

Abstract
For most paramyxoviruses, virus type-specific interaction between fusion (F) protein and attachment protein (hemagglutinin-neuraminidase [HN], hemagglutinin [H], or glycoprotein [G]) is a prerequisite for mediating virus-cell fusion and cell-cell fusion. Our previous cell-cell fusion assay using the chimeric F proteins of human parainfluenza virus 2 (HPIV2) and simian virus 41 (SV41) suggested that the middle region of the HPIV2 F protein contains the site(s) that determines its specificity for the HPIV2 HN protein. In the present study, we further investigated the sites of the F protein that could be critical for determining the HN protein specificity. By analyzing the reported structure of the F protein of parainfluenza virus 5 (PIV5), we found that four major domains (M1, M2, M3, and M4) and five minor domains (A to E) in the middle region of the PIV5 F protein were exposed on the trimer surface. We then replaced these domains with the SV41 F counterparts individually or in combination and examined whether the resulting chimeras could mediate cell-cell fusion when coexpressed with the SV41 HN protein. The results showed that a chimera designated M(1+2), which harbored SV41 F-derived domains M1 and M2, mediated cell-cell fusion with the coexpressed SV41 HN protein, suggesting that these domains are involved in determining the HN protein specificity. Intriguingly, another chimera which harbored the SV41 F-derived domain B in addition to domains M1 and M2 showed increased specificity for the SV41 HN protein compared to that of M(1+2), although it was capable of mediating cell-cell fusion by itself.
The members of genus Rubulavirus of the subfamily Paramyxovirinae, as well as those of genera Respirovirus and Avulavirus in the same subfamily, have two kinds of glycoprotein spikes on the envelope: hemagglutinin-neuraminidase (HN) protein tetramers and fusion (F) protein trimers (11). The attachment protein HN is responsible for binding to the sialoconjugate receptors on the cell surface and for enzymatic destruction of the receptors, while the F protein mediates membrane fusion, such as cell-cell fusion or virus-cell fusion. Cleavage of the F precursor (F0) by cellular proteases into subunits F1 and F2 is a prerequisite for its fusion activity, similar to the other class I viral fusion proteins.
Unlike most of the other class I fusion proteins, however, the F protein does not have an apparent receptor-binding function. Moreover, most F proteins require a fusion-promoting function of the attachment protein HN in a virus type-specific manner (4, 5). Although it is not well known how the HN protein promotes the F protein-mediated membrane fusion, it is believed that the fusion is induced through a series of conformational changes of the F protein that is initiated by its specific interaction with the homologous HN protein (6, 10, 16). The fusion-promoting function of the HN protein appears to depend on it being bound to its cellular receptors (13, 17, 18, 25), and it seems that the HN protein promotes the F protein-mediated fusion dependently on the balance between its inherent F-triggering efficiency and receptor-attachment regulatory functions (binding and destruction), as suggested by Porotto and colleagues (23). Furthermore, the presumptive signal-transducing activity of the HN protein may be required for the induction of cell-cell fusion (36).
The stalk region of the HN protein is inferred to contain the site that determines the F protein specificity in promoting fusion (3, 28, 35), while the head region carries both the receptor-binding and -destroying activities (15, 30). Indeed, involvement of the HN stalk region of an avulavirus Newcastle disease virus (NDV) in the physical interaction with the F protein has been certified by coimmunoprecipitation analysis (14). On the other hand, there is inadequate information concerning the site of the F protein that determines the HN protein specificity. Previously, Yao and Compans demonstrated that a cytoplasmic tail truncation mutant of the F protein of a rubulavirus human parainfluenza virus 2 (HPIV2) was transported to the cell surface and induced cell-cell fusion when coexpressed with the HPIV2 HN protein (37), indicating that the HPIV2 F protein needs only the ectodomain and transmembrane domain to undergo transport and induce cell-cell fusion. Thus, the cytoplasmic domain of the HPIV2 F protein is involved neither in the interaction with the HN protein nor in the induction of fusion. Consistently, our previous chimeric analyses of the F proteins of HPIV2 and another closely related rubulavirus, simian virus 41 (SV41), suggested that a region (amino acids [aa] 227 to 370) in the ectodomain of the HPIV2 F protein contains the site(s) that determines its specificity for the HPIV2 HN protein in the induction of cell-cell fusion (34). Since this region, designated the middle region, was considerably large (occupying about 27% of the total molecule), we tried to narrow it down, but it was very difficult to create additional chimeras in the absence of information about the three-dimensional (3-D) structure. Furthermore, the fact that the HPIV2 HN protein was able to weakly promote the SV41 F-mediated cell-cell fusion sometimes made the interpretation of the results of the fusion assay complicated.
Recently, the 3-D structure of the F protein of a rubulavirus parainfluenza virus 5 (PIV5) has been reported (38). In order to identify more precisely the sites of the F protein that determine the HN protein specificity and would be responsible for the physical interaction with the HN protein, we decided to carry out chimeric analyses of the F proteins of PIV5 and SV41, because these F proteins are phylogenetically close to each other (31) and because they induced cell-cell fusion only when coexpressed with their homologous HN proteins. To begin with, we adopted the F protein of PIV5 strain WR, which can induce cell-cell fusion only when coexpressed with the HN protein (7). It is known, on the other hand, that the F protein of PIV5 strain W3A can mediate fusion in the absence of the HN protein (21). The middle region of the PIV5 F protein was then mapped on the 3-D structure of the F trimer, and it was proved that four major domains and five minor domains in the middle region were exposed on the trimer surface. Consequently, chimeric analyses of these domains have indicated that the two SV41 F-derived major domains, which harbor 16 noncontiguous amino acids that are not conserved between the F proteins, make the PIV5 F protein to induce cell-cell fusion when coexpressed with the SV41 HN protein.
MATERIALS AND METHODS
Cells and recombinant plasmids.
Monolayers of HeLa cells were maintained in Eagle's minimum essential medium (MEM) supplemented with 5% fetal calf serum (9, 32). The recombinant SRα plasmid encoding the HN or F protein of SV41 or that of NDV was described previously (34, 35). The recombinant SRα plasmid encoding the HN protein of PIV5 strain W3A and that encoding the F protein of PIV5 strain WR were described elsewhere (7, 8). The PIV5 W3A HN protein was used instead of the PIV5 WR HN protein, because the former exhibited higher fusion-promoting activity than the latter when coexpressed with the PIV5 F protein (data not shown). The cleavage site mutant of the F protein of PIV5 strain WR, Se-WR F, whose cleavage site was replaced with that of a respirovirus Sendai virus (SeV) and thus cleavable with exogenously added trypsin (acetylated trypsin; Sigma), was reported more recently (32, 33). To generate the cleavage site mutants, Se-M(1+2) and Se-M(2+4), chimeric recombination was performed by using the cleavage site mutant, Se-WR F, and newly created chimeric proteins, M(1+2) and M(2+4), respectively.
To generate the chimeric HN protein, CH5-41, in which the amino terminal 90-amino-acid region of the SV41 HN protein was replaced with the corresponding 89-amino-acid region of the PIV5 WR HN protein, the restriction enzyme site for ScaI in the SV41 HN protein-encoding plasmid was used for chimeric recombination, as reported previously (35); an ScaI site was introduced into the corresponding position in the PIV5 WR HN protein-encoding plasmid using an U.S.E. mutagenesis kit (Amersham Pharmacia Biotech).
Generation of chimeric F proteins.
We showed previously that the middle region (aa 227 to 370) of the HPIV2 F protein harbors the site(s) that determines the HN protein specificity (34). In order to investigate further the sites of the F protein that determine the HN protein specificity, the middle region (aa 223 to 366) of the PIV5 F protein was identified by amino acid sequence alignment with the HPIV2 F protein. Since the interacting sites of the F protein with the HN protein would be exposed on the trimer surface, the side chain of every amino acid in the middle region was visually inspected whether it is exposed on the 3-D structure of the PIV5 F cleavage site mutant, FR3 (Protein Data Bank identifier code [PDB ID] 2B9B), with the aid of the DeepView Swiss-PdbViewer program (GlaxoSmithKline R&D and the Swiss Institute of Bioinformatics). Then, the exposed residues were grouped into four major domains and five minor domains by visual inspection.
To generate chimeric F proteins of SV41 and PIV5, the desired domain of the SV41 F protein-encoding plasmid was amplified by PCR, while both the franking regions of the corresponding domain of the PIV5 F protein-encoding plasmid were amplified, simultaneously introducing the restriction enzyme site for SpeI or PstI at the distal end. These three fragments were then combined stepwise by fusion PCR and inserted into the PIV5 F protein-encoding plasmid by utilizing restriction enzyme sites SpeI and PstI. In principle, each domain was fixed so that it starts and ends with an amino acid that is shared by the two F proteins. The positions of cysteine and proline residues were also taken into consideration for fixing the start and end of each domain.
Quantification of cell-cell fusion.
Subconfluent HeLa cell monolayers in six-well culture plates were transfected with 2 μg/well of each recombinant plasmid as described previously (32). After 24 h of incubation at 37°C, the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS), washed three times with PBS, and stained with Giemsa solution. The photomicrographs of 10 randomly chosen fields were subjected to morphometric measurement of cell-cell fusion, and the average fusion index (%) and standard deviation were determined as described previously (35).
Western blot analysis.
Subconfluent HeLa cell monolayers were transfected with the recombinant plasmid encoding each F protein. After 24 h of incubation at 37°C, the cells were lysed with 500 μl/well of lysis buffer (25 mM HEPES [pH 7.6], 1% Triton X-100, 3 mM β-glycerophosphate, 3 mM EDTA, 137 mM NaCl, and protease inhibitors [Complete; Roche Applied Science]). An aliquot (15 μl) of each cell lysate was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the separated proteins were electroblotted to a nitrocellulose membrane (Whatman). The membrane was then successively treated with monoclonal antibody (MAb) 1D1, which recognizes the amino acid region (aa 448 to 452) immediately upstream of the heptad repeat B domain of the PIV5 F protein (33), biotinylated horse immunoglobulin specific for mouse IgG (Vector Laboratories), and streptoavidin-biotin-peroxidase complex (Vector Laboratories). The F protein bands were then visualized by enhanced chemiluminescence (ECL) using the Western blotting luminol reagent (Santa Cruz Biotechnology), followed by exposure to X-ray film (Konica, Tokyo, Japan).
Cell surface biotinylation.
Subconfluent HeLa cell monolayers were transfected with recombinant plasmid encoding each F protein. After 24 h of incubation at 37°C, the cells were treated with 0.3 mg/ml of NHS-LC-biotin (Pierce) or thiocleavable sulfo-NHS-LC-biotin (Pierce) solution in PBS, which contained 0.1 mM CaCl2 and 1 mM MgCl2, at 23°C for 30 min and lysed with 500 μl/well of lysis buffer as described previously (33). The biotinylated proteins in the cell lysates were immunoprecipitated with MAb 1D1 or with rabbit antiserum specific for the PIV5 F2 subunit (8) and subjected to SDS-PAGE, followed by blotting to a polyvinylidene difluoride (PVDF) membrane (Hybond-P; Amersham Biosciences). The biotinylated F proteins on the membrane were detected by ECL after being treated with the streptoavidin-biotin-peroxidase complex as described above and quantified with the aid of a graphics software, NIH Image version 1.60. The results were then normalized by the value given by the PIV5 F protein M(1+2) or Se-WR F.
RESULTS
The amino terminal region of the PIV5 HN protein determines the F protein specificity in promoting cell-cell fusion.
We reported previously that the F protein of PIV5 strain WR did not mediate detectable cell-cell fusion when it was coexpressed with the heterologous SV41 HN protein in BHK cells (7). We confirmed this observation by using HeLa cells in the current study (Fig. 1 A). Similarly, cell-cell fusion was not detected when the SV41 F protein was coexpressed with the heterologous PIV5 HN protein (Fig. 1A). We also reported previously, on the other hand, that the 90 amino terminal residues of the SV41 HN protein, which include the 53 membrane proximal amino acids of the stalk region, determine its specificity for the SV41 F protein in promoting cell-cell fusion (35). As shown in Fig. 1, when this 90-amino-acid region was replaced with the PIV5 HN counterpart (89 amino acids), the resultant chimera, CH5-41, promoted cell-cell fusion when coexpressed with the PIV5 F protein but not with the SV41 F protein. The fusion-promoting activity of CH5-41 was much higher than that of the PIV5 HN protein (Fig. 1B), most likely because the cell surface expression level of the PIV5 HN protein was remarkably low compared to that of CH5-41 (data not shown). These results indicate that the HN proteins of PIV5 and SV41 promote cell-cell fusion only when coexpressed with their homologous F proteins and that the chimeric HN protein, whose membrane proximal part of the stalk region is derived from the PIV5 HN protein while the remaining part of the ectodomain is derived from the SV41 HN protein, specifically promotes PIV5 F protein-mediated fusion.
FIG. 1.

(A) Cell-cell fusion induced by coexpressed HN and F proteins. Subconfluent HeLa cell monolayers in six-well culture plates were transfected with the recombinant plasmid encoding the F protein of SV41 or PIV5 (WR) together with the recombinant plasmid encoding the SV41 HN protein, PIV5 (W3A) HN protein, or the chimeric HN protein, CH5-41. After 24 h of incubation, the cells were fixed with 4% paraformaldehyde and stained with Giemsa solution. Scale bar, 100 μm. (B) Schematic diagram of CH5-41. TM, transmembrane domain.
Identification of domains in the middle region of the PIV5 F protein that are exposed on the trimer surface.
Our previous chimeric analyses of the F proteins of HPIV2 and SV41 suggested that a 144-amino-acid region (aa 227 to 370), which is located in the middle of the primary structure of the HPIV2 F protein, harbors the site(s) that determines the HN protein specificity (34). In order to investigate further the sites of the F protein that could determine the HN protein specificity, the middle region (aa 223 to 366) of the PIV5 F protein was identified by amino acid sequence alignment with the HPIV2 F protein (Fig. 2 A). The side chain of every amino acid in the middle region was then examined for its exposure on the trimer surface of the PIV5 F protein by analyzing the reported 3-D structure, and 84 out of 144 side chains per one monomer proved to be exposed on the trimer surface as three distinct patches, I, II, and III (Fig. 2B and C). We then focused on the largest patch (II) and found that it could be split into four major domains (M1, M2, M3, and M4) and two minor domains (B and D) (Fig. 2B and C). On the other hand, patch III was divided into two minor domains (C and E), while patch I contained only one domain, designated A (Fig. 2B and C). To see which domain would determine the HN protein specificity, we decided to carry out chimeric analyses of the F proteins of PIV5 and SV41, because they are phylogenetically close to each other (31) and because these F proteins show specificity for their homologous HN proteins as described above.
FIG. 2.

Identification of domains in the middle region of the PIV5 F protein that are exposed on the trimer surface. (A) Schematic diagram of the PIV5 F protein. Positions of four major domains (M1, M2, M3, and M4) in the middle region are indicated. FP, fusion peptide; TM, transmembrane domain; HRA and HRB, heptad repeat regions A and B. (B) Amino acid sequence alignment of the middle regions of the F proteins of PIV5 and SV41. Positions of the four major domains and five minor domains (A to E) are indicated. Amino acid residues of the PIV5 F protein that are exposed on the trimer surface are underlined. Dashes in the SV41 F sequence indicate amino acids identical to those of the PIV5 F protein. (C) Location of the middle region and exposed domains on the 3-D structures of the PIV5 F trimer. For simplicity, the major and minor domains of only one monomer are shown, as capital letters; some of the minor domains of the remaining two monomers are shown by small letters. The positions of Ala-47, Thr-97, and Ala-111, which correspond to Arg-164, Gly-219, and Val-233 of the F protein of CDV strain Onderstepoort, respectively, are indicated in the side view of the PIV5 F trimer. The figures were depicted with the aid of the DeepView Swiss-PdbViewer program on the basis of the crystal structure of the PIV5 F cleavage site mutant.
The chimeric PIV5 F protein harboring the SV41 F-derived domains M1 and M2 mediates cell-cell fusion with the coexpressed SV41 HN protein.
First, to examine whether the four major domains (M1, M2, M3, and M4) on the PIV5 F trimer surface could determine the HN protein specificity, each domain of the PIV5 F protein was individually replaced with the SV41 F counterpart; the resulting chimeras were designated M1, M2, M3, and M4 (Fig. 3 A, top). The amounts of these chimeric F proteins produced in the transfected cells were comparable to or even more than that of the wild type, the PIV5 F protein, as analyzed by Western blot analysis (Fig. 3B, left). However, except for chimera M2, these chimeric F proteins were not efficiently cleaved: the F1 bands of chimeras M1 and M4 were faint, and that of chimera M3 was below the detection level. Since we could not detect chimera M3 on the cell surface (Fig. 3C, left) despite the fact that it was present in the cells as the uncleaved F0 precursor (Fig. 3B), it seemed that chimera M3 was not cleaved due to its severe defect in membrane transport: the cleavage site of the PIV5 F protein is multibasic and thus considered to be digested by subtilisin-like proteases in the trans-Golgi network (TGN) (11, 19, 20). Nonetheless, when coexpressed with the PIV5 HN protein or with CH5-41, the chimeric F proteins, except for chimera M3, mediated prominent cell-cell fusion (Fig. 3C). However, none of them was able to mediate fusion with the SV41 HN protein.
FIG. 3.

The chimeric PIV5 F protein harboring the SV41 F-derived domains M1 and M2 mediates cell-cell fusion with the coexpressed SV41 HN protein. (A) Schematic diagram of chimeric F proteins. Dark boxes represent amino acid regions derived from the PIV5 F protein, while light boxes represent those derived from the SV41 F protein. (B) Western blot of chimeric F proteins. Subconfluent HeLa cell monolayers in six-well culture plates were transfected with the recombinant plasmid encoding each F protein and lysed at 24 h posttransfection. The cell lysates were subjected to SDS-PAGE under reducing conditions, followed by Western blot analysis with MAb 1D1 that is specific for the PIV5 F1 subunit. (C) Detection of cell surface-expressed chimeric F proteins by cell surface biotinylation and comparison of their fusion activity. For detection of the F proteins on the cell surface, subconfluent HeLa cell monolayers in six-well culture plates were transfected with the recombinant plasmid encoding each F protein, biotinylated with thiocleavable sulfo-NHS-SS-biotin at 24 h posttransfection, and the biotinylated proteins in the cell lysates were immunoprecipitated with rabbit antiserum specific for the PIV5 F2 subunit. The immunoprecipitates were subjected to SDS-PAGE under nonreducing conditions, followed by blotting onto a PVDF membrane. The biotinylated proteins on the membrane were detected by the avidin-biotin-peroxidase complex and visualized by enhanced chemiluminescence (ECL). The F protein bands were then quantified, normalized by the value given by the PIV5 F protein, and expressed as cell surface expression (CSE) levels. The average of results from three independent experiments was determined; error bars indicate standard deviations. For analyzing fusion activity of the F proteins, subconfluent HeLa cell monolayers in six-well culture plates were transfected with the recombinant plasmid encoding each F protein together with recombinant plasmid encoding the PIV5 (W3A) HN protein, SV41 HN protein, or CH5-41. After 24 h, the cells were fixed with 4% paraformaldehyde, and the average fusion index was determined as described in Materials and Methods; error bars indicate standard deviations.
We then created six chimeric F proteins, in which two of the four major domains of the PIV5 F protein were replaced with the SV41 F counterparts in various combinations (Fig. 3A, bottom). The three double chimeras, M(1+2), M(2+3), and M(2+4), were expressed on the cell surface and cleaved as efficiently as did the PIV5 F protein, whereas the other three chimeras were neither efficiently expressed on the cell surface nor efficiently cleaved (Fig. 3B and C). We found that the double chimera M(1+2), which harbored the SV41 F-derived domains M1 and M2, was capable of mediating cell-cell fusion when it was coexpressed with the SV41 HN protein (Fig. 3C). In contrast, M(2+4) did not mediate fusion with the SV41 HN protein, whereas it mediated fusion with the PIV5 HN protein or with CH5-41 as efficiently as did M(1+2). On the other hand, M(2+3) did not induce fusion with any of the HN proteins. These results indicate that the SV41 F-derived domains M1 and M2, which harbor 16 noncontiguous amino acids that are not conserved between the F proteins (Fig. 2B), are both needed for the PIV5 F protein to mediate fusion with the SV41 HN protein, as demonstrated by the double chimera M(1+2). However, since this chimera also mediated fusion with the PIV5 HN protein or with CH5-41, an additional region(s) seemed to be involved in determining the HN protein specificity. It was as expected, on the other hand, that CH5-41 behaved toward the chimeric F proteins the same as the PIV5 HN protein in terms of fusion promotion, except that it induced more extensive fusion compared to the PIV5 HN protein.
Replacement of domains M3 and M4 with the SV41 F counterparts does not increase the specificity of M(1+2) for the SV41 HN protein.
On the basis of the above findings, we created five chimeric F proteins, in which three or all of the major domains were replaced with the SV41 F counterparts (Fig. 4 A). Among the four triple chimeras, M(1+2+3) and M(1+2+4) harbored the SV41 F-derived domains M1 and M2 and mediated fusion with the SV41 HN protein (Fig. 4C). However, they also induced fusion either with the PIV5 HN protein or with CH5-41; it seemed that their specificities for the SV41 HN protein did not greatly differ from that of the double chimera M(1+2). Notably, M(1+2+4) exhibited remarkably high fusion activity when coexpressed with every HN protein compared to that of M(1+2) (Fig. 4C) and was able to mediate very weak fusion (0.7% ± 0.5%) in the absence of the HN protein (Table 1). In order to see whether M(1+2+4)-induced fusion was nonspecifically promoted by the coexpressed HN proteins, the NDV HN protein was employed as the negative control, because we previously found that it does not induce fusion when coexpressed with the F protein of PIV5 or SV41 (7, 34) and because NDV belongs to the genus Avulavirus and thus is phylogenetically distinct from PIV5 and SV41. Since it turned out that the NDV HN protein could not promote M(1+2+4)-mediated fusion while it induced fusion with its homologous F protein (Table 1), we concluded that M(1+2+4)-mediated fusion was specifically promoted by the HN proteins of PIV5 and SV41 or by CH5-41. On the other hand, the triple chimera M(2+3+4) did not induce fusion with the SV41 HN protein, whereas it induced weak fusion with the PIV5 HN protein or with CH5-41, similar to that of M(1+2+3), reinforcing the significance of domain M1 in determining the HN protein specificity. In the case of the quadruple chimera M(1+2+3+4), very weak fusion was observed when coexpressed with the SV41 HN protein, but its specificity for the SV41 HN protein did not seem to exceed that of M(1+2+3). These results indicate that replacement of domains M3 and M4 with the SV41 F counterparts does not increase the specificity of M(1+2) for the SV41 HN protein and that the PIV5 F protein cannot be fully converted to an SV41 HN-specific protein even if all the major domains have been replaced with the SV41 F counterparts.
FIG. 4.

Replacement of domains M3 and M4 with the SV41 F counterparts does not increase the specificity of M(1+2) for the SV41 HN protein. (A) Schematic diagram of chimeric F proteins. (B) Western blotting of chimeric F proteins was performed as described in the legend to Fig. 3B. (C) Cell surface expression levels and fusion activity of the chimeric F proteins were quantified as described in the legend to Fig. 3C, except that the cell surface expression levels were normalized by the value given by M(1+2).
TABLE 1.
HN-independent fusion activity of two chimeric F proteins
| F protein | % (±SD) cell-cell fusiona with: |
||
|---|---|---|---|
| SV41 HN | NDV HN | No HN | |
| M(1+2+4) | 10.6 ± 1.6 | 1.1 ± 0.8 | 0.7 ± 0.5 |
| B+M(1+2) | 13.4 ± 1.7 | 5.0 ± 1.1 | 4.2 ± 1.0 |
| NDV F | - | 14.3 ± 2.1 | - |
Cell-cell fusion (%) of the transfected HeLa cells was morphometrically quantified at 24 h posttransfection as described in Materials and Methods. -, cell-cell fusion was not found even when all of the well was observed.
Replacement of domain B with the SV41 F counterpart increases the specificity of M(1+2) for the SV41 HN protein.
We then turned our interest to the minor domains of the middle region (Fig. 2B and C). To see whether the minor domains could be involved in determining the specificity for the SV41 HN protein, we created five chimeras in which each minor domain of M(1+2) was individually replaced with the SV41 F counterpart (Fig. 5 A). As shown in Fig. 5C, it turned out that the triple chimera harboring the three SV41 F-derived domains (M1, M2, and B) exhibited increased specificity for the SV41 HN protein. However, this chimera, designated B+M(1+2), was able to mediate evident fusion (4.2% ± 1.0%) even in the absence of the HN protein (Table 1). It should be pointed out in this context that the fusion activity of B+M(1+2) was not significantly promoted by the NDV HN protein as it was the case with M(1+2+4) described above (Table 1) and that the chimeras created in this study, except for B+M(1+2) and M(1+2+4), did not induce fusion in the absence of the HN protein. On the one hand, replacement of the minor domain A or C of M(1+2) with the SV41 F counterpart resulted in complete loss of fusion activity, presumably due to a defect in transport (Fig. 5). On the other hand, intriguingly, replacement of the minor domain D or E of M(1+2) with the SV41 F counterpart resulted in specific loss of fusion activity with the SV41 HN protein (Fig. 5).
FIG. 5.

Replacement of minor domain B with the SV41 F counterpart increases the specificity of M(1+2) for the SV41 HN protein. (A) Schematic diagram of chimeric F proteins. (B) Western blotting of chimeric F proteins was performed as described in the legend to Fig. 3B. (C) Cell surface expression levels and fusion activity of the chimeric F proteins were quantified as described in the legend to Fig. 3C, except that the cell surface expression levels were normalized by the value given by M(1+2).
These results, together with the results obtained so far, indicate that replacement of domains M1 and M2 with the SV41 F counterparts is necessary for the PIV5 F protein to mediate fusion with the SV41 HN protein and that further replacement of domain B increases the specificity for the SV41 HN protein.
Fusion activity of cleavage site mutants.
As described above, the chimeric F proteins, M(1+2) and M(2+4), induced fusion with the PIV5 HN protein or with CH5-41 to a similar extent, but only the former chimera exhibited prominent fusion activity with the SV41 HN protein (Fig. 3C). Importantly, these chimeras could not induce fusion in the absence of the HN protein. To ascertain these findings, we created cleavage site mutants of M(1+2) and M(2+4), whose cleavage site sequences were replaced with that of the SeV F protein. Since the cleavage site of the SeV F protein is monobasic, it is not cleaved in the TGN but can be cleaved by exogenously added trypsin. It was anticipated that these cleavage site mutants would show increased fusion activity after treatment with trypsin compared to that of their parents, as we have reported previously for a PIV5 F protein mutant that has HN-independent fusion activity (33). When the resultant cleavage site mutants, Se-M(1+2) and Se-M(2+4), or the cleavage site mutant of the PIV5 (WR) F protein, Se-WR F (32), were coexpressed with the SV41 HN protein in HeLa cells and treated with trypsin, they were efficiently cleaved (Fig. 6), and the cleaved Se-M(1+2) induced prominent fusion within 1 h, while the cleaved Se-WR F did not (Table 2). Unexpectedly, the cleaved Se-M(2+4) was able to mediate very weak fusion with the SV41 HN protein (Table 2). It should be pointed out in this context that when coexpressed with CH5-41 and treated with trypsin, all three cleavage mutants induced extensive and indistinguishable fusion within 30 min, and none of the cleavage site mutants showed HN-independent fusion activity up to 8 h after trypsin treatment (Table 2). These results thus confirmed the importance of domains M1 and M2 in determining the HN protein specificity. Furthermore, it was found that domain M4 is also involved in determining the HN protein specificity, although to a lesser extent than domains M1 and M2.
FIG. 6.
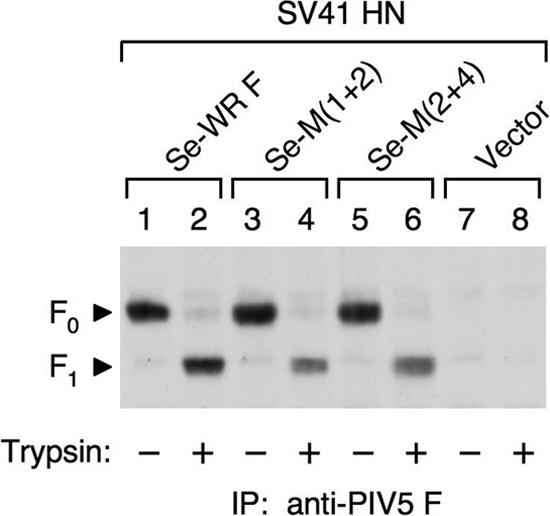
Effect of trypsin on the cleavage site mutants. HeLa cell monolayers were cotransfected with plasmids encoding the SV41 HN protein and each cleavage site mutant. After 22 h, the cells were treated or mock treated with 5 μg/ml of trypsin at 37°C for 15 min and biotinylated with NHS-LC-biotin. Then, the cell lysates were subjected to immunoprecipitation with MAb 1D1, and the immunoprecipitates were separated by SDS-PAGE under reducing conditions, followed by electroblotting onto a PVDF membrane. The biotinylated proteins on the membrane were visualized as described in the legend to Fig. 3C.
TABLE 2.
Fusion activity of cleavage site mutants
| F protein | Relative cell surface expressiona | % (±SD) cell-cell fusionb with |
|||
|---|---|---|---|---|---|
| SV41 HN |
CH5-41 (30 min) | No HN (8 h) | |||
| 1 h | 4 h | ||||
| Se-WR F | 1.0 | - | - | 31.3 ± 3.6 | - |
| Se-M(1+2) | 1.2 ± 0.1 | 10.2 ± 1.0 | 14.9 ± 3.0 | 27.7 ± 3.3 | - |
| Se-M(2+4) | 1.1 ± 0.2 | 0.3 ± 0.4 | 1.7 ± 1.6 | 30.4 ± 2.2 | - |
The relative amount of the F proteins on the cell surface of the HeLa cells coexpressing each cleavage site mutant and the SV41 HN protein was measured by cell surface biotinylation as described in the legend to Fig. 3C at 22 h posttransfection.
After treatment with 5 μg/ml of trypsin (at 37°C for 15 min) at 22 h posttransfection, cell-cell fusion (%) of HeLa cells coexpressing each cleavage site mutant and one of the HN proteins was morphometrically quantified at the times indicated. -, cell-cell fusion was not found even when all of the well was observed.
DISCUSSION
In the present study, we have shown that replacement of domains M1 and M2 of the PIV5 F protein with the SV41 F counterparts results in the creation of the double chimera M(1+2), which can mediate cell-cell fusion with the coexpressed SV41 HN protein. We have also shown that the triple chimera B+M(1+2), which harbors the SV41 F-derived domain B in addition to domains M1 and M2, induces cell-cell fusion with the SV41 HN protein more efficiently than does M(1+2), whereas these chimeras induce fusion with the PIV5 HN protein to a similar extent. We thus conclude that domain B, together with domains M1 and M2, plays a significant role in determining HN protein specificity. This domain in each F monomer forms a cluster on the lateral surface of the F head region and most likely mediates physical interaction with the HN protein; verification of this hypothesis is our future task.
Our present findings do not exclude the possibility that other domains of the F protein are also involved in the interaction with the SV41 HN protein. Indeed, the cleavage site mutant Se-M(2+4) can induce very weak cell-cell fusion when coexpressed with the SV41 HN protein, indicating that domain M4 plays a subsidiary role in determining the HN protein specificity. Intriguingly, M(2+4) does not induce detectable fusion with the SV41 HN protein, presumably because a substantial amount of this chimera may undergo premature conformational change after cleavage in TGN and thus would be inactivated before reaching the neighboring cells. On the contrary, the uncleaved Se-M(2+4) may be relatively stable, and a number of the molecules may be in close proximity to the neighboring cells, which would then efficiently induce fusion upon cleavage by trypsin treatment. This idea could be supported by our previous report, where the cleavage site mutant Se-L22P induced HN-independent fusion far more efficiently than its parent, L22P (33). On the other hand, domains D and E do not seem to contribute to the HN protein specificity. Intriguingly, however, D+M(1+2) and E+M(1+2) do not induce fusion with the SV41 HN protein, while M(1+2) does. A plausible possibility is that the SV41 F-derived domains D and E hamper the interaction of D+M(1+2) and E+M(1+2) with the SV41 HN protein, respectively, although these domains are not involved in the interaction between the F and HN proteins of SV41. In all likelihood, the conformation and/or position of the SV41 F-derived domain D or E in the context of the chimeric F protein may be different from that in the SV41 F protein; this difference may be subtle but may be enough to sterically inhibit the HN-F interaction between the chimeric F protein and the SV41 HN protein, because domain D and domain e (domain E of a different monomer) are located in the vicinity of domain M2 (Fig. 2C).
It has been reported for NDV that the membrane proximal part of the HN stalk region is involved in the physical interaction with the F protein (14). Being consistent with this result, the amino terminal region of the HN protein of HPIV2 or SV41, which includes the 53 membrane proximal amino acids of the stalk region, determines the specificity for the homologous F protein in promoting cell-cell fusion (35). Similarly, our current study has shown that the chimeric HN protein CH5-41, in which the aforementioned amino terminal region of the SV41 HN protein has been replaced with the PIV5 HN counterpart, exhibits clear specificity for the PIV5 F protein. Although the 3-D structure of the HN stalk region remains to be clarified, the secondary structure prediction of the PIV5 HN protein in the absence of bound receptor has indicated that the stalk region is predominantly composed of α helix (39). If that were the case, the length of the 53 membrane proximal amino acids of the stalk region could be calculated as about 8.0 nm, while the length would be about 18.6 nm provided that it is in β sheet conformation. On the other hand, the heights of the identified domains (M1, M2, and B) of the PIV5 F protein from the membrane surface is 5.8 to 9.7 nm as deduced from the total length (about 12 nm) of the ectodomain reported by Connolly and colleagues (2). Thus, presumptive physical interaction of the identified domains in the F head region with the HN stalk region could become reality, dependent on the conformation of the stalk region, which might be transfigured after receptor binding.
Interestingly, the F protein of canine distemper virus (CDV) strain Onderstepoort is able to induce cell-cell fusion either with the homologous attachment protein (hemagglutinin [H]) or with the heterologous measles virus (MV) H protein, while the F protein of CDV strain Lederle induces fusion only with the CDV H protein (12). CDV and MV are the closely related members of the genus Morbillivirus of the subfamily Paramyxovirinae (11). It has been shown that mutations of four amino acids (Arg-164, Gly-219, Val-233, and Arg-317) of the CDV Onderstepoort F protein impairs the ability to induce fusion with the MV H protein without significantly affecting the ability to induce fusion with the CDV H protein (12). Among the identified four amino acids, Arg-164, Gly-219, and Val-233 are exposed on the F trimer surface (12) and thus could be directly involved in the interaction with the MV H protein. Importantly, these observations indicate that an unidentified amino acid(s) of the CDV Onderstepoort F protein is responsible for the interaction with the homologous CDV H protein. Similarly, our present data suggest that an unidentified domain(s) of the PIV5 F protein is responsible for the interaction with the homologous PIV5 HN protein, since the PIV5 F protein cannot be fully converted to an SV41 HN-specific protein even if all the three domains (M1, M2, and B) have been replaced with the SV41 F counterparts (Fig. 5C). It may be that the SV41 HN-derived domains M1, M2, and B are enough for B+M(1+2) to interact with the SV41 HN protein, while the unidentified domain(s) of B+M(1+2) can still mediate its interaction with the PIV5 HN protein. This hypothesis seems to depend on a premise that the F-interacting site of the HN protein is flexible; such flexibility has recently been reported for the PIV5 HN stalk region (39). In all likelihood, domains M1, M2, and B of the PIV5 F protein and amino acid residues Arg-164, Gly-219, and Val-233 of the CDV F protein, whose PIV5 F counterparts are Ala-47, Thr-97, and Ala-111, respectively (Fig. 2C), are the common constituents of the presumptive multiple domains of the F protein that mediate HN-F and H-F interaction. Alternatively, it is possible that the domains of the F protein that are involved in the HN-F interaction may not be identical to those involved in the H-F interaction.
Surprisingly, B+M(1+2) has proved to induce prominent cell-cell fusion even in the absence of coexpressed HN protein. It is known that the F protein of PIV5 strain W3A induces cell-cell fusion by itself (21), while the F proteins of most parainfluenza viruses, including PIV5 strain WR, require their homologous HN proteins for the induction of cell-cell fusion (7, 11). Interestingly, the L22P mutant of the PIV5 WR F protein, in which the leucine at position 22 has been replaced with the PIV5 W3A F counterpart, mediates extensive cell-cell fusion in the absence of the HN protein (7). Similarly, the G3A or G7A mutation in the fusion peptide renders the PIV5 WR F protein HN independent in terms of cell-cell fusion-inducing activity (24), and the L539A or L548A mutation in the cytoplasmic domain of the F protein of PIV5 strain SER eliminates the HN protein requirement for cell-cell fusion (27). In the case of NDV, on the other hand, the N211A, L289A, I463A, or I463F mutation in the F protein eliminates the HN protein requirement for cell-cell fusion (1, 26). These findings suggest that substitution of a single amino acid at various positions of the parainfluenza virus F protein bestows the HN-independent fusion activity on the F protein, although a given amino acid may confer an HN-independent phenotype only on certain F protein backgrounds (8, 9, 29). It has not been clarified, however, how substitution of these amino acids mediates destabilization of the F proteins and whether the destabilized F proteins require an interaction with some cellular molecule in order to undergo the conformational changes. Interestingly, the PIV5 WR F protein can mediate cell-cell fusion by itself when it is heated at 53°C, while the S443P mutant of the PIV5 W3A F protein, which is functionally equivalent to the PIV5 WR F mutant L22P, can do so even at 22°C, suggesting that the presence of proline residues at positions 22 and 443 destabilizes the F protein and thereby decrease the energy required for triggering the conformational changes in the absence of the HN protein (22). It is conceivable in this context that the role of the HN protein in promoting fusion is to provide activating energy to the F protein via physical HN-F interaction. As the result, the F protein would be destabilized and would undergo the conformational changes that lead to fusion. Provided that physical interaction between the HN protein stalk region and the HN-interacting domains of the F protein results in destabilization of the F protein, then mutation or chimeric recombination of the HN-interacting domains might result in destabilization of the F molecule in some cases. We therefore envisage that replacement of domain B of M(1+2) with the SV41 F counterpart somehow destabilizes the resultant B+M(1+2), which then undergoes the conformational changes in the absence of the HN protein but less efficiently than it does in the presence of the HN protein. It is of interest to note in this context that the PIV5 F counterpart (Leu-275) of the Leu-289 of the NDV F protein is located at the carboxyl terminus of domain B (Fig. 2B). The L289A mutation eliminates the HN protein requirement for cell-cell fusion as described above.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (grant 20590470) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Footnotes
Published ahead of print on 26 January 2011.
REFERENCES
- 1.Ayllón, J., E. Villar, and I. Muñoz-Barroso. 2010. Mutations in the ectodomain of Newcastle disease virus fusion protein confer a hemagglutinin-neuraminidase-independent phenotype. J. Virol. 84:1066-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Connolly, S. A., G. P. Leser, H. S. Yin, T. S. Jardetzky, and R. A. Lamb. 2006. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 103:17903-17908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng, R., A. M. Mirza, P. J. Mahon, and R. M. Iorio. 1997. Functional chimeric HN glycoproteins derived from Newcastle disease virus and human parainfluenza virus-3. Arch. Virol. 13(Suppl.):115-130. [DOI] [PubMed] [Google Scholar]
- 4.Heminway, B. R., Y. Yu, and M. S. Galinski. 1994. Paramyxovirus mediated cell fusion requires co-expression of both the fusion and hemagglutinin-neuraminidase glycoproteins. Virus Res. 31:1-16. [DOI] [PubMed] [Google Scholar]
- 5.Hu, X., R. Ray, and R. W. Compans. 1992. Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainfluenza viruses. J. Virol. 66:1528-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iorio, R. M., V. R. Melanson, and P. J. Mahon. 2009. Glycoprotein interactions in paramyxovirus fusion. Future Virol. 4:335-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito, M., et al. 1997. Role of a single amino acid at the amino terminus of the simian virus 5 F2 subunit in syncytium formation. J. Virol. 71:9855-9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito, M., M. Nishio, H. Komada, Y. Ito, and M. Tsurudome. 2000. An amino acid in the heptad repeat 1 domain is important for the haemagglutinin-neuraminidase-independent fusing activity of simian virus 5 fusion protein. J. Gen. Virol. 81:719-727. [DOI] [PubMed] [Google Scholar]
- 9.Ito, M., et al. 2009. Effects of multiple amino acids of the parainfluenza virus 5 fusion protein on its haemagglutinin-neuraminidase-independent fusion activity. J. Gen. Virol. 90:405-413. [DOI] [PubMed] [Google Scholar]
- 10.Lamb, R. A., and T. S. Jardetzky. 2007. Structural basis of viral invasion: lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 17:427-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamb, R. A., and G. D. Parks. 2007. Paramyxoviridae: the viruses and their replication, p. 1449-1496. In D. M. Knipe, P. M. Howley, D. E. Griffin, M. A. Martin, R. A. Lamb, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 12.Lee, J. K., et al. 2008. Functional interaction between paramyxovirus fusion and attachment proteins. J. Biol. Chem. 283:16561-16572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGinnes, L. W., and T. G. Morrison. 2006. Inhibition of receptor binding stabilizes Newcastle disease virus HN and F protein-containing complexes. J. Virol. 80:2894-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melanson, V. R., and R. M. Iorio. 2006. Addition of N-glycans in the stalk of the Newcastle disease virus HN protein blocks its interaction with the F protein and prevents fusion. J. Virol. 80:623-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirza, A. M., J. P. Sheehan, L. W. Hardy, R. L. Glickman, and R. M. Iorio. 1993. Structure and function of a membrane anchor-less form of the hemagglutinin neuraminidase glycoprotein of Newcastle disease virus. J. Biol. Chem. 268:21425-21431. [PubMed] [Google Scholar]
- 16.Morrison, T. G. 2003. Structure and function of a paramyxovirus fusion protein. Biochim. Biophys. Acta 1614:73-84. [DOI] [PubMed] [Google Scholar]
- 17.Moscona, A., and R. W. Peluso. 1991. Fusion properties of cells persistently infected with human parainfluenza virus type 3: participation of hemagglutinin-neuraminidase in membrane fusion. J. Virol. 65:2773-2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moscona, A., and R. W. Peluso. 1992. Fusion properties of cells infected with human parainfluenza virus type 3: receptor requirements for viral spread and virus-mediated membrane fusion. J. Virol. 66:6280-6827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortmann, D., et al. 1994. Proteolytic cleavage of wild type and mutants of the F protein of human parainfluenza virus type 3 by two subtilisin-like endoproteases, furin and Kex2. J. Virol. 68:2772-2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paterson, R. G., T. J. Harris, and R. A. Lamb. 1984. Fusion protein of the paramyxovirus simian virus 5: nucleotide sequence of mRNA predicts a highly hydrophobic glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 81:6706-6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paterson, R. G., S. W. Hiebert, and R. A. Lamb. 1985. Expression at the cell surface of biologically active fusion and hemagglutinin/neuraminidase proteins of the paramyxovirus simian virus 5 from cloned cDNA. Proc. Natl. Acad. Sci. U. S. A. 82:7520-7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paterson, R. G., C. J. Russell, and R. A. Lamb. 2000. Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology 270:17-30. [DOI] [PubMed] [Google Scholar]
- 23.Porotto, M., M. Murrell, O. Greengard, L. Doctor, and A. Moscona. 2005. Influence of the human parainfluenza virus 3 attachment protein's neuraminidase activity on its capacity to activate the fusion protein. J. Virol. 79:2383-2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell, C. J., T. S. Jardetzky, and R. A. Lamb. 2004. Conserved glycine residues in the fusion peptide of the paramyxovirus fusion protein regulate activation of the native state. J. Virol. 78:13727-13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell, C. J., T. S. Jardetzky, and R. A. Lamb. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024-4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sergel, T. A., L. M. McGinnes, and T. G. Morrison. 2000. A single amino acid change in the Newcastle disease virus fusion protein alters the requirement for HN protein in fusion. J. Virol. 74:5101-5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seth, S., A. Vincent, and R. W. Compans. 2003. Mutations in the cytoplasmic domain of a paramyxovirus fusion glycoprotein rescue syncytium formation and eliminate the hemagglutinin-neuraminidase protein requirement for membrane fusion. J. Virol. 77:167-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanabayashi, K., and R. W. Compans. 1996. Functional interaction of paramyxovirus glycoproteins: identification of a domain in Sendai virus HN which promotes cell fusion. J. Virol. 70:6112-6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terrier, O., et al. 2009. Engineering of a parainfluenza virus type 5 fusion protein (PIV-5 F): development of an autonomous and hyperfusogenic protein by a combinational mutagenesis approach. Virus Res. 146:115-124. [DOI] [PubMed] [Google Scholar]
- 30.Thompson, S. D., and A. Portner. 1987. Localization of functional sites on the hemagglutinin-neuraminidase glycoprotein of Sendai virus by sequence analysis of antigenic and temperature-sensitive mutants. Virology 160:1-8. [DOI] [PubMed] [Google Scholar]
- 31.Tsurudome, M., et al. 1991. Transcripts of simian virus 41 (SV41) matrix gene are exclusively dicistronic with the fusion gene which is also transcribed as a monocistron. Virology 184:93-100. [DOI] [PubMed] [Google Scholar]
- 32.Tsurudome, M., et al. 2001. Hemagglutinin-neuraminidase-independent fusion activity of simian virus 5 fusion (F) protein: difference in conformation between fusogenic and nonfusogenic F proteins on the cell surface. J. Virol. 75:8999-9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsurudome, M., et al. 2006. A mutant fusion (F) protein of simian virus 5 induces hemagglutinin-neuraminidase-independent syncytium formation despite the internalization of the F protein. Virology 347:11-27. [DOI] [PubMed] [Google Scholar]
- 34.Tsurudome, M., et al. 1998. Identification of regions on the fusion protein of human parainfluenza virus type 2 which are required for haemagglutinin-neuraminidase proteins to promote cell fusion. J. Gen. Virol. 79:279-289. [DOI] [PubMed] [Google Scholar]
- 35.Tsurudome, M., et al. 1995. Identification of regions on the hemagglutinin-neuraminidase protein of human parainfluenza virus type 2 important for promoting cell fusion. Virology 213:190-203. [DOI] [PubMed] [Google Scholar]
- 36.Tsurudome, M., et al. 2008. Effects of hemagglutinin-neuraminidase protein mutations on cell-cell fusion mediated by human parainfluenza type 2 virus. J. Virol. 82:8283-8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao, Q., and R. W. Compans. 1995. Differences in the role of the cytoplasmic domain of human parainfluenza virus fusion proteins. J. Virol. 69:7045-7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin, H. S., X. Wen, R. G. Paterson, R. A. Lamb, and T. S. Jardetzky. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan, P., G. P. Leser, B. Demeler, R. A. Lamb, and T. S. Jardetzky. 2008. Domain architecture and oligomerization properties of the paramyxovirus PIV 5 hemagglutinin-neuraminidase (HN) protein. Virology 378:282-291. [DOI] [PMC free article] [PubMed] [Google Scholar]