

Abstract
The African swine fever virus (ASFV)-encoded CD2v transmembrane protein is required for the hemadsorption of red blood cells around infected cells and is also required for the inhibition of bystander lymphocyte proliferation in response to mitogens. We studied the expression of CD2v by expressing the gene with a V5 tag downstream from the signal peptide near the N terminus and a hemagglutinin (HA) tag at the C terminus. In ASFV-infected cells, a full-length glycosylated form of the CD2v protein, which migrated mainly as a 89-kDa product, was detected, as well as an N-terminal glycosylated fragment of 63 kDa and a C-terminal nonglycosylated fragment of 26 kDa. All of these forms of the protein were localized in the membrane fraction of cells. The 26-kDa C-terminal fragment was also produced in infected cells treated with brefeldin A. These data indicate that the CD2v protein is cleaved within the luminal domain and that this occurs in the endoplasmic reticulum or Golgi compartments. Confocal microscopy showed that most of the expressed CD2v protein was localized within cells rather than at the cell surface. Comparison of the localization of full-length CD2v with that of a deletion mutant lacking all of the cytoplasmic tail apart from the 12 membrane-proximal amino acids indicated that signals within the cytoplasmic tail are responsible for the predominant localization of the full-length and C-terminal 26-kDa fragment within membranes around the virus factories, which contain markers for the Golgi compartment. Processing of the CD2v protein was not observed in uninfected cells, indicating that it is induced by ASFV infection.
African swine fever (ASF) is a severe hemorrhagic fever of domestic pigs that results in major economic losses in affected countries. The disease is endemic in many sub-Saharan African countries and Sardinia. Following its introduction to Georgia, in the Caucasus region, in 2007, ASF spread to neighboring countries, including the Russian Federation, where outbreaks have been reported from 9 states (International Office of Epizootics World Animal Health Database [OIE WAHID]) (8). Wild suids, including warthogs and bushpigs, are infected but show few if any disease signs. Soft ticks of the Ornithodoros spp. act as vectors and reservoirs and can remain infected for long periods (1, 10).
African swine fever virus (ASFV) is a large cytoplasmic DNA virus and is the only member of the family Asfarviridae. The virus carries between 151 and 167 open reading frames (ORFs) depending on the isolate. Many of these are not essential for replication in host cells but play important roles in virus-host interactions, and they include genes encoding proteins involved in evading the host's defense system. The target cells for ASFV replication are those of monocyte-macrophage lineage, and by manipulation of their functions, ASFV can effectively modulate the host response to infection (9, 28, 31).
The protein encoded by the ASFV EP402R ORF (designated CD2v, CD2-like, 5HL, or PEP402R in the literature) is a transmembrane protein with an N-terminal signal peptide and a single membrane-spanning domain that has similarities in its extracellular or luminal domain to the host CD2 protein (5, 26). The virus CD2v protein shares with host CD2 the two predicted Ig superfamily (IgSF) domains, including an NH2-terminal V-like domain lacking the usual disulfide bridge and a membrane-proximal C-like domain. CD2v and host CD2 also share the function of mediating the formation of red blood cell rosettes around cells expressing the protein. The sequence of the luminal domain of the virus CD2v protein varies considerably between isolates from about 50% to 100% amino acid identity. This ASFV protein is required for the hemadsorption (HAD) of red blood cells around ASFV-infected cells and also for the association of extracellular virions with red blood cells (5, 23, 26). One report showed that deleting the ORF EP153R, which encodes a C-type lectin, reduced the numbers of red blood cells binding to infected cells, suggesting that EP153R was also involved in HAD (13). Although most isolates of ASFV cause HAD, some field isolates that are non-HAD have been characterized, and these include both virulent and nonvirulent isolates (4, 20, 30). Deletion of the EP402R gene from the genome of a virulent isolate was shown to have no impact on mortality caused by infection in pigs, but there was a delay in the onset of viremia and of virus dissemination to tissues (5). Infection in vitro of peripheral blood leukocytes with ASFV was shown to inhibit the ability of lymphocytes to proliferate in response to mitogens (5). This inhibition was abrogated when the EP402R gene was deleted, suggesting that the CD2v protein is required for this function. Expression of the CD2v protein was also shown to enhance virus infection in the Ornithodoros erraticus tick vector by increasing uptake of virus across the tick gut (27).
The cytoplasmic tail of the CD2v protein does not share significant similarity with this domain of the host CD2 protein. The domain is well conserved in sequence between the EP402R ORFs of different ASFV isolates, apart from a region encoding variable numbers of proline-rich tandem repeats of the sequence PPPKPC. These proline-rich repeats are required for binding of the CD2v protein to the host actin-binding adaptor protein SH3P7/mabp1 (17).
In the current study, we determined the expression and localization of the CD2v protein in infected and uninfected cells using antibodies to different epitope tags fused near the N terminus and C terminus. In infected cells, we identified, in addition to full-length protein, fragments of 63 kDa and 26 kDa containing the N and C termini of the CD2v protein, respectively. These fragments are predicted to be produced by cleavage within the extracellular or luminal domain, and both localized to membrane compartments. These N- and C-terminus-containing CD2v fragments were not detected in uninfected cells, showing that a virus-induced processing event is involved. The data suggest that these smaller fragments of the CD2v protein may have a function in infected cells, possibly related to the immunomodulatory role of the protein.
MATERIALS AND METHODS
Infection and transfection.
Vero cells were seeded into 35-mm wells of a 6-well plate at a density of 1 × 106 in Dulbecco's modified Eagle's medium (DMEM)-10% fetal calf serum (FCS)-50 U ml penicillin−1-50 μg ml streptomycin−1 and incubated at 37°C and 5% CO2 for 24 h. The cells were transfected with 2.5 μg of plasmid DNA using TransIT-LTI (Mitus Bio LLC) according to the manufacturer's recommendations. After 5 h at 37°C and 5% CO2, the transfection reagent was replaced with an ASFV BA71V isolate at a multiplicity of infection of 3 to 5, and incubation was continued for 1 h. The virus was removed and replaced with 2 ml DMEM-2% FCS and incubated at 37°C for various times. Tunicamycin and brefeldin were added in some experiments at concentrations of 1 μg ml−1 and 15 μg ml−1, respectively.
Plasmids.
A fragment containing the EP402R (CD2v) gene from the Malawi LIL20/1 isolate, without the translation stop codon and with a 120-bp upstream sequence containing the promoter region, was amplified by PCR from clone LMw8 (11). The primers used were 5′-ATAAAGCTTGATATTATAAAAAACAAAAAC-3′ (forward) and 5′-ATAGGATCCAATAATTCTATCTACGTGAATAAGCG-3′ (reverse). This fragment was cloned into the HindIII and BamHI sites in the pcDNA3 vector (Clontech) to generate plasmid pcDNA3CD2v. A double-stranded oligonucleotide encoding the influenza virus hemagglutinin (HA) epitope tag and including a 3′ stop codon, with upstream BamHI and downstream XbaI sites, was cloned downstream and in frame with the CD2v gene. The oligonucleotides were 5′-ATAGGATCCATGGCTTACCCATACGACGTACCAGACTACGCATCACTATGATCTAGAATA-3′ and 5′-TATTCTAGATCATAGTGATGCGTAGTCTGGTACGTCGTATGGGTAAGCCATGGATCCTAT-3′. The plasmid derived (CD2vHA) was used as the template to construct mutant versions of the gene, one with the sequences encoding proline-rich repeats deleted (CD2v-proHA) and the other with all of the cytoplasmic tail deleted, apart from a sequence encoding 12 amino acids proximal to the transmembrane domain (CD2v-cytoHA). The primers used to amplify a fragment from upstream of the CD2v promoter to upstream of the sequence encoding the proline-rich repeats were as follows: forward primer, 5′-ATAAAGCTTGATATTATAAAAAACAAAAAC-3′, and reverse primer, 5′-TATGAATTCTTTGGGTAGGGGAGATGG-3′. This fragment was cloned into the HindIII and EcoRI sites in the pcDNA3 vector. A fragment from downstream of the sequence encoding proline-rich repeats to the 3′ end of the gene, including the HA tag, was amplified using the primers 5′-TATGAATTCTCATCTGAATCATGTTCT-3′ and 5′-TATGCGGCCGCTCATAGTGATGCGTAGTCTGGT-3′, which contained an EcoRI site at the 5′ end and a NotI site at the 3′ end. This fragment was cloned in the pcDNA3 vector containing the fragment encoding sequences upstream of the proline-rich repeats to give plasmid CD2v-proHA. To construct the mutant gene CD2v-cytoHA, which lacks most of the cytoplasmic tail, the same forward primer was used as for the CDv2-pro plasmid, and the reverse primer, 5′-TAGATATCAACATGTTTTTTTCTTTTTCG-3′, was from the sequence encoding 12 amino acids downstream of the transmembrane region. The fragment amplified was cloned into the pcDNA3 vector at the HindIII and EcoRV sites. A double-stranded oligonucleotide encoding the HA tag and including a 3′ stop codon, with EcoRV and NotI sites, was cloned downstream of the CD2v gene sequences to generate plasmid CD2v-cytoHA. Two plasmids were constructed with a sequence encoding a V5 epitope tag downstream of the predicted signal peptide, full-length EP402R (SV5CD2vHA) and the mutant gene with most of the cytoplasmic tail deleted (SV5CD2v-cytoHA). The promoter and the signal peptide were amplified using the following primers: forward, 5′-ATAAAGCTTGATATTATAAAAAACAAAAAC-3′, containing a HindIII site, and reverse, 5′-ATAGGATCCCTAAAATTATTGTTTTATTAAA-3′, containing a BamHI site. For both these plasmids, the same primers were used to amplify the remaining part of the gene: forward, 5′-ATAGAATTCGATAGTAATATTACTAATGAT-3′, containing an EcoRI site, and reverse, 5′-TATGCGGCCGCTCATAGTGATGCGTAGTCTGGT-3′, containing a NotI site. The templates used to amplify these fragments were CD2vHA and CD2v-cytoHA. A double-stranded oligonucleotide encoding the V5 epitope tag was inserted between the upstream and downstream parts of the gene using BamHI and EcoRI sites. These oligonucleotides were 5′-ATAGGATCCATGGGAAAGCCGATCCCAAACCCTTTGCTGGGATTGGACTCCACCCTCGAGGAATTCATA-3′ and 5′-TATGAATTCCTCGAGGGTGGAGTCCAATCCCAGCAAAGGGTTTGGGATCGGCTTTCCCATGGATCCTAT-3′. To clone the EP402R (CD2v) gene downstream from a cellular promoter, the SV5CD2vHA plasmid was used as the template to amplify the gene from the ATG start site. The primers used were as follows: forward, 5′-TACCCCGGGTGTGCATAAAATGATAATAATAC-3′, and reverse, 5′-TATGCGGCCGCTCATAGTGATGCGTAGTCTGGT-3′. The amplified fragment was cloned into the vector pTriex 1.1 neo (Novagen) at the SmaI and NotI sites to give plasmid TriexSV5CD2vHA. All PCRs were carried out using AccuPrime pfx Supermix (Invitrogen). Inserts were verified by PCR and sequencing.
Immunoprecipitation and Western blotting.
Cells were washed with cold phosphate-buffered saline and lysed in 500 μl of RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 1 mM EDTA, 5 μg ml aprotinin−1, 5 μg ml leupeptin−1, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]). The cell lysate was centrifuged for 10 min at 13,000 rpm to remove nuclei and debris. The supernatants were incubated overnight with rotation at 4°C with either anti-HA matrix (11815016001; Roche) or anti-V5 agarose affinity gel (A7345; Sigma). The beads were washed with ice-cold RIPA buffer and resuspended in SDS loading buffer containing dithiothreitol (DTT). Endo-β-N-acetylglucosaminidase H (endo H) and N-glycosidase F (PNGase F) (P0702S and P0704S, respectively; NEB) digestion was performed on the cell lysates, according to the manufacturer's instructions, before immunoprecipitation. The immunoprecipitates were resolved on SDS-PAGE before being transferred to Hybond ECL membranes (RPN303D; GE Healthcare). The membranes were blocked for 20 min in PBS with 5% milk powder and 0.2% Tween 20 and then incubated in the diluted antibodies overnight at 4°C. The membranes were washed with PBS containing 0.2% Tween 20, and then the bound antibodies were detected using LumiGlo reagent (7003; Cell Signaling Technology) following exposure to X-ray films.
Confocal microscopy.
Cell monolayers were grown on 19-mm glass coverslips in 12-well plates and fixed for 20 min in phosphate-buffered saline containing EDTA (PBSe) containing 4% paraformaldehyde. The cells were permeabilized in 0.2% Triton X-100 in PBSe for 5 min and then incubated in 0.2% gelatin in PBSe for 10 min to inhibit nonspecific antibody binding. They were incubated with primary antibody diluted in 0.2% gelatin for 20 min at room temperature (RT) and washed 4 times. Secondary antibody diluted in 0.2% gelatin was incubated for 20 min. After being washed, the cells were mounted onto glass slides using VectaShield mounting medium with DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories). The cells were visualized using a Leica TCS SP2 confocal microscope with a 63× lens.
Cell fractionation.
At 18 h postinfection, the cells were washed with cold phosphate-buffered saline. Five hundred microliters of cold cytosolic fractionation buffer (250 mM sucrose, 4 mM HEPES, pH 7.4, 2 mM EDTA, 0.1% [vol/vol] Triton X-100, protease inhibitors) was added to the cells, and incubation was continued at room temperature for 5 min. The supernatant was transferred to a fresh tube, and 500 μl of cold membrane fractionation buffer was added to the cells (150 mM Tris-HCl, pH 8.8, 10 mM KCl, 1 mM EDTA, 0.2% [vol/vol] NP-40, 10% [vol/vol] glycerol, protease inhibitors). After a 5-min incubation at room temperature, the supernatants were transferred to a fresh tube and clarified by centrifugation in a microcentrifuge for 10 min. SDS-PAGE gel-loading buffer was added to the supernatants.
Antibodies.
The antibodies used included mouse monoclonal antibody against the Golgi 58,000-molecular-weight (58K) protein; formiminotransferase cyclodeaminase (FTCD) (G2404; Sigma), used at a dilution of 1:400 for Western blotting; and the mouse monoclonal antibody against annexin 1 (sc-12740; Santa Cruz), used at 1:500. The horseradish peroxidise (HRP)-conjugated V5 tag antibody (MCA1360P; AbD Serotec) and the HRP-conjugated HA tag antibody (11022100; Roche) were both used at 1:2,000. Anti-mouse-HRP and anti-rabbit-HRP antibodies (P0260 and P0448, respectively; Dako) were used at 1:1,000. Mouse anti-V5 (MCA1360; AbD Serotec) and rabbit anti-HA (Sc-805; Santa Cruz) were both used at 1:300 for confocal microscopy. Rabbit anti-endoplasmic reticulum (ER) (ERp60) (26a) antibody was used at 1:300, mouse anti-Golgi GM130 (BD Biosciences) at 1:50, and mouse anti-VP72 (4H3 [6]) at 1:20. The secondary antibodies used for confocal microscopy were from Molecular Probes and were used at 1:500: anti-mouse 488 (A11001), anti-rabbit 488 (A11006), anti-mouse 568 (A11061), and anti-rabbit 568 (A11077).
RESULTS
The CD2v protein is expressed in different molecular weight forms in ASFV-infected cells.
In order to detect the full-length CD2v protein, as well as fragments that lack either the C terminus or N terminus of the protein, a dual-epitope-tagged CD2v gene was constructed. A sequence encoding the V5 tag was inserted near the 5′ end of the CD2v gene downstream from the signal sequence. A sequence encoding the HA tag was fused at the 3′ end of the gene. This dual-tagged CD2v gene was cloned, under the control of its own ASFV promoter, in the plasmid SV5CD2vHA, so that it could be expressed in ASFV-infected cells. Typical red blood cell rosettes were formed around porcine macrophages that were transfected with the SV5CD2vHA plasmid and infected with a non-HAD ASFV isolate, OURT88/3 (data not shown). This demonstrates that addition of the SV5 and HA tags did not alter the ability of the CD2v protein to cause HAD. Extracts from ASFV-infected Vero cells that had been transfected with SV5CD2vHA and harvested at 18 h postinfection were immunoprecipitated with anti-HA or anti-SV5 antibodies, and then the immunoprecipitated proteins were separated by SDS-PAGE and Western blots were probed with anti-V5 antibody or with anti-HA antibody. Probing with the anti-HA antibody detected bands of 89 and 26 kDa (Fig. 1A, lane 1). The 89-kDa band is predicted to be a full-length glycosylated CD2v. The 26-kDa band is likely to be a C-terminal cleavage fragment. Probing with the anti-V5 antibody to detect N-terminally tagged forms of the CD2v protein identified 89- and 63-kDa bands (Fig. 1A, lane 7, and 1B, lane 1). A more diffuse band of about 105 to 110 kDa was detected with both anti-V5 and anti-HA antibodies. The 63-kDa band detected with anti-V5 is of the predicted size to represent a form of the protein from which the C-terminal 26-kDa fragment, detected with anti-HA antibody, has been cleaved.
FIG. 1.

Western blot showing CD2v protein expression in ASFV-infected Vero cells. Vero cells were transfected with plasmid SV5CD2vHA expressing the full-length EP402R (CD2v) protein tagged at the C terminus with the HA epitope tag and near the N terminus downstream from the signal peptide with the SV5 tag. The cells were then infected with the ASFV BA71V isolate and lysed at 18 h postinfection. (A) Lanes 1 to 6, proteins precipitated from lysates with anti-HA matrix, separated by SDS-PAGE, and blotted onto membranes. Immunoprecipitated proteins were detected by probing with anti-HA coupled to HRP, followed by enhanced chemiluminescence (ECL). In lanes 7 to 12, the lysates were immunoprecipitated using anti-V5 agarose, and the V5-tagged proteins were detected by probing Western blots of the immunoprecipitated proteins using anti-V5 coupled to HRP. Lanes 1 and 7, untreated lysates; lanes 2 and 8, lysates from cells treated with tunicamycin overnight at 1 μg ml−1 before being harvested; lanes 3 and 9, lysates from cells treated with brefeldin overnight at 15 μg ml−1 before being harvested. In lanes 4 and 10, cell lysates were digested with endo H prior to immunoprecipitation, and in lanes 5 and 11, the lysates were treated with PNGase F prior to immunoprecipitation. Lanes 6 and 12 show lysates from mock-transfected cells. (B) Cells transfected with a plasmid expressing SV5CD2vHA were infected with the ASFV BA71V isolate, and SV5-tagged proteins were detected as described above. Lane 1 shows untreated lysate, and lane 2 shows lysates from cells treated overnight with tunicamycin at 1 μg ml−1. The positions of molecular weight markers run in parallel are shown. The lanes shown in panel A were run on the same gel, and those in panel B were run on a different gel.
Cells were incubated with tunicamycin to inhibit glycosylation and to determine the effect on production of the N- and C-terminal fragments detected using antibodies against V5 and HA tags (Fig. 1A, lanes 2 and 8, and 1B, lane 2). Extracts were separated by SDS-PAGE, blotted onto membranes, and then probed with anti-V5 or anti-HA antibody. Probing blots with the anti-HA antibody did not detect the 89-kDa band, but two additional bands of about 42 kDa were detected. The 26-kDa C-terminal fragment was detected and migrated with the same mobility as that in cells that had not been treated with tunicamycin (Fig. 1A). In extracts from tunicamycin-treated cells, the 89- and 63-kDa bands were not detected using the anti-V5 antibody, but two additional bands around 42 kDa were detected (Fig. 1A, lane 8). A smaller band migrating at 19 kDa was detected in blots from gels that had been run for a shorter time, and this is expected to be the nonglycosylated N-terminal 69-kDa fragment of CD2v (Fig. 1B, lane 2). Samples were digested with endoglycosidases H and F before separation by SDS-PAGE and blotting (Fig. 1A, lanes 4, 5, 10, and 11). Probing blots with anti-HA antibodies detected bands of 47 kDa and 26 kDa and a faint smear at 105 to 110 kDa in samples digested with endoglycosidase H (Fig. 1A, lane 4) and 50 and 26 kDa in samples digested with endoglycosidase F (Fig. 1A, lane 5). Probing with anti-V5 detected bands of 47 and 29 kDa in samples digested with endoglycosidase H and a small amount of a diffuse band of about 105 to 110 kDa (Fig. 1A, lane 10) and bands of 50 and 32 kDa in samples digested with endoglycosidase F (Fig. 1A, lane 11). These data show that glycosylated products of 89 kDa and a more diffuse band of 105 to 110 kDa are expressed from the full-length CD2v ORF, as well as two additional products, a 26-kDa nonglycosylated product containing the C terminus and a 63-kDa glycosylated product containing the N terminus. These products are most likely derived from the CD2v protein by proteolytic cleavage.
The cellular compartment in which this processing takes place was investigated by treatment of cells with brefeldin A to inhibit transport of proteins beyond the Golgi compartment (Fig. 1A, lanes 3 and 9). In these experiments, the 26-kDa fragment was detected in blots probed with the anti-HA antibody (Fig. 1A, lane 3) but, as expected, not in those treated with anti-V5 antibody (Fig. 1A, lane 9). These results suggest that the processing of CD2v to produce the 26-kDa fragment takes place within the ER or Golgi compartment.
Deletion of most of the cytoplasmic tail of the CD2v protein does not interfere with production of N- and C-terminal fragments of the protein.
In order to determine if mutations introduced into the cytoplasmic tail of the CD2v protein interfere with production of the C-terminal products, mutant forms of the CD2v gene tagged at the 3′ end with the HA tag were constructed (Fig. 2B). The mutations introduced into the 3′ end of the CD2v gene included deletion of the sequence encoding the proline-rich repeats (CD2v-proHA) and deletion of most of the cytoplasmic tail apart from the sequences encoding 12 amino acids proximal to the transmembrane domain (CD2v-cytoHA). Plasmids expressing full-length CD2v-HA, CD2v-proHA, and CD2v-cytoHA were transfected into Vero cells, which were infected with the ASFV BA71V isolate. Cell extracts were prepared 18 h postinfection and immunoprecipitated with anti-HA antibodies, and then the immunoprecipitated proteins were separated by SDS-PAGE, and Western blots were probed with anti-HA antibodies. This detected the full-length CD2v and fragments containing the C terminus (Fig. 2C). As expected, fragments of 89 and 26 kDa were detected in extracts from cells transfected with the plasmid expressing wild-type CD2vHA (Fig. 2C, lane 1). In extracts from cells expressing mutant forms of the protein (Fig. 2C, lanes 2 and 3), shorter fragments were detected, as expected. In extracts from cells transfected with the mutant CD2v-proHA gene, which does not contain sequences encoding the proline repeats, fragments of 20 kDa and a fragment that migrates slightly faster than the 89-kDa band were detected (Fig. 2C, lane 2). In extracts from cells transfected with the CD2v-cytoHA plasmid, bands of 13 kDa and a band migrating slightly faster than 89 kDa were detected (Fig. 2C, lane 3). In parallel, cells were treated with tunicamycin before being harvested. In extracts from these cells (Fig. 2C, lanes 5, 6, and 7), the smaller fragments detected were the same size as those detected in untreated cells (26, 20, or 13 kDa). In contrast, the larger fragments were reduced in size to about 42 kDa in cells transfected with the plasmid CD2vHA expressing the wild-type CD2v (Fig. 2C, lane 5), 40 kDa in cells transfected with the CD2v-proHA plasmid (Fig. 2C, lane 6), and 26 kDa in cells transfected with CD2v-cytoHA (Fig. 2C, lane 7). These larger fragments were the sizes predicted for the full-length nonglycosylated products of the wild-type or mutant genes. These results show that shorter C-terminal fragments were produced from each of the mutant proteins, and therefore, the amino acids deleted do not contain sequences required for any processing event. The sizes of these smaller fragments, estimated from SDS-PAGE, suggest that they include sequences upstream of the transmembrane domain. These estimates, based on the molecular weight of the C-terminal fragments, suggest that up to 90 amino acids upstream of the transmembrane domain may be present in the C-terminal fragments.
FIG. 2.

Western blot showing expression of mutant forms of CD2v. (A) Diagram indicating the locations of domains in the CD2v protein. The scale in amino acids is shown at the top. Domains indicated by boxes or underlined include the signal peptide (SP), immunoglobulin superfamily-like domains (IG), transmembrane domain (TM), acidic domain, and proline-rich domain. (B) Sequences (numbered on the right) of mutant forms of the CD2v protein. Sequence 1 is the sequence of the N-terminal 245 residues encoding the extracellular/luminal and transmembrane domains of the CD2v protein of the Malawi LIL 20/1 isolate. The sequence of the inserted SV5 tag is underlined. Sequence 1 is included in all of the mutant proteins. The signal peptide and transmembrane domains are in boldface. Sequences 2 to 4 are the sequences of the cytoplasmic domains of the full-length CD2v cytoplasmic tail, SV5CD2vHA (sequence 2); the deletion mutant lacking proline-rich repeats, SV5CD2v-proHA (sequence 3); and the deletion mutant lacking most of the cytoplasmic tail, SV5CD2v-cytoHA (sequence 4). The sequence of the C-terminal HA tag is underlined. Proline-rich-repeat sequences are in green, and acid-rich sequences are in blue. (C) Vero cells were transfected with plasmids expressing the full-length or deleted forms of the CD2v protein and infected with the ASFV BA71V isolate for 18 h. Cell lysates were immunoprecipitated using anti-HA matrix, and the immunoprecipitated proteins were separated by SDS-PAGE and blotted onto membranes. The blots were probed with anti-HA-HRP, and bound antibodies were detected by ECL. Lanes 1 and 5, extracts from cells expressing CD2vHA; lanes 2 and 6, extracts from cells expressing CD2v-proHA; lanes 3 and 7, extracts from cells expressing CD2v-cytoHA. Lanes 5, 6, and 7 show extracts from cells treated with tunicamycin (1 μg ml−1) overnight. Lane 4 shows extracts from mock-transfected Vero cells. The positions of molecular weight markers run in parallel are shown.
The data presented above suggest that processing occurs within the extracellular or luminal domain of the CD2v protein. To assess the localization of the cleaved products, cells were transfected with the plasmid expressing SV5CD2vHA and infected with ASFV. After 18 h, cell extracts were prepared and fractionated into membrane and cytosolic compartments and then separated by SDS-PAGE and probed by Western blotting with anti-HA antibody. This showed (Fig. 3) that both the 89-kDa and 26-kDa products of SV5CD2vHA were localized almost entirely in the membrane fraction rather than the cytosolic fraction (Fig. 3, lanes 2 and 3). The anti-V5 antibody detected the 63-kDa and 89-kDa fragments in total cell extracts and the membrane fraction (Fig. 3, lanes 1 and 3). In parallel, blots were probed with antibodies against annexin, as a control for the cytosolic compartment, and the Golgi 58-kDa protein FTCD as a control for the membrane compartment. As expected, annexin was not detected in the membrane compartment, and the Golgi protein was detected largely in the membrane fraction (Fig. 3, lanes 2 and 3).
FIG. 3.
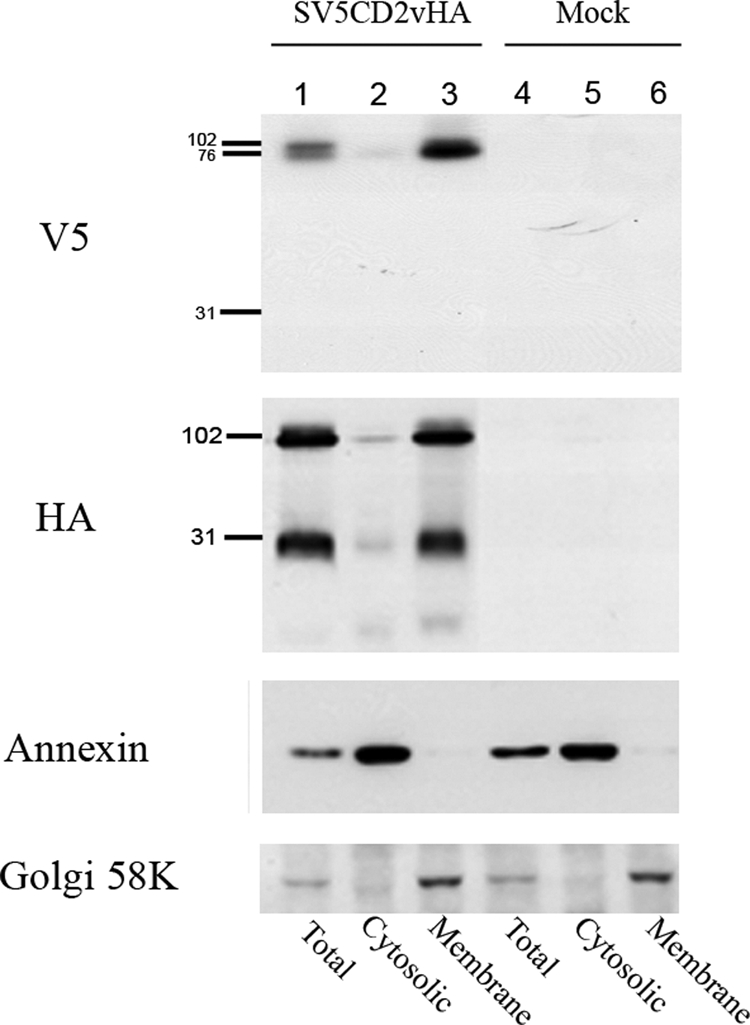
Western blot showing the localization of CD2v protein fragments in membrane or cytosolic cell fractions in ASFV-infected cells. Vero cells were transfected with plasmids expressing SV5CD2vHA and infected with the ASFV BA71V isolate. At 18 h postinfection, the cells were lysed and separated into cytosolic or membrane fractions. Lanes 1, 2, and 3, extracts from cells expressing the SV5CD2vHA protein; lanes 4, 5, and 6, extracts from mock-transfected cells. Lanes 1 and 4 show total cell extracts prepared by lysis in RIPA buffer, and lanes 2 to 6 show extracts fractionated into the cytosolic (lanes 2 and 5) and membrane (lanes 3 and 6) fractions. Extracts were separated by SDS-PAGE and blotted onto membranes. The blots were probed as indicated on the left with anti-V5-HRP, anti-HA-HRP, anti-annexin followed by anti-mouse-HRP, and anti-Golgi 58K protein followed by anti-mouse-HRP. Bound antibodies were detected by ECL.
Since the N-terminal 63-kDa fragment of the CD2v protein does not contain the transmembrane domain, it may be secreted from cells. However, analysis of supernatants from infected cells expressing the SV5CD2vHA protein by immunoprecipitation with anti-SV5 antibody, followed by Western blotting, did not detect a secreted form of the protein (data not shown). Therefore, we assume that this fragment of the protein is retained in the membrane fraction of cells through an uncleaved signal peptide that remains inserted into the membrane or is associated with the luminal faces of membranes by association with membrane proteins, or possibly both of these options.
Localization of CD2v in infected cells.
To determine if signals in the cytoplasmic tail are involved in the localization of the CD2v protein, plasmids expressing the mutant protein CD2v-cytoHA and the full-length protein CD2vHA were transfected into Vero cells, which were then infected with the ASFV BA71V isolate. Cells were fixed and permeabilized and then stained with anti-HA antibody, which detects the C-terminal HA tag, and with an antibody against the major capsid protein of ASFV, anti-VP72. Most of the full-length CD2vHA protein (Fig. 4A, row 1, shown in red) was localized around the cytoplasmic virus factories identified by staining with anti-VP72 (shown in green), as described previously (17). CD2v-cytoHA had a more dispersed cytoplasmic pattern (Fig. 4A, row 2). These results suggest that sequences in the cytoplasmic tail of the CD2v protein are required for the predominant localization of the CD2v forms detected using anti-HA antibodies around virus factories.
FIG. 4.

Localization of CD2V in ASFV-infected cells. Vero cells were grown on coverslips and transfected with plasmids expressing full-length CD2v or deletion mutants and then infected with the ASFV BA71V isolate. The cells were fixed with paraformaldehyde at 18 h postinfection, permeabilized, and then stained with primary and secondary antibodies as indicated at the bottom and visualized by confocal microscopy. (A) Images of cells probed with anti-VP72 (green) and anti-HA (red), followed by appropriate Alexa Fluor-stained secondary antibodies. Row 1 shows cells expressing CD2vHA, and row 2 shows cells expressing CD2v-cytoHA. (B) Rows 1, 2, and 3 show different cells expressing SV5CD2vHA. (C) Rows 1, 2, and 3 show different cells expressing SV5CD2v-cytoHA. The SV5CD2vHA and SV5CD2v-cytoHA proteins were detected using anti-V5 tag (green) and anti-HA tag (red) antibodies, followed by the appropriate Alexa Fluor-conjugated secondary antibodies, and visualized by confocal microscopy. Merged images with DNA stained with DAPI are on the right.
The localization of full-length CD2v protein and fragments containing the N-terminal SV5 tag was compared with that of the fragments containing the C-terminal HA tag by transfecting the wild-type SV5CD2vHA plasmid or a plasmid expressing mutant SV5CD2v-cytoHA into cells that were then infected with ASFV. The cells were fixed 18 h postinfection, permeabilized, and then stained to detect either the SV5 tag (Fig. 4B and C, shown in green) or the HA tag (shown in red). As expected, anti-HA antibody stained cells transfected with the full SV5CD2vHA (Fig. 4B, shown in red) mainly around the cytoplasmic virus factories. Staining with the anti-V5 antibody showed some differences in localization in different cells (Fig. 4B). In some cells, staining was largely colocalized with the anti-HA staining around factories (Fig. 4B, row 3), whereas in other cells, more staining was detected around the nucleus and in a more dispersed pattern throughout the cytoplasm (Fig. 4B, rows 1 and 2). In these cells, various amounts of colocalization were detected with the staining observed using the anti-HA antibody.
The full-length SV5CD2vHA protein must be localized in areas where staining is detected with both the SV5 and HA antibodies, whereas areas detected only by staining with either anti-V5 or anti-HA antibody must contain either the N- or C-terminal fragment of SV5CD2vHA protein. The data are consistent with predominant localization of the full-length SV5CD2vHA protein and the C-terminal 26-kDa fragment in areas around the virus factories and localization of at least some of the N-terminal fragment in a more dispersed pattern in the cytoplasm and around the nucleus.
In Vero cells transfected with the plasmid expressing SV5CD2v-cytoHA mutant protein and then infected with ASFV strain BA71V, staining detected with both the anti-HA and anti-V5 antibodies was predominantly in a more dispersed pattern than in cells expressing the full-length SV5CD2vHA protein (Fig. 4C, rows 1 and 2). However, in some cells (Fig. 4C, row 3), a distribution of the anti-HA staining around virus factories similar to that in cells expressing the full-length SV5CD2vHA protein was observed. Varying proportions of the anti-SV5 and anti-HA staining were colocalized in cells.
To determine in which membrane compartments SV5CD2vHA or SV5CD2v-cytoHA might be present, cells were transfected and then infected and fixed at 18 h postinfection. Staining of permeabilized cells expressing the SV5CD2vHA and the SV5CD2v-cytoHA proteins with anti-V5 antibody showed partial colocalization with the staining observed using an antibody against a marker for the ER compartment (Fig. 5A and B). In contrast, most of the SV5CD2vHA protein detected with the anti-HA antibody was colocalized in areas around the virus factories with a marker for the Golgi compartment (Fig. 5C). However, most staining of the SV5CD2v-cytoHA protein with anti-HA antibody did not colocalize with the antibody against the Golgi compartment (Fig. 5D). These data suggest that the sequences deleted from the cytoplasmic tail of the SV5CD2v-cytoHA protein are required for the predominant localization, detected using the anti-HA antibody, of the SV5CD2vHA protein in areas around the virus factories, which costain with an antibody against a Golgi-localized protein.
FIG. 5.

Colocalization of SV5CD2vHA and SV5CD2v-cytoHA proteins within cellular compartments. Vero cells were grown on coverslips, transfected with plasmids expressing SV5CD2vHA or SV5CD2v-cytoHA, and infected with the BA71V ASFV isolate. The cells were fixed with paraformaldehyde at 18 h postinfection, permeabilized, stained with primary and secondary antibodies as indicated, and visualized by confocal microscopy. (A and C) Cells expressing SV5CD2vHA. (B and D) Cells expressing SV5CD2v-cytoHA. Panels A and B show staining with anti-V5 (green) and an anti-ERp60 marker (red). Panels C and D show staining with the anti-Golgi marker GM130 (green) and anti-HA (red), followed by appropriate Alexa Fluor-stained secondary antibodies. Merged images with DNA stained with DAPI are on the right.
Expression of CD2v at the cell surface.
Since the CD2v protein is required for the HAD of red blood cells around ASFV-infected cells, at least some of the SV5CD2vHA and SV5CD2v-cytoHA proteins must be expressed at the cell surface. To detect the cell surface expression of CD2v protein, Vero cells were transfected with plasmids expressing SV5CD2vHA or SV5CD2v-cytoHA and infected with the ASFV BA71V isolate. At 18 h postinfection, cells were fixed with paraformaldehyde and SV5CD2vHA or SV5CD2v- cytoHA proteins were detected by staining them with anti-SV5 antibodies, either on nonpermeabilized cells to detect cell surface protein or on permeabilized cells to also detect internal SV5CD2vHA protein. In a typical experiment on individual coverslips, more than 1,400 cells that expressed SV5CD2vHA and SV5CD2v-cytoHA were detected by staining of fixed and permeabilized cells. In contrast, staining of the same number of nonpermeabilized cells with anti-SV5 antibody detected only about 60 cells expressing either SV5CD25HA or SV5CD2v-cytoHA. These results indicate that most of the SV5CD2vHA and SV5CD2v-cytoHA proteins are present in the cytoplasm of cells rather than on the cell surface. The predominantly cytoplasmic localization of these proteins may result from their retention in the secretory system during transport to the cell surface so that they do not reach the cell surface, from recycling from the cell surface, or a combination of these two possibilities.
To determine if the CD2v protein is internalized from the cell surface, we transfected cells with the plasmids expressing SV5CD2vHA or SV5CD2v-cytoHA and infected them with the ASFV BA71V isolate. After 17 h, the cells were incubated with anti-V5 antibody for 60 min, and then they were either fixed immediately or washed to remove antibody, and incubation continued at 37°C for a further 10 min before they were fixed. Anti-V5 antibodies were detected in permeabilized or nonpermeabilized cells by staining them with Alexa Fluor 488-conjugated secondary antibody. On individual coverslips incubated with anti-V5 antibody for 60 min and then fixed, surface staining was detected on a number of cells transfected with the plasmid expressing full-length SV5CD2vHA similar to that on cells transfected with SV5CD2v-cytoHA (data not shown). In contrast, when cells were washed to remove antibody and incubation was continued for a further 10 min before they were fixed, no surface staining was detected in those cells transfected with SV5CD2vHA (data not shown), whereas some cells expressing SV5CD2v-cytoHA showed surface staining (Fig. 6C). In parallel, internalized anti-SV5 antibodies were detected by using Alexa Fluor-conjugated secondary antibodies on permeabilized cells, and similar numbers of positive cells were detected in cells transfected with the full-length SV5CD2vHA and SV5CD2v-cytoHA (Fig. 6A and B). These results suggest that cell surface-expressed SV5CD2vHA and SV5CD2v-cytoHA are internalized but that this process is more rapid for the full-length SV5CD2vHA protein.
FIG. 6.

Localization of cell surface and internalized SV5CD2vHA and SV5CD2v-cytoHA in ASFV-infected cells. Vero cells were grown on coverslips, transfected with plasmids expressing SV5CD2vHA or SV5CD2v-cytoHA, and then infected with the ASFV isolate BA71V. At 17 h postinfection, anti-V5 antibody was added to the medium (10-μg ml−1 final concentration), and incubation was continued for 1 h at 37°C. The cells were washed, fresh medium was added, and incubation continued at 37°C for 10 min before the cells were fixed with paraformaldehyde. (A and B) Cells transfected with SV5CD2vHA and SV5CD2v-cytoHA, respectively, that were permeabilized before being stained with Alexa Fluor-conjugated secondary antibody. These cells were also stained with anti-HA (red). (C) Cells transfected with SV5CD2v-cytoHA that were not permeabilized before being stained to detect the anti-V5 antibody. Merged images with DNA stained with DAPI are on the right.
Permeabilized cells containing the internalized anti-V5 antibody were also costained with the anti-HA antibody. A proportion of the internalized SV5CD2vHA colocalized with the anti-HA staining around the virus factories, indicating that at least a portion of the protein in this location is SV5CD2vHA that has been recycled from the cell surface (Fig. 6A and B). Control experiments detected no staining with secondary antibody alone and no staining of nontransfected cells with anti-SV5 antibodies.
CD2v expressed in uninfected cells is not processed.
To determine which forms of CD2v were expressed in uninfected cells, the SV5CD2vHA full-length gene was cloned under the control of a eukaryotic promoter in the Triex vector and transfected into uninfected Vero cells. After 24 h, cell extracts were prepared, separated by SDS-PAGE, and blotted. Probing with the anti-V5 antibodies detected a single band of 89 kDa in uninfected cells (Fig. 7A, lane 2), whereas in infected cells, the lower band of 63 kDa was also detected (Fig. 7A, lane 1). The anti-HA antibodies detected bands of 89 kDa in both uninfected cells and infected cells, and as expected, a band of 26 kDa was also detected in extracts from infected cells but not in uninfected cells (Fig. 7A, lanes 4 and 5). The data suggest that infection with ASFV is necessary for production of the N- and C-terminal fragments of CD2v. Uninfected cells transfected with a plasmid expressing SV5CD2VHA were also fixed, permeabilized, and stained with anti-V5 or anti-HA antibody. This showed that almost all of the staining with anti-HA and anti-V5 antibodies colocalized (Fig. 7B). This was expected, since fragments containing the N- and C termini were not produced. The staining was mainly cytoplasmic and in a perinuclear localization, although some more dispersed staining through the cytoplasm was also detected.
FIG. 7.

Expression and localization of full-length CD2v in uninfected cells. (A) Vero cells were transfected with plasmid TriexSV5CD2vHA, and at 24 h posttransfection, lysates were prepared, or cells were transfected with plasmid SV5CD2vHA and infected with the ASFV BA71V isolate, and then lysates were prepared at 18 h postinfection. The lysates were immunoprecipitated using anti-HA matrix or anti-V5 agarose. The immunoprecipitates were separated by SDS-PAGE, transferred to membranes, and blotted using anti-HA antibody conjugated to HRP (lanes 1, 2, and 3) or anti-V5 antibody conjugated to HRP (lanes 4, 5, and 6). Lanes 1 and 4, lysates from infected cells expressing SV5CD2vHA; lanes 2 and 5, lysates from uninfected cells transfected with plasmid TriexSV5CD2vHA; lanes 3 and 6, lysates from mock-transfected cells. The positions of molecular weight markers run in parallel are indicated. (B) Cells were grown on coverslips, transfected with plasmid TriexSV5CD2vHA, and then fixed with paraformaldehyde at 24 h posttransfection. The cells were permeabilized and stained with anti-V5 antibody (green) and anti-HA antibody (red), followed by appropriate Alexa Fluor-conjugated secondary antibodies, and then visualized by confocal microscopy. A merged image with DNA stained with DAPI is on the right.
DISCUSSION
The results presented show that the ASFV CD2v protein is expressed in different forms in ASFV-infected cells. In addition to the full-length glycosylated form, which was detected predominantly as a band of 89 kDa, two processed forms of the protein were detected. These were a 26-kDa nonglycosylated product detected with an antibody directed against an HA tag fused to the C terminus of the protein and a 63-kDa glycosylated product detected with an antibody directed against an SV5 tag fused at the N terminus. These products are most likely to be derived by proteolytic cleavage. Alternative explanations, such as initiation from internal ATG codons, are less likely, since only two internal ATGs are present in the open reading frame and both are present within the transmembrane domain. Translation from these ATG codons is not predicted to produce a peptide of the observed 26-kDa size. These N-terminal and C-terminal fragments were present in approximately equimolar quantities in comparison to the unprocessed SV5CD2v HA protein, suggesting they are stable and likely to have a function.
The processing site(s) has not been defined precisely. However, the sizes of the C-terminal products expressed from the full-length SV5CD2vHA gene, a mutant gene lacking the proline-rich repeats, and the mutant expressing all of the cytoplamsic tail apart from 12 amino acids adjacent to the transmembrane domain suggest that the processing site is within the extracellular or luminal domain and possibly within the hinge region between the IgSF domains or within the membrane-proximal IgSF domain. The N-terminal Ig-V domain would be contained in the N-terminal 63-kDa glycosylated fragment. The ligand for the virus CD2v protein is not known, but the extracellular domain shares some of the conserved residues with host CD2, which has been shown to be important for ligand interaction. The ligands for host CD2 are CD58 and CD59 in humans and CD48 in rodents (2, 16). Most of the critical residues in host CD2 involved in ligand interaction are within the N-terminal Ig-V domain (3, 22, 24, 29). Expression of the extracellular domain of human CD2 revealed that a relatively stable 15-kDa N-terminal fragment was generated by protease digestion with papain. This single Ig-V domain-containing fragment bound to LFA-3 on B cells with a dissociation constant similar to those of the two Ig domain-containing extracellular domains. The 15-kDa fragment retained the ability to inhibit sheep erythrocyte rosette formation with human T cells (24). Thus, the 63-kDa N-terminal fragment of CD2v may retain the ability to bind to its ligand and to red blood cells. Evidence in support of this was provided by our preliminary experiments, which showed that expression of the first 114 amino acids of the CD2v protein in cells infected with an ASFV isolate lacking CD2v induced hemadsorbtion of red blood cells around the infected cells. The first 114 amino acids include the signal peptide and the N-terminal IgSF domain (L. C. Goatley and L. K. Dixon unpublished data). The C-terminal 26-kDa fragment is predicted to contain all or part of the membrane-proximal Ig-C domain, and it may also bind to a cellular or virus ligand. Incubation of cells with brefeldin A to inhibit anterograde protein transport from the endoplasmic reticulum to the Golgi apparatus did not inhibit production of the C-terminal product, suggesting that the processing occurs at an early stage in the secretory pathway. Since the N-terminal 63-kDa fragment does not contain the transmembrane region, it may be secreted from infected cells; however, since we did not detect it in cell supernatants, this suggests that is not secreted. The 63-kDa fragment was associated with the membrane fraction, showing that it is retained on the luminal side of internal membranes by another mechanism. Possibly, this may involve retention by an uncleaved N-terminal signal peptide, or it may occur by association with other membrane components. An attractive hypothesis is that association of CD2v fragments with cellular membrane proteins may modify their functions and contribute to the immunomodulatory functions of CD2v. Previous reports identified an immunomodulatory protein, p36, with a molecular mass of 36 kDa secreted from ASFV-infected macrophages (25, 32). This is very different in size from any of the CD2v fragments we have detected and indicates that p36 is not derived from CD2v.
Using an antibody directed against a C-terminal HA tag on the CD2v protein, the SV5CD2vHA protein was detected mainly in areas surrounding the virus factories, and these were also stained with an antibody against a Golgi marker protein. The anti-HA antibody detects both the full-length CD2v protein and the C-terminal 26-kDa protein. In contrast, using the antibody directed at the SV5 tag downstream of the signal peptide at the N terminus of the CD2v protein, a more dispersed staining was detected throughout the cytoplasm in some of the cells. Some of the CD2v protein detected with the anti-V5 antibody colocalized with that detected using the anti-HA antibody, some colocalized with an antibody against an endoplasmic reticulum marker, and some did not colocalize with either of these. Areas where staining of SV5CD2vHA with both anti-V5 and anti-HA colocalized contain the full-length protein and possibly some of the N- or C-terminal fragments, whereas areas stained only with anti-SV5 or anti-HA contain the N-terminal or C-terminal fragment, respectively. The data suggest that the 26-kDa C-terminal fragment and the full-length protein are mainly concentrated in membrane areas around the virus factories. These areas were also stained with an anti-Golgi marker. In contrast, the N-terminal 69-kDa fragment was detected in a more dispersed cytoplasmic pattern.
Deletion of the sequences encoding all except the membrane-proximal 12 amino acids from the cytoplasmic tail of CD2v resulted in a different distribution of the SV5CD2v-cytoHA protein. When detected using the antibody directed against the C-terminal HA tag, the distribution was more dispersed throughout the cytoplasm and colocalization of staining with that detected by the anti-Golgi marker protein was very much reduced. This suggests that signals within the cytoplasmic tail are involved in targeting of the SV5CD2vHA protein and the C-terminal 26-kDa fragment to a predominant localization around the perinuclear virus factories in membranes containing a Golgi marker protein. The signals in the CD2v cytoplasmic domain involved in targeting of the protein have not been defined. Potential sorting motifs present in the cytoplasmic domain include dileucine motifs and an acid-rich domain that resemble PACS-1 binding domains. PACS-1 is involved in recycling between the cell surface and the Golgi compartment.
We have previously shown that the proline-rich repeats in the cytoplasmic domain are required for binding of the CD2v protein to the SH3P7/mabp1 protein. In infected cells, SH3P7/mabp1 is localized in areas surrounding virus factories and in areas peripheral to the plasma membrane. SH3P7/mabp1 is an actin binding adaptor protein that contains an actin depolymerization domain and has been implicated in transport of proteins through the Golgi apparatus, in clathrin-mediated endocytosis, in formation of membrane ruffles, and in modulating signaling pathways (7, 12, 14, 15, 18, 19, 21). The binding of SH3P7 to the CD2v cytoplasmic tail has no apparent effect on localization of CD2v, but possibly, the interaction may modulate or target the function of SH3P7. The 26-kDa C-terminal fragment of the CD2v protein also contains this region of proline-rich repeats and would therefore also be predicted to bind to SH3P7.
The amounts of full-length CD2v and N-terminal 69-kDa and C-terminal 26-kDa fragments we detected were approximately equimolar, suggesting these fragments, as well as the full-length protein, are likely to have a function in infected cells. Our hypothesis is that they contribute to the immunomodulatory functions of CD2v by binding to cellular membrane proteins on the luminal surfaces of internal membranes, thus modifying their trafficking within cells and/or binding to ligands.
Acknowledgments
We acknowledge funding from BBSRC and DEFRA.
We thank Pippa Hawes for help with confocal microscopy and Chris Netherton, Fuquan Zhang, and Pam Lithgow for helpful discussions.
Footnotes
Published ahead of print on 19 January 2011.
REFERENCES
- 1.Arias, M., and J. M. Sanchez-Vizcaino. 2002. African swine fever, p. 119-124. In A. Morilla Gonzaìlez, K.-J. Yoon, and J. J. Zimmerman (ed.), Trends in emerging viral infections of swine. Iowa State Press, Ames, IA.
- 2.Barclay, A. N. 2001. Biochemical analysis of the lymphocyte cell surface—from alloantisera to the role of membrane proteins. Immunol. Rev. 184:69-81. [DOI] [PubMed] [Google Scholar]
- 3.Bodian, D. L., E. Y. Jones, K. Harlos, D. I. Stuart, and S. J. Davis. 1994. Crystal structure of the extracellular region of the human cell-adhesion molecule cd2 at 2.5-Angstrom resolution. Structure 2:755-766. [DOI] [PubMed] [Google Scholar]
- 4.Boinas, F. S., G. H. Hutchings, L. K. Dixon, and P. J. Wilkinson. 2004. Characterization of pathogenic and non-pathogenic African swine fever virus isolates from Ornithodoros erraticus inhabiting pig premises in Portugal. J. Gen. Virol. 85:2177-2187. [DOI] [PubMed] [Google Scholar]
- 5.Borca, M. V., et al. 1998. Deletion of a CD2-like gene, 8-DR, from African swine fever virus affects viral infection in domestic swine. J. Virol. 72:2881-2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cobbold, C., J. T. Whittle, and T. Wileman. 1996. Involvement of the endoplasmic reticulum in the assembly and envelopment of African swine fever virus. J. Virol. 70:8382-8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cortesio, C. L., B. J. Perrin, D. A. Bennin, and A. Huttenlocher. 2010. Actin-binding protein-1 interacts with WASp-interacting protein to regulate growth factor-induced dorsal ruffle formation. Mol. Biol. Cell 21:186-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costard, S., et al. 2009. African swine fever: how can global spread be prevented? Philos. Trans. R. Soc. Lond. B Biol. Sci. 364:2683-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon, L. K., C. C. Abrams, D. G. Chapman, and F. Zhang. 2008. African swine fever virus, p. 457-521. In T. C. Mettenleiter and F. Sobrino (ed.), Animal viruses: molecular biology. Caister Academic Press, Norfolk, United Kingdom.
- 10.Dixon, L. K., et al. 2005. Asfarviridae, p. 135-143. In C. M. Fauquet, M. A. Mayo, J. Maniloff, U. Desselberger, and L. A. Ball (ed.), Virus taxonomy. VIIIth report of the ICTV. Academic Press, London, United Kingdom.
- 11.Dixon, L. K., and P. J. Wilkinson. 1988. Genetic diversity of African swine fever virus isolates from soft ticks (Ornithodoros-Moubata) inhabiting warthog burrows in Zambia. J. Gen. Virol. 69:2981-2993. [DOI] [PubMed] [Google Scholar]
- 12.Fenster, S. D., et al. 2003. Interactions between Piccolo and the actin/dynamin-binding protein Abp1 link vesicle endocytosis to presynaptic active zones. J. Biol. Chem. 278:20268-20277. [DOI] [PubMed] [Google Scholar]
- 13.Galindo, I., F. Almazan, M. J. Bustos, E. Vinuela, and A. L. Carrascosa. 2000. African swine fever virus EP153R open reading frame encodes a glycoprotein involved in the hemadsorption of infected cells. Virology 266:340-351. [DOI] [PubMed] [Google Scholar]
- 14.Han, J., et al. 2003. The SH3 domain-containing adaptor HIP-55 mediates c-Jun N-terminal kinase activation in T cell receptor signaling. J. Biol. Chem. 278:52195-52202. [DOI] [PubMed] [Google Scholar]
- 15.Han, J., et al. 2005. HIP-55 is important for T-cell proliferation, cytokine production, and immune responses. Mol. Cell. Biol. 25:6869-6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato, K., et al. 1992. CD48 is a counter-receptor for mouse CD2 and is involved in T-cell activation. J. Exp. Med. 176:1241-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kay-Jackson, P. C., et al. 2004. The CD2v protein of African swine fever virus interacts with the actin-binding adaptor protein SH3P7. J. Gen. Virol. 85:119-130. [DOI] [PubMed] [Google Scholar]
- 18.Kessels, M. M., A. E. Y. Engqvist-Goldstein, and D. G. Drubin. 2000. Association of mouse actin-binding protein 1 (mAbp1/SH3P7), an Src kinase target, with dynamic regions of the cortical actin cytoskeleton in response to Rac1 activation. Mol. Biol. Cell 11:393-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kessels, M. M., A. E. Y. Engqvist-Goldstein, D. G. Drubin, and B. Qualmann. 2001. Mammalian Abp1, a signal-responsive F-actin-binding protein, links the actin cytoskeleton to endocytosis via the GTPase dynamin. J. Cell Biol. 153:351-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leitão, A., et al. 2001. The non-haemadsorbing African swine fever virus isolate ASFV/NH/P68 provides a model for defining the protective anti-virus immune response. J. Gen. Virol. 82:513-523. [DOI] [PubMed] [Google Scholar]
- 21.Mise-Omata, S., B. Montagne, M. Deckert, J. Wienands, and O. Acuto. 2003. Mammalian actin binding protein 1 is essential for endocytosis but not lamellipodia formation: functional analysis by RNA interference. Biochem. Biophys. Res. Commun. 301:704-710. [DOI] [PubMed] [Google Scholar]
- 22.Peterson, A., and B. Seed. 1987. Monoclonal-antibody and ligand-binding sites of the T-cell erythrocyte receptor (CD2). Nature 329:842-846. [DOI] [PubMed] [Google Scholar]
- 23.Quintero, J. C., R. D. Wesley, T. C. Whyard, D. Gregg, and C. A. Mebus. 1986. Invitro and invivo association of African swine fever virus with swine erythrocytes. Am. J. Vet. Res. 47:1125-1131. [PubMed] [Google Scholar]
- 24.Recny, M. A., E. A. Neidhardt, P. H. Sayre, T. L. Ciardelli, and E. L. Reinherz. 1990. Structural and functional characterization of the CD2 immunoadhesion domain. Evidence for includion of CD2 in an alpha-beta protein folding class. J. Biol. Chem. 265:8542-8549. [PubMed] [Google Scholar]
- 25.Ribeiro, A. D., M. P. Aralachaves, M. Vilanova, M. T. Porto, and A. Coutinho. 1991. Roles of lymphocyte-B and lymphocyte-T in the specific immunosuppression induced by a protein released by porcine monocytes infected with African swine fever virus. Int. Immunol. 3:165-174. [DOI] [PubMed] [Google Scholar]
- 26.Rodríguez, J. M., R. J. Yanez, F. Almazan, E. Vinuela, and J. F. Rodriguez. 1993. African swine fever virus encodes a Cd2 homolog responsible for the adhesion of erythrocytes to infected cells. J. Virol. 67:5312-5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26a.Rouiller, I., S. M. Brooks, A. D. Hyatt, M. Windsor, and T. Wileman. 1998. African swine fever virus is wrapped by the endoplasmic reticulum. J. Virol. 72:2373-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rowlands, R. J., M. M. Duarte, F. Boinas, G. Hutchings, and L. K. Dixon. 2009. The CD2v protein enhances African swine fever virus replication in the tick vector, Ornithodoros erraticus. Virology 393:319-328. [DOI] [PubMed] [Google Scholar]
- 28.Salas, J., and M. L. Salas. 2003. Current perspectives in African swine fever virus infection and evasion of host defenses. Curr. Top. Virol. 3:155-163. [Google Scholar]
- 29.Somoza, C., P. C. Driscoll, J. G. Cyster, and A. F. Williams. 1993. Mutational analysis of the CD2/CD58 interaction. The binding site for CD58 lies on one face of the 1st domain of human CD2. J. Exp. Med. 178:549-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomson, G. R., M. D. Gainaro, and A. F. Van Dellen. 1979. African swine fever: pathogenicity and immunogenicity of two non-hemadsorbing viruses. Onderstepoort J. Vet. Res. 46:149-154. [PubMed] [Google Scholar]
- 31.Tulman, E. R., G. A. Delhon, B. K. Ku, and D. L. Rock. 2009. African swine fever virus. Curr. Top. Microbiol. Immunol. 328:43-87. [DOI] [PubMed] [Google Scholar]
- 32.Vilanova, M., P. Ferreira, A. Ribeiro, and M. Arala-Chaves. 1999. The biological effects induced in mice by p36, a proteinaceous factor of virulence produced by African swine fever virus, are mediated by interleukin-4 and also to a lesser extent by interleukin-10. Immunology 96:389-395. [DOI] [PMC free article] [PubMed] [Google Scholar]