

Abstract
Ebola virus (EBOV) causes severe hemorrhagic fever, for which therapeutic options are not available. Preventing the entry of EBOV into host cells is an attractive antiviral strategy, which has been validated for HIV by the FDA approval of the anti-HIV drug enfuvirtide. To identify inhibitors of EBOV entry, the EBOV envelope glycoprotein (EBOV-GP) gene was used to generate pseudotype viruses for screening of chemical libraries. A benzodiazepine derivative (compound 7) was identified from a high-throughput screen (HTS) of small-molecule compound libraries utilizing the pseudotype virus. Compound 7 was validated as an inhibitor of infectious EBOV and Marburg virus (MARV) in cell-based assays, with 50% inhibitory concentrations (IC50s) of 10 μM and 12 μM, respectively. Time-of-addition and binding studies suggested that compound 7 binds to EBOV-GP at an early stage during EBOV infection. Preliminary Schrödinger SiteMap calculations, using a published EBOV-GP crystal structure in its prefusion conformation, suggested a hydrophobic pocket at or near the GP1 and GP2 interface as a suitable site for compound 7 binding. This prediction was supported by mutational analysis implying that residues Asn69, Leu70, Leu184, Ile185, Leu186, Lys190, and Lys191 are critical for the binding of compound 7 and its analogs with EBOV-GP. We hypothesize that compound 7 binds to this hydrophobic pocket and as a consequence inhibits EBOV infection of cells, but the details of the mechanism remain to be determined. In summary, we have identified a novel series of benzodiazepine compounds that are suitable for optimization as potential inhibitors of filoviral infection.
Ebola viruses (EBOV) are enveloped, single-stranded, negative-sense RNA viruses and have been classified as category A pathogens by the Centers for Disease Control and Prevention (CDC). Together with Marburg virus (MARV), they constitute the filovirus family. There are five species of EBOV, namely, Zaire, Sudan, Ivory Coast, Bundibugyo, and Reston (61). EBOV infection causes severe viral hemorrhagic fevers (VHFs) in humans and nonhuman primates, with a mortality rate of up to 90% (55). These outbreaks are infrequent and so far have been restricted to small pockets of population in Africa. The natural reservoir for the virus is still not known, but fruit bats have been implicated (27, 34).
The EBOV genome contains seven genes that encode eight viral proteins, NP, VP35, VP40, glycoprotein (GP), sGP, VP30, VP24, and RNA-dependent RNA polymerase (L) (44, 56). Transcriptional editing of the fourth gene results in expression of a 676-residue EBOV envelope glycoprotein (EBOV-GP), as well as a 364-residue secreted glycoprotein (sGP1) (44). EBOV-GP mediates the viral entry into host cells and provides a potential target for the design of vaccines and entry inhibitors. EBOV-GP is posttranslationally cleaved by furin, to yield disulfide-linked GP1 and GP2 subunits (63). GP1 is involved in attachment of EBOV to host cells, whereas GP2 mediates fusion of viral and host membranes (18, 59). EBOV is believed to enter host cells by receptor-mediated endocytosis (44), where further processing by endosomal cathepsin L (cat L) and/or cathepsin B (cat B) (11, 31, 46) is required for entry. A cellular receptor(s) for EBOV has not yet been identified, but DC-SIGN/L-SIGN, hMGL, β-integrins, folate receptor-α, and Tyro family receptors have all been implicated as cellular factors in entry (10, 51, 52). EBOV-GP, apart from its role in virus entry, also plays an important role in the pathogenicity of infection. Expression of EBOV-GP induces a cytopathic effect (CPE) in cell lines and human blood vessel explants (53, 62). This cytopathic effect was mapped to the mucin-like region present in the C terminus of GP1(62). EBOV-GP, when overexpressed, also downregulates molecules involved in cell adhesion and causes anoikis (39). Virus-like particles (VLPs) containing EBOV-GP on the surface activate macrophages to secrete several proinflammatory cytokines (6, 54).
Virus entry is an essential component of the viral life cycle and an attractive target for therapy because inhibition of this step can block the propagation of virus at an early stage, minimizing the chance for the virus to evolve and acquire drug resistance. Anti-infective drug discovery for EBOV presents significant logistical and safety challenges due to the requirement for biosafety level 4 (BSL-4) containment and procedures. The advent of replication-incompetent pseudotype viruses, which utilize the replication machinery of vesicular stomatitis virus (VSV) (16, 48), murine leukemia virus (MLV) (37), or human immunodeficiency virus (HIV) (29, 30) but package the EBOV-GP on the virion surface, offers an opportunity to safely screen libraries of small molecules for antiviral properties in a BSL-2 environment. In this study, we report the discovery of a novel small-molecule entry inhibitor with specific inhibitory activity against both EBOV and MARV. A benzodiazepine derivative (compound 7) was identified from a high-throughput screen (HTS) of small-molecule compound libraries utilizing the EBOV pseudotype virus. Compound 7 also specifically inhibited cell culture-grown EBOV in vitro, and the potential binding site was mapped to a specific area within a hydrophobic pocket at the EBOV GP1-GP2 interface.
(Part of this study was presented in 2009 at the 28th Annual Meeting of the American Society for Virology, Vancouver, Canada.)
MATERIALS AND METHODS
Cell lines, chemicals, and plasmids.
293T, Vero E6, A549, HeLa, MDCK, and BHK cell lines were procured from ATCC. The cell lines were maintained in either Dulbecco modified Eagle medium (DMEM) or minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (50 units/ml). Plasmid vectors expressing the envelope proteins of EBOV Zaire (GenBank accession number L11365) (EBOV-GP), MARV (Lake Victoria strain) (MARV-GP), vesicular stomatitis virus (VSV-G), lymphocytic choriomeningitis virus (LCMV), Lassa virus (LASV), and Machupo virus (MACV) and the hemagglutinin [HA(H5)] gene from a highly pathogenic H5N1 avian influenza virus (Goose/Qinghai/59/05) were described earlier (14, 29, 30, 37).
Pseudotyping.
Ebola pseudotype viruses (HIV/EBOV-GP) were produced by cotransfecting 12 μg of wild-type (wt) or mutant EBOV-GP with 12 μg replication-defective HIV vector (pNL4.3.Luc-R-E-) (15) into 293T cells (90% confluent) in 10-cm plates with Lipofectamine 2000 (Invitrogen) according to the supplier's protocol (4, 29). The supernatants containing the pseudotype viruses were collected at 48 h posttransfection, pooled, clarified from floating cells and cell debris by low-speed centrifugation, and filtered through a 0.45-μm-pore-size filter (Nalgene). The culture supernatants containing HIV/EBOV-GP were either used immediately or flash frozen in aliquots and stored at −80°C until use. Each aliquot was thawed only once for use in a single round of replication. Mutant HIV/EBOV-GP pseudotype viruses, along with pseudotype viruses bearing VSV envelope protein (HIV/VSV-G), LASV envelope protein (HIV/LASV-GP), LCMV envelope protein (HIV/LCMV-GP), MACV envelope protein (HIV/MACV-GP), and HA(H5) from avian influenza A H5N1 virus (Quinghai strain) [HIV/HA(H5)], were prepared in a similar fashion, using the same Env-deficient HIV vector as previously described (4, 29, 37, 42).
Chemical libraries.
The chemical libraries screened represent broad and well-balanced collections of over 52,500 compounds. They were purchased from Chembridge (San Diego, CA) and Timtec (Newark, DE), diluted in 96-well master plates at 2.5 mM in dimethyl sulfoxide (DMSO), and stored at −20°C. Compounds were selected in the molecular mass range of 200 to 500 Da and are within the size range of molecules considered small and “drug-like.” Compounds were evaluated using numerous chemical filters, including Lipinski's “rule of 5” (28), to remove unwanted and known cytotoxic scaffolds, such as metal complexes, highly conjugated ring systems, oxime esters, nitroso groups, and strong Michael acceptors. The compounds have favorable cLogP values (calculated logarithm of n-octanol/water partition coefficient), and encompass over 200 chemotypes.
HTS of chemical libraries.
High-throughput screening (HTS) of chemical libraries using pseudotype virus was performed in 96-well plates as described previously (22, 24). The final concentration of test compound was 25 μM, while the final concentration of DMSO was maintained at 1% in all wells. Low-passage 293T cell monolayers were infected with 100 μl of p24-normalized HIV/EBOV-GP pseudotype virus containing 8 μg/ml Polybrene in the presence of test compounds. After 2 h, the inoculum was removed, the cells were washed briefly, fresh medium was added, and the plates were incubated for 72 h. Prior to each screening, each batch of the viral preparation was titrated to determine the amount of virus required to infect the target cells, so that a relatively high luciferase activity could be recorded while still remaining in a linear response range (105 to 106 relative luciferase units [RLU]). Infection was quantified from the luciferase activity of the infected 293T cells using the Britelite Plus assay system (Perkin-Elmer, Waltham, MA) in a Wallac EnVision 2102 multilabel reader (Perkin-Elmer). In the absence of compound, the assay showed an average luciferase signal of 1.2 × 106 ± 0.6 × 106 RLU, a signal-to-background ratio of >103, and a calculated screening window coefficient (Z′ factor) (63) of 0.5 ± 0.2. The luciferase signal standard error was ±50%, and greater than 90% inhibition of luciferase activity at 25 μM was used as the criterion for designating a compound a “hit.” Test compounds were in DMSO solutions, with 80 compounds per plate. Controls were also included in each plate: eight wells for 0% inhibition (DMSO only, maximum signal) (positive control) and eight wells for 100% inhibition (e.g., E64 for EBOV, minimum signal) (negative control). The percent inhibition was calculated as 100 × [1 − (RLU in the presence of compound − RLU of negative control)/(RLU of positive control [without any inhibitor] − RLU of negative control)].
Cell viability assay.
Cells were plated in a 96-well format (4 × 103 cells/well) in the presence or absence of serial dilutions of the test compound. Following 3 days of incubation in growth medium at 37°C, cell viability was determined using the CellTiter 96 aqueous nonradioactive cell proliferation assay (Promega, Madison, WI) as previously described (5).
Infection assay with cell culture-grown recombinant GFP-ZEBOV.
All experiments with cell culture-grown Zaire EBOV expressing green fluorescent protein (GFP-ZEBOV) were performed under BSL-4 conditions. Cell culture-grown GFP-ZEBOV was used to screen compounds for inhibition of EBOV replication using methods described previously (1, 33). Briefly, Vero E6 cells (40 × 103 cells/well in a 96-well plate format) were infected with the GFP-ZEBOV at a multiplicity of infection (MOI) of 5 in the presence of compounds. Cultures were incubated for 48 h before fixation (10% neutral-buffered formalin, 72 h) and removal from the BSL-4 facility. After nuclear (Hoechst dye) and cytoplasm/nuclear (high-content screening [HCS] cell mask deep red stain) staining, viral infection was imaged and quantified using an automated Opera confocal reader (model 3842-quadruple excitation high sensitivity [QEHS]; Perkin-Elmer). Images were analyzed within the Opera environment using standard Acapella scripts. This procedure identifies the total number of adherent cells and the fraction of infected cells, thereby providing an immediate assessment of efficacy and toxicity. Cell viability assays were conducted as detailed previously (1, 33). All assays were repeated at least three times, and representative findings are shown. Percent inhibition values were calculated as follows: 100 × [1 − (average GFP fluorescence from compound-treated wells/average fluorescence from wells containing medium only)].
Antiviral assays.
The activity of compound 7 against hepatitis C virus (HCV) was evaluated using cell culture-grown HCV genotype 1a (H77) and immortalized human hepatocytes (IHH) as previous described (4). The HCV titer was calculated as ∼105 focus-forming units (FFU)/ml, and antiviral activity was measured in a dose-dependent manner (4). Cells were incubated with the virus in the presence of compounds for 3 days, and the fluorescent-focus formation was determined by immunofluorescence using NS4-fluorescein isothiocyanate (FITC)-conjugated monoclonal antibody (Biodesign). Nuclear staining was performed with TO-PRO-3 (Molecular Probes). This procedure identifies the total number of adherent cells and the fraction of infected cells, thereby providing an immediate assessment of efficacy and toxicity. The percent inhibition was calculated based on the number of fluorescent foci in the treated sample compared to the number in the untreated positive control. A selectivity index (SI) (50% cytotoxic concentration [CC50]/50% inhibitory concentration [IC50]) of ≥3 was used as an indication of selective antiviral activity.
The activities of compound 7 against Tacaribe virus (strain TRVL 11573), Rift valley fever virus (strain MP-12), Venezuelan equine encephalitis (VEE) virus (strain TC-83), cowpox virus (strain Brighton), vaccinia virus (strains WR and IHD), influenza A (H1N1) virus (strain California/07/2009), influenza A (H3N2) virus (strain Brisbane/10/2007), and severe acute respiratory syndrome (SARS) virus (strain Urbani) were determined in collaboration with NIAID's Antimicrobial Acquisition and Coordinating Facility (AACF) using standard procedures (2, 3, 49, 64). For toxicity studies different methods, including (i) visual observation of the viable cells and (ii) neutral red uptake assay, were used to determine the number of viable cells in the well. This procedure provided an immediate assessment of efficacy and toxicity. An SI (CC50/IC50) of ≥3 was used as an indication of selective antiviral activity.
Time-of-addition experiment.
The “time-of-addition” experiment was designed to determine the mechanism of action of the antiviral compounds, and the procedure is shown schematically in Fig. 2A. 293T cells were incubated with a 25 μM concentration of the test compound at 1 h before infection (−1 h), during infection (0 h), and 1 h following infection (+1 h). Duplicate wells were used for each time point. Control infected-cell cultures were treated with drug vehicle (DMSO) only. At 72 h postinfection, cells were developed and infectivity was measured as described above.
Compound-virus binding assay.
The compound-virus binding assay, with slight modifications, was performed as described earlier (13, 60). Microcon Ultracell YM3 centrifugal devices (Millipore), which are capable of separating small molecules from large macromolecules, were used to measure the binding of compound 7 to EBOV-GP, as follows. HIV/EBOV-GP and HIV/VSV-G pseudotype viruses were ultracentrifuged using a 10% sucrose cushion at 35,000 rpm at 4°C in a Beckman SW41 rotor for 60 min. The viruses were resuspended in DMEM, and the concentration of HIV capsid protein p24 was measured by enzyme-linked immunosorbent assay (ELISA). Fifty microliters of either HIV/EBOV-GP or HIV/VSV-G pseudotype virus supernatant (containing 300 ng/ml of p24) was incubated with 25 μM compound 7 at room temperature for 60 min. The virus-compound mixture was then loaded onto the Microcon Ultracell YM3 centrifugal device, equilibrated for 5 min, and centrifuged for 2 min. After centrifugation, the eluent was analyzed by high-pressure liquid chromatography to measure the quantity of unbound compound 7. Signals from a reaction mix containing no pseudotype virus were used as a positive control. The percent binding was calculated based on the quantity of compound 7 in the eluent in the presence of pseudotype viruses compared to that in the positive control.
EBOV-GP RBD cell binding assay.
The EBOV-GP receptor binding domain (RBD) cell binding assay was performed according to a protocol described by Kuhn et al. (20, 21). The RBD of EBOV-GP (residues 54 to 201) was fused with human IgG Fc to generate a fusion protein (GP-hIgG). The fusion protein gene was expressed and the protein purified as previously described (20, 21). This RBD fusion protein has been demonstrated to bind to cells that are permissive for EBOV infection but not to lymphocyte cell lines, which are nonpermissive for EBOV infection (20, 21). 293T cells were detached by treatment with phosphate-buffered saline (PBS)-5 mM EDTA, resuspended in PBS-1% bovine serum albumin (BSA), and kept on ice for 30 min. Then, 10 μg of GP-hIgG protein was incubated with 0.5 × 106 cells on ice for 1 h in the presence of different concentrations of compound. Following the incubation, cells were washed with PBS-1% BSA two times. Anti-human IgG antibody conjugated with FITC was incubated with cells on ice for 45 min at a dilution of 1:100. Cells were washed three times with PBS-1% BSA and two times with PBS and subjected to flow cytometry. In this experiment, a SARS virus spike fusion protein (S1-hIgG) that does not bind to Vero or 293T cells was used as a negative control.
Computational methods.
A published crystal structure of EBOV-GP, at 3.4-Å resolution, in its metastable trimeric, prefusion conformation in complex with the neutralizing human antibody KZ52 (25), was used for the computational examination. To keep regions between protein monomers from generating unnatural putative binding sites, which exist only in the crystal lattice, only the protein monomer was retained and the antibody was edited out of the structure. The protein structure was prepared in Maestro (Schrödinger, San Diego, CA) using the protein preparation wizard. The OPLS 2005 force field was employed as the force field for the study. Appropriate bond orders and formal charges were assigned using Maestro. To remove interactions between nonrelevant contacts, the “impref” procedure was employed to relax the protein structure. This procedure carries out a series of short refinements until the root mean square deviation (RMSD) is <0.3 Å. Next, binding site prediction was performed using the SiteMap (Schrödinger, San Diego, CA) package (default setting). SiteMap assigns numerical descriptors to evaluate predicted binding sites by a series of physical parameters such as size, degree of enclosure/exposure, tightness, hydrophobic/hydrophilic character, and hydrogen bonding possibilities. A weighted average of these measurements is then assigned to prioritize possible binding sites.
RESULTS
HTS for the identification of EBOV entry inhibitors.
A chemically diverse small-molecule library (52,500 compounds) was screened for ≥90% inhibition of HIV/EBOV-GP infection to identify viral entry inhibitors. As shown in Table 1, the initial high-throughput screening (HTS) hit rate from the primary screen was 2.2%, with a total of 1,146 “hit” compounds inhibiting the HIV/EBOV-GP. To assess the target specificity of the HTS hits, we counterscreened these hits against a second pseudotype virus (HIV/VSV-G) having the same HIV backbone but expressing unrelated vesicular stomatitis virus envelope glycoprotein (VSV-G). Based on this counterscreen, only 57 compounds were demonstrated to block EBOV-GP-mediated infection specifically with little or no effect on infectivity of HIV/VSV-G (Table 1). These EBOV-GP-specific hits were tested for cytotoxicity, and 18 compounds exhibited CC50 values of >25 μM (Table 1). These 18 specific hits were subsequently reordered from different batches from the original vendors and retested. The reordered compounds were determined to have the correct mass and to be >95% pure by LC-MS. All 18 compounds were reconfirmed against HIV/EBOV-GP, dose-response curves were generated, and 90% inhibitory concentration (IC90) and CC50 values of compounds were determined. Table 2 shows results of the eight most potent inhibitors.
TABLE 1.
Results of the screening of compound libraries
| Parameter | No. (%) |
|---|---|
| Compounds screened | 52,500 |
| Primary hits (inhibition of HIV/EBOV-GP | |
| in the HTS screen)a | 1,146 (2.2) |
| Primary hits inhibiting HIV/VSV-Ga,b | 1,089 (2.07) |
| Specific hits (primary hits inhibiting HIV/ | |
| EBOV-GP only) | 57 (0.13) |
| Specific hits displaying a CC50 of >25 μMc | 18 |
HIV/EBOV-GP and HIV/VSV-G were generated by transfection of 293T cells with pNL4.3.Luc.R-E- as the HIV-l expression vector and with EBOV-GP or VSV-G, respectively.
A total of 1,089 primary hits inhibited HIV/VSV-G by more than 90% at 25 μM, whereas the RLU values of the controls varied by ≤50%.
CC50 values were determined using the CellTiter 96 aqueous nonradioactive cell proliferation assay (Promega).
TABLE 2.
Anti-EBOV activities of the hit compounds
 |
HIV/EBOV-GP was generated by transfection of 293T cells with pNL4.3.Luc.R-E- as the HIV-l expression vector and with EBOV-GP. IC90 values were determined using reordered compounds as different batches from the original vendors. For determination of CC50 values, 293T cells were treated with compound alone, and values were determined from linear portions of the dose-response curve. SI, selectivity index (CC50/IC90).
All experiments with GFP-ZEBOV were performed under biosafety level 4 conditions. GFP-ZEBOV was incubated with Vero E6 cells at a multiplicity of infection of 5 for 1 h in the presence or absence of inhibitor compounds. Virus was removed after 1 h, cells were washed in PBS and incubated for 48 h, and the percentage of GFP-expressing cells was measured. IC50 and IC90 values were determined from the linear portion of the full dose-response curve. For determination of CC50 values, Vero E6 cells were treated with compound alone, and values were determined from linear portions of the dose-response curve. SI, selectivity index (CC50/IC50).
Validation of HTS hits against cell culture-grown GFP-ZEBOV.
The anti-EBOV activities of the 18 HIV/EBOV-GP-specific inhibitors were also evaluated against cell culture-grown GFP-ZEBOV in a BSL-4 containment facility at USAMRIID (Frederick, MD). As shown in Table 2, eight of the HIV/EBOV-GP-specific hits, all from different chemical families, were found to inhibit GFP-ZEBOV with IC50s of ≤20 μM, as calculated from the linear portions of full dose-response curves. Cytotoxicity was determined in the same assay. Compounds 2 and 7 were found to be of most interest, with CC50 values of >50 μM and with calculated selectivity index (SI = CC50/IC50) values of >5. SI values for the remaining active compounds were ≤4, and several exhibited CC50 values tracking close to their IC50s (Table 2). Based upon its overall potency, significant selectivity for antiviral activity versus cytotoxicity (Fig. 1 A), and chemically tractable benzodiazepine backbone, compound 7 was selected as a suitable starting point for further viral specificity studies, mode-of-action (MOA) evaluation, and medicinal chemistry optimization.
FIG. 1.

Specificity of inhibition by compound 7. (A) Comparison of antiviral activity and cell toxicity of compound 7. GFP-ZEBOV was incubated with Vero E6 cells at an MOI of 5 for 1 h in the presence or absence of compound 7 in a dose-dependent manner as described in Materials and Methods. The blue diamonds represent anti-GFP-EBOV activity, while the red squares represent cytotoxicity. (B) The inhibitory effect of compound 7 on virus infectivity was investigated using HIV/MARV-GP pseudotype and infectious cell culture-grown MARV as described in Materials and Methods. Three independent experiments were performed to determine the effect of compound 7 on virus infectivity, and standard deviations are indicated. (C) Retroviral pseudotypes of LASV (HIV/LASV-GP), LCMV (HIV/LCMV-GP), and MACV (HIV/MACV-GP) were added to 293T cells, and influenza H5 [HIV/HA(H5)] pseudotype virus was added to A549 cells, in the presence of the indicated concentrations of compound 7. Pseudotype infection was assessed after 72 h by luciferase reporter assay. Three independent experiments were performed to determine the effect of compound 7 on virus infectivity, and standard deviations are indicated.
Compound 7 exhibits antifilovirus activity.
EBOV and MARV are the two members of the filovirus family. The critical amino acid residues in EBOV-GP that are important for virus entry are highly conserved in EBOV and MARV (21, 29). Compound 7 displayed a concentration-dependent inhibitory activity against both HIV/MARV-GP pseudotype virus (IC90 = 3.1 μM) and infectious MARV (IC50 = 12.5 μM) (Fig. 1B). These overall results demonstrate that compound 7 displays filovirus-inhibitory activity, inhibiting both infectious EBOV and MARV with similar potencies (EBOV IC50 = 10 μM) (Table 2; Fig. 1A).
Antiviral spectrum of compound 7.
We next investigated the activity of compound 7 against arenaviruses and avian influenza H5N1 virus since they bear type 1 envelope proteins similar to that of EBOV. The pseudotype platform was used because it can be performed in a BSL-2 facility and it provides a direct comparison of the activities of compound 7 against these viruses and the HIV/EBOV-GP. Compound 7 inhibited HIV/LASV-GP, HIV/LMCV-GP, and HIV/MACV-GP weakly, in a dose-dependent manner, exhibiting inhibition of ∼30 to 40% at 25 μM (Fig. 1C), while almost no inhibition (∼10%) was observed against HIV/HA(H5) at 25 μM in A549 cells (Fig. 1C). In contrast, compound 7 inhibited HIV/EBOV-GP and HIV/MARV-GP infection by ∼90% at 12.1 μM and 3.1 μM, respectively (Table 2 and Fig. 1B).
The effect of compound 7 on several RNA and DNA viruses was further investigated by the Antimicrobial Acquisition and Coordinating Facility (AACF) of the National Institute of Allergy and Infectious Diseases (NIAID). As shown in Table 3, compound 7 did not inhibit cell culture-grown infectious Tacaribe virus, influenza A H1N1 virus (California/07/2009), influenza A H3N2 virus (Brisbane/10/2007), or SARS virus (Urbani strain) at concentrations of up to 32 μM, 25 μM, 33 μM, and 36 μM, respectively (the 50% cytotoxic concentration [CC50] in each assay). The inhibitory activity of compound 7 against other infectious RNA viruses, including Rift Valley fever virus, Venezuelan equine encephalitis (VEE) virus, and hepatitis C virus (HCV), and DNA viruses, including cowpox virus and vaccinia virus, was also investigated. None of these viruses was inhibited by compound 7 at or below its 50% cytotoxic concentration (Table 3). Clearly, compound 7 inhibits EBOV and MARV significantly more potently than it does any of the other viruses tested, indicating that it is primarily selective for filoviruses.
TABLE 3.
Inhibitory activity of compound 7 against different viruses
| Family | Virus | Assay type | IC50 (μM)a | CC50 (μM)b | Cells used |
|---|---|---|---|---|---|
| Filovirus | GFP-ZEBOV | GFP | 10 | 55 | Vero E6 |
| MARV | GFP | 12.5 | 55 | Vero E6 | |
| Arenavirus | Tacaribe virus | Neutral red | >32 | 32 | Vero 76 |
| Biodefense-related virus | Rift Valley fever virus | Neutral red | >16 | >16 | Vero 76 |
| VEE virus | Visual | >18 | 18 | Vero | |
| DNA virus | Cowpox virus | CPE | >300 | 300 | HFF |
| Vaccinia virus | CPE | >300 | >300 | HFF | |
| Flavivirus | HCV | PFU | >50 | >50 | IHH |
| Respiratory virus | Influenza A (H1N1) virus California/07/2009 | Neutral red | >25 | 24 | MDCK |
| Influenza (H3N2) virus Brisbane/10/2007 | Neutral red | >33 | 33 | MDCK | |
| SARS virus Urbani | Neutral red | >36 | 36 | Vero 76 |
IC50 values were generated for cell culture-grown viruses from the linear portion of the dose response curve.
CC50 values were determined from linear portions of the dose-response curve.
Compound 7 binds to HIV/EBOV-GP.
EBOV entry can broadly be divided into two major steps: (i) virus attachment and receptor binding, followed by (ii) receptor-mediated endocytosis, pH-dependent GP processing, and membrane fusion. Entry inhibitors can act at any stage of the entry process. A time-of-addition experiment was performed with HIV/EBOV-GP to determine the target stage of EBOV entry that is blocked by compound 7 (Fig. 2). Compound 7 was added at 1 h before infection (−1 h), during infection (0 h), and at 1 h postinfection (+1 h). Control infected cultures were treated with either DMSO (no inhibition) or E64, an EBOV entry inhibitor (complete inhibition) (11, 46). Compound 7, when added either before (−1 h) or after (+1 h) virus infection, displayed less than 18% and 27% inhibition of HIV/EBOV-GP infection, respectively (Fig. 2B). Interestingly, when compound 7 was present during the virus adsorption process (0 h) at either 37°C or 0°C, it inhibited more than 75% of the HIV/EBOV-GP infection (Fig. 2B). These results suggest that compound 7 is acting early during the virus attachment and receptor binding stage, possibly by binding to HIV/EBOV-GP.
FIG. 2.

Compound 7 binds to EBOV-GP and inhibits virus infection. (A) Schematic diagram of a “single-cycle time-of-addition experiment” with HIV/EBOV-GP to determine the stage of virus entry blocked by compound 7. This experiment was designed to characterize the mechanism of action of the antiviral compounds. (B) A single-cycle time-of-addition experiment was done with HIV/EBOV-GP to determine the stage of EBOV entry blocked by compound 7. 293T cells were infected with 100 μl of p24-normalized HIV/EBOV-GP. Compound 7 was added and left for 1 h before infection (−1 h), for 1 h during adsorption (0 h), and for 1 h after infection (+1 h). Infected monolayers were washed with PBS and incubated for 72 h. Inhibition of HIV/EBOV-GP pseudotype infection was detected as a reduced luciferase signal. Error bars indicate standard deviations. (C) A Microcon Ultracell YM3 centrifugal device (Millipore) was used to study the binding of compound 7 to HIV/EBOV-GP and HIV/VSV-G pseudotype viruses. Pseudotype viruses (or a no-virus buffer control) were incubated with 25 μM compound 7 at room temperature for 60 min, and the virus-compound mixture was then loaded onto the Microcon Ultracell YM3 centrifugal device. After centrifugation, the flowthrough was analyzed for unbound compound by high-pressure liquid chromatography. The percentage of bound compound was calculated based on the peak area compared to that of a no-virus control. Error bars indicate standard deviations.
We further addressed the binding of compound 7 with HIV/EBOV-GP directly by using a modification of the gel filtration method of the compound-virus binding assay (13, 60). We used a Microcon Ultracell YM3 centrifugal device (Millipore) that is capable of separating small molecules from large macromolecules. In the absence of virus, compound 7 will flow into the lower chamber and will be detected in the flowthrough fraction when analyzed by liquid chromatography-mass spectrometry. In contrast, virus-bound compound 7 will not pass through a Microcon YM3 centrifugal filter device upon centrifugation and will not be collected in the flowthrough. As shown in Fig. 2C, only 30% of compound 7 (compared to untreated compound 7) was observed in the flowthrough when it was preincubated with HIV/EBOV-GP. When the experiment was repeated using HIV/VSV-G, 73% of compound 7 was observed in the flowthrough. The results suggest that compound 7 exhibits some affinity for both HIV/EBOV-GP and HIV/VSV-G pseudotype virus. However, there is a higher degree of binding of compound 7 to HIV/EBOV-GP under similar experimental conditions. Since HIV/EBOV-GP and HIV/VSV-G have similar HIV backbones but different envelope glycoproteins at their surfaces, the results along with the time-of-addition studies indirectly suggest that compound 7 is binding with EBOV-GP.
Computational prediction of potential EBOV-GP binding sites.
Recently, Lee et al. (25) published a crystal structure (3.4 Å) of EBOV-GP in its metastable trimeric, prefusion conformation. Using this published crystal structure of EBOV-GP and the Schrödinger SiteMap program, we were able to examine the EBOV-GP structure and identify potential small-molecule binding sites on the basis of contiguous groups of hydrophobic amino acids positioned in an orientation that defined a hydrophobic pocket. For unbiased results, we set the program parameters to cover the entire EBOV-GP structure so that potential binding sites could be discovered.
The computational studies identified three potential binding sites (S1 to S3) with site scores of >0.9 (Fig. 3 A). S1 and S3 are hydrophilic in nature and seem unlikely binding sites for compound 7, a small hydrophobic molecule which bears two hydrophobic groups (cLogP, >5). Therefore, the hydrophobic binding site S2, with a hydrophobic/hydrophilic ratio of 4.75, appears to be the most likely compound 7 binding site. The S2 site is created by clamping of the GP1 base subdomain with the GP2 internal fusion loop and the heptad repeat 1A helix (Fig. 3A and B). At site S2, amino acid residues Val66, Leu68, Asn69, Leu70, Leu184, and Leu186 in the base and head subdomains of GP1 and residues Tyr517, Met548, Leu554, and Leu558 on the GP2 internal fusion loop are predicted to be the residues most likely involved in compound 7 binding to EBOV-GP. Close to this hydrophobic pocket, amino acids Arg64, Glu523, and Thr520 may act as potential hydrogen bond donors and acceptors for ligand binding (Fig. 3B). An examination of the residues in the GP1 sequence suggests that (i) these residues are located in and around the conserved charged and hydrophobic residues of GP1 (Fig. 3C) that are important for GP1 structure, folding, and receptor binding (29, 30) and (ii) except for Lys190 and Lys191, all other amino acid residues potentially involved in binding of compound 7 to EBOV-GP are either nonpolar or aromatic and are involved in maintaining the structural integrity of GP1 for receptor binding or fusion. Therefore, single-amino-acid mutations of these nonpolar or aromatic residues would be expected to generate escape mutants. In summary, the computational studies using the Schrödinger SiteMap program identified a putative binding site on the basis of contiguous groups of hydrophobic amino acids positioned in an orientation that defines a hydrophobic pocket.
FIG. 3.

Computational studies locate a hydrophobic pocket at or near the GP1-GP2 interface in the GP prefusion crystal structure. (A) The three proposed binding sites S1 to S3, calculated with the Schrödinger Sitemap program, are shown in colored grids. Hydrophobic, ligand donor, and ligand acceptor maps are shown in yellow, blue, and red, respectively. EBOV-GP1 is shown in yellow and red ribbons, and GP2 is displayed in blue ribbons. (B) Magnified view of the S2 binding site circled in panel A. The putative binding site S2 was identified using Schrödinger SiteMap. Selected residues which are proposed to make contacts within the binding site are displayed as stick models. (C) The amino acid residues in the EBOV-GP1 sequence that are important for viral entry are shown. The charged residues are underlined, while hydrophobic residues are shown as gray shaded regions. The arrows indicate the positions of mutations in EBOV-GP1 of the HIV/EBOV-GP mutant pseudotype viruses utilized in this study.
Confirmation of binding of compound 7 in the hydrophobic S2 site by mutational analysis.
We performed mutational studies to confirm and validate the computational results. Work with EBOV requires a BSL-4 facility, making it challenging to generate EBOV escape mutants that are resistant to compound 7. Moreover a selectivity index of less than 10 also suggests that we may not be able to generate mutants without considerable cytotoxicity. Therefore, to support the S2 site binding hypothesis generated by the computational studies, we examined HIV/EBOV-GP mutants carrying single-amino-acid mutations in the S2 region for inhibition of infectivity by compound 7. Previous studies by Manicassamy et al. (29) have shown that single-amino-acid alterations at positions R64K, N69A, L70A, D78A, K84A, R172A, L184A, I185A, L186A, K190A/K191A, and Y225A cause no defect in viral entry. The same study also indicated that mutations at amino acid positions Val66, and Leu68 in EBOV-GP1 generate noninfectious virus. Mutants carrying K84A, R172A, and Y225A were used as controls in the study, since these residues are unlikely to be involved in binding with compound 7 but are conserved among filoviruses. As shown in Fig. 4 A, compound 7, even at concentrations of as high as 50 μM, displayed little or no inhibition of HIV/EBOV-GP N69A and L70A mutants. The HIV/EBOV-GP L184A, I185A, L186A, and K190A/K191A mutants were also less susceptible to inhibition, displaying ≤30% inhibition at a 25 μM concentration of compound 7 (Fig. 4B). In contrast, compound 7 inhibited the infectivity of the R64K, D78A, K84A, R172A, and Y225A mutants by ≥75% at a concentration of 25 μM (Fig. 4A and B). Together, these results suggest that compound 7 interacts with the amino acid residues Asn69, Leu70, Leu184, Ile185, Leu186, Lys190, and Lys191 and therefore likely binds to EBOV-GP at or near the S2 hydrophobic site.
FIG. 4.

Effect of compound 7 on the infectivity of HIV/EBOV-GP mutants. 293T cells were infected with mutant or wild-type HIV/EBOV-GP pseudotype virus in the presence of increasing concentrations of compound 7. Inhibition of HIV/EBOV-GP pseudotype infection was detected as a reduced luciferase signal. Error bars indicate standard deviations. (A) Effect of compound 7 on the infectivities of R64K, N69A, L70A, D78A, and K84A HIV/EBOV-GP mutants. (B) Effect of compound 7 on the infectivities of R172A, L184A, I185A, L186A, K190A/K191A, and Y225A HIV/EBOV-GP mutants.
We further investigated whether compound 7 binds to the RBD of EBOV-GP and inhibits its binding to host receptors and cofactors. We used a previously described fusion protein that contains EBOV-GP1 RBD (amino acid residues 54 to 201) fused with the human IgG Fc region (21). As a negative control, we used the SARS virus spike protein (S1-hIgG), which was previously shown not to bind to 293T and Vero cells due to a very low level of the SARS virus receptor (20). We found that compound 7 had no effect on the binding of the EBOV RBD domain to 293T or Vero cells (data not shown). Therefore, we hypothesize that compound 7 binds to the S2 hydrophobic pocket and not to the RBD of EBOV-GP.
Preliminary SAR analysis.
We performed a preliminary structure-activity relationship (SAR) analysis of the benzodiazepine structure without altering the benzodiazepine backbone. The results are shown in Table 4 along with those for the screening hit, compound 7. The initial SAR study was designed to explore the effect of electronic properties and to examine the effect of the sizes of substituents of the benzodiazepine backbone. As shown in Table 4, a dichloro substitution on the benzene ring of the benzodiazepine core (compound 12) increased activity against the anti-HIV/EBOV-GP pseudotype virus 3-fold. In contrast, a bulky fused benzo ring (compound 9) or dimethyl substitution (compound 10) decreased the potency. Compound 12 was also found to inhibit GFP-ZEBOV in a dose-dependent manner, providing over 90% inhibition at a concentration (5 μM) that displayed no cytotoxicity (Fig. 5 B). Overall, small substitutions on the diazepine ring of the benzodiazepine are tolerated, while addition of a second aromatic or heteroaromatic group on the diazepine ring is detrimental to the antiviral activity (Fig. 5A). These preliminary SAR results are consistent with a specific target-inhibitor interaction in which the bulky aromatic substitutions on the azepine ring introduce some size constraints on the inhibitor and prevent it from occupying the binding pocket. Because compound 12 exhibits increased antiviral potency and a higher selectivity index than compound 7 (Table 4), it will be the starting point for optimization of the benzodiazepine series for identification of lead compounds using medicinal chemistry.
TABLE 4.
IC90 values for the benzodiazepine analogs
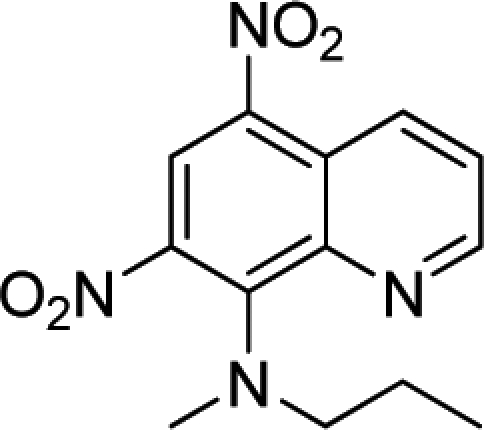 
|
HIV/EBOV-GP was generated by transfection of 293T cells with HIV-l expression vector (pNL4.3.Luc.R-E-) and with EBOV-GP. IC90 values were determined using reordered compounds as different batches from the original vendors or freshly synthesized compounds.
293T cells were treated with compound alone. CC50 values were determined from linear portions of the dose-response curve.
SI, selectivity index (CC50/IC90).
cLogP values were calculated with ChemDraw v12.0.
ND, not done.
FIG. 5.
Analysis of analogs of compound 7. (A) Summary of preliminary SAR results for benzodiazepine analogs of HIV/EBOV-GP compound 7. (B) Comparison of antiviral activity and cell toxicity of compound 12, as described in Materials and Methods. The blue diamonds represent anti-GFP-ZEBOV activity, while the red squares represent cytotoxicity. (C) Effect of compound 12 on the infectivities of N69A, L70A, L184A, I185A, L186A, and K190A/K191A HIV/EBOV-GP mutants. 293 T cells were infected with HIV/EBOV-GP pseudotype virus in the presence of increasing concentrations of compound 12. Inhibition of HIV/EBOV-GP infection was detected as a reduced luciferase signal. Error bars indicate standard deviations.
As shown in Fig. 5C, compound 12 also failed to inhibit the same HIV/EBOV-GP mutants, carrying N69A, L70A, L184A, I185A, L186A, and K190A/K191A, that were resistant to compound 7, suggesting that compound 12 localizes in the same hydrophobic pocket (S2) in EBOV-GP as compound 7. Taken together, these results confirm our earlier findings that these novel benzodiazepine filovirus inhibitors bind to the S2 hydrophobic pocket and interact with amino acid residues Asn69, Leu70, Leu184, Ile185, Leu186, Lys190, and Lys191.
DISCUSSION
In recent years, entry inhibitors have emerged as a new class of antiviral drugs, with the HIV fusion-blocking peptide enfuvirtide being the first available fusion inhibitor applied in clinical treatment of a viral infection in humans (57). Several small-molecule entry inhibitors against other important viruses, such as measles virus (35, 36), influenza virus (43), respiratory syncytial virus (12), and arenaviruses (22, 24), are currently in development. EBOV-GP mediates binding of EBOV with its cell surface receptor(s)/coreceptor(s) and subsequent entry, involving endocytosis of virus and fusion between viral and host cell membranes (44). Therefore, we hypothesized that blocking of EBOV-GP-mediated viral entry will lead to inhibition of infection. Moreover, the aggressive nature of EBOV infections, in particular the rapid and overwhelming viral burdens in infected patients and EBOV-GP-related cell cytotoxicity (44, 45), justify preferentially targeting the entry process over subsequent downstream stages of viral replication.
In this study, we used HIV/EBOV-GP pseudotype virus as a surrogate model to screen libraries of 52,500 small-molecule compounds for inhibitors based on the following rationale. Since viral entry is determined solely by interaction of the virus envelope proteins with cell receptors (32), the replication-incompetent HIV/EBOV-GP mimics the EBOV entry process (29, 30) and can be used in HTS of compound libraries in a BSL-2 laboratory. The HTS “hits” include at least four possible categories of inhibitors: (i) inhibitors of HIV/EBOV-GP entry, (ii) inhibitors of HIV replication, (iii) inhibitors of luciferase enzyme activity, and (iv) cytotoxic compounds. Therefore, to eliminate off-target hits such as the inhibitors of HIV replication and/or luciferase enzyme activity, we counterscreened the HTS hits with HIV/VSV-G pseudotype virus. HIV/VSV-G has the same HIV backbone as HIV/EBOV-GP but expresses VSV-G, a member of a different class of viral fusion-active protein (24, 40, 41), on its surface. The counterscreen with HIV/VSV-G proved to be particularly useful since it eliminated the off-target hits that compromise 95% of the total HTS hits (Table 1). It is possible that the counterscreen may have eliminated some HIV/EBOV-GP entry inhibitors, since the cell receptors of both EBOV-GP and VSV-G are not known and the wild-type EBOV and VSV infect a wide range of cells. Nevertheless, the counterscreen is an important step to eliminate undesired off-target HTS hits, and it allows us to focus on the most specific hits. Moreover, both EBOV and VSV enter cells by endocytosis followed by pH-dependent membrane fusion in endosomes (11, 31, 46). Therefore, this counterscreen also eliminated compounds that modulate membrane trafficking or endosomal pH and identified compounds that bind to either EBOV-GP or specific components of the EBOV entry pathway that are not shared with VSV.
We identified eight novel inhibitors of EBOV entry. The IC90 values for six of the eight compounds against GFP-ZEBOV were within ∼2- to 3-fold of the values for HIV/EBOV-GP, while IC90 values for compounds 3 and 4 against GFP-ZEBOV are significantly higher (>20-fold difference) (Table 2). At this time we do not know the reasons for the differences in the IC90 values between HIV/EBOV-GP and GFP-ZEBOV, but they may be due to differences in the (i) virus morphology (EBOV is cylindrical, while HIV/EBOV-GP is spherical), (ii) GP density at the cell surface, (iii) GP modification (e.g., producer cell type-specific glycosylation patterns), and (iv) target cells (293T versus VeroE6). Compound 7 was selected for further chemical optimization and mechanism studies because (i) the benzodiazepine structure is a suitable “drug-like” starting point for medicinal chemistry, (ii) the compound exhibited good potency in both the HIV/EBOV-GP and cell culture grown-infectious EBOV assay, and (iii) its selectivity index (SI of >5) indicates that the compound exhibits significant selectivity for antiviral activity versus cytotoxicity (Fig. 1A). The preliminary SAR evaluation so far has identified several additional benzodiazepine analogs that exhibit anti-EBOV activity, including one more potent (3 times more) analog (compound 12) that also displays a selectivity index of greater than 10. Although the sample size for the SAR analysis is small, the increased activity of compound 12 suggests that more potent inhibitors can be designed using the benzodiazepine backbone. In addition, with a selectivity index of >10 and higher antiviral potency, compound 12 will be the starting point for optimization of the benzodiazepine series to identify a lead inhibitor using medicinal chemistry. The systematic synthesis of a larger library of compounds (see the supplemental material), with the substituents carefully controlled, will be necessary to identify informative SAR trends, and this will be the first priority of the synthetic plan for further development of the benzodiazepine series to obtain lead compounds.
Virus entry is a multistep process, and target identification for compound 7 may be challenging. Three approaches have been taken so far to determine the antiviral target. First, we examined the antiviral spectrum of compound 7 against various filoviruses and nonfiloviruses (Table 3). It is reasonable to hypothesize that a compound capable of inhibiting multiple viruses from different families most likely inhibits or blocks either host receptors or other host factors involved in virus entry. However, compound 7 inhibited primarily only EBOV and MARV, the two members of the filovirus family. The critical amino acid residues that are important for virus entry are conserved in both EBOV-GP and MARV-GP (29, 30). Compound 7 exhibited very weak activity against other viruses bearing similar type 1 envelope proteins but, in general, exhibited no activity against other RNA and DNA viruses. The results suggest that compound 7 is not inhibiting host factors such as the endosomal pathways involved in infection of a number of enveloped viruses.
Second, we performed “time-of-addition” studies, which indicated that compound 7 blocks at an early stage of virus entry into cells, possibly by binding to EBOV-GP. Preincubation of HIV/EBOV-GP with compound 7 also blocked its infection of 293T cells (data not shown), further suggesting that compound 7 binds to HIV/EBOV-GP. Similarly, the differential binding of HIV/EBOV-GP and HIV/VSV-G by compound 7 in the virus-compound binding assay suggests that compound 7 binds EBOV-GP, since both the viruses have the same HIV core. Taken together, these studies indicate that compound 7 acts early in the infection process, probably by binding to EBOV-GP. From this experiment, it is not possible to stoichiometrically measure the binding of EBOV-GP and compound 7. Since the pseudotype virus randomly acquires EBOV-GP from the surfaces of infected cells and since it is competent for only one round of replication, it has not been feasible to determine precisely the number of mature trimeric EBOV-GP molecules on the viral surface, the number of viral particles carrying the EBOV-GP, or the stoichiometry of the compound 7-EBOV-GP interaction. Nuclear magnetic resonance (NMR) studies are planned to better understand the interactions of compound 7 with EBOV-GP.
EBOV-GP is heavily glycosylated by both N-linked and O-linked glycans. As a result, only a few sites are left exposed and accessible for binding interactions. These sites include (i) a region at the base of the chalice where GP1 meets GP2, (ii) short linear stretches of polypeptide between glycans in the mucin-like domain, and (iii) the HR2 region. Studies using an EBOV-GP1 RBD peptide (amino acids [aa] 54 to 201) showed that compound 7 has no effect on the binding of EBOV-GP to 293T and Vero cell lines. In addition, compound 7 inhibited both wt HIV/EBOV-GP and HIV/EBOV-GPΔmucin with similar potencies (data not shown). In HIV/EBOV-GPΔmucin, the mucin domain is removed to expose the EBOV-GP1 RBD. Taken together, these results indicate that the primary mechanism of action of compound 7 is not direct blocking of virus attachment to the cells by binding the RBD of GP1. Instead, we hypothesize that compound 7 binds to a region of the EBOV-GP at the base of the chalice where GP1 meets GP2, since this region is exposed in both EBOV-GP and EBOV-GPΔmucin.
Supporting evidence for this hypothesis comes from our computational studies using the recently published crystal structure (3.4 Å) of EBOV-GP in its metastable trimeric, prefusion conformation (25), as confirmed by our mutational studies. The computational studies using the Schrödinger SiteMap program identified a putative hydrophobic binding pocket (S2) for small-molecule ligand binding at the junction of the GP1 base subdomain with the GP2 internal fusion loop and the heptad repeat 1A helix (HR1A). Amino acid residues Val66, Leu68, Asn69, Leu70, Leu184, and Leu186 from the base and head subdomains of GP1 together with residues Tyr517, Met548, Leu554, and Leu558 on the GP2 internal fusion loop in the hydrophobic S2 binding site appear to be involved in interacting with bound ligands, according to the model. Indeed, a significant reduction in the potency of compound 7 against HIV/EBOV-GP mutants N69A, L70A, L184A, I185A, L186A, and K190A/K191A further implicates the S2 hydrophobic pocket of EBOV-GP as a potential binding site for small-molecule ligands such as the benzodiazepine compound 7.
The EBOV-GP1 base subdomain contains four discontinuous sections (residues 33 to 69, 95 to 104, 158 to 167, and 176 to 189) which form two mixed beta-sheets, with strands β3 and β13 shared between the two beta-sheets. The head subdomain is composed of residues from four discontinuous segments, i.e., residues 70 to 94, 105 to 157, 168 to 175, and 214 to 226, and forms a four-stranded, mixed beta-sheet supported by an alpha-helix and a smaller, two-stranded antiparallel beta-sheet (25, 26). As shown from the mutation studies, the HIV/EBOV-GP N69A and L70A mutants are not inhibited by compound 7, suggesting that the amino acid residues at the junction between the head and base subdomains of GP1 are important for binding. Compound 7 weakly inhibited HIV/LASV-GP, HIV/LMCV-GP, HIV/MACV-GP, and HIV/HA(H5) at higher concentrations. These pseudotype viruses also contain type 1 membrane proteins similar to EBOV-GP. The type 1 membrane protein carries a similar cavity formed by the clamping of the N-terminal membrane-proximal base of the receptor binding subunit over the internal fusion loop through hydrophobic interactions, including an interchain disulfide bond (37, 38). Therefore, our results suggest that the size and the conformation of the S2 hydrophobic pocket of EBOV-GP are important and that compound 7 selectively binds in this pocket to prevent viral activity. This hypothesis is supported by limited preliminary SAR analysis of compound 7, which showed that bulky aromatic substitutions on the diazepine ring introduce some size constraints on the inhibitor and reduce its antiviral activity (Fig. 5A; Table 4). The lack of activity for compounds with a bulky substituent(s) implies a binding site with well-defined boundaries.
A limited number of small-molecule inhibitors of EBOV have been discovered (7, 8). However, none of these inhibitors are used in clinical settings, and none of them resemble compound 7 structurally. The existing small-molecule inhibitors of EBOV infections can be characterized by three general modes of action: (i) impairment of viral replication, (ii) stimulation of innate antiviral mechanisms, and (iii) prevention of virus entry into the cells.
The carbocyclic adenosine analog 3-deazaadenosine (C-c3Ado), inhibits cellular S-adenosylhomocysteine hydrolase and inhibits the replication of EBOV Zaire in vitro, with an IC50 of 30 μM (7, 9). The activity of this antiviral agent has been attributed to diminished methylation of the 5′ cap of viral mRNA by (guanine 7) methyltransferase, which impairs the translation of viral transcripts. Administration of C-c3Ado to EBOV-infected mice has also been found to dramatically increase production of alpha interferon (IFN-α), which may serve to counteract the virus suppression of the innate antiviral response. Unfortunately, C-c3Ado failed to promote IFN-α production in Ebola virus-infected monkeys (7, 9). More recently, other small-molecule EBOV inhibitors, such as FGI-103, FGI-104, and FGI-106, were discovered (1, 50, 61) by HTS screening. These inhibitors were found to be potent and to provide protection against EBOV and Marburg virus in vitro, and in vivo. While FGI-103 inhibits infection by an unknown mechanism, FGI-104 targets EBOV vp40-tgs101-mediated budding. In contrast, FGI-106 displays potent and broad-spectrum inhibition of lethal viral hemorrhagic fever pathogens, including Ebola, Rift Valley and dengue fever viruses, in cell-based assays, suggesting that it interferes with a common pathway utilized by different viruses (1).
The glycodendritic structure BH30sucMan (23, 58), which contains 32 individual α-mannose units linked to the hyperbranched dendrimer BH30 through succinyl spacers, has recently been shown to block the interaction between dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN) (58) and EBOV-GP. However, this compound is not specific for EBOV infection. Endosomal proteases CatB and CatL mediate viral entry by carrying out proteolysis of the EBOV-GP1 subunit (11). Recently, the CatL inhibitor tetrahydroquinoline oxocarbazate was reported to inhibit EBOV infection at nanomolar concentrations (47). It also inhibits other viruses that use CatL for entry. Unfortunately, given the demonstrated hypersensitivity of EBOV-GP1 to digestion by other proteases (17, 46), such as thermolysin (17), the clinical prospects for antiviral agents that solely target CatB and CatL are not encouraging. Compound 7 and its analogs were found to have no activity against CatL in in vitro enzymatic assays (data not shown). Compound 7 and its analogs differ from these previously reported small-molecule inhibitors by the specificity exhibited for filoviruses and the apparent mechanism of action. Unlike the other entry inhibitors, the benzodiazopenes may bind directly to EBOV-GP within a hydrophobic pocket at the EBOV GP1-GP2 interface. Moreover, blocking of propagation of EBOV at an early stage will minimize the chance for the virus to evolve and acquire drug resistance.
We conclude that compound 7 acts at an early stage of viral entry, apparently by binding to a hydrophobic pocket (S2) in the prefusion conformation of EBOV-GP and preventing infection by an unknown mechanism. An analogy can be made with several different classes of small-molecule HIV entry inhibitors, including maraviroc, that are thought to bind within a pocket created by four transmembrane domains of CCR5, an important HIV coreceptor (19). Furthermore, computational and mutational studies suggest that the S2 hydrophobic binding pocket is a well-defined small-molecule binding site, which may serve as a viable target for future antifilovirus drug discovery.
Supplementary Material
Acknowledgments
We thank Krishna Kota, Dutch Boltz, Julie Tran, and Xiaoli Chi from USAMRIID for assays using BSL-4 containment and procedures.
This research was supported by DHHS/NIH grants 1R43AI071450-01, 1R01AI089590-01, and 5U01AI077767-02 from the National Institutes of Health.
Footnotes
Published ahead of print on 26 January 2011.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Aman, M. J., et al. 2009. Development of a broad-spectrum antiviral with activity against Ebola virus. Antiviral Res. 83:245-251. [DOI] [PubMed] [Google Scholar]
- 2.Barnard, D. L., et al. 1999. 2-5A-DNA conjugate inhibition of respiratory syncytial virus replication: effects of oligonucleotide structure modifications and RNA target site selection. Antiviral Res. 41:119-134. [DOI] [PubMed] [Google Scholar]
- 3.Barnard, D. L., et al. 2002. Coumarins and pyranocoumarins, potential novel pharmacophores for inhibition of measles virus replication. Antivir. Chem. Chemother. 13:39-59. [DOI] [PubMed] [Google Scholar]
- 4.Basu, A., et al. 2007. Sulfated homologues of heparin inhibit hepatitis C virus entry into mammalian cells. J. Virol. 81:3933-3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basu, A., et al. 2006. Stellate cell apoptosis by a soluble mediator from immortalized human hepatocytes. Apoptosis 11:1391-1400. [DOI] [PubMed] [Google Scholar]
- 6.Bosio, C. M., et al. 2004. Ebola and Marburg virus-like particles activate human myeloid dendritic cells. Virology 326:280-287. [DOI] [PubMed] [Google Scholar]
- 7.Bray, M. 2003. Defense against filoviruses used as biological weapons. Antiviral Res. 57:53-60. [DOI] [PubMed] [Google Scholar]
- 8.Bray, M., and J. Paragas. 2002. Experimental therapy of filovirus infections. Antiviral Res. 54:1-17. [DOI] [PubMed] [Google Scholar]
- 9.Bray, M., J. L. Raymond, T. Geisbert, and R. O. Baker. 2002. 3-Deazaneplanocin A induces massively increased interferon-alpha production in Ebola virus-infected mice. Antiviral Res. 55:151-159. [DOI] [PubMed] [Google Scholar]
- 10.Chan, S. Y., et al. 2001. Folate receptor-alpha is a cofactor for cellular entry by Marburg and Ebola viruses. Cell 106:117-126. [DOI] [PubMed] [Google Scholar]
- 11.Chandran, K., N. J. Sullivan, U. Felbor, S. P. Whelan, and J. M. Cunningham. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cianci, C., et al. 2004. Orally active fusion inhibitor of respiratory syncytial virus. Antimicrob. Agents Chemother. 48:413-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo, Q., et al. 2003. Biochemical and genetic characterizations of a novel human immunodeficiency virus type 1 inhibitor that blocks gp120-CD4 interactions. J. Virol. 77:10528-10536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo, Y., et al. 2009. Analysis of hemagglutinin-mediated entry tropism of H5N1 avian influenza. Virol. J. 6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He, J., et al. 1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69:6705-6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito, H., S. Watanabe, A. Takada, and Y. Kawaoka. 2001. Ebola virus glycoprotein: proteolytic processing, acylation, cell tropism, and detection of neutralizing antibodies. J. Virol. 75:1576-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jane-Valbuena, J., L. A. Breun, L. A. Schiff, and M. L. Nibert. 2002. Sites and determinants of early cleavages in the proteolytic processing pathway of reovirus surface protein sigma3. J. Virol. 76:5184-5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeffers, S. A., D. A. Sanders, and A. Sanchez. 2002. Covalent modifications of the Ebola virus glycoprotein. J. Virol. 76:12463-12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kondru, R., et al. 2008. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol. Pharmacol. 73:789-800. [DOI] [PubMed] [Google Scholar]
- 20.Kuhn, J. H., W. Li, S. R. Radoshitzky, H. Choe, and M. Farzan. 2007. Severe acute respiratory syndrome coronavirus entry as a target of antiviral therapies. Antivir. Ther. 12:639-650. [PubMed] [Google Scholar]
- 21.Kuhn, J. H., et al. 2006. Conserved receptor-binding domains of Lake Victoria marburgvirus and Zaire ebolavirus bind a common receptor. J. Biol. Chem. 281:15951-15958. [DOI] [PubMed] [Google Scholar]
- 22.Larson, R. A., et al. 2008. Identification of a broad-spectrum arenavirus entry inhibitor. J. Virol. 82:10768-10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lasala, F., E. Arce, J. R. Otero, J. Rojo, and R. Delgado. 2003. Mannosyl glycodendritic structure inhibits DC-SIGN-mediated Ebola virus infection in cis and in trans. Antimicrob. Agents Chemother. 47:3970-3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee, A. M., et al. 2008. Unique small molecule entry inhibitors of hemorrhagic fever arenaviruses. J. Biol. Chem. 283:18734-18742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee, J. E., et al. 2008. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454:177-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, J. E., et al. 2008. Complex of a protective antibody with its Ebola virus GP peptide epitope: unusual features of a V lambda x light chain. J. Mol. Biol. 375:202-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leroy, E. M., et al. 2009. Human Ebola outbreak resulting from direct exposure to fruit bats in Luebo, Democratic Republic of Congo, 2007. Vector Borne Zoonotic Dis. 9:723-728. [DOI] [PubMed] [Google Scholar]
- 28.Lipinski, C. A., F. Lombardo, B. W. Dominy, and P. J. Feeney. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv Rev. 46:3-26. [DOI] [PubMed] [Google Scholar]
- 29.Manicassamy, B., J. Wang, H. Jiang, and L. Rong. 2005. Comprehensive analysis of Ebola virus GP1 in viral entry. J. Virol. 79:4793-4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manicassamy, B., et al. 2007. Characterization of Marburg virus glycoprotein in viral entry. Virology 358:79-88. [DOI] [PubMed] [Google Scholar]
- 31.Martinez, O., et al. 2010. Zaire Ebola virus entry into human dendritic cells is insensitive to cathepsin L inhibition. Cell. Microbiol. 12:148-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer, K., A. Basu, and R. Ray. 2000. Functional features of hepatitis C virus glycoproteins for pseudotype virus entry into mammalian cells. Virology 276:214-226. [DOI] [PubMed] [Google Scholar]
- 33.Panchal, R. G., K. P. Kota, K. B. Spurgers, G. Ruthel, J. P. Tran, R. C. Boltz, and S. Bavari. Development of high-content imaging assays for lethal viral pathogens. J. Biomol. Screen. 15:755-765. [DOI] [PubMed]
- 34.Peterson, A. T., D. S. Carroll, J. N. Mills, and K. M. Johnson. 2004. Potential mammalian filovirus reservoirs. Emerg. Infect. Dis. 10:2073-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plemper, R. K., et al. 2005. Design of a small-molecule entry inhibitor with activity against primary measles virus strains. Antimicrob. Agents Chemother. 49:3755-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plemper, R. K., et al. 2004. A target site for template-based design of measles virus entry inhibitors. Proc. Natl. Acad. Sci. U. S. A. 101:5628-5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radoshitzky, S. R., et al. 2007. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446:92-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radoshitzky, S. R., et al. 2008. Receptor determinants of zoonotic transmission of New World hemorrhagic fever arenaviruses. Proc. Natl. Acad. Sci. U. S. A. 105:2664-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray, R. B., et al. 2004. Ebola virus glycoprotein-mediated anoikis of primary human cardiac microvascular endothelial cells. Virology 321:181-188. [DOI] [PubMed] [Google Scholar]
- 40.Roche, S., S. Bressanelli, F. A. Rey, and Y. Gaudin. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187-191. [DOI] [PubMed] [Google Scholar]
- 41.Roche, S., F. A. Rey, Y. Gaudin, and S. Bressanelli. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843-848. [DOI] [PubMed] [Google Scholar]
- 42.Rumschlag-Booms, E., Y. Guo, J. Wang, M. Caffrey, and L. Rong. 2009. Comparative analysis between a low pathogenic and a high pathogenic influenza H5 hemagglutinin in cell entry. Virol. J. 6:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Russell, C. A., et al. 2008. Influenza vaccine strain selection and recent studies on the global migration of seasonal influenza viruses. Vaccine 26(Suppl. 4):D31-D34. [DOI] [PubMed] [Google Scholar]
- 44.Sanchez, A., et al. 2001. Filoviridae: Marburg and Ebola viruses, p. 1279-1304. In D. M. Knipe and P. M. Howley (ed.), Fields virology. Lippincott, Williams & Wilkins, Philadelphia PA.
- 45.Sanchez, A., et al. (ed.). 2001. Filoviridae: Marburg and Ebola viruses, vol. 1. Lippincott, Williams & Wilkins, Philadelphia, PA.
- 46.Schornberg, K., et al. 2006. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 80:4174-4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shah, P. P., et al. 2010. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and Ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 78:319-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimojima, M., et al. 2006. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 80:10109-10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sidwell, R. W., and D. F. Smee. 2000. In vitro and in vivo assay systems for study of influenza virus inhibitors. Antiviral Res. 48:1-16. [DOI] [PubMed] [Google Scholar]
- 50.Silvestri, L. S., et al. 2007. Involvement of vacuolar protein sorting pathway in Ebola virus release independent of TSG101 interaction. J. Infect. Dis. 196(Suppl. 2):S264-S270. [DOI] [PubMed] [Google Scholar]
- 51.Simmons, G., et al. 2003. DC-SIGN and DC-SIGNR bind Ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 305:115-123. [DOI] [PubMed] [Google Scholar]
- 52.Simmons, G., et al. 2003. Folate receptor alpha and caveolae are not required for Ebola virus glycoprotein-mediated viral infection. J. Virol. 77:13433-13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simmons, G., R. J. Wool-Lewis, F. Baribaud, R. C. Netter, and P. Bates. 2002. Ebola virus glycoproteins induce global surface protein down-modulation and loss of cell adherence. J. Virol. 76:2518-2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stroher, U., et al. 2001. Infection and activation of monocytes by Marburg and Ebola viruses. J. Virol. 75:11025-11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sullivan, N. J., A. Sanchez, P. E. Rollin, Z. Y. Yang, and G. J. Nabel. 2000. Development of a preventive vaccine for Ebola virus infection in primates. Nature 408:605-609. [DOI] [PubMed] [Google Scholar]
- 56.Takada, A., and Y. Kawaoka. 2001. The pathogenesis of Ebola hemorrhagic fever. Trends Microbiol. 9:506-511. [DOI] [PubMed] [Google Scholar]
- 57.Tilton, J. C., and R. W. Doms. 2010. Entry inhibitors in the treatment of HIV-1 infection. Antiviral Res. 85:91-100. [DOI] [PubMed] [Google Scholar]
- 58.van Kooyk, Y., A. Engering, A. N. Lekkerkerker, I. S. Ludwig, and T. B. Geijtenbeek. 2004. Pathogens use carbohydrates to escape immunity induced by dendritic cells. Curr. Opin. Immunol. 16:488-493. [DOI] [PubMed] [Google Scholar]
- 59.Volchkov, V. E., et al. 1995. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 214:421-430. [DOI] [PubMed] [Google Scholar]
- 60.Wang, Q. Y., et al. 2009. A small-molecule dengue virus entry inhibitor. Antimicrob. Agents Chemother. 53:1823-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Warren, T. K., et al. 2010. Antiviral activity of a small-molecule inhibitor of filovirus infection. Antimicrob. Agents Chemother. 54:2152-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang, Z. Y., et al. 2000. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 6:886-889. [DOI] [PubMed] [Google Scholar]
- 63.Zhang, J. H., T. D. Chung, and K. R. Oldenburg. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Scree.n. 4:67-73. [DOI] [PubMed] [Google Scholar]
- 64.Zhang, N., et al. 2002. In vitro inhibition of the measles virus by novel ring-expanded (′fat') nucleoside analogues containing the imidazo[4,5-e]diazepine ring system. Bioorg. Med. Chem. Lett. 12:3391-3394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.