

Abstract
Vaccines against Neisseria meningitidis group C are based on its α-2,9-linked polysialic acid capsular polysaccharide. This polysialic acid expressed on the surface of N. meningitidis and in the absence of specific antibody serves to evade host defense mechanisms. The polysialyltransferase (PST) that forms the group C polysialic acid (NmC PST) is located in the cytoplasmic membrane. Until recently, detailed characterization of bacterial polysialyltransferases has been hampered by a lack of availability of soluble enzyme preparations. We have constructed chimeras of the group C polysialyltransferase that catalyzes the formation α-2,9-polysialic acid as a soluble enzyme. We used site-directed mutagenesis to determine the region of the enzyme necessary for synthesis of the α-2,9 linkage. A chimera of NmB and NmC PSTs containing only amino acids 1 to 107 of the NmB polysialyltransferase catalyzed the synthesis of α-2,8-polysialic acid. The NmC polysialyltransferase requires an exogenous acceptor for catalytic activity. While it requires a minimum of a disialylated oligosaccharide to catalyze transfer, it can form high-molecular-weight α-2,9-polysialic acid in a nonprocessive fashion when initiated with an α-2,8-polysialic acid acceptor. De novo synthesis in vivo requires an endogenous acceptor. We attempted to reconstitute de novo activity of the soluble group C polysialyltransferase with membrane components. We found that an acapsular mutant with a defect in the polysialyltransferase produces outer membrane vesicles containing an acceptor for the α-2,9-polysialyltransferase. This acceptor is an amphipathic molecule and can be elongated to produce polysialic acid that is reactive with group C-specific antibody.
Neisseria meningitidis groups B and C are the most common causes of meningococcal meningitis in adolescents and adults in Canada, Europe, and the United States. In the United States, 95% to 97% of cases of meningococcal disease are sporadic; however, since 1991, the frequency of localized outbreaks has increased (12, 13). Most of these outbreaks have been caused by serogroup C. Several vaccines based on the meningococcal capsular polysaccharides have been licensed. A tetravalent vaccine consisting of meningococcal groups A, C, Y, and W-135 has also been licensed. Subsequently, a conjugate vaccine of the same serogroup polysaccharide was licensed in the United States (3, 28). In addition there are two meningococcal group C conjugate vaccines licensed in Europe. These meningococcal capsular polysaccharides are polysialic acids. The N. meningitidis group B polysaccharide is an α-2,8-linked polysialic acid, while the group C polysaccharide is an α-2,9-linked polysialic acid (see Fig. S1 in the supplemental material).
The gene clusters responsible for the synthesis of these polysialic acids have been identified and characterized (5, 7, 10, 20, 24, 27). The glycosyltransferase genes of meningococcal gene clusters have been useful targets for the development of epidemiological tools. For instance, PCR assays based on the polysialyltransferase (PST) genes are routinely used for the detection and identification of serogroups (15). The polysialic acids are polymerized by a single polysialyltransferase in the case of each serogroup. The group B polysialyltransferase (NmB PST) is encoded by synD, and the group C PST is encoded by synE. Several investigators have cloned and begun to characterize the group B polysialyltransferase (9, 34). Both enzymes are membrane associated and are presumed to transfer sialic acid from the donor, CMP-NeuNAc, to the growing chain on the internal face of the cytoplasmic membrane of the bacterium (19, 21, 33).
Much of the work on the biosynthesis of polysialic acid capsules has been done on the Escherichia coli K1 and K92 polysialyltransferases (21, 22). Like the meningococcal enzymes, the E. coli polysialyltransferases are associated with the cytoplasmic membrane and transfer sialic acid to the nonreducing end of the acceptor chain. Neither E. coli nor meningococcus can initiate synthesis de novo; they require an oligosialic acid or an endogenous acceptor (9, 17). The endogenous acceptor for these enzymes has yet to be identified. The meningococcal polysialyltransferase has a longer carboxyl-terminal sequence than the E. coli enzymes. The bacterial polysialyltransferases do not share motifs or sequence homologies with other sialyltransferases. E. coli and meningococcal polysialyltransferases belong to the CAZy glycosyltransferase family GT-38 (6).
Until recently the characterization of the bacterial polysialyltransferases has been limited to studies with membrane fragments of cells harboring the polysialyltransferase genes or in vivo experiments (17, 23, 27, 29, 30). Soluble enzyme was not available for structure-function studies due to resistance of the membrane-associated enzymes to extraction in active form with detergents (17). The expression of some soluble membrane proteins has been achieved without detergents by fusion to proteins that promote solubilization. Recently, Freiberger et al. and Willis et al. (9, 34) demonstrated the ability to produce soluble group B polysialyltransferase as a chimera of synD and the malE or nusA gene.
In our study, we constructed several soluble chimeras of the group C polysialyltransferase. The chimeric enzymes were expressed in E. coli and purified. The activity of the purified enzymes clearly demonstrated that only a single protein is required for elongation of polysialic acid acceptors.
MATERIALS AND METHODS
DNA manipulations.
Recombinant DNA techniques were carried out using standard methods and commercially available materials. PCR amplifications were performed using either Taq Ready-to-Go beads (GE Healthcare) or a proofreading DNA polymerase, Phusion HF, purchased from New England BioLabs. Transformants were screened by either restriction digestion of plasmid minipreps or directly by PCR. Freshly picked colonies for PCR screening were boiled in diethyl pyrocarbonate water for 5 min and centrifuged. The supernatant was mixed with 1 μl of appropriate primers and Ready-to-Go PCR beads and then amplified in a thermocycler and analyzed on agarose gels.
Construction of expression plasmids.
Plasmids and primers used for our constructions are described in Table 1.
TABLE 1.
Plasmid constructs
| Plasmid | Gene(s) | Primer | Sequence |
|---|---|---|---|
| pWV234 | synE-nusA | CPST LIC for | GACGACGACAAGATGTTGCAGAAAATAAGA |
| CPST LIC rev | GAGGAGAAGCCCGGTTTAATTGGTTACAAAGGC | ||
| pWV235 | synE-SUMO | CPST-SUMO Met for | ATGTTGCAGAAAATAAGAAAA |
| CPST-SUMO rev | TTATTGGTTACAAAGGCTATA | ||
| pWV241 | synE | NmCPST fwd pENTR TOPO | CACCATGTTGCAGAAAATAAGAAAAGCT |
| NmCPST BamHI rev | TTTGGATCCTTATTGGTTACAAAGGCTATATTT | ||
| pWV237 | synD | NmB directional for | CACCATGCTAAAGAAAATAAAAAAAGC |
| NmB rev HindIII | AAGCTTTTATCTATCTCTACCAATTCT | ||
| pWV238 | synD | NmB directional for | CACCATGCTAAAGAAAATAAAAAAAGC |
| NmB rev HindIII | AAGCTTTTATCTATCTCTACCAATTCT | ||
| pWV245 | synD-synE | DSnmB FOR | GAGGAAAACCTGTATTTTCAGGGCT |
| DSnmB REV | TAAATGAGTCGTAATATTCTTTTTTTTTGC | ||
| pWV246 | synE-Halo tag | NmC PST Flexivector forward | AGGAGCGATCGCCATGTTGCAGAAAATAAGAAA |
| NmC PST Flexivector reverse | AACTGTTTAAACTTATTGGTTACAAAGGCTAT | ||
| pWV248 | synE | NmCPST fwd pENTR TOPO | CACCATGTTGCAGAAAATAAGAAAAGCT |
| MengC PST rev primer | TTATTGGTTACAAAGGC | ||
| pWV249 | synD-synE | B322 primer | GCGGGTCTCTTTTTTAGGAGTTATATTATT |
| C322 primer | GCGGGTCTCAAAAAATTCCTATATCCAGC | ||
| 151 primer C | GCGGGTCTCCATTCTACGGAAAACCTGTA | ||
| 151 primer D | CCCAGTGCTGCAATGATACCGCGAGA | ||
| pWV250 | synD-synE | QCL NmC322 sense primer | CCTAATAATATAACTCCTAAAAAATTCCTATATATCCAGCGTGGATATAAGA |
| QCL NmC322 antisense primer | TCTTATATCCACGCTGGATATATAGGAATTTTTTAGGAGTTATATTATTAGG |
pWV234 NusA-PST.
The pWV234 NusA-PST plasmid encodes an amino-terminal NusA fusion with the NmC PST (synE). The synE gene was amplified from chromosomal DNA isolated from N. meningitidis group C strain P2181 with the forward primer CPST LIC FOR and the reverse primer CPST LIC REV and Taq Ready-to-Go PCR beads. The amplified fragment was purified and ligated into the expression vector pET44 Ek/LIC containing the nusA gene (Novagen) as described in the manufacturer's instructions.
pWV235 SUMO-PST.
For WV235 SUMO-PST, the synE gene was amplified as described above by using plasmid pWV234 as template with forward primer CPST-SUMO Met FOR and reverse primer CPST-SUMO REV. The amplified fragment was ligated in the pETSUMO TOPO plasmid as recommended by Invitrogen.
pWV237.
For pWV237, N. meningitidis group B synD was amplified from chromosomal DNA isolated from strain H355 using primers NmB rev HindIII and NmB directional for. The resulting fragment was ligated into the pET151 TOPO vector from Invitrogen.
pWV241.
For pWV241, the Gateway system of Invitrogen was used to construct a chimera of the maltose binding protein (MBP) gene, malE, and the NmC PST gene, synE. The synE gene was introduced into an entry vector as follows. The synE gene was amplified from pWV234 with the forward primer NmC PST FWD pENTR TOPO, the reverse primer NmC PST BamHI REV, and the thermostable polymerase reagent Phusion HF master mix from Finnzymes. The purified fragment was ligated into the Invitrogen pENTR/TEV/D-TOPO plasmid as recommended by the manufacturer.
pWN602 malE Gateway destination vector.
The plasmid pN-WWW was a gift of Warrren Wakarchuk, National Research Council, Canada, and is based on pCWOri+ from the same source. pN-WWW was digested with NdeI and HindIII and converted to blunt ends with mung bean nuclease and calf intestine alkaline phosphatase. The Gateway cassette B was inserted into the blunt end site as described by Invitrogen. The resulting plasmid, pWN602, contained the Gateway B cassette at the 3′ end of the malE gene.
pWV243 MBP-PST.
For pWV243 MBP-PST, the syn E (NmC PST) gene in pWV241 was used as the insert gene for the LR clonase (Invitrogen) reaction. pWN602 was used as the destination vector for pWV243. The protein expressed by this plasmid has an MBP at the amino-terminal end of NmC PST separated by a tobacco etch virus (TEV) protease cleavage site.
pWV246.
For pWV246, the HaloTag fusion plasmid was constructed by inserting the synE gene into the expression plasmid pFN22K HaloTag Flexivector (Promega). The synE gene was amplified from the plasmid pWV243 with the Phusion Hi-Fidelity enzyme mix (New England BioLabs) using the primers NmC PST Flexivector forward and NmC PST Flexivector reverse.
pWV239.
For pWV239, the NmB PST gene, synD, was cloned into the malE-containing Gateway vector pWN602 in a fashion similar to that described for pWV243 above. The synD gene was first amplified using primers NmB directional for and NmB rev HindIII and then inserted in pENTR/TEV-TOPO. The resulting plasmid, designated pWV238, was combined with pWN602 in an in vitro recombination reaction mixture to yield pWV239. The resulting plasmid expresses a MalE-PST chimera with MalE and the amino terminus of NmCPST separated by a TEV protease cleavage site.
pWV248.
For pWV248, the NmC PST synE gene was amplified with pWV234 as PCR template and NmCPST fwd pENTR TOPO and MengC PST rev as primers. The resulting NmC PST synE was ligated into pET151TOPO as described by the manufacturer.
Mutagenesis.
The domain swap mutants of NmB and NmC PST were constructed by two methods. The plasmid pWV245 was constructed with the QuikChange II XL kit (Stratagene). The sequence encoding the amino terminus of SynD (nucleotides 1 to 439) was amplified using the primers DSnmB FOR and DSnmB REV with the NmB PST plasmid pWV239 as a template. The resulting fragment was purified and used as a mutagenic primer with the QuikChange II XL kit to mutate the NmC PST expression plasmid pWV243. This procedure requires significant areas of homology in the regions to be swapped. Because of this requirement for homology, the QuikChange method was not suitable for swapping the desired shorter region of synD. A modification of the method of Stemmer and Morris (25) was used to construct the other synD-synE chimera, although inverse PCR was not performed (4). Briefly, nucleotides 1 to 322 were amplified with a primer that included nucleotides 322 of synD in a Bsa1 cleavage site and a pET151 vector-based primer, including its Bsa1 site from pWV237. Similarly, nucleotides 322 to 1479 of synE were amplified with a primer that included nucleotides 322 of synE in a Bsa1 cleavage site and a synE primer that included its Bsa1 site from pWV248. The fragments were isolated, cut with Bsa1, and ligated together. The resulting hybrid fragment was ligated into the Bsa1 sites of pWV248 to yield plasmid pWV249. Plasmid pWV249 was sequenced and found to have a deletion error in the region of one of the primers used in its construction. The error was corrected using a QuikChange Lightning mutagenesis kit to give pWV250. The sequences of all plasmids were confirmed by sequencing in the CBER Core Facility.
The plasmids pWV245 and pWV250 were transformed into E. coli BL21(DE3) cells, and the polysialyltransferase activities of cell lysates were determined as described below.
Protein analysis.
The protein concentration was determined using the Bio-Rad dye binding reagent. Purified enzyme fractions were analyzed by SDS-PAGE (Invitrogen) and stained with Coomassie blue. HaloTag proteins were labeled as described above, with a tetramethylrhodamine (TAMR)-HaloTag, and analyzed by SDS-PAGE with fluorescence detection.
Polysialyltransferase expression and purification.
The NusA polysialyltransferase chimera was expressed by transformation of plasmid pWV234 into BL21(DE3) Star and preparation of an overnight inoculum in 25 ml of LB containing 200 μg/ml ampicillin. The culture in LB broth (1.5 liters) containing 50 μg/ml ampicillin was inoculated and shaken at 30°C until the absorbance at 600 nm reached 1.2. The culture was induced by addition of 360 mg isopropyl-β-d-thiogalactopyranoside (IPTG) and shaking at 30°C for an additional 2 h 45 min. The cells were harvested by centrifugation at 10,000 × g for 15 min, at 4 to 10°C, and stored at −80°C. Frozen cells were thawed and suspended in 10 ml 50 mM Tris, 10 mM MgCl2 in which was dissolved one EDTA-free protease inhibitor tablet (pH 7.5; Roche). The cell suspension was lysed in a French pressure cell and centrifuged for 12 min at 10,000 × g, at 4 to 10°C. Membranes were separated from the cytosolic fraction by ultracentrifugation for 1 h at 114,000 × g at 4°C. The membrane fraction was resuspended in 1.5 ml of cryo buffer (31). The supernatant fraction was adjusted to 10% glycerol and stored at −80°C.
Polysialyltransferase constructs with a His tag epitope were purified from the supernatant by tumbling overnight at 2 to 8°C with 4 ml of Ni-nitrilotriacetic acid-agarose resin (Qiagen) in 50 mM Tris, 10 mM imidazole, pH 7.5. The suspension was poured into a column and washed at 0.5 ml/min with 50 mM Tris, 10 mM imidazole, 200 mM NaCl, pH 7.5. The protein was eluted with the same buffer containing 60 mM imidazole and then 250 mM imidazole, collecting 3-ml fractions. Fractions were assayed for activity using the paper chromatography assay described below. Peak activity fractions were combined and concentrated using a Centriplus YM10 apparatus from 4 ml to 1 ml. The concentrated enzymes was adjusted to 10% glycerol and stored at −80°C.
The SUMO-polysialyltransferase chimeric enzyme was purified from cells harboring the plasmid pWV235 by a similar procedure, except the culture was grown in LB with kanamycin.
The MalE-NmC polysialyltransferase was purified as follows. Plasmid pWV243 was transformed into AD202 cells, and the fresh transformants from the entire plate were suspended in a few milliliters of medium and used to inoculate 1.5 liters of rich broth (consisting of 15 g Bacto tryptone, 7.5 g yeast extract, 7.5 g NaCl) plus 3 g glucose and 100 μg/ml ampicillin (pH 7.2). Cells were shaken at 30°C for 4 h and then induced for 3 h with 400 mg IPTG. A cytosolic fraction was prepared as described above, and the enzyme content was purified on a 12-ml amylose resin (New England BioLabs) column. The amylose column was washed with 50 mM Tris, 200 mM NaCl, 1 mM EDTA, pH 7.4, and the protein was eluted with 10 mM maltose in the same buffer.
Preparation of fluorescent-tagged polysialyltransferase.
HaloTag-NmC PST was prepared by transforming pWV246 in BL21(DE3) STAR cells. The fresh transformants were used to inoculate 2 × 1.5 liters of LB-kanamycin and grown to an A600 of 0.7, then induced with 1 mM IPTG for 3 h. The cells were harvested and frozen in 25 ml 50 mM Tris, 25 mM MgCl2 (pH 8) overnight. The cell suspension was lysed in a French pressure cell, and cellular debris was removed by centrifugation at 10,000 × g for 15 min.
The lysate (25 ml) was labeled with 250 μl of TAMR-HaloTag substrate (10 to 20 μg/ml) as described by Promega. The membranes were separated from the soluble fraction by ultracentrifugation at 114,000 × g. The membrane pellet was resuspended in 4 ml cryoprotective buffer.
Gel filtration of HaloTag-NmC PST.
The soluble fraction described above was adjusted to 25% saturated ammonium sulfate and stirred at 4°C for 1 h. The pellet was collected by centrifuging at 15,000 rpm for 30 min and dissolved in 3 ml of 50 mM Tris, 25 mM MgCl2 (pH 8.0). Most of the enzyme activity in the soluble fraction was precipitated at 25% (NH4)2SO4 saturation. The enzyme fraction was analyzed on a Superose 12 fast-protein liquid chromatography (FPLC) column (GE) on a Dionex Summit high-performance liquid chromatograph (HPLC) equipped with a Hitachi model L-7485 fluorescence detector. TAMR-labeled fractions were detected at an excitation wavelength of 555 nm and emission at 585 nm. Peak fractions were concentrated on Nanosep microconcentrators and assayed for polysialyltransferase activity as described previously (17).
Assay for polysialyltransferase.
Polysialyltransferase activity was measured using a paper chromatography assay described previously (17).
The linkage of the product of the chimera plasmids pWV245 and pWV250 was determined as follows. In a typical assay, the membrane fraction was incubated in a solution containing 22 mM Tris, 11 mM MgCl2, pH 8, 10 μg polysaccharide acceptor (NmC polysaccharide, colominic acid, or K92 polysaccharide) in 110 μl and 3 μl 100-μCi/ml 14C-labeled CMP-sialic acid (American Radiolabeled Chemicals). After a 1-h incubation, α-2,8-endoneuraminidase was added, and incubation at 37°C continued for 3 h, prior to analysis by paper chromatography. Control reaction mixtures were incubated for 3 h without endoneuraminidase.
NmC PST competition assay with unlabeled acceptors.
Lactosyl-boron dipyrromethene label (BODIPY) was sialylated with the bifunctional sialyltransferase CST II in a large-scale reaction volume (500 μl), and oligosialylated products were separated by anion exchange HPLC as described before (16). Product peaks were collected and desalted using Sep Pak chromatography (Waters). The extent of sialylation was confirmed by matrix-assisted laser desorption ionization-mass spectrometery (data not shown). Trisialylated lactosyl-BODIPY (2.5 μM) was incubated with NmC PST (30 μg/ml) in the presence of CMP-NeuNAc (5 mM) and MnCl (40 mM) in 200 mM sodium cacodylate buffer (pH 8.0) at 37°C for 40 min. For competition experiments, the reaction was run in the presence of sialic acid trimer, hexamer, or GD3 (Calbiochem) at a concentration of 5 μM. All reactions were quenched by the addition of ethanol to 25%. Products were dried by using a Speedvac, brought up to 50 μl with H2O, and subjected to HPLC analysis as previously described (18).
Preparation of outer membrane vesicles.
Outer membrane vesicles (OMV) were prepared from cultures of mutant N. meningitidis group B strains according to the procedure described by Frasch and Peppler (8). Cultures were grown overnight on tryptic soy broth supplemented with yeast extract as described by Arakere et al. (2). N. meningitidis strain M7 lacking the ability to synthesize sialic acid and a synD mutant lacking an active polysialyltransferase were obtained from David Stephens, Emory University (14).
Detection of polysialic acid formation by ELISA.
NmC PST (13 μl) was incubated with 10 μl OMV and CMP-NeuNAc (33, 330, 1,000, or 2,000 μM) in a total of 30 μl as described previously (17) for 1 h at 37°C. The assay mixture was then diluted to 400 μl with enzyme-linked immunosorbent assay (ELISA) coating buffer (2), which was then used to coat Immulon 1B plates (100 μl/well) at room temperature overnight. The plates were washed and then incubated with mouse monoclonal IgG antibody Mcl2075 (against OAc+) or Mcl2016 (against OAc−) group C polysaccharide at 1:400 dilution (kindly donated by Marjorie Shapiro, FDA) (11). Plates were developed with goat anti-mouse IgG-alkaline phosphatase conjugate (Sigma) diluted 1:2,000.
RESULTS AND DISCUSSION
Distribution and purification of PST activity.
The polysialyltransferases of Neisseria meningitidis groups B and C are encoded by synD and synE, respectively. Experiments by Freiberger et al. (9) demonstrated that a soluble and active group B α-2,8-polysialyltransferase could be expressed by some chimeras of synD. We prepared several chimeras of synE that were capable of expressing soluble N. meningitidis group C α-2,9-polysialyltransferase. As shown in Table 2, most of the chimeras that we constructed expressed group C α-2,9-polysialyltransferase activity equally in the membrane and supernatant fractions of cell lysates. The expression levels of the chimeric protein in the membrane and supernatant fractions were confirmed by Western blot analysis of the MalE-PST chimera (Fig. 1). Membrane and cytosolic fractions of pWV243 lysates were subjected to SDS-PAGE, transferred to nitrocellulose, and immunostained with anti-MalE antibody. Bands with a mass of 84 kDa were observed in both fractions. Although several chimeras of the NmC PST yielded a significant amount of enzyme activity in the soluble fraction, we obtained a high yield of purified enzyme with the maltose binding protein chimera. The maltose binding protein chimera was stable when stored at −80°C in 10% glycerol and was therefore used for most of the characterization experiments described below.
TABLE 2.
Distribution of polysialyltransferase activity
| Plasmid | Expression chimera | Activity (cpm/min/mg of protein) in: |
|
|---|---|---|---|
| Membranes | Supernatant | ||
| pWV234 | NusA-NmC | 1,800 | 1,868 |
| pWV243 | MalE-NmC | 2,277 | 2,669 |
| pWV235 | SUMO-NmC | 2,453 | 91 |
| pWV245 | HaloTag-NmC | 4,148 | 5,671 |
FIG. 1.
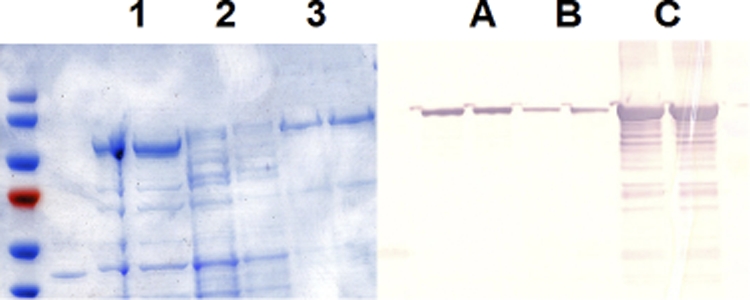
Distribution of the MalE-NmC PST chimera, after PAGE of purified MalE-NmC PST. The ultracentrifugation pellet (lane 1), supernatant of a cell lysate of AD202(pWV243) (lane 2), and the major protein fractions eluted from the amylase column (lane 3) were loaded onto a 4-to-20% polyacrylamide SDS-PAGE gel. For Western blot analysis another gel was loaded with membrane fraction (lane A), the ultracentrifuge supernatant fraction of the BL21(pWV243) lysate (lane B), and the amylose-purified MalE-NmC PST (lane C). The gel was transferred to nitrocellulose, and the immunoblot was developed with anti-MalE antibody.
Acceptor domain.
The NmC and NmB polysialyltransferases synthesize different polysialic acid homopolymers and share 75% sequence homology and 64% identity. The product of the NmC enzyme is α-2,9-polysialic acid, while that of NmB is the more common α-2,8 polymer. E. coli and N. meningitidis polysialyltransferases are only about 40% homologous, yet the E. coli K1 and N. meningitidis group B polysialyltransferases synthesize identical polysialic acids. This leads one to wonder which amino acid differences in the NmC and NmB polysialyltransferases are associated with the linkage differences in the polysaccharide products. Steenbergen et al. (22) showed that the region of the E. coli K1 and K92 enzymes responsible for the synthesis of different polysialic acid linkages is contained within the amino acid sequence of residues 1 to 100 of the E. coli polysialyltransferases. Similarly, we reasoned that we could localize the amino acids responsible for the link differences in the polysialic acid products by domain swapping of the group B and C polysialyltransferases. We used site-directed mutagenesis to determine the region of the Neisseria polysialyltransferases required to synthesize α-2,8 or α-2,9 linkages. Chimeras were constructed with nucleotides 1 to 439 of synD, nucleotides 440 to 1479 of synE, nucleotides 1 to 322 of synD, and nucleotides 323 to 1479 of synE (Fig. 2). The chimeric enzymes were expressed, and the linkage of their polysialic acid products was determined. The products of the reaction with CMP-[14C]NeuNAc were sensitive to the α-2,8-specific endoneuraminidase (1, 22), suggesting that the enzymes now had α-2,8 linkage specificity (Fig. 2). Both synE and the synD-synE chimera encoded enzymes that utilized α-2,8-polysialic acid, α-2,9-polysialic acid, and α-2,8 α-2,9 polysialic acids as acceptors. Both chimeras synthesized α-2,8-polysialic acid, suggesting that like the E. coli enzyme the amino acid sequence sufficient to determine the linkage of the product is contained within residues 1 to 107.
FIG. 2.

Domain swap of NmB PST and NmC PST. Chimeras were constructed with nucleotides 1 to 439 of synD, 440 to 1479 of synE (pWV250), 1 to 322 of synD, and 323 to 1479 of synE (pWV245) as described in Materials and Methods. The chimeric enzymes were expressed and assayed, and the linkages of their polysialic acid products were determined by the sensitivity of the enzymatic reaction products to the α-2,8-NeuNAc-specific endoneuraminidase as described in Materials and Methods.
Active molecular weight.
It has been proposed that bacterial glycosyltransferases responsible for synthesis of some capsular polysaccharides function as members of a complex (31, 33). Thus, we investigated the active molecular weight of NmC PST. The HaloTag-PST chimera can be fluorescently labeled with selective reagents (16), and it therefore provided a means of detecting the molecular species of polysialyltransferase present in a cell lysate without extensive purification. We prepared a crude ammonium sulfate fraction of fluorescently labeled HaloTag-PST for analysis by HPLC size exclusion chromatography. We constructed a plasmid that expressed the group C polysialyltransferase fused to a HaloTag protein, which reacts covalently with specific chloroalkane substrates. The chimeric polysialyltransferase isolated from a capsule-free E. coli background was labeled with TAMR-HaloTag substrate prior to ammonium sulfate fractionation to determine which molecular sizes were active in the native state. Two major fluorescent species were observed by HPLC, a high-molecular-weight (MW) aggregate and a monomer. Both species were active in the PST assay when meningococcal group C polysaccharide was used as the acceptor. Most of the activity was observed in the high-MW peak. Gel electrophoresis of the peak fraction suggested that the high-molecular-weight fraction contained mostly the PST chimera (Fig. 3), while the low-molecular-weight fraction contained mostly HaloTag. Little or no fluorescent chimera was detected by SDS-PAGE gels in the low-molecular-weight fraction. These results suggest that the meningococcal group C polysialyltransferase is active as a monomer but has a tendency to form high-MW aggregates. This result is consistent with the enzyme being a membrane-associated protein and having exposed hydrophobic sites.
FIG. 3.

Native molecular size of the HaloTag-NmC PST chimera. A crude 25% saturation ammonium sulfate fraction of cell supernatant containing HaloTag-NmC polysialyltransferase was labeled as described in Materials and Methods. (A) The labeled lysate was applied to a Superose 12 FPLC column, and peak fractions were assayed for activity as described in Materials and Methods. (B) The labeled lysate (lane 4) was also applied to an SDS-PAGE gel, and labeled proteins (lanes 2 and 3) were detected under UV light.
Metal ion requirement, pH optimum, and kinetics.
The NmC PST-MalE chimera exhibits a broad pH dependence of activity, with a maximum activity obtained at pH 8.0. Polysialyltransferase activity is stimulated by the divalent cations Mg2+ and Mn2+; however, the NmC PST does not require a metal ion cofactor. Enzyme fractions were active after treatment with the metal chelators EDTA and dipicolinic acid (26). The Km for CMP-NeuNAc was determined using a radiolabel assay with NmC polysaccharide. The Km for CMP-NeuNAc was 432 μM for NmC PST, which is very close to the value reported for the MalE-NmB PST chimera, 420 μM (9).
Acceptor activity of polysaccharides.
The NmC PST can extend polysialic acid and oligosialic acid acceptors. We tested the ability of the NmC PST to use E. coli K1 and K92 polysaccharides as well as the meningococcal group C polysialic acid as acceptors. All polysaccharides supported sialic acid transfer in the radiolabel assay. We previously showed that the E. coli and meningococcal group B polysialyltransferases are able to extend the fluorescent disialylated oligosaccharide to form high-molecular-weight polysialylated products in a nonprocessive fashion (9). Figure 4 shows the time-dependent extension of GD3-FCHASE to form high-molecular-weight polymer. The product was confirmed to be α-2,9 linked by the resistance to the α-2,8-specific endoneuraminidase. The high-MW fluorescent product was isolated and analyzed by HPLC size exclusion chromatography. The results of this experiment suggested a species with a mass greater than the 670-kDa marker.
FIG. 4.

Time course of GD3-FCHASE elongation. Purified MalE-NmC PST was incubated with the fluorescent acceptor GD3-FCHASE and analyzed by ion exchange HPLC as previously described (32).
The fact that the disialylated GD3 analogue is a substrate is consistent with the enzyme using α-2,8-NeuNAc K1 polysaccharide as an acceptor. This result implies that although NmC PST requires at least a disialylated acceptor, it does not require 2,9-linked acceptors to form 2,9-polysialic acid. In Fig. 5 the disialylated ganglioside GD3 and a hexasaccharide of K92 possessing α-2,9-linked disialic acid at the nonreducing end are compared. In the radioactivity assay, the ganglioside GD3 was a much better acceptor than the K92 oligosaccharide. Although the K92 oligosaccharide has a 2,9-disialic acid at the nonreducing end, at least 10-fold less sialic acid was incorporated when it was used as an acceptor. This suggests that at least for short oligosaccharides the NmC PST prefers a hydrophobic region in addition to the oligosialic acid. The preference for hydrophobic acceptors was tested further in a competition experiment. In this experiment we measured the ability of unlabeled acceptor to compete with the fluorescent acceptor trisialyllactosyl-BODIPY by HPLC (18). As can be seen in Fig. 6, the disialyl ganglioside GD3 was a better inhibitor of elongation of the fluorescent substrate than the trisialyl oligosaccharide. The observation that the elongation reaction can be inhibited by competition with other acceptor substrates is consistent with the notion that the in vitro elongation reaction is not processive.
FIG. 5.

Comparison of acceptor activity of GD3 and sialic acid hexasaccharide. The ganglioside GD3 and a hexasaccharide isolated from the K92 polysaccharide were incubated as acceptors with purified MalE-NmC PST and CMP-[14C]sialic acid in an assay described in Materials and Methods. The hexasaccharide was prepared by digestion of K92 polysaccharides and purification by ion exchange and gel filtration as described previously (17, 32).
FIG. 6.
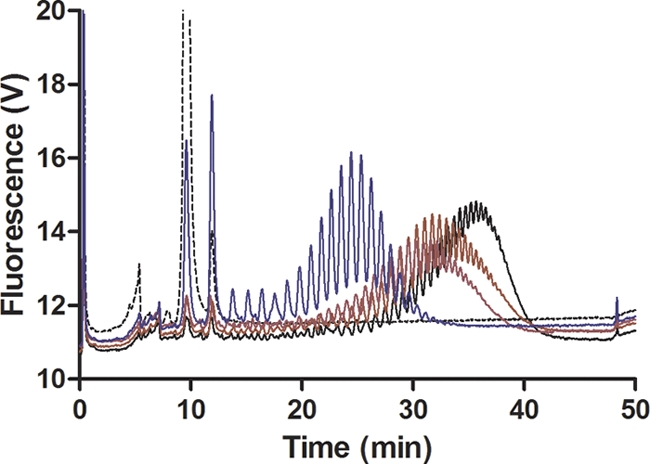
Competition of the PST elongation reaction with unlabeled acceptors. Trisialylated lactosyl-BODIPY (2.5 μM; dotted trace) was incubated with NmC PST (30 μg/ml) in the absence (black trace) or presence of 5 μM α-2,8-sialic acid trimer (maroon trace), α-2,8-hexamer (purple trace), or GD3 (blue trace).
NmC PST can transfer to a hydrophobic acceptor in outer membrane vesicles.
We prepared outer membrane vesicles from a strain of N. meningitidis group B lacking sialyltransferase activity and therefore unable to synthesize a polysialic acid. We tested these vesicles for their level of an endogeneous polysialyltransferase acceptor. We confirmed that these preparations were indeed outer membranes, based on their SDS-gel electrophoresis profiles and sucrose density gradient fractionation. The protein profile detected by SDS-gel electrophoresis was characteristic of N. meningitidis outer membranes. The OMV fraction was also loaded onto a sucrose density gradient and assayed for the cytoplasmic membrane marker enzyme lactate dehydrogenase. No lactate dehydrogenase activity was detected, and thus no detectable cytoplasmic membranes were present.
These vesicles were tested for the presence of an acceptor by addition of a polysialyltransferase assay mixture containing the PST-MalE chimera and, as the donor substrate, radiolabeled CMP-sialic acid. The acceptor activity observed in these assays was similar to that expected for the addition of homologous meningococcal polysaccharides (Table 3). To demonstrate that a polysaccharide was being formed on these vesicles, the ELISA plates were coated with these reaction mixtures and assayed for the presence of α-2,9-polysialic acid with meningococcal group C-specific antibody (11). As is seen in Fig. 7, the reaction product was reactive with monoclonal antibodies specific for the de-O-acetylated 2,9-polysialic acid (11) in a CMP-NeuNAc concentration-dependent manner.
TABLE 3.
Acceptor activity of OMV (synD) and phenol-extracted OMV
| Acceptor | Polysialyltransferase | Acceptor activity (cpm)a |
|---|---|---|
| NmC PS | NmC PST | 36,460 |
| NmC PS | Boiled PST | 165 |
| OMV (synD) | NmC PST | 64,320 |
| OMV (phenol extract) | 20,884 | |
| C18 elution solvent | ||
| Aqueous | 920 | |
| 10% CH3CN | 18,282 | |
| 30% CH3CN | 19,092 | |
| 50% CH3CN | 1,451 |
Acceptors were incubated with the polysialyltransferase, and the formation of 14C-labeled polysialic acid was assayed as described in Materials and Methods.
FIG. 7.

CMP-NeuNAc dependence of outer membrane acceptor elongation. A reaction mixture containing MalE-NmC PST, CMP-sialic acid, and OMV as described in Materials and Methods was applied to ELISA plates. The presence of meningococcal group C polysaccharide was detected with monoclonal antibodies to O-acetylated (closed triangles) and de-O-acetylated (closed circles) group C polysaccharides as described in Materials and Methods.
This acceptor could be extracted from the membrane vesicles by using a buffer phenol-water method. This acceptor is active with N. meningitidis B and E. coli K1 and K92 polysialyltransferases (data not shown). That this acceptor is hydrophobic is demonstrated by its adsorption to C18 reverse-phase resin. The acceptor activity contained in the aqueous phase of the phenol extraction was retained on the C18 resin and could be eluted with 30% acetonitrile (Table 3). The fraction eluted from the C18 resin did not stain in silver-stained SDS-PAGE gels and therefore probably was not lipopolysaccharide.
In summary, the results described above show that the NmC polysialyltransferase can be expressed and purified as a soluble active enzyme. The soluble enzyme exists in its active form primarily as a high-molecular-weight aggregate. Like the E. coli polysialyltransferases, the linkage of the polysialic acid product is determined by the amino terminus of the enzyme. This would suggest that at least part of the acceptor site resides in the amino terminus of the enzyme. While the structure of the natural acceptor remains unknown, we have learned from these experiments that it probably contains a hydrophobic moiety. This is inferred from the acceptor preference of the NmC polysialyltransferase and from the nature of the acceptor fractions extracted from Neisseria membranes.
Supplementary Material
Acknowledgments
We thank the following investigators for the generous donation of strains and plasmids, Warren Warkachuk, National Research Council, Canada; Eric Vimr, University of Illinois, Urbana, IL; David Stephens, Emory University, Atlanta, GA. We also thank Eric Vimr for helpful discussions.
Footnotes
Published ahead of print on 28 January 2011.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Andreishcheva, E. N., and W. F. Vann. 2006. Gene products required for de novo synthesis of polysialic acid in K1 Escherichia coli. J. Bacteriol. 188:1786-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arakere, G., A. L. Lee, and C. E. Frasch. 1994. Involvement of phospholipid end groups of group C Neisseria meningitidis and Haemophilus influenzae type B polysaccharides in association with isolated outer membranes and in immunoassays. J. Bacteriol. 176:691-695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Astronomo, R. D., and D. R. Burton. 2010. Carbohydrate vaccines: developing sweet solutions to sticky situations? Nat. Rev. Drug Discov. 9:308-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, Q., K. B. Decker, P. E. Boucher, D. Hinton, and S. Stibitz. 2010. Novel architectural features of Bordetella pertussis fimbrial subunit promoters and their activation by the global virulence regulator BvgA. Mol. Microbiol. 77:1326-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claus, H., U. Vogel, M. Muhlenhoff, R. Gerardy-Schahn, and M. Frosch. 1997. Molecular divergence of the sia locus in different serogroups of Neisseria meningitidis expressing polysialic acid capsules. Mol. Gen. Genet. 257:28-34. [DOI] [PubMed] [Google Scholar]
- 6.Coutinho, P. M., E. Deleury, G. J. Davies, and B. Henrissat. 2003. An evolving hierarchical family classification of glycosyltransferases. J. Mol. Biol. 328:307-317. [DOI] [PubMed] [Google Scholar]
- 7.Echarti, C., et al. 1983. Cloning and analysis of the K1 capsule biosynthesis genes of Escherichia coli: lack of homology with Neisseria meningitidis group B DNA sequences. Infect. Immun. 41:54-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frasch, C. E., and M. S. Peppler. 1982. Protection against group B Neisseria meningitidis disease: preparation of soluble protein and protein-polysaccharide immunogens. Infect. Immun. 37:271-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freiberger, F., et al. 2007. Biochemical characterization of a Neisseria meningitidis polysialyltransferase reveals novel functional motifs in bacterial sialyltransferases. Mol. Microbiol. 65:1258-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frosch, M., C. Weisgerber, and T. F. Meyer. 1989. Molecular characterization and expression in Escherichia coli of the gene complex encoding the polysaccharide capsule of Neisseria meningitidis group B. Proc. Natl. Acad. Sci. U. S. A. 86:1669-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Ojeda, P. A., S. Hardy, S. Kozlowski, K. E. Stein, and I. M. Feavers. 2004. Surface plasmon resonance analysis of antipolysaccharide antibody specificity: responses to meningococcal group C conjugate vaccines and bacteria. Infect. Immun. 72:3451-3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girard, M. P., M. P. Preziosi, M. T. Aguado, and M. P. Kieny. 2006. A review of vaccine research and development: meningococcal disease. Vaccine 24:4692-4700. [DOI] [PubMed] [Google Scholar]
- 13.Jackson, L. A., A. Schuchat, M. W. Reeves, and J. D. Wenger. 1995. Serogroup C meningococcal outbreaks in the United States. An emerging threat. JAMA 273:383-389. [PubMed] [Google Scholar]
- 14.Kahler, C. M., et al. 1998. The (α2→8)-linked polysialic acid capsule and lipooligosaccharide structure both contribute to the ability of serogroup B Neisseria meningitidis to resist the bactericidal activity of normal human serum. Infect. Immun. 66:5939-5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis, C., M. A. Diggle, and S. C. Clarke. 2003. Nucleotide sequence analysis of the sialyltransferase genes of meningococcal serogroups B, C, Y and W135. J. Mol. Microbiol. Biotechnol. 5:82-86. [DOI] [PubMed] [Google Scholar]
- 16.Los, G. V., and K. Wood. 2007. The HaloTag: a novel technology for cell imaging and protein analysis. Methods Mol. Biol. 356:195-208. [DOI] [PubMed] [Google Scholar]
- 17.McGowen, M. M., J. Vionnet, and W. F. Vann. 2001. Elongation of alternating α2,8/2,9 polysialic acid by the Escherichia coli K92 polysialtransferase. Glycobiology 11:613-620. [DOI] [PubMed] [Google Scholar]
- 18.Mosley, S. L., et al. 2010. Chemoenzymatic synthesis of conjugatable oligosialic acids. Biocatal. Biotransformation 28:41-50. [Google Scholar]
- 19.Roberts, I. S. 1996. The biochemistry and genetics of capsule polysaccharide production in bacteria. Annu. Rev. Microbiol. 50:285-315. [DOI] [PubMed] [Google Scholar]
- 20.Silver, R. P., et al. 1981. Molecular cloning of the K1 capsular polysaccharide genes of Escherichia coli. Nature 289:696-698. [DOI] [PubMed] [Google Scholar]
- 21.Steenbergen, S. M., and E. R. Vimr. 2008. Biosynthesis of the Escherichia coli K1 group 2 polysialic acid capsule occurs within a protected cytoplasmic compartment. Mol. Microbiol. 68:1252-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steenbergen, S. M., and E. R. Vimr. 2003. Functional relationships of the sialyltransferases involved in expression of the polysialic acid capsules of Escherichia coli K1 and K92 and Neisseria meningitidis groups B or C. J. Biol. Chem. 278:15349-15359. [DOI] [PubMed] [Google Scholar]
- 23.Steenbergen, S. M., and E. R. Vimr. 1990. Mechanism of polysialic acid chain elongation in Escherichia coli K1. Mol. Microbiol. 4:603-611. [DOI] [PubMed] [Google Scholar]
- 24.Steenbergen, S. M., T. J. Wrona, and, and E. R. Vimr. 1992. Functional analysis of the sialyltransferase complexes in Escherichia coli K1 and K92. J. Bacteriol. 174:1099-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stemmer, W. P., and S. K. Morris. 1992. Enzymatic inverse PCR: a restriction site independent, single-fragment method for high-efficiency, site-directed mutagenesis. Biotechniques 13:214-220. [PubMed] [Google Scholar]
- 26.Sundaram, A. K., et al. 2004. Characterization of N-acetylneuraminic acid synthase isoenzyme 1 from Campylobacter jejuni. Biochem. J. 383:83-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swartley, J. S., J. H. Ahn, L. J. Liu, C. M. Kahler, and D. S. Stephens. 1996. Expression of sialic acid and polysialic acid in serogroup B Neisseria meningitidis: divergent transcription of biosynthesis and transport operons through a common promoter region. J. Bacteriol. 178:4052-4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan, L. K., G. M. Carlone, and R. Borrow. 2010. Advances in the development of vaccines against Neisseria meningitidis. N. Engl. J. Med. 362:1511-1520. [DOI] [PubMed] [Google Scholar]
- 29.Troy, F. A., II. 1992. Polysialylation: from bacteria to brains. Glycobiology 2:5-23. [DOI] [PubMed] [Google Scholar]
- 30.Vimr, E. R., R. Bergstrom, S. M. Steenbergen, G. Boulnois, and I. Roberts. 1992. Homology among Escherichia coli K1 and K92 polysialyltransferases. J. Bacteriol. 174:5127-5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vionnet, J., E. S. Kempner, and W. F. Vann. 2006. Functional molecular mass of Escherichia coli K92 polysialyltransferase as determined by radiation target analysis. Biochemistry 45:13511-13516. [DOI] [PubMed] [Google Scholar]
- 32.Vionnet, J., and W. F. Vann. 2007. Successive glycosyltransfer of sialic acid by Escherichia coli K92 polysialyltransferase in elongation of oligosialic acceptors. Glycobiology 17:735-743. [DOI] [PubMed] [Google Scholar]
- 33.Whitfield, C., and I. S. Roberts. 1999. Structure, assembly and regulation of expression of capsules in Escherichia coli. Mol. Microbiol. 31:1307-1319. [DOI] [PubMed] [Google Scholar]
- 34.Willis, L. M., M. Gilbert, M.-F. Karwaski, M.-C. Blanchard, and W. W. Wakarchuk. 2008. Characterization of the α-2,8-polysialyltransferase from Neisseria meningitidis with synthetic acceptors, and the development of a self-priming polysialyltransferase fusion enzyme. Glycobiology 18:177-186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.