMADpfo, respectively. These plasmids were then digested by AflII and BamHI, respectively, and end filled with T4 DNA polymerase. A 1.5-kb SmaI fragment containing the entire spectinomycin resistance gene spc (30) was purified from pDIA (a generous gift from I. Martin-Verstraete). This purified DNA fragment was ligated into the digested pMADldhA and pMADpfo plasmids. The resulting plasmids were introduced into B. cereus strains by electroporation, and the ldhA and pfo genes were deleted by a double-crossover event as described by Arnaud et al. (4). Chromosomal allele exchanges were confirmed by PCR with oligonucleotide primers located upstream and downstream of the DNA regions used for allelic exchange. To complement the ldhA gene in trans, the 1,767-bp ldhA locus and the 2,748-bp ldhA-pfo locus were first PCR amplified using the primer pairs ldhA1compF-ldhA1compR and ldhA2compF-ldhA2compR, respectively (see Table S1 in the supplemental material) and then cloned into the pCRXL-TOPO plasmid (Invitrogen). The PCR fragments were then cut with HindIII and PstI and ligated to similarly digested pHT304 (3). The integrity of the inserts of the recombinant vectors (pHT304ldhA and pHT304ldhApfo) was verified by sequencing, and the vectors were then used to transform the B. cereus mutant strain.
The ldhB and ldhC mutants were also constructed using pMAD as described above for ldhA and pfo, with the following modifications. The ldhB gene was PCR amplified using oligonucleotide pair ldhBmutF-ldhBmutR (Table S1) and then cloned into pMAD using BamHI and SalI. The recombinant vector was then digested with Csp45I and ligated with the 1.5-kb spc-containing fragment to obtain the vector used for ldhB replacement. The ldhC gene was PCR amplified using oligonucleotide pair ldhCmutF-ldhCmutR (Table S1) and then cloned into pUC18 by using PstI and BamHI. The pUC18ldhC plasmid was then digested with SnaBI and ligated with the 1.5-kb spc-containing SmaI fragment. The NcoI-BglII fragment containing the disrupted ldhC gene was then isolated and cloned into pMAD.
Wild-type strains F4430/73, F4810/72, F837/76, and PA and ldhA and pfo mutants were grown in synthetic MOD medium supplemented with 30 mM glucose as a carbon source (34). A 2-liter bioreactor (Discovery 100; Inceltech, Toulouse, France) was used for all cultivations, and the working volume was maintained at 1.3 liters. The temperature was maintained at 37°C, and the pH was kept at a controlled 7.2 by automatic addition of 5 M KOH. The regulated batch was equipped with a Mettler Toledo polarographic oxygen electrode coupled with feedback regulation to maintain the set point dissolved oxygen tension value (pO2) using air sparging and agitation speed (10). Sparging the bioreactor with air alone set a pO2 value of 100%. A pO2 of 0% was obtained by continuously flushing the medium at 20 ml·h−1 with pure N2 gas previously passed through a Hungate column. Each bioreactor was inoculated with a subculture grown overnight under anaerobiosis in such a way that the initial optical density at 560 nm (OD560) was equal to 0.02. For ldhA mutant complementation assays and analyses of ldhB and ldhC mutant F4430/73 strains, uncontrolled aerobic and anaerobic cultures were performed as previously described (34).
Analytical procedures and physiological parameters.
B. cereus growth was monitored spectrophotometrically at 560 nm and calibrated against cell dry weight measurements as previously described (10). The specific growth rate (μ) was determined using the modified Gompertz equation (18, 44). For substrate, by-product, and Nhe measurements, a 4-ml sample was centrifuged at 10,000 × g for 5 min at 4°C and the supernatants were frozen at −80°C until analysis. Glucose, lactate, ethanol, formate, acetate, and succinate concentrations were determined using commercial enzymatic kits (from Diffchamb, Lyon, France, from R-Biopharm, Saint-Didier au Mont-d'Or, France, and from Roche, Meylan, France). The specific glucose consumption rate, defined as the differential change in glucose concentration with time, was calculated from the equation qglucose = μ/Yx, where μ is the specific growth rate (h−1) and Yx is the biomass yield (g·mol carbon substrate−1). Amounts of Nhe were estimated by measuring the optical density at 420 nm (one unit was defined as one OD unit at 420 nm) as previously described (42). Specific Nhe production was defined as the amount of Nhe produced per gram of cell dry mass (U·g−1) or per OD560 unit (U·OD−1).
Enzyme assay.
Lactate dehydrogenase activity was assayed with pyruvate on purified enzymes. The assay mixture (1 ml) contained phosphate buffer at pH 6.6, 0.3 mM NADH, 3 mM fructose 1,6-diphosphate, 5 mM MgCl2, and 5 to 10 μl of purified protein (10 μg/μl). Sodium pyruvate (20 mM) was added to start the reaction. NADH oxidation was followed by the decrease in absorbance at 340 nm. One unit of lactate dehydrogenase activity is 1 A340 unit per min.
In vitro lactate dehydrogenase synthesis.
Proteins were synthesized by using the rapid-translation system RTS 500 E. coli HY kit (Roche Diagnostics) according to the manufacturer's instructions. Template DNA fragments were prepared using a two-step PCR protocol. The first step (PCR 1) was used to obtain specific fragments (primer pairs are listed in Table S1 in the supplemental material). The second step (PCR 2) was used to obtain the products with regulatory elements and His tag at the C terminus. The PCR products were purified by use of a PCR purification kit (Roche). The purified products were sequenced; after the sequence was confirmed to be correct, these products were used for expressing corresponding proteins. The protein products were rapidly purified using a Ni-nitrilotriacetic acid (NTA) column. The expressed protein was confirmed by Western blotting using anti-His antibody as the first antibody.
DNA sequencing and bioinformatics tools.
Sequencing was performed by Beckman Coulter Genomics (United Kingdom). Homology and conserved domain searches were performed with the NCBI database by using the BLAST algorithm. Pairwise alignment was carried out using ClustalW software. The presence of transmembrane helices in proteins was predicted using TMpred and TMHMM version 2.0 software via the ExPASy proteomics server.
RNA isolation, operon mapping, and expression analysis.
To investigate the transcriptional organization and expression of the ldhA-pfo locus, reverse transcription-PCR (RT-PCR) was performed with total RNA isolated from B. cereus F4430/73 as previously described (11). Portions of ldhA, pfo, and the ldhA-pfo intergenic region were amplified by RT-PCR using the Titan one-tube RT-PCR system following the manufacturer's protocol (Roche). The primer pairs used for ldhA, pfo, and the ldhA-pfo intergenic region are listed in Table S1 in the supplemental material. To check whether contaminant genomic DNA was present, each sample was tested in a control reaction that did not contain reverse transcriptase.
The 5′ end of ldhA-pfo mRNA was mapped from a 5′ rapid amplification of cDNA ends (RACE) PCR product obtained with the 3′/5′ RACE kit (Roche). Briefly, the first-strand cDNA was synthesized from total RNA with the ldhA-pfo-specific SP1 primer (Table S1), avian myeloblastosis virus reverse transcriptase, and the deoxynucleotide mixture of the 3′/5′ RACE kit by following the manufacturer's instructions. After purifying and deoxyribosyladenine (dA) tailing the cDNA, PCR with the deoxyribosylthymine (dT) anchor oligonucleotide primer and the ldhA-pfo-specific SP2 primer (Table S1) followed by a nested PCR with the SP3 primer (Table S1) led to a PCR product of approximately 140 bp, as revealed by 2% agarose gel electrophoresis. This PCR product was purified and sequenced.
Real-time RT-PCR was performed using SYBR green staining on a LightCycler instrument (Roche) as previously described (11). Sequences specifically used in the present study are given in Table S1.
Nucleotide sequence accession number.
Given that the B. cereus F4430/73 strain belongs to the same genetic group as the ATCC 14579 strain (21), we designed primers (Table S1) corresponding to the ATCC 14579 locus to clone and sequence its homolog from strain F4430/73. The DNA sequence of the 2,748-bp cloned fragment corresponding to the ldhA-pfo locus was deposited in GenBank under accession no. HQ336744.
RESULTS
Fermentative and respiratory capacities of strains F4810/72, F837/76, and PA relative to those of strain F4430/73.
B. cereus strains F4430/73, F4810/72, F837/76, and PA were grown in glucose-limited and pH-regulated batch culture (pH 7.2) under anaerobiosis (pO2 = 0%; generated under an N2 atmosphere) and aerobiosis (pO2 = 100%). Table 1 summarizes the growth features of the four strains. Under anaerobiosis, strains F4810/72, F837/76, and PA grew significantly more slowly and reached the stationary growth phase at a lower final biomass than strain F4430/73. This probably resulted from both lower levels and lower specific rates of glucose uptake. Interestingly, strain F4810/72 exhibited the highest biomass yield on glucose. Under aerobiosis, growth rates for the four strains were similar, although those for strains F837/76 and PA were slightly lower than those for the two other strains. However, final biomass and biomass yields of strains F4810/72, F837/76, and PA were significantly higher than those of strain F4430/73, while the glucose uptake rates for these three strains were lower. Taken together, these results indicate that glucose supports lower glycolytic fluxes (or glucose uptake rates) in strains F4810/72, F837/76, and PA than in strain F4430/73 under both anaerobiosis and aerobiosis, leading to lower fermentative growth and more efficient respiratory growth, respectively.
TABLE 1.
Growth parameters determined from controlled batch cultures (pH 7.2) of strains F4430/73, F4810/72, F837/76, and PAa
| Parameter | Anaerobic growth |
Aerobic growth |
||||||
|---|---|---|---|---|---|---|---|---|
| F4430/73 | F4810/72 | F837/76 | PA | F4430/73 | F4810/72 | F837/76 | PA | |
| μmax (h−1) | 0.9 | 0.5 | 0.5 | 0.5 | 1.4 | 1.3 | 1.1 | 1.1 |
| Final biomass (g·liter−1)b | 0.8 | 0.3 | 0.3 | 0.3 | 2.6 | 2.9 | 3.7 | 3.0 |
| Glucose consumption (%)b | 100 | 28 | 42 | 35 | 100 | 100 | 100 | 100 |
| Yglucose (g of cells·mol of glucose−1) | 26 | 34 | 27 | 24 | 86 | 97 | 123 | 100 |
| Maximal specific glucose consumption rate (mmol·mg−1·h−1) | 35 | 6 | 11 | 7 | 16 | 13 | 9 | 11 |
Cells were grown under N2-generated anaerobiosis (pO2 = 0%) and full aerobiosis (pO2 = 100%). Data are the means of duplicate cultures. For clarity, standard deviations (below 10%) are not shown.
Final biomass and glucose consumption were determined at the end of growth, i.e., 1.5 h after the OD560 reached its maximal value.
Figure 1A depicts the main catabolic pathways used by B. cereus strains. Glucose was converted predominantly into lactate in all four strains (Fig. 1B). However, F4810/72, F837/76, and PA cells produced smaller amounts of lactate and concomitantly larger amounts of formate, acetate (except for PA), and ethanol than F4430/73 cells. This suggests that the carbon flux from pyruvate to lactate was lower in strains F4810/72, F837/76, and PA than in strain F4430/73 under anaerobic fermentative conditions and that the carbon flux through pyruvate formate lyase (Pfl) at the pyruvate node and then through the NADH-recycling alcohol dehydrogenase (AdhE-AdhA) pathway at the acetyl-CoA node was higher (Fig. 1A). When oxygen was present, no detectable ethanol, formate, or succinate was produced in any of the four strains. The amounts of secreted lactate in all four strains were similar, while strains F4810/72, F837/76, and PA secreted smaller amounts of acetate than strain F4430/73 (Fig. 1B). Since aerobic acetate generation results from an overflow metabolism in B. cereus (34), glucose could support a lower overflow metabolism in strains F4810/72, F837/76, and PA than in strain F4430/73 under aerobic respiratory conditions.
Table 2 shows the catabolic gene expression profiles in strains F4810/72, F837/76, and PA relative to those in strain F4430/73 from cells cultured under fermentative anaerobic and respiratory aerobic growth conditions. The data revealed that strains F4810/72, F837/76, and PA exhibited a pattern of catabolic gene expression very different from that of strain F4430/73. Interestingly, we observed that the mRNA profiles of the three genes predicted to encode l-lactate dehydrogenase (ldhA, ldhB, and ldhC) in strains F4810/72, F837/76, and PA differed significantly from those of these genes in strain F4430/73 in the absence and presence of oxygen. They differed in the following ways: (i) ldhA expression was moderately reduced in strain F4810/72 and severely reduced in strains F837/76 and PA under both anaerobiosis and aerobiosis, (ii) ldhB expression levels differed between strains and between condition sets, and (iii) ldhC expression was inversely correlated to ldhA expression (r = −0.63, P < 0.05) in the three strains F4810/72, F837/76, and PA, whatever the growth conditions. In addition, concomitantly with ldhA, adhE, which encodes a key enzyme of the ethanol-generating pathway, and citZ, which encodes an enzyme that governs carbon entry into the TCA cycle, were significantly downregulated in strains F4810/72, F837/76, and PA compared to strain F4430/73 under both aerobiosis and aerobiosis.
TABLE 2.
mRNA levels of genes involved in glucose catabolism and enterotoxin synthesis in B. cereus strains F4810/72, F837/76, and PA relative to those of strain F4430/73a
| Gene | Difference in mRNA level (n-fold)b under the indicated condition |
|||||
|---|---|---|---|---|---|---|
| Anaerobic growth |
Aerobic growth |
|||||
| F4810/72 | F837/76 | PA | F4810/72 | F837/76 | PA | |
| Glucose catabolism gene | ||||||
| ldhA | −2.5 | −100 | −100 | −2.0 | −100 | −100 |
| ldhB | +2.5 | +2.0 | −5.0 | −50 | −100 | −5.0 |
| ldhC | +3.2 | +6.2 | +6.5 | +2.2 | +12.3 | +5.4 |
| pfl | −4.0 | −1.2 | −2.2 | −1.4 | −3.7 | −3.1 |
| pdhA | +1.4 | +1.9 | +1.6 | −1.1 | +1.0 | +1.0 |
| pta | −5.0 | −3.3 | −3.8 | −2.5 | −2.0 | +1.5 |
| ackA | −3.3 | −2.3 | +1.6 | −1.7 | −1.5 | +2.0 |
| adhE | −7.7 | −4.8 | −7.7 | −4.5 | −33.3 | −3.7 |
| adhA | −1.1 | +1.7 | +1.7 | +2.3 | −1.8 | +1.1 |
| sdhA | −2.2 | +1.0 | −2.5 | +3.0 | +2.9 | +2.4 |
| citZ | −100 | −100 | −16.0 | −50.0 | −33.0 | −2.7 |
| citB | +1.1 | −12.5 | −10.0 | +2.0 | +1.5 | −2.3 |
| mdh | +1.6 | +4.8 | +4.9 | −1.1 | −1.1 | +1.2 |
| Enterotoxin gene nhe | −10 | −100 | −100 | −3.1 | −500 | −14.3 |
| Regulator genes | ||||||
| plcR | +3.0 | +1.4 | +4.2 | +2.9 | +1.1 | −1.1 |
| resD | +4.0 | +3.2 | +5.6 | +2.2 | +1.9 | +2.3 |
| fnr | +6.7 | +6.2 | +7.5 | −1.8 | +2.4 | +1.5 |
| ccpA | −1.4 | −1.1 | −2.3 | −3.6 | −1.7 | −1.2 |
B. cereus cells were grown under oxic and anoxic conditions in controlled batch cultures (pH 7.2) on MOD medium with 30 mM glucose as the carbon source.
Each change (n-fold) represents the mean value of the mRNA levels of one strain sample in relation to that of the F4430/73 sample. For each experiment, two measurements from two independent RNA samples taken from the mid-exponential growth phase (μmax) of the same culture were analyzed in parallel. Each data point is an average of the results of the combined experiments. Only ratios of ≤−2 and ≥+2 were considered significant (i.e., P ≤ 0.05) according to the precision of the method. + and − indicate up- and downregulation of genes, and significant values for upregulation and downregulation are underlined and boldface, respectively.
Nhe secretion capacities of strains F4810/72, F837/76, and PA compared to that of strain F4430/73.
Strains F4810/72, F837/76, and PA secreted smaller amounts of Nhe than strain F4430/73 under both anaerobiosis and aerobiosis (Fig. 1C). Among strains F4810/72, F837/76, and PA, F4810/72 was clearly the greatest producer of Nhe (1.1 and 0.4 U·g−1 of dry cells under anaerobiosis and aerobiosis, respectively, compared to 0.2 and 0.002 for strain F837/76 and 0.5 and 0.01 for strain PA, respectively).
Table 2 shows that nhe expression was strongly reduced in strains F4810/72, F837/76, and PA compared to strain F4430/73 under both anaerobiosis and aerobiosis. This may explain why Nhe was secreted less in these strains. Transcript levels of nhe, regulatory genes, and genes encoding catabolic enzymes that were ≥2-fold upregulated or downregulated under both anaerobiosis and aerobiosis were subjected to Pearson's r correlation analysis. The results indicated that only the ldhA, adhE, and citZ mRNA levels were significantly correlated (P < 0.05) with nhe levels (r values of +0.62, +0.91, and +0.77, respectively). This suggests a regulatory link between these catabolic genes and nhe. To validate such a link, we studied the ldhA gene in relation to nhe in strain F4430/73.
Analysis of the ldhA locus in B. cereus F4430/73.
As in the case for reference strain ATCC 14579 (GenBank accession no. AE016877), the stop codon of B. cereus F4430/73 ldhA is located 14 bp upstream of the start codon of an ORF, annotated pfo by Ivanova et al. (24), suggesting that these genes are cooperonic (Fig. 2A). RT-PCR experiments demonstrated that ldhA and pfo were effectively cotranscribed under both aerobiosis and anaerobiosis (Fig. 2B). A transcriptional start site (G) located 48 bp from the ldhA start codon was identified by 5′ RACE PCR (Fig. 2C). We identified upstream of this start site a potential σA-type −10 sequence, TATAAT, that is not preceded by a typical −35 sequence (TTGACA) (22). A review of the ldhA promoter regions of the other B. cereus strains confirmed the absence of the typical −35 region. Inspection of the 5′ untranscribed region (UTR) sequence revealed the presence of a palindromic sequence spanning positions −64 to −41. Each symmetrical arm contains highly conserved potential binding sites for the redox regulators Fnr and ResD (8, 11, 23, 43). No CcpA binding site was found (39). Furthermore, the ldhA-pfo locus appeared to be followed by an inverted repeat (change in Gibbs free energy [ΔG], −23.1 kcal/mol) that may be a transcriptional terminator, confirming that ldhA and pfo could form a transcriptional unit.
FIG. 2.

Operon mapping and expression of the B. cereus F4430/73 ldhA-pfo operon. (A) Large arrows represent the ldhA and pfo ORFs. The small arrowheads flag the positions and directions of primers used in RT-PCR. Lengths of expected PCR products are also shown for every primer pair. (B) RT-PCR analyses for detecting ldhA, pfo, and the intergenic (IG) region between ldhA and pfo in strain F4430/73. M, molecular mass marker. (C) The nucleotide sequence of the promoter region of F4430/73 ldhA is shown. The transcriptional start site (+1) and the putative −10 sequence are in bold and underlined. Putative regulatory sequences are highlighted in gray, the start codon is boxed, and inverted repeat sequences are indicated by arrows.
Expression of the B. cereus ldhA gene in E. coli.
To demonstrate that B. cereus ldhA encodes lactate dehydrogenase, ldhA was expressed as a His-tagged protein by using a cell-free Escherichia coli translation system in the presence of GroES and GroEL chaperone proteins. Figure 3 shows that the LdhA peptide (panel A), like LdhB and LdhC peptides (panel B), was successfully expressed in vitro. The purified LdhA protein has a specific Ldh activity of 40 ± 4 U·mg−1, using pyruvate as the substrate and NADH as the cosubstrate. In comparison, LdhB and LdhC exhibited specific Ldh activities of 50 ± 6 U·mg−1 and 83 ± 10 U·mg−1, respectively. Taken together, these results demonstrate that B. cereus ldhA, like ldhB and ldhC, encodes functional lactate dehydrogenase.
FIG. 3.
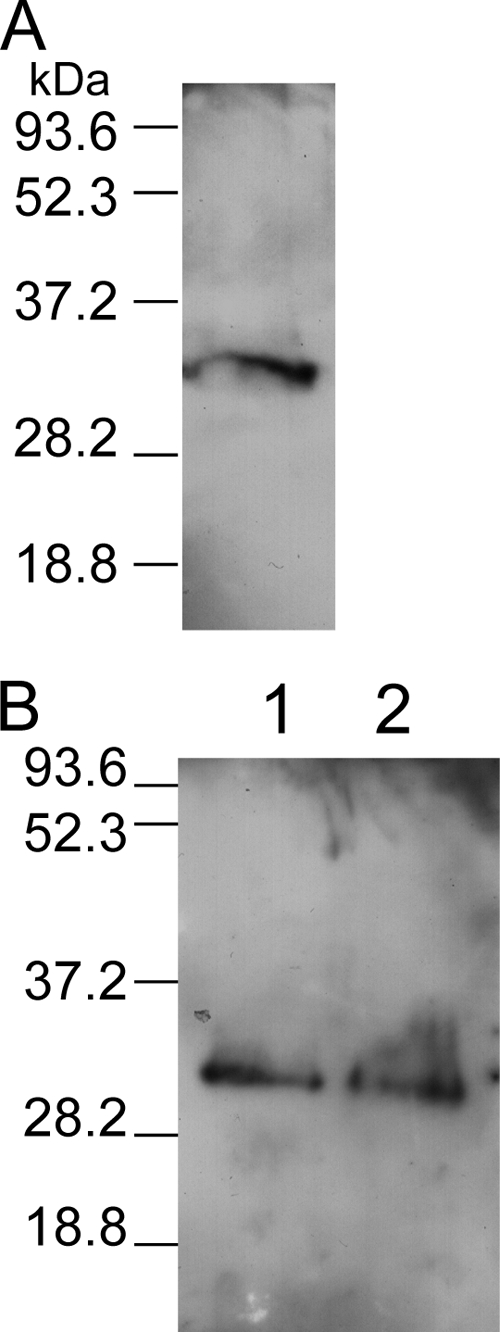
Cell-free expression of ldhA, ldhB, and ldhC. Lactate dehydrogenases were expressed as His-tagged proteins by using the RTS 500 E. coli HY kit in the presence of GroE supplement. Purified LdhA, LdhB, and LdhC were visualized by Western blotting using anti-His antibodies. (A) Purified LdhA (34.6 kDa). (B) Lane 1, purified LdhB (34.5 kDa); lane 2, purified LdhC (34.7 kDa). The sizes of the molecular mass markers (in kDa) are indicated on the left.
Primary structure analysis of LdhA and PfoR.
Although it displays all the characteristics of the lactate dehydrogenase/malate dehydrogenase (LDH/MDH) superfamily, LdhA shares no more than 50% identity over 316 amino acids with the two other B. cereus lactate dehydrogenases, LdhB and LdhC (Fig. 4). Unlike LdhB and LdhC, LdhA is produced from a bicistronic mRNA. The second coding sequence of this mRNA is predicted to produce a transmembrane protein. This protein was proposed to function as a perfringolysin O regulator in strain ATCC 14579 and named PfoR (24). Our analysis of B. cereus F4430/73 PfoR failed to detect any significant similarity to this regulator (5). However, we found that PfoR could display the hydrophobic characteristics of a transport protein, although no homology between this protein and known bacterial permeases was found (5, 9).
FIG. 4.

Amino acid sequence alignment of the three B. cereus l-lactate dehydrogenases. Black and gray boxes correspond to identical and similar amino acid residues, respectively. The tyrosine residue reported as acting in both dehydrogenase activity and ssDNA binding in eukaryotic cells is indicated with an asterisk (40). Dashes indicate gaps.
Physiological characteristics of the B. cereus F4430/73 ldhA mutant.
A B. cereus F4430/73 ldhA mutant was created by introducing a spectinomycin resistance gene deprived of its transcription initiation and termination sequence, as described in Materials and Methods. To prove the nonpolar effect of ldhA disruption, we also constructed a pfo mutant and compared the growth features of the ldhA and pfo mutants to those of the parental F4430/73 strain under anaerobiosis and aerobiosis. Table 3 shows that neither ldhA nor pfo disruption significantly affected the parameters of B. cereus F4430/73 growth under aerobiosis. However, ldhA disruption severely impaired the growth rate of fermentative F4430/73 cells under anaerobiosis, probably due to a lower specific rate of glucose consumption. Analysis of glucose by-product spectra at the end of growth showed only a significant decrease in formate secretion in the ldhA mutant strain compared to formate secretion of the parental strain. This lower level of formate generation was also observed for the pfo mutant concomitantly with higher- level acetate generation. More importantly, the ethanol-to-acetate ratio of the ldhA mutant strain was higher than those of the parental strain and the pfo mutant, while NADH recovery levels were similar. This indicates an effective NAD+ regeneration in spite of greater NADH availability in ldhA mutant cells (35). To determine if this greater NADH availability could be due to a defect of lactate secretion at the beginning of the exponential growth phase (corresponding to the maximal specific growth rate [μmax]), we compared the amounts of lactate produced by the ldhA mutant and the parental strain. The results showed that the ldhA mutant strain produced less lactate than the parental strain (0.15 versus 0.23 mol/mol of glucose, respectively), indicating a defect in the NADH-recycling lactate pathway. Table 4 shows that ldhB expression was upregulated by the disruption of ldhA under both anaerobiosis and aerobiosis, although upregulation occurred more strongly under anaerobiosis. Based on these data, we hypothesized that overexpression of ldhB would be sufficient for maintaining redox balance (NADH/NAD+ ratio) and that greater NADH availability rather than ineffective NAD+ regeneration limits glycolytic flow and subsequently fermentative growth.
TABLE 3.
Results from controlled batch cultures (pH 7.2) of ldhA and pfo mutants and their parent strain, B. cereus F4430/73a
| Growth parameter | Anaerobic growth |
Aerobic growth |
||||
|---|---|---|---|---|---|---|
| WTb | ldhA | pfo | WT | ldhA | pfo | |
| μmax (h−1) | 0.9 | 0.5 | 1.0 | 1.4 | 1.4 | 1.1 |
| Final biomass (g·liter−1) | 0.8 | 1.0 | 0.9 | 2.6 | 2.5 | 2.6 |
| Glucose consumption (%) | 100 | 100 | 100 | 100 | 100 | 100 |
| Yglucose (g of cells·mol of glucose−1) | 26 | 33 | 30 | 86 | 83 | 86 |
| Maximal specific glucose consumption rate (mmol·mg−1·h−1) | 35 | 15 | 34 | 16 | 17 | 13 |
| Yields of end products (mol·mol glucose−1) | ||||||
| Lactate | 1.50 | 1.60 | 1.70 | 0.03 | 0.04 | NZc |
| Acetate | 0.29 | 0.26 | 0.37 | 1.0 | 2.0 | 2.0 |
| Formate | 0.34 | 0.24 | 0.19 | NZ | NZ | NZ |
| Ethanol | 0.06 | 0.07 | 0.08 | NZ | NZ | NZ |
| Succinate | 0.01 | 0.01 | 0.01 | NZ | NZ | NZ |
| Ethanol/acetate | 0.21 | 0.27 | 0.21 | NDd | ND | ND |
| NADH recoverede | 1.1 | 1.1 | 1.2 | ND | ND | ND |
| Total Nhe level (U·g−1 of dry cells) | 5.30 | 1.25 | 4.35 | 1.41 | 0.09 | 1.29 |
Cells were grown under N2 anaerobiosis (pO2 = 0%) and full aerobiosis (pO2 = 100%). Data are the means of triplicate measures obtained from two independent cultures. For clarity, standard deviations (below 10%) are not shown.
WT, wild-type parent strain B. cereus F4430/73.
NZ, yield was below 0.01 mol·mol glucose−1.
ND, not determined.
NADH recovery was calculated as the ratio of pathways producing NADH versus those consuming NADH.
TABLE 4.
Comparison of the mRNA levels of genes involved in glucose catabolism and enterotoxin synthesis in ldhA and pfo mutant strainsa
| Gene | Difference in mRNA levels (n-fold)b under the indicated conditions |
|||
|---|---|---|---|---|
| Anaerobic growth |
Aerobic growth |
|||
| ldhA | pfo | ldhA | pfo | |
| Glucose catabolism genes | ||||
| ldhA | ND | +1.2 | ND | +1.5 |
| pfo | −4.0 | ND | −1.1 | ND |
| ldhB | +32.0 | −3.4 | +2.4 | +1.1 |
| ldhC | −1.4 | +1.2 | +1.0 | −1.2 |
| pfl | +1.4 | −1.1 | +2.8 | +3.5 |
| pdhA | −1.5 | +1.5 | +1.0 | +1.2 |
| pta | −2.0 | +1.3 | −2.4 | +1.9 |
| ackA | +2.4 | +5.8 | +2.3 | +10.8 |
| adhE | +3.9 | −1.7 | +5.8 | +2.6 |
| adhA | +2.7 | −2.4 | +2.6 | +1.9 |
| sdhA | +1.0 | +1.5 | +2.1 | +3.4 |
| citZ | −2.7 | +3.2 | +1.4 | +2.4 |
| citB | +1.4 | +1.0 | +2.6 | +1.6 |
| mdh | −1.3 | +1.1 | +2.1 | +1.3 |
| Enterotoxin gene nhe | −166 | +1.8 | −12.6 | +2.4 |
| Regulator genes | ||||
| plcR | −2.2 | +1.2 | −1.6 | +1.6 |
| resD | −1.3 | +1.2 | +1.2 | +2.6 |
| fnr | −1.2 | −1.1 | +1.4 | +1.1 |
| ccpA | −1.1 | +1.1 | +1.1 | +4.8 |
B. cereus cells were grown under aerobic and anaerobic conditions in controlled batch cultures (pH 7.2) on MOD medium with 30 mM glucose as the carbon source.
Each change (n-fold) represents the mean value of the mRNA levels of one mutant strain sample in relation to that of the parent strain, F4430/73. For each experiment, two measurements from two independent RNA samples taken from the mid-exponential growth phase (μmax) of the same culture were analyzed in parallel. Each data point is an average of the results of the combined experiments. Only ratios of ≤−2 and ≥2 were considered significant (i.e., P ≤ 0.05) according to the precision of the method. + and − indicate up- and downregulation of genes, and significant values for upregulation and downregulation are underlined and boldface, respectively. ND, not detectable.
Table 3 shows that ldhA disruption, but not pfo disruption, significantly reduced Nhe secretion under both aerobiosis and anaerobiosis. Table 4 shows that transcription of nhe was strongly downregulated under these two conditions. Taken together, these data indicate that the main control of Nhe production in the ldhA mutant was oxygen independent and occurred mainly at the transcriptional level. Interestingly, no significant correlation between the variations in nhe transcript levels and those of other tested genes could be highlighted.
Table 4 also shows that pfo mRNA levels were not affected in aerobic ldhA cells, while pfo transcripts were significantly decreased in fermentative anaerobic cells when ldhA was disrupted. The same results were obtained with ldhA-pfo intergenic region mRNA (data not shown). This highlights the nonpolar effect of spectinomycin resistance cassette insertion and suggests a possible role of ldhA in ldhA-pfo transcription, i.e., in its self-regulation. Finally, to definitively confirm that the defect in fermentative growth and enterotoxin production was specifically ascribable to the disruption of ldhA, wild-type copies of ldhA and ldhA-pfo loci were cloned into the pHT304 vector (see Materials and Methods) and each resulting vector was used to complement the ldhA mutant strain. Whatever the vector used, trans-complementation only partially restored the fermentative growth of the ldhA mutant and enabled the mutant to produce 2-fold-higher Nhe levels (data not shown).
Validation of ldhA as a specific lactate dehydrogenase gene of pathogenic relevance.
To compare the role played by LdhA with the roles of LdhB and LdhC in glucose catabolism and Nhe secretion, we disrupted the ldhB and ldhC genes in B. cereus strain F4430/73 as described in Materials and Methods. The abilities of the mutants to grow and produce Nhe under both anaerobiosis and aerobiosis in uncontrolled batch cultures were assessed. Unlike ldhA disruption, ldhB and ldhC disruptions did not induce fermentative growth deficiency or lower-level Nhe secretion (Table 5). This suggests that LdhA, unlike LdhB and LdhC, could play a major role in both physiology and Nhe production in B. cereus F4430/73. Because ldhA disruption caused the same effects in strains ATCC 14579 and F837/76 and in strain F4430/73 (data not shown), we cannot rule out the possibility that LdhA could play a major role in several species of the B. cereus group.
TABLE 5.
Growth parameters and Nhe production determined from uncontrolled batch cultures of ldhA, pfo, ldhB, and ldhC mutants and their parent strain, F4430/73a
| Parameter | Anaerobic growth |
Aerobic growth |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WTb | ldhA | pfo | ldhB | ldhC | WT | ldhA | pfo | ldhB | ldhC | |
| μmax (h−1) | 0.90 | 0.45 | 0.82 | 0.94 | 0.96 | 1.40 | 1.33 | 1.43 | 1.48 | 1.35 |
| Final OD560 | 0.42 | 0.17 | 0.42 | 0.37 | 0.42 | 1.49 | 1.56 | 1.51 | 1.51 | 1.39 |
| Total Nhe level (U·OD560−1) | 12.90 | 0.13 | 10.10 | 12.40 | 13.90 | 6.10 | 0.52 | 5.20 | 5.00 | 5.10 |
Cells were grown under anaerobiosis and aerobiosis. Data are the means of duplicate cultures. For clarity, standard deviations (below 15%) are not shown.
WT, wild-type parent strain B. cereus F4430/73.
In addition to expressing the toxin encoded by nhe, B. cereus expresses several other toxins or putative toxins (7). We determined the effect of ldhA disruption on the mRNA levels of these enterotoxin genes. Table 6 reports that all these genes were downregulated in ldhA mutant cells, suggesting that ldhA should participate in the regulatory network of toxinogenesis in B. cereus F4430/73.
TABLE 6.
Differences in mRNA levels of toxin and putative toxin genes in the ldhA mutant compared to those of the parental F4430/73 straina
| Toxin gene | Difference in mRNA level (n-fold)b under the indicated conditions |
|
|---|---|---|
| Anaerobic growth | Aerobic growth | |
| hbl | −15.4 | −4.1 |
| cytK | −123 | −18.5 |
| hlyI | −3.7 | −8.9 |
| entFM | −2.0 | −2.7 |
| entA | −3.4 | −3.4 |
| entB | −8.1 | −2.0 |
| entC | −6.7 | −4.3 |
B. cereus cells were grown under aerobic and anaerobic conditions in controlled batch cultures (pH 7.2) on MOD medium with 30 mM glucose as the carbon source.
Each change (n-fold) represents the mean value of the mRNA levels of the ldhA mutant strain sample in relation to that of the parent strain, F4430/73. For each experiment, two measures from two independent RNA samples taken from the mid-exponential growth phase (μmax) ) of the same culture were analyzed in parallel. Each data point is an average of the results of the combined experiments. All ratios were ≤−2 and thus considered significant (i.e., P ≤ 0.05) according to the precision of the method. − indicates downregulation of genes.
DISCUSSION
Our exploration of the physiology and nhe expression profile of different B. cereus strains revealed that the ldhA gene, encoding l-lactate dehydrogenase A (LdhA), is required for B. cereus fermentative metabolism and toxinogenesis under both aerobiosis and anaerobiosis. This makes LdhA the first classical catabolic enzyme of both physiological relevance and pathogenic relevance in B. cereus.
B. cereus group species contain three high-conserved genes proposed to encode l-lactate dehydrogenase (http://www.ncbi.nlm.nih.gov/nuccore/29899096). The results obtained in the current study showed that these three genes (ldhA, ldhB, and ldhC) were responsible for active fermentative lactate dehydrogenase. ldhA expression, like ldhB and ldhC expression, was detectable in both fermenting and respiring B. cereus cells according to their ability to secrete lactate under these two conditions, which is in line with the results of our previous studies (28). Low-level expression of ldhA was coupled with high-level expression of ldhC in strains F4810/72, F837/76, and PA, whereas disruption of ldhA induced ldhB overexpression in strain F4430/73 under both anaerobiosis and aerobiosis. This suggests that the regulation network that coordinates the differential expression of ldhA, ldhB, and ldhC in B. cereus could function independently of the presence or absence of oxygen in a strain-dependent manner. The existence of strain-dependent ldh gene regulation in aerobic respiring cells where lactate dehydrogenase activity is not necessary to maintain redox homeostasis indicates that fine regulation of lactate dehydrogenase synthesis could confer upon B. cereus a metabolic advantage when grown under aerobiosis, as is the case for Staphylococcus aureus (33). Expression of ldh genes is affected by the redox sensors ResDE and Fnr, although direct binding of the regulators and their binding sites have not been elucidated (11, 43). ResD and Fnr were showed to regulate each other and their own synthesis at the transcriptional level (13, 14). Since no correlation between the mRNA levels of these two regulators and those of ldhA was observed, we conclude that strain-dependent regulation of ldh genes cannot be simply related to ResDE and Fnr.
The importance of LdhA, unlike LdhB and LdhC, in maintaining redox homeostasis in fermentative cells was demonstrated by (i) the inability of low-level ldhA-expressing F4810/72, F837/76, and PA cells to sustain rapid anaerobic growth compared to high-level ldhA-expressing F4430/73 cells and (ii) the fermentative anaerobic growth defect of the F4430/73 ldhA mutant. The importance of LdhA for enterotoxin gene expression in fermentative anaerobic and respiring aerobic B. cereus cells was highlighted by (i) the capacity for low-level ldhA-expressing F4810/72, F837/76, and PA cells to express nhe mRNA being lower than that for high-level ldhA-expressing F4430/73 cells and (ii) the downregulation of nhe and other putative enterotoxin genes in strain F4430/73 lacking LdhA. A key question is how LdhA impacts enterotoxin gene expression. A detailed analysis of this impact was beyond the scope of this study. However, we speculate that under fermentative conditions, the defect in LdhA synthesis may indirectly downregulate enterotoxin gene expression through its impact on NADH availability and subsequently on redox potential of the cytoplasm and on growth rate (31). Activation of nhe expression in fermentative anaerobiosis has been shown to involve ResDE and Fnr regulators (11, 43). These redox regulators activate their own transcription in response to redox potential changes (13, 14). Unlike nhe mRNA levels, resD and fnr mRNA levels did not differ significantly between the ldhA mutant strain and the parental strain. This suggests that ResDE and Fnr are not the key regulators of nhe expression in ldhA mutant cells. This is probably also the case for the autoregulatory PlcR transcriptional regulator (16). In Bacillus subtilis (25), as in other Gram-positive bacteria (36), changes in NADH availability are sensed by the transcriptional regulator Rex. A Rex homologue is encoded in the genome of B. cereus (www.ncbi.nlm.nih.gov and unpublished data). Interestingly, we found Rex binding motifs (32), overlapping ResD and Fnr binding motifs in ldhA, ldhB (but not ldhC), and nhe (data not shown). It remains to be examined whether these sequences are really targets for this redox regulator that could thus regulate directly and simultaneously nhe and ldhA.
Previous results showed that ResDE, Fnr, and PlcR also activate nhe expression in aerobic respiring cells (11, 17, 43). Our transcriptomic data suggest that downregulation of nhe expression in ldhA mutant cells could not be simply controlled by these three regulators under aerobiosis as they were under anaerobiosis. Under aerobiosis, involvement of Rex seems more questionable, since no NADH-dependent physiological change in the ldhA mutant strain relative to the parental strain was observed. We thus speculate that LdhA could directly control nhe expression under aerobiosis and maybe under anaerobiosis in addition to its probable indirect control. In accordance with the latter hypothesis, we observed that a lack of LdhA led to a greater downregulation of nhe expression under anaerobiosis than that seen under aerobiosis (Table 4). The idea that LdhA has a regulatory role is not unfounded, because (i) our results indicate that LdhA regulates its own expression, (ii) LdhA is structurally very different from other B. cereus classical fermentative lactate dehydrogenases (2, 15), and (iii) lactate dehydrogenases have been identified as single-stranded DNA (ssDNA) binding proteins in eukaryotic cells and speculated to be components of transcriptional coactivator complexes (6, 19). Lactate dehydrogenase binding to ssDNA involved a tyrosine residue located near the coenzyme binding site that was regulated by the NADH/NAD+ ratio (40). LdhA possesses this Tyr residue; its position and structural environment (2, 15) differ from those encountered in LdhB and LdhC (Fig. 4). It is thus tempting to speculate that LdhA could be a partner regulator of a complex involving at least ResD and Fnr (and possibly Rex) to regulate its own transcription as well as nhe transcription. Such a complex could also activate the expression of other toxin genes, since we demonstrated that a lack of LdhA downregulated their expression (Table 6). Finally, LdhA could function as both a main fermentative enzyme and a virulence-associated transcriptional regulator, although further analyses are necessary to confirm and further define its nonenzymatic role.
In conclusion, this study exploring the catabolism and nhe expression of different B. cereus strains was a successful approach for the discovery of a fermentative enzyme of pathogenic relevance. In addition, this study indicates that links between metabolic sensors and virulence factor transcription could be established directly through enzymes that participate in carbohydrate metabolism in B. cereus like in other bacteria (41). These roles further underscore the need to consider the nonenzymatic functions of central metabolism enzymes in studies aiming to determine the virulence potentials of a B. cereus strain and, more widely, of any other pathogenic bacterial strain. Finally, LdhA, like adapted glycolytic enzymes in eukaryotes, may act as a link between environmental cues and regulatory circuits, ensuring B. cereus adaptation and propagation in anoxic and oxic conditions, such as those found in the human intestine (27).
Supplementary Material
Acknowledgments
S.L. received a fellowship from the Provence Alpes Côte d'Azur region and INRA, and K.M. received a fellowship from the Franco-Algerian intergovernmental program.
We thank Tefisoa Randrianarivony and Chahla Louldji for technical assistance.
Footnotes
Published ahead of print on 4 February 2011.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Agaisse, H., M. Gominet, O. A. Okstad, A. B. Kolsto, and D. Lereclus. 1999. PlcR is a pleiotropic regulator of extracellular virulence factor gene expression in Bacillus thuringiensis. Mol. Microbiol. 32:1043-1053. [DOI] [PubMed] [Google Scholar]
- 2.Arai, K., et al. 2010. Active and inactive state structures of unliganded Lactobacillus casei allosteric L-lactate dehydrogenase. Proteins 78:681-694. [DOI] [PubMed] [Google Scholar]
- 3.Arantes, O., and D. Lereclus. 1991. Construction of cloning vectors for Bacillus thuringiensis. Gene 108:115-119. [DOI] [PubMed] [Google Scholar]
- 4.Arnaud, M., A. Chastanet, and M. Debarbouille. 2004. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70:6887-6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Awad, M. M., and J. I. Rood. 2002. Perfringolysin O expression in Clostridium perfringens is independent of the upstream pfoR gene. J. Bacteriol. 184:2034-2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cattaneo, A., S. Biocca, N. Corvaja, and P. Calissano. 1985. Nuclear localization of a lactic dehydrogenase with single-stranded DNA-binding properties. Exp. Cell Res. 161:130-140. [DOI] [PubMed] [Google Scholar]
- 7.Clair, G., S. Roussi, J. Armengaud, and C. Duport. 2010. Expanding the known repertoire of virulence factors produced by Bacillus cereus through early secretome profiling in three redox conditions. Mol. Cell Proteomics 9:1486-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cruz Ramos, H., et al. 2000. Fermentative metabolism of Bacillus subtilis: physiology and regulation of gene expression. J. Bacteriol. 182:3072-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong, J. M., J. S. Taylor, D. J. Latour, S. Iuchi, and E. C. Lin. 1993. Three overlapping lct genes involved in L-lactate utilization by Escherichia coli. J. Bacteriol. 175:6671-6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duport, C., S. Thomassin, G. Bourel, and P. Schmitt. 2004. Anaerobiosis and low specific growth rates enhance hemolysin BL production by Bacillus cereus F4430/73. Arch. Microbiol. 182:90-95. [DOI] [PubMed] [Google Scholar]
- 11.Duport, C., A. Zigha, E. Rosenfeld, and P. Schmitt. 2006. Control of enterotoxin gene expression in Bacillus cereus F4430/73 involves the redox-sensitive ResDE signal transduction system. J. Bacteriol. 188:6640-6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ehling-Schulz, M., et al. 2005. Emetic toxin formation of Bacillus cereus is restricted to a single evolutionary lineage of closely related strains. Microbiology 151:183-197. [DOI] [PubMed] [Google Scholar]
- 13.Esbelin, J., J. Armengaud, A. Zigha, and C. Duport. 2009. ResDE-dependent regulation of enterotoxin gene expression in Bacillus cereus: evidence for multiple modes of binding for ResD and interaction with Fnr. J. Bacteriol. 191:4419-4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esbelin, J., Y. Jouanneau, J. Armengaud, and C. Duport. 2008. ApoFnr binds as a monomer to promoters regulating the expression of enterotoxin genes of Bacillus cereus. J. Bacteriol. 190:4242-4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eventoff, W., et al. 1977. Structural adaptations of lactate dehydrogenase isozymes. Proc. Natl. Acad. Sci. U. S. A. 74:2677-2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gohar, M., et al. 2008. The PlcR virulence regulon of Bacillus cereus. PLoS One 3:e2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gohar, M., et al. 2005. A comparative study of Bacillus cereus, Bacillus thuringiensis and Bacillus anthracis extracellular proteomes. Proteomics 5:3696-3711. [DOI] [PubMed] [Google Scholar]
- 18.Gompertz, B. 1925. On the nature of the function expressive of the law of human mortality, and on a new mode of determining the value of life contingencies. Philos. Trans. R. Soc. 115:513-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grosse, F., H. P. Nasheuer, S. Scholtissek, and U. Schomburg. 1986. Lactate dehydrogenase and glyceraldehyde-phosphate dehydrogenase are single-stranded DNA-binding proteins that affect the DNA-polymerase-alpha-primase complex. Eur. J. Biochem. 160:459-467. [DOI] [PubMed] [Google Scholar]
- 20.Guinebretière, M. H., V. Broussolle, and C. Nguyen-The. 2002. Enterotoxigenic profiles of food-poisoning and food-borne Bacillus cereus strains. J. Clin. Microbiol. 40:3053-3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guinebretière, M. H., et al. 2008. Ecological diversification in the Bacillus cereus group. Environ. Microbiol. 10:851-865. [DOI] [PubMed] [Google Scholar]
- 22.Haldenwang, W. G. 1995. The sigma factors of Bacillus subtilis. Microbiol. Rev. 59:1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartig, E., et al. 2004. Bacillus subtilis ResD induces expression of the potential regulatory genes yclJK upon oxygen limitation. J. Bacteriol. 186:6477-6484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivanova, N., et al. 2003. Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature 423:87-91. [DOI] [PubMed] [Google Scholar]
- 25.Larsson, J. T., A. Rogstam, and C. von Wachenfeldt. 2005. Coordinated patterns of cytochrome bd and lactate dehydrogenase expression in Bacillus subtilis. Microbiology 151:3323-3335. [DOI] [PubMed] [Google Scholar]
- 26.Lund, T., and P. E. Granum. 1996. Characterisation of a non-haemolytic enterotoxin complex from Bacillus cereus isolated after a foodborne outbreak. FEMS Microbiol. Lett. 141:151-156. [DOI] [PubMed] [Google Scholar]
- 27.Marteyn, B., F. B. Scorza, P. J. Sansonetti, and C. Tang. 2011. Breathing life into pathogens: the influence of oxygen on bacterial virulence and host responses in the gastrointestinal tract. Cell. Microbiol. 13:171-176. [DOI] [PubMed] [Google Scholar]
- 28.Messaoudi, K., T. Clavel, P. Schmitt, and C. Duport. 2010. Fnr mediates carbohydrate-dependent regulation of catabolic and enterotoxin genes in Bacillus cereus F4430/73. Res. Microbiol. 161:30-39. [DOI] [PubMed] [Google Scholar]
- 29.Moravek, M., et al. 2006. Determination of the toxic potential of Bacillus cereus isolates by quantitative enterotoxin analyses. FEMS Microbiol. Lett. 257:293-298. [DOI] [PubMed] [Google Scholar]
- 30.Murphy, E. 1985. Nucleotide sequence of a spectinomycin adenyltransferase AAD(9) determinant from Staphylococcus aureus and its relationship to AAD(3") (9). Mol. Gen. Genet. 200:33-39. [DOI] [PubMed] [Google Scholar]
- 31.Ouhib, O., T. Clavel, and P. Schmitt. 2006. The production of Bacillus cereus enterotoxins is influenced by carbohydrate and growth rate. Curr. Microbiol. 53:222-226. [DOI] [PubMed] [Google Scholar]
- 32.Pagels, M., et al. 2010. Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol. Microbiol. 76:1142-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richardson, A. R., S. J. Libby, and F. C. Fang. 2008. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 319:1672-1676. [DOI] [PubMed] [Google Scholar]
- 34.Rosenfeld, E., C. Duport, A. Zigha, and P. Schmitt. 2005. Characterization of aerobic and anaerobic vegetative growth of the food-borne pathogen Bacillus cereus F4430/73 strain. Can. J. Microbiol. 51:149-158. [DOI] [PubMed] [Google Scholar]
- 35.Sánchez, A. M., G. N. Bennett, and K. Y. San. 2005. Effect of different levels of NADH availability on metabolic fluxes of Escherichia coli chemostat cultures in defined medium. J. Biotechnol. 117:395-405. [Epub ahead of print.] [DOI] [PubMed] [Google Scholar]
- 36.Somerville, G. A., and R. A. Proctor. 2009. At the crossroads of bacterial metabolism and virulence factor synthesis in staphylococci. Microbiol. Mol. Biol. Rev. 73:233-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spira, W. M., and J. M. Goepfert. 1975. Biological characteristics of an enterotoxin produced by Bacillus cereus. Can. J. Microbiol. 21:1236-1246. [DOI] [PubMed] [Google Scholar]
- 38.Stenfors Arnesen, L. P., A. Fagerlund, and P. E. Granum. 2008. From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Rev. 32:579-606. [DOI] [PubMed] [Google Scholar]
- 39.van der Voort, M., O. P. Kuipers, G. Buist, W. M. de Vos, and T. Abee. 2008. Assessment of CcpA-mediated catabolite control of gene expression in Bacillus cereus ATCC 14579. BMC Microbiol. 8:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams, K. R., S. Reddigari, and G. L. Patel. 1985. Identification of a nucleic acid helix-destabilizing protein from rat liver as lactate dehydrogenase-5. Proc. Natl. Acad. Sci. U. S. A. 82:5260-5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yesilkaya, H., et al. 2009. Pyruvate formate lyase is required for pneumococcal fermentative metabolism and virulence. Infect. Immun. 77:5418-5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zigha, A., E. Rosenfeld, P. Schmitt, and C. Duport. 2006. Anaerobic cells of Bacillus cereus F4430/73 respond to low oxidoreduction potential by metabolic readjustments and activation of enterotoxin expression. Arch. Microbiol. 185:222-233. [DOI] [PubMed] [Google Scholar]
- 43.Zigha, A., E. Rosenfeld, P. Schmitt, and C. Duport. 2007. The redox regulator Fnr is required for fermentative growth and enterotoxin synthesis in Bacillus cereus F4430/73. J. Bacteriol. 189:2813-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zwietering, M., I. Jongenburger, F. Rombouts, and K. Van't Riet. 1990. Modeling of the bacterial growth curve. Appl. Environ. Microbiol. 56:1875-1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.