

Abstract
Staphylococcus aureus encodes the Sec-independent Ess secretion pathway, an ortholog of mycobacterial T7 secretion systems which is required for the virulence of this Gram-positive microbe. The Ess (ESX secretion) pathway was previously defined as a genomic cluster of eight genes, esxA, esaA, essA, essB, esaB, essC, esaC, and esxB. essABC encode membrane proteins involved in the stable expression of esxA, esxB, and esaC, genes specifying three secreted polypeptide substrates. esaB, which encodes a small cytoplasmic protein, represses the synthesis of EsaC but not that of EsxA and EsxB. Here we investigated a hitherto uncharacterized gene, esaD, located downstream of esxB. Expression of esaD is activated by mutations in esaB and essB. EsaD, the 617-amino-acid product of esaD, is positioned in the membrane and is also accessible to EsaD-specific antibodies on the bacterial surface. S. aureus mutants lacking esaD are defective in the secretion of EsxA. Following intravenous inoculation of mice, S. aureus esaD mutants generate fewer abscesses with a reduced bacterial load compared to wild-type parent strain Newman. The chromosomes of Listeria and Bacillus species with Ess pathways also harbor esaD homologues downstream of esxB, suggesting that the contributory role of EsaD in Ess secretion may be shared among Gram-positive pathogens.
Staphylococcus aureus, a Gram-positive bacterium and commensal of the human skin and nares, is also the most common cause of soft tissue infections (22). Following invasion of deeper tissues, staphylococci secrete proteins that trigger the establishment of abscess lesions and enable this pathogen to persist in host tissues without eliciting protective immune responses (9, 21). During the initial stages of infection, staphylococci are surrounded by massive infiltrates of immune cells, predominantly polymorphonuclear leukocytes (PMNs). Within 4 to 5 days, these lesions are programmed for the replication of staphylococcal abscess communities, which are enclosed by a pseudocapsule of fibrin deposits and surrounded by layers of immune cells (9). Staphylococcal abscesses are dynamic lesions where immune cells respond to the many secreted products of this pathogen. Eventually, these lesions rupture on the surface of organ epithelia, thereby releasing staphylococci into body fluids and promoting the formation of new abscesses (9). Recent work has shown that the S. aureus Ess pathway (ESX or type VII-like secretion system) is an important contributor to this developmental program of abscess formation and staphylococcal persistence in host tissues (7).
ESAT-6 and its homologue CFP-10 are small, α-helical polypeptides and founding members of the WXG100 motif family (27, 30). Both proteins are secreted by Mycobacterium tuberculosis, the causative agent of tuberculosis (18, 29, 32). A hallmark of M. tuberculosis infection is the formation of a granuloma, where bacteria replicate within macrophages (11). Following drainage of necrotic debris from a granuloma, mycobacteria can disseminate throughout the tissues of an infected host (10). ESAT-6 and CFP-10 are crucial for the replication of M. tuberculosis in macrophages and presumably also for the pathogen's ability to suppress innate and adaptive immune responses (11, 24, 28, 31, 32). In agreement with this model, the live-attenuated vaccine strain Mycobacterium bovis Bacille Calmette-Guérin (BCG) harbors a deletion of the ESAT-6 secretion gene cluster (23, 28). Upon vaccination, BCG elicits antimycobacterial immune responses that reduce the incidence of military tuberculosis and of M. tuberculosis meningitis in children (17).
In addition to the WXG100 proteins EsxA and EsxB, the ess gene clusters of S. aureus harbor essC, a homologue of the FtsK/SpoIIIE ATPase superfamily. In M. tuberculosis, the products of orthologous genes are thought to fuel protein transport by the type VII secretion system and contribute to substrate recognition (8, 27). The chromosome of M. tuberculosis harbors multiple ESX gene clusters, as well as genomic islands that encode non-WXG100 secretion substrates and regulatory factors for these secretion pathways (1, 5, 14). In S. aureus, ess was initially identified as a single locus flanked by two genes for WXG100 proteins, esxA esaA essA essB esaB essC esaC esxB. essAB encode membrane proteins involved in the stable expression of secreted polypeptides; these genes are not conserved in M. tuberculosis (8). The same applies to esaB, which encodes a small cytoplasmic protein that represses the synthesis of the staphylococcal secreted protein EsaC but not the synthesis and secretion of EsxA and EsxB (7).
Here we investigated a hitherto uncharacterized gene, esaD, which is located downstream of esxB. Expression of esaD is activated by mutations in esaB and essB. EsaD, the 617-amino-acid product of esaD, is positioned in the membrane and is also accessible to EsaD-specific antibodies on the bacterial surface. S. aureus mutants lacking esaD display a reduction in EsxA secretion. Following intravenous inoculation of mice, S. aureus Newman mutants lacking esaD display defects in the unique ability of staphylococci to establish persistent abscess lesions. The genomes of Listeria and Bacillus species also harbor esaD homologues downstream of esxB in their ess cluster, suggesting that the contributory role of EsaD in Ess secretion may be shared among Gram-positive pathogens.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
S. aureus cells were grown in tryptic soy broth (TSB) at 37°C. Chloramphenicol and erythromycin were used at 10 mg liter−1 for plasmid and allele selection. S. aureus strain USA300 was obtained through the Network on Antimicrobial Resistance in S. aureus (NARSA; National Institute of Allergy and Infectious Diseases [NIAID]). All of the mutants used in this study, with the exception of the esxA mutant, were obtained from the Phoenix (ΦNΞ) library (3) or have been previously described (7). Each Phoenix isolate carries a mapped bursa aurealis (a minitransposon carrying the ermB marker) insertion in the chromosome of the clinical isolate Newman (3, 15). All bursa aurealis insertions were crossed back into strain Newman or USA300 using bacteriophage φ85. Transductants were validated by PCR analysis and sequencing of the transposon insertion site using inverse PCR as described previously (4a). Most, but not all, USA300 isolates harbor three plasmids, one of which contains the ermC gene (13). This plasmid can be lost readily, permitting the construction of mutants marked with the ermC allele. For deletion of esxA, a 2-kbp DNA fragment flanking the esxA gene but carrying only the first and last four codons of the esxA gene was amplified by PCR with abutted BamHI-EcoRI restriction sites. The DNA fragment was cloned into pKOR1 for allelic replacement performed as described earlier (4). The Escherichia coli-S. aureus shuttle vector pOS1, which carries the hprK promoter and Shine-Dalgarno sequence (275 bp upstream of the hprK lgt yvoF yvcD translational start site) and three cloning sites, NdeI, XhoI, and BamHI, as described earlier (6), was used for complementation studies. All cloning procedures were carried out with E. coli, and ampicillin was used at 100 mg liter−1 for plasmid selection. The complementation plasmids pOS-esaB, pOS-essB, and pOS-esaD were generated by amplifying the minimal coding sequence of each gene using primers AACTCGAGATGAATCAGCACGTAAAAGTAAC and AAGGATCCCTATAGTAACTTCAAAATATCTCC for esaB, AACTCGAGATGGTTAAAAATCATAACCCTAAAAATG and AAGGATCCCTATTTTTTTCTTTCAGCTTCTTGGCG for essB, and AACATATGACAAAAGATATTGAATATC and AAGGATCCTAGTTATTTAATATTCTTCTAATATTTC for esaD. Cloning of the first 100 amino acids of EsaD (His6-EsaD1-100) was achieved in vector pET16b using primers with the nucleotide sequences AACATATGACAAAAGATATTGAATAT and AACTCGAGTTAATCTATATCTTCTATA.
Analysis of Ess secretion in staphylococcal cultures.
Bacterial strains were grown overnight from isolated colonies in TSB at 37°C with shaking. Cultures were diluted 1:100 in fresh broth and shaken at 37°C until they reached an A600 of 1. For whole-culture lysates (see Fig. 2 and 3B), cultures (6 ml) were incubated in the presence of lysostaphin (100 μg/ml) for 30 min at 37°C. To assess protein secretion (see Fig. 4), cultures (6 ml) were spun (10,000 × g for 4 min) and bacteria in the cell pellets were washed with phosphate-buffered saline (PBS) and suspended in 6 ml of PBS containing lysostaphin (100 μg/ml). Six milliliters of cell extracts (referred to as cell lysate) was used for precipitation of proteins. Supernatants of spun cultures (6 ml) were transferred to a fresh tube (referred to as medium). For subcellular localization of EsaD (see Fig. 3C), lysostaphin-treated cell extracts (referred to as cell lysate) were spun at 4°C and 100,000 × g for 40 min, and the supernatant containing soluble proteins was transferred to a new tube while pellets containing nonsoluble membrane proteins were suspended in 6 ml PBS buffer. Proteins in all fractions (cell lysate, supernatant, and membrane) were precipitated with 6 ml of a methanol-chloroform mixture (4:1). All precipitates were washed with methanol and dried overnight. Precipitates were solubilized in 100 μl of 0.5 M Tris-HCl (pH 8.0)-4% sodium dodecyl sulfate (SDS) and heated at 90°C for 10 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidene difluoride membrane for immunoblot analysis with appropriate polyclonal antibodies. Immunoreactive signals were revealed by using a secondary antibody coupled to IRDye 680. Quantification of Western blot assays was conducted using a Li-Cor Biosciences Odyssey imager. Statistical analysis was gained through three independent experiments.
Immunofluorescence microscopy.
Microscopic observation of intact cells was performed as previously described (12). Briefly, overnight cultures of S. aureus cells were diluted 1:100 into fresh growth medium and grown to mid-log phase (A600 of 1). The culture (0.5 ml) was centrifuged for 3 min at 8,000 × g. Bacterial cell pellets were washed twice with PBS, suspended in 0.5 ml of PBS, vortexed for 15 s, and fixed with 14% paraformaldehyde-0.006% glutaraldehyde in 30 mM PBS (pH 7.4) for 20 min at room temperature. Cells were washed three times and suspended in a final volume of 0.5 ml PBS. Cell suspensions were applied to l-polylysine-coated coverslips for 5 min and then washed three times with 60 μl H2O. Sixty microliters of 3% bovine serum albumin (BSA) in PBS was added as a blocking reagent for nonspecific antibody binding, and the cells were incubated for 1 h at room temperature. Primary incubation was followed by incubation with an Alexa Fluor 647-conjugated IgG secondary antibody (Invitrogen Molecular Probes). Cells were washed 10 times with 60 μl PBS containing 0.025% Tween 20 (PBS-T) and then incubated for 1 h in the dark with 60 μl of 1:250 goat anti-rabbit Alexa Fluor 647-conjugated secondary antibody in 3% BSA-PBS. Cells were again washed 10 times with 60 μl PBS-T. Slides were mounted with N-propyl gallate, sealed, and viewed, and images were collected with an Olympus AX70 microscope.
Protein purification and affinity purification of EsaD antibodies.
The N-terminal domain of EsaD (the first 100 amino acids) was produced as a histidine-tagged protein His6-EsaDN in E. coli BL21(DE3) as previously described (33). Purified EsaDN (5 mg protein) was covalently linked to HiTrap N-hydroxysuccinimide-activated HP columns (GE Healthcare) that were used for affinity chromatography of 10 ml of rabbit serum at 4°C. Charged matrix was washed with 50 column volumes of PBS prior to the elution of antibodies with elution buffer (1 M glycine [pH 2.5], 0.5 M NaCl), followed by rapid neutralization with 1 M Tris-HCl, pH 8.5. The purified antibodies were dialyzed overnight against PBS at 4°C and used for immunoblotting and immunofluorescence microscopy experiments.
Animal infections and histopathology.
BALB/c mice (10 animals per group, 6-week-old female animals; Charles River) were infected by retro-orbital injection with a suspension of staphylococci. S. aureus strains were grown overnight in TSB, diluted 1:100 in fresh medium, and incubated with rotation at 37°C for 2 h. Staphylococci were sedimented by centrifugation (7,000 × g for 10 min), suspended and washed once in PBS, and diluted in PBS to an A600 of 0.4 (0.5 × 107 to 1 × 107 CFU/100 μl). Each bacterial inoculum was quantified by colony formation on agar plates. Mice were anesthetized by intraperitoneal injection of 80 to 120 mg/kg ketamine and 3 to 6 mg/kg xylazine and infected by lateral injection of ∼1 × 107 CFU in 100 μl into the retro-orbital plexus of the right eye. On day 5 or 15 following infection, mice were euthanized by compressed CO2 inhalation. Kidneys were removed and homogenized in 1% Triton X-100; aliquots were diluted and plated on agar medium for triplicate determination of CFU. The experiment was reproduced twice. Student's t test was performed for statistical analysis by using the software Analyze-it (Analyze-it Software, Leeds, United Kingdom), and P values of less than 0.05 were considered significant. For histology, kidney tissues were incubated at room temperature in 10% formalin for 24 h. Tissues were embedded in paraffin, thin sectioned, hematoxylin-eosin stained, and then examined by microscopy.
RESULTS
Identification of EsaD.
BLAST searches using S. aureus Newman ess and esxAB sequences as queries against the genomic sequences of other Gram-positive bacteria identified ess clusters, as well as their flanking genes. S. aureus Newman NWMN_0228 is located downstream of esxB, and its homologues assume similar positions in Listeria and Bacillus species with Ess pathways (Fig. 1A). In agreement with the possibility that NWMN_0228 is a component of the S. aureus type VII-like secretion pathway, Gram-positive organisms that lack ess gene clusters also do not harbor a homologue of NWMN_0228. The product of NWMN_0228, a 617-residue polypeptide, or of its homologues, harbors the ancient conserved domain COG5444, which is also found in EsxL, a putative WXG motif secretion substrate of Bacillus anthracis (Fig. 1B). Some homologues of NWMN_0228 harbor a C-terminal domain, either DUF600 (a domain of unknown function) or VIP2 (Bacillus-produced vegetative insecticidal proteins related to a family of actin-ADP-ribosylating toxins) (Fig. 1B). Thus, depending on the host genome and unique attributes of each Ess pathway, the homologues of NWMN_0228 may have acquired specific functions that are not shared by all of the members of this gene family. Here we designated NWMN_0228 esaD for Ess-associated gene D.
FIG. 1.

Schematic of Ess loci and EsaD-like proteins in Gram-positive bacteria. (A) Comparison of the S. aureus Ess (type VII-like) secretion locus with L. monocytogenes and “Bacillus cytotoxicus” ess loci, as well as the B. anthracis ess gene cluster (16). Genes showing sequence homology are depicted in the same color. (B) Domain organization of S. aureus EsaD and protein homologues showing the conserved COG5444 domain. With the exception of B. subtilis YeeE, all COG5444 proteins lie within an Ess-like locus. The colors of some conserved proteins and domains indicate WXG100 proteins (red), FtsK SpoIIIE-like ATPases (yellow), EsaD and COG5444 (dark blue), proteins with a DUF600 domain (light blue), and VIP2 (pink).
To characterize esaD and its product, we expressed codons 1 to 100 of its open reading frame as a translational hybrid with an N-terminal hexahistidyl tag via the T7 promoter in E. coli BL21(DE3) and purified the recombinant product His6-EsaD1-100 by affinity chromatography on Ni-nitrilotriacetic acid resin. Purified His6-EsaD1-100 was injected into rabbits to raise polyclonal antibodies that were subsequently affinity purified on an antigen matrix (anti-EsaD antibodies) and then used for immunoblotting experiments. Growth of S. aureus Newman in TSB does not induce expression of the Ess pathway, and anti-EsaD antibodies failed to detect immunoreactive species. As a control, antibodies against sortase A (anti-SrtA antibodies) readily detected the constitutively expressed transpeptidase in staphylococcal extracts. The product of S. aureus esaB functions as a repressor of ess expression, and we therefore tested whether anti-EsaD antibodies detected a translational product in extracts of esaB mutant staphylococci. Indeed, anti-EsaD antibodies identified a polypeptide in esaB mutant staphylococci that migrated with the expected mass of 62 kDa on SDS-PAGE (Fig. 2A). An anti-EsaD antibody-immunoreactive species with the same mobility was detected in extracts of S. aureus Newman harboring phrpK-esaD, a plasmid that drives esaD expression from the constitutive hprK promoter, but not in mutants with a bursa aurealis insertion at nucleotide 23 of the esaD open reading frame (Fig. 2A). Mutations in esxA, esxB, essA, essC, or esaC did not affect esaD expression. Surprisingly, a bursa aurealis insertion in essB, which encodes a membrane protein that is required for Ess secretion, also induced the expression of esaD (Fig. 2A). The esaD regulatory phenotypes of esaB and essB mutants could be complemented in trans by plasmids expressing either wild-type esaB or essB, respectively (Fig. 2B). Thus, the regulatory phenotypes of esaB and essB mutants are due to the corresponding genetic lesions of their bursa aurealis insertions and are not caused by polar effects on other ess or esa genes.
FIG. 2.

EsaB and EssB regulate EsaD production in the staphylococcal Ess pathway. (A) Whole-culture lysates of S. aureus Newman or its variants with bursa aurealis insertions (esxA, essA, esaB, essB, essC, esaC, esxB, and esaD) were examined for production of EsaD. Staphylococci were grown in TSB to an A600 of 1.0 and treated with lysostaphin. Proteins in whole-culture lysates were precipitated with methanol-chloroform, separated by SDS-PAGE, and detected by immunoblotting with specific antibodies (anti-EsaD antibodies, as well as anti-SrtA antibodies as a loading control). As a control, an extract of S. aureus Newman carrying pOS-esaD, a plasmid expressing esaD under the control of the constitutive hprK promoter, was examined for EsaD production. (B) Plasmid complementation analysis of esaB and essB mutants via immunoblotting of total cell extracts derived from S. aureus Newman or esaB and essB mutants harboring the vector alone (pOS) or plasmids pOS-esaB and pOS-essB. (C) Whole-culture lysates of S. aureus USA300 and its esaD variant were examined for EsaD production. Extracts of USA300 cultures were compared to strain Newman extracts. Samples were prepared and analyzed as described for panel A. (D) Whole-culture lysates of S. aureus Newman and its esaD variant were prepared and analyzed as described for panel A, except that cultures were grown in TSB with (+) or without (−) 5% horse serum.
We wondered whether other S. aureus isolates express EsaD and examined USA300, a strain responsible for the current North American epidemic of community-acquired methicillin-resistant S. aureus infections. Earlier work on the secretion substrate EsaC showed that, when grown in laboratory medium, S. aureus USA300 produces machine components and transport reactions of the Ess pathway. In agreement with this, an anti-EsaD antibody-immunoreactive product was detected in lysostaphin extracts of USA300 but not in those of S. aureus Newman (Fig. 2C). EsaD was not detectable by immunoblotting in extracts of the USA300 esaD variant with bursa aurealis inserted at nucleotide 23 of its open reading frame (Fig. 2C).
To test whether S. aureus Newman can express esaD under growth conditions that more closely resemble infected host tissues, we inoculated bacteria into TSB broth supplemented with fetal calf serum. When grown under these conditions, S. aureus Newman, but not its isogenic esaD mutant, produced EsaD polypeptide (Fig. 2D). Taken together, these results indicate that esaD is expressed in clinical isolates of methicillin-sensitive and methicillin-resistant S. aureus strains.
EsaD is displayed on the staphylococcal surface.
When analyzed with the Kyte-Doolittle hydrophobicity plot, EsaD is predicted to harbor an α-helical hydrophobic transmembrane segment at amino acids 217 to 250 of the 617 residue polypeptide. As positively charged amino acids (three Lys residues and one Arg residue) are positioned near the C-terminal end of the transmembrane segment, we predicted that the N-terminal portion of EsaD (residues 1 to 216) may be positioned on the staphylococcal surface, whereas the C-terminal domain (residues 251 to 617) may be located in the cytoplasm. To test this, affinity-purified rabbit anti-EsaD antibodies were added to S. aureus Newman Δspa mutants, which are unable to bind immunoglobulin by nonimmune mechanisms. When analyzed by immunoblotting, lysostaphin extracts of Δspa mutants harboring plasmid pesaD revealed the presence of EsaD, whereas Δspa mutants with the corresponding pOS1 vector control did not (Fig. 3A and B). Antibody binding to the staphylococcal surface was detected with goat anti-rabbit secondary antibodies conjugated to Alexa Fluor 488 (green) and fluorescence microscopy. Anti-EsaD antibodies bound to staphylococci harboring pesaD but not to cells with the pOS1 vector control. Further, irrelevant anti-V10 antibodies, which recognize the plague-protective antigen LcrV, failed to generate fluorescent signals on the staphylococcal surface. Taken together, these data indicate that EsaD is displayed on the staphylococcal surface.
FIG. 3.
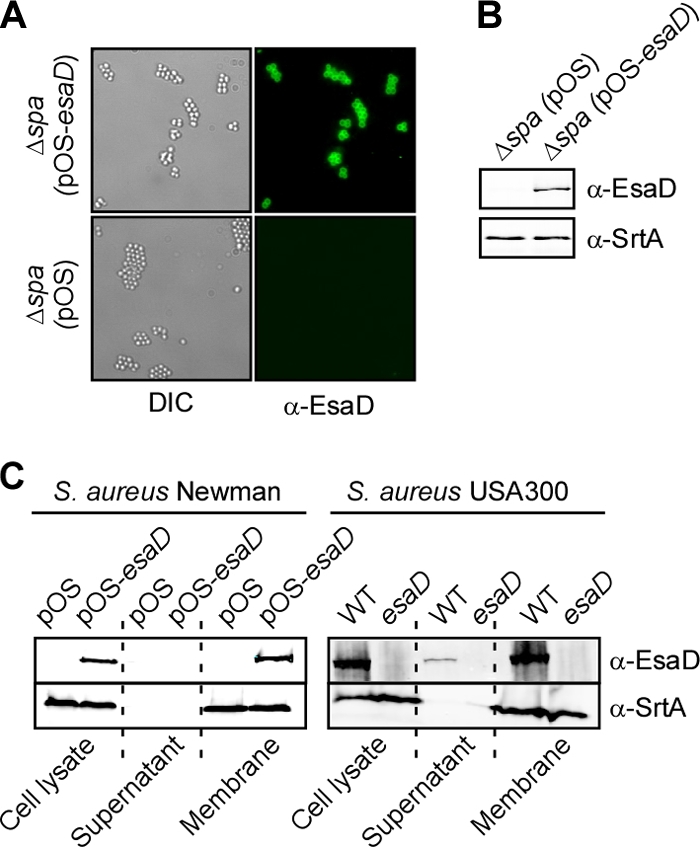
EsaD is displayed on the staphylococcal surface and anchored in the membrane. (A) An S. aureus Newman variant lacking protein A (Δspa) was transformed with plasmid vector pOS or pOS-esaD, and bacteria were viewed by differential interference contrast or fluorescence microscopy using EsaD-specific antibodies, followed by goat anti-rabbit secondary antibodies conjugated to Alexa Fluor 647 (green). Color and differential interference contrast images were acquired sequentially with separate laser lines and merged using ImageJ software. (B) Whole-culture lysates of staphylococcal strains in panel A were examined for the production of EsaD as described in the legend to Fig. 2A. (C) Subcellular fractionation of S. aureus Newman or USA300 cultures was achieved by growing cells as described in the legend to Fig. 2A. Cultures were centrifuged, and staphylococci were washed in buffer and treated with lysostaphin. Samples were split to examine proteins in the total bacterial lysate or in sedimented membrane and cytoplasm (supernatant) following ultracentrifugation at 4°C and 100,000 × g for 40 min. Left, S. aureus Newman carrying either pOS or pOS-esaD. Right, S. aureus USA300 wild-type (WT) and esaD mutant cultures. Proteins in all fractions were precipitated with methanol-chloroform, separated by SDS-PAGE, and detected by immunoblotting with specific antibodies (anti-EsaD antibodies and anti-SrtA antibodies as a control for a membrane protein).
EsaD fractionates with staphylococcal membranes.
Membrane proteins of S. aureus, for example, sortase A, are generally not accessible to antibodies on the surface of these bacteria. We wondered whether the surface display of EsaD requires the posttranslational processing of this polypeptide, for example, cleavage of the transmembrane segment so that the N-terminal domain of EsaD may be transported to the surface. To examine this, S. aureus Newman, as well as USA300, cells were sedimented by centrifugation, peptidoglycan was removed with lysostaphin, and the resulting protoplasts were lysed in hypotonic buffer. Protoplast lysates were subjected to ultracentrifugation at 100,000 ×g, and membrane sediment was separated from soluble proteins in the supernatant. As a control, immunoblotting of fractionation samples demonstrated the sedimentation of sortase A with staphylococcal membranes (Fig. 3C). Immunoblotting of samples derived from S. aureus strain USA300 or Newman carrying either pesaD or the pOS1 vector control revealed that most of the EsaD products sedimented with staphylococcal membrane proteins. Further, immunoblotting with anti-EsaD antibodies failed to detect soluble EsaD products with altered mobility on SDS-PAGE. These results suggest that the surface display of EsaD is a unique feature of this membrane protein and is likely not due to the generation of soluble EsaD fragments.
EsaD is required for the efficient secretion of EsxA.
esaD expression is controlled by esaB and essB, two genes that are involved in regulating either the expression of specific Ess secretion substrates or the transport of EsxA, EsxB, and EsaC by this pathway. We therefore wondered whether EsaD is also involved in Ess secretion. To test this, we analyzed isogenic variants of S. aureus USA300 for the ability to express the EssB machine component or secrete EsxA into the extracellular medium. Staphylococcal cultures were grown to mid-log phase and then centrifuged to separate proteins secreted into the culture medium with the supernatant from proteins in the bacterial sediment. Following lysis of staphylococci with lysostaphin, proteins in both fractions were precipitated with a methanol-chloroform mixture (4:1), washed in acetone, and then analyzed by immunoblotting (Fig. 4A). As controls, S. aureus USA300 and its isogenic variants expressed equal amounts of the ribosomal protein L6, which was found only in bacterial cells and not in the culture supernatant. Further, the culture supernatant of all of the strains examined harbored equal amounts of α-hemolysin, a protein that is secreted via the canonical Sec pathway of staphylococci. Mutations in S. aureus USA300 esxA or esaD did not affect the expression of the EssB machine component (Fig. 4A). Nevertheless, bursa aurealis insertion in esaD reduced the secretion of EsxA, which accumulated in the cytoplasm of mutant staphylococci. This defect in EsxA secretion was restored by transformation of esaD mutant staphylococci with the plasmid encoding wild-type esaD (pesaD) (Fig. 4A and B).
FIG. 4.

S. aureus Ess secretion requires esaD expression. Wild-type S. aureus USA300 and isogenic esxA and esaD mutant variants, with or without the complementing pOS-esaD plasmid, were examined for the production and secretion of EsxA. Staphylococci were grown in TSB, and cultures were centrifuged to separate proteins in lysostaphin-lysed cells (cell lysate) from the extracellular medium. (A) Proteins in both extracts were separated by SDS-PAGE and detected by immunoblotting with specific antibodies (anti-EsaC antibodies, anti-L6 antibodies as a control for cytoplasmic proteins, and anti-HLA antibodies as a control for a secreted protein). (B) Quantification of EsxA secretion in medium fractions was performed using a Li-Cor Biosystems Odyssey imager. Representative data of four independent experiments are shown.
Virulence defects of esaD mutant staphylococci.
Previous experiments suggested that Ess secreted products EsxA, EsxB, and EsaC are important for S. aureus Newman to replicate in organ tissues of infected mice (7, 8). Further, Ess secretion is also required for the ability of staphylococci to establish abscess lesions wherein this pathogen can persist and evade host immune responses. To ascertain the contribution of esaD to staphylococcal pathogenesis, cohorts of 10 BALB/c mice (6-week-old female animals) were infected by retro-orbital injection of 1 × 107 CFU S. aureus Newman or its isogenic esaD mutant into the bloodstream. At 5 or 15 days following infection, animals in each cohort were euthanized and necropsied to remove both kidneys. One kidney from each animal was homogenized and spread on agar plates. Following incubation and enumeration of staphylococcal colonies, the bacterial load was calculated as the number of CFU per organ (Fig. 5A). As expected, the average load of S. aureus Newman in kidney tissues was 6.8 log10 CFU on day 5 and 6.3 log10 CFU on day 15. The average load of the isogenic esaD mutant was reduced by 1.5 log10 CFU on day 5 (P < 0.02) and 2.3 log10 CFU on day 15 (P < 0.01), revealing defects in the ability of the esaD variants to replicate and persist in mouse organ tissues. This ability was restored to wild-type levels when esaD mutants were transformed with a plasmid carrying the wild-type esaD allele (pOS-esaD) but not with the plasmid vector alone (pOS) (Fig. 5C).
FIG. 5.

esaD mutants exhibit a virulence defect. (A) Staphylococcal replication and persistence in kidney tissue were measured at days 5 and 15 following retro-orbital infection with 107 CFU of wild-type S. aureus strain Newman or its isogenic variant lacking esaD. Kidneys were removed from infected mice during necropsy; tissue was homogenized and plated on agar medium for colony formation and enumeration. (B) Survival rates of animals infected with 1 × 107 CFU of wild-type S. aureus strain Newman or the esaD mutant were plotted as Kaplan-Meier curves. The data represent the average of three experiments. (C) Complementation studies for the esaD mutant in the animal model. Staphylococcal replication and persistence in kidney tissue were recorded at day 5 following retro-orbital infection as shown in panel A. Cells (107 CFU) of wild-type S. aureus strain Newman carrying the control vector (pOS) or its isogenic variant lacking esaD carrying either the control vector pOS or the complementing vector pOS-esaD were used for this study.
Following necropsy, the kidneys of infected mice were analyzed for the presence or absence of surface abscess lesions (Fig. 6). One of the two kidneys was fixed in formalin, embedded in paraffin, thin sectioned (four sagittal sections 200 μm apart), and tissue stained with hematoxylin-eosin. Renal tissue slides were analyzed by microscopy for immune cell infiltrates and abscess formation, which allowed us to assess the number of internal lesions on four slides per kidney. S. aureus Newman infection caused an average of 3.8 surface abscesses per kidney (Fig. 6A and B), as well as an average of 2.2 internal abscesses (Fig. 6C and D). By comparison, esaD mutant staphylococci produced an average of 1.8 surface abscesses (Fig. 6A and B; P = 0.03, wild type versus esaD mutant) and 0.7 internal abscess per infected organ (Fig. 6C and D; P = 0.002, wild type versus esaD mutant).
FIG. 6.

EsaD is required for abscess formation and staphylococcal persistence in host tissues. Kidneys of BALB/c mice (cohorts of 10 animals) that had been infected with S. aureus Newman or the isogenic esaD mutant were removed during necropsy of animals on day 5 following inoculation. (A) Photographs of whole kidneys showing surface abscesses in animals infected with S. aureus Newman. (B) Surface abscesses were enumerated by visual examination of organs and recorded as the average number of surface abscesses per kidney. (C) Kidneys were fixed in formalin, embedded, thin sectioned, and stained with hematoxylin and eosin. Histopathology images were acquired by light microscopy at ×12.5 (top frames) and ×200 (lower frames). (D) Enumeration of internal abscesses is reported as the average number of internal abscesses per kidney.
Although considered sublethal, an intravenous injection of 1 × 107 staphylococci leads to some mortality in 6-week-old BALB/c mice. We noticed that infection with the isogenic esaD mutant does not cause mortality, unlike infection with wild-type strain Newman. To assess the significance of this finding, we examined animal survival with Kaplan-Meier plots (Fig. 5B) for three independent experiments and found a significant difference for mice challenged with either wild-type or esaD mutant staphylococci (P = 0.037). Taken together, these data provide strong support for the role of EsaD and the Ess secretion pathway during S. aureus infection of mice.
DISCUSSION
In mice, as in humans, S. aureus causes infections that persist with reiterative cycles of bacterial deposition in multiple organ tissues, abscess formation, and maturation of purulent lesions. Abscesses rupture, thereby permitting staphylococcal dissemination via lymph and blood circulation and the formation of new infectious lesions. The establishment of abscesses, the pathological-anatomical substrates of staphylococcal disease, can be thought of as a developmental program with four discernible stages (9). Following the entry of S. aureus into the bloodstream, the microbes escape innate immune mechanisms and are disseminated via the vasculature to peripheral organ tissues (stage I). Once within organ tissue, staphylococci attract massive infiltrates of PMNs, as well as other immune cells (stage II). The infectious lesions are transformed into a unique structure enabling staphylococcal abscess communities enclosed by fibrin deposits (eosinophilic pseudocapsule) to replicate at the center, while a surrounding mantle of PMNs, as well as other immune cells, organized into zones of apoptotic and live cells, generates the purulent lesions (stage III). Eventually, these lesions erupt on the surface of organ epithelia and enable staphylococcal dissemination (stage IV).
We have previously shown that the S. aureus Ess (type VII-like) secretion pathway is required for the establishment of abscesses (7, 8). In agreement with these reports, esaD mutants with defects in EsxA secretion do survive in mouse blood at rates similar to those of their wild-type parent (stage I) (data not shown). However, esaD mutants are less able to seed abscesses or replicate in these lesions than their wild-type parent is, suggesting that EsaD is required during stages II and III of this developmental program.
EsaD is located in the staphylococcal membrane and may contribute to the selection of secretion substrates and/or interact with the Ess secretion machine. The production of EsaD is regulated by the ess gene cluster, which responds to environmental conditions in particular elements of the infected host such as serum proteins (Y.-H. Chen, unpublished data). Further, both esaB and essB contribute to the regulation of esaD. Although the molecular underpinnings of this regulation are not yet known, current experimental evidence is in agreement with the possibility that the synthesis of the Ess machine and substrate components may be ordered in a manner that involves the esaB essB regulatory arm of this pathway. A similar trait has been reported for EsaC (7). In S. aureus Newman, very little EsaC is expressed; however, expression is induced by the addition of serum to growth medium or by a genetic lesion in esaB (7). Highly virulent strains such as S. aureus USA300 synthesize greater amounts of EsaC and EsaD than the less virulent isolates do (7) (Fig. 2). Together, these findings indicate that EsaD is part of the Ess pathway and that the polypeptide supports the secretion of EsxA and is thereby involved in the pathogenesis of staphylococcal infections.
Bioinformatic searches revealed that EsaD belongs to the cluster of orthologous group of proteins COG5444 without known function (25). These proteins are typically associated with esx- or ess-like genes in Firmicutes (Fig. 1A). Some COG5444 proteins carry additional domains; for example, Listeria monocytogenes lmo0066 carries a VIP2 domain otherwise found in the family of toxins that ADP-ribosylate actin. In bacilli, the YeeE and YeeF proteins (COG5444 cluster) are annotated as uncharacterized transposase_30; however, an alignment of COG5444 and transposase_30 failed to identify overlaps for the transposase catalytic residues. The YeeE and YeeF proteins carry a partial COG5444 domain followed by the sequence for a domain of unknown function named DUF600. Of note, genes located downstream of S. aureus esaD also harbor DUF600 domains. Depending on the strain examined, staphylococci carry 10 to 12 tandem repeats of DUF600 genes. The significance of a Rosetta stone association between the esaD and DUF600 genes in bacilli is still unclear (26). Intriguingly, mycobacterial chromosomes carry tandem repeats of PE and PPE repeat genes whose products are secreted via the Esx-5 pathway (2). Thus, it seems plausible that DUF600 polypeptides could also represent S. aureus substrates for the Ess pathway, analogously to PE/PPE in M. tuberculosis. Finally, BLAST searches identified homology between the last 140 amino acids of EsaD and the C-terminal end of filamentous-hemagglutinin-type outer membrane proteins in Gram-negative bacteria (expect = 1e-26; identities = 59/145 = 40% between NM_0228 and HSM_1651, for example). These very large proteins, often exceeding 2,500 amino acids, are part of a two-partner secretion system that promotes the transport of polypeptides across the outer membrane (19, 20). It is interesting that EsaD shares some sequence similarity with these proteins as both represent components of Sec-independent secretion systems.
While mycobacterial ESX (type VII) secretion systems share little in sequence homology with the Ess pathway of S. aureus, both organisms reside in abscesses and granulomas in a manner that appears to prevent the acquisition of protective immunity in infected hosts (9, 34). Thus, these secretion systems may have evolved to transport their immunosuppressive factors to unique locations during infection, thereby enabling the establishment of characteristic infectious lesions and disease pathologies.
Acknowledgments
We thank Andrea DeDent and Hwan Keun Kim for help with the microscopy and antibody purification experiments, Ann Elmer for technical help, Olaf Schneewind for careful reading of the manuscript, and members of the Schneewind and Missiakas laboratories for discussions.
Mark Anderson and Emily K. Butler were supported by the Biodefense Training Grant in Host-Pathogen Interactions T32 AI065382 at the University of Chicago. This work was supported by the NIAID Infectious Diseases Branch (AI 75258 to D.M.). Dominique Missiakas acknowledges membership in the Region V Great Lakes Regional Center of Excellence in Biodefense and Emerging Infectious Diseases Consortium and technical support from the GLRCE Animal Research and Immunology Core (GLRCE, NIAID award U54-AI-057153).
Footnotes
Published ahead of print on 28 January 2011.
REFERENCES
- 1.Abdallah, A. M., et al. 2007. Type VII secretion—mycobacteria show the way. Nat. Rev. Microbiol. 5:883-891. [DOI] [PubMed] [Google Scholar]
- 2.Abdallah, A. M., et al. 2009. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol. Microbiol. 73:329-340. [DOI] [PubMed] [Google Scholar]
- 3.Bae, T., et al. 2004. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc. Natl. Acad. Sci. U. S. A. 101:12312-12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bae, T., and O. Schneewind. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58-63. [DOI] [PubMed] [Google Scholar]
- 4a.Bae, T., E. M. Glass, O. Schneewind, and D. Missiakas. 2008. Generating a collection of insertion mutations in the Staphylococcus aureus genome using bursa aurealis. Methods Mol. Biol. 416:103-116. [DOI] [PubMed] [Google Scholar]
- 5.Bitter, W., et al. 2009. Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog. 5:e1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bubeck Wardenburg, J., W. A. Williams, and D. Missiakas. 2006. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 103:13831-13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burts, M. L., A. C. DeDent, and D. M. Missiakas. 2008. EsaC substrate for the ESAT-6 secretion pathway and its role in persistent infections of Staphylococcus aureus. Mol. Microbiol. 69:736-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burts, M. L., W. A. Williams, K. DeBord, and D. M. Missiakas. 2005. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc. Natl. Acad. Sci. U. S. A. 102:1169-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng, A. G., et al. 2009. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 23:3393-3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cosma, C. L., D. R. Sherman, and L. Ramakrishnan. 2003. The secret lives of the pathogenic mycobacteria. Annu. Rev. Microbiol. 57:641-676. [DOI] [PubMed] [Google Scholar]
- 11.Davis, J. M., and L. Ramakrishnan. 2009. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136:37-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeDent, A. C., M. McAdow, and O. Schneewind. 2007. Distribution of protein A on the surface of Staphylococcus aureus. J. Bacteriol. 189:4473-4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diep, B. A., H. A. Carleton, R. F. Chang, G. F. Sensabaugh, and F. Perdreau-Remington. 2006. Roles of 34 virulence genes in the evolution of hospital- and community-associated strains of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 193:1495-1503. [DOI] [PubMed] [Google Scholar]
- 14.DiGiuseppe Champion, P. A., and J. S. Cox. 2007. Protein secretion systems in mycobacteria. Cell. Microbiol. 9:1376-1384. [DOI] [PubMed] [Google Scholar]
- 15.Duthie, E. S., and L. L. Lorenz. 1952. Staphylococcal coagulase: mode of action and antigenicity. J. Gen. Microbiol. 6:95-107. [DOI] [PubMed] [Google Scholar]
- 16.Garufi, G., E. Butler, and D. Missiakas. 2008. ESAT-6-like protein secretion in Bacillus anthracis. J. Bacteriol. 190:7004-7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hawkridge, A. 2009. Clinical studies of TB vaccines. Hum. Vaccin. 5:773-776. [DOI] [PubMed] [Google Scholar]
- 18.Hsu, T., et al. 2003. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. U. S. A. 100:12420-12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacob-Dubuisson, F., C. Locht, and R. Antoine. 2001. Two-partner secretion in Gram-negative bacteria: a thrifty, specific pathway for large virulence proteins. Mol. Microbiol. 40:306-313. [DOI] [PubMed] [Google Scholar]
- 20.Jacob-Dubuisson, F., V. Villeret, B. Clantin, A. S. Delattre, and N. Saint. 2009. First structural insights into the TpsB/Omp85 superfamily. Biol. Chem. 390:675-684. [DOI] [PubMed] [Google Scholar]
- 21.Kim, H. K., A. G. Cheng, H. Y. Kim, D. M. Missiakas, and O. Schneewind. 2010. Nontoxigenic protein A vaccine for methicillin-resistant Staphylococcus aureus infections in mice. J. Exp. Med. 207:1863-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowy, F. D. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520-532. [DOI] [PubMed] [Google Scholar]
- 23.Mahairas, G. G., P. J. Sabo, M. J. Hickey, D. C. Singh, and C. K. Stover. 1996. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J. Bacteriol. 178:1274-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Majlessi, L., et al. 2005. Influence of ESAT-6 secretion system 1 (RD1) of Mycobacterium tuberculosis on the interaction between mycobacteria and the host immune system. J. Immunol. 174:3570-3579. [DOI] [PubMed] [Google Scholar]
- 25.Marchler-Bauer, A., et al. 2009. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 37:D205-D210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcotte, C. J., and E. M. Marcotte. 2002. Predicting functional linkages from gene fusions with confidence. Appl. Bioinformatics 1:93-100. [PubMed] [Google Scholar]
- 27.Pallen, M. J. 2002. The ESAT-6/WXG100 superfamily—and a new Gram-positive secretion system? Trends Microbiol. 10:209-212. [DOI] [PubMed] [Google Scholar]
- 28.Pym, A. S., P. Brodin, R. Brosch, M. Huerre, and S. T. Cole. 2002. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol. Microbiol. 46:709-717. [DOI] [PubMed] [Google Scholar]
- 29.Pym, A. S., et al. 2003. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat. Med. 9:533-539. [DOI] [PubMed] [Google Scholar]
- 30.Renshaw, P. S., et al. 2005. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J. 24:2491-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stanley, S. A., J. E. Johndrow, P. Manzanillo, and J. S. Cox. 2007. The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 178:3143-3152. [DOI] [PubMed] [Google Scholar]
- 32.Stanley, S. A., S. Raghavan, W. W. Hwang, and J. S. Cox. 2003. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc. Natl. Acad. Sci. U. S. A. 100:13001-13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tam, C., B. Collinet, G. Lau, S. Raina, and D. Missiakas. 2002. Interaction of the conserved region 4.2 of sigma(E) with the RseA anti-sigma factor. J. Biol. Chem. 277:27282-27287. [DOI] [PubMed] [Google Scholar]
- 34.Volkman, H. E., et al. 2010. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science 327:466-469. [DOI] [PMC free article] [PubMed] [Google Scholar]