

Abstract
Reovirus cell entry is initiated by viral attachment to cell surface glycans and junctional adhesion molecule A. Following receptor engagement, reovirus is internalized into cells by receptor-mediated endocytosis using a process dependent on β1 integrin. Endocytosed virions undergo stepwise disassembly catalyzed by cathepsin proteases, followed by endosomal membrane penetration and delivery of transcriptionally active core particles into the cytoplasm. Cellular factors that mediate reovirus endocytosis are poorly defined. We found that both genistein, a broad-spectrum tyrosine kinase inhibitor, and PP2, a specific Src-family kinase inhibitor, diminish reovirus infectivity by blocking a cell entry step. Although neither inhibitor impedes internalization of reovirus virions, both inhibitors target virions to lysosomes. Reovirus colocalizes with Src during cell entry, and reovirus infection induces phosphorylation of Src at the activation residue, tyrosine 416. Diminished Src expression by RNA interference reduces reovirus infectivity, suggesting that Src is required for efficient reovirus entry. Collectively, these data provide evidence that Src kinase is an important mediator of signaling events that regulate the appropriate sorting of reovirus particles in the endocytic pathway for disassembly and cell entry.
Viral replication is initiated by engagement of target cell receptors by viral capsid components. This initial contact elicits alterations in the virus, cell, or both that promote viral entry. For some viruses, receptor binding alone appears to activate the membrane-penetration machinery required to invade at the cell surface. For others, receptor-linked signaling events lead to internalization, which allows exposure to acidic pH or host enzymes required for viral penetration into the cytosol. How viruses induce cellular uptake and traffic in the endocytic compartment is important for an understanding of viral tissue tropism and may foster the development of antiviral therapeutics that target critical nodes in the viral entry process.
Mammalian orthoreoviruses (reoviruses) are nonenveloped double-stranded RNA (dsRNA) viruses that belong to the Reoviridae family, which includes the human pathogen rotavirus and the livestock pathogens African horse sickness virus and bluetongue virus. Reoviruses have a broad host range and infect most mammalian species (58). In newborn mice, reoviruses infect the intestine, heart, liver, lung, and central nervous system (67). Junctional adhesion molecule A (JAM-A) serves as a receptor for all reovirus serotypes (6, 13, 28). Following attachment to JAM-A, reovirus utilizes β1 integrins (38, 39) to enter cells, likely by clathrin-dependent endocytosis (9, 26, 39, 52, 61). After internalization, reovirus undergoes proteolytic disassembly mediated by endosomal cathepsin proteases (25, 40, 61). Cathepsin proteolysis results in removal of outer capsid-protein σ3 and cleavage of μ1 protein into particle-associated fragments δ and Φ (4, 10), yielding infectious subvirion particles (ISVPs). The σ1 attachment protein is subsequently shed, and the μ1 cleavage products mediate endosomal membrane penetration and release of transcriptionally active core particles into the cytoplasm (14, 15, 21, 43, 44).
The intracellular compartment in which reovirus disassembly occurs has not been conclusively identified. Late endosomes or lysosomes likely serve as disassembly sites, as these organelles are acidic and contain cathepsins (64). How reovirus is targeted to intracellular compartments used for disassembly also is poorly understood. Asparagine-proline-any residue-tyrosine (NPXY) motifs in the β1 integrin cytoplasmic tail are required for efficient reovirus infection. Moreover, mutation of the NPXY tyrosine residues to phenylalanine targets the virus to lysosomes for degradation (38). However, the mechanism by which β1 integrin NPXY motifs promote reovirus entry is not known.
The Src family of kinases contains eight members, Blk, Fgr, Fyn, Hck, Lck, Lyn, Src, and Yes, three of which, Fyn, Src, and Yes, are expressed in most cell types (62). Src is the prototype member of the Src family initially identified to be the oncoprotein of Rous sarcoma virus (12, 49). Src-family kinases contain six distinct functional domains: a myristylation domain that mediates interaction with the plasma membrane; a unique domain; Src homology (SH) domains 2 and 3, which regulate protein-protein interactions; a kinase domain that contains an autophosphorylation site (Y416 in Src); and a carboxy-terminal domain that includes a regulatory tyrosine (Y527 in Src) (56). Src activity is regulated by phosphorylation at residues Y416 and Y527. Phosphorylation of Y527 by the cytoplasmic kinase Csk maintains Src in an inactive conformation (18, 42, 45). Dephosphorylation of residue Y527, in parallel with Y416 autophosphorylation and conformational rearrangement, results in Src activation (11, 18, 47).
Src-family kinases regulate numerous cellular processes, including proliferation, differentiation, migration, adhesion, and cytoskeletal rearrangements (62). Src kinases transduce signals from a variety of receptors, including the epithelial growth factor receptor, fibroblast growth factor receptor, and vascular endothelial growth factor receptor (23, 31, 41, 54). These enzymes also mediate cell entry of both enveloped viruses, e.g., human immunodeficiency virus (HIV) (63) and Kaposi's sarcoma-associated herpesvirus (KSHV) (65), and nonenveloped viruses, e.g., coxsackievirus (19) and avian reovirus (48). However, mechanisms used by Src kinases to promote viral entry are not fully understood.
In this study, we found that genistein, a broad-spectrum tyrosine kinase inhibitor, and PP2, a specific Src-family kinase inhibitor, diminish reovirus infectivity by inhibiting an early step in the viral life cycle resulting in improper targeting of incoming virions to lysosomes. At early times following infection, Src colocalizes with reovirus virions and is phosphorylated at Y416, suggesting that reovirus induces activation of Src during entry. Reduction of Src expression by RNA interference (RNAi) results in decreased reovirus infectivity. These findings suggest that Src coordinates a signaling network that targets reovirus to endocytic organelles for viral disassembly and thus promotes functional entry into host cells.
MATERIALS AND METHODS
Cells, viruses, chemical inhibitors, and antibodies.
Spinner-adapted murine L929 cells were grown in either suspension or monolayer cultures in Joklik's modified Eagle's minimal essential medium (SMEM; Lonza) supplemented to contain 5% fetal bovine serum (FBS; Invitrogen), 2 mM l-glutamine (Invitrogen), 100 U of penicillin per ml, 100 μg of streptomycin per ml (Invitrogen), and 0.25 mg amphotericin B per ml (Sigma). HeLa CCL2 cells (obtained from Carolyn Coyne, University of Pittsburgh) were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented to contain 10% FBS, minimal essential medium nonessential amino acid solution (Sigma), 0.11 mg sodium pyruvate (Sigma) per ml, penicillin, streptomycin, and amphotericin B. Human embryonic kidney 293T (293T) cells were maintained in DMEM supplemented to contain 10% FBS, penicillin, streptomycin, and amphotericin B.
Reovirus type 1 Lang (T1L) is a laboratory stock strain. Working stocks of virus were prepared by plaque purification and passage using L929 cells (68). Purified virions were generated from second-passage L929 cell lysate stocks. Virus was purified from infected cell lysates by Freon extraction and CsCl gradient centrifugation as described previously (30). The band corresponding to the density of reovirus particles (1.36 g/cm3) was collected and dialyzed exhaustively against virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]). The reovirus particle concentration was determined from the equivalence of 1 unit of optical density at 260 nm to 2.1 × 1012 particles (59). Viral titers were determined by plaque assay using L929 cells (68). ISVPs were generated by treating 2 × 1011 particles with 20 μg of α-chymotrypsin (Sigma) in a 100-μl volume of virion storage buffer at 37°C for 60 min (3). Reactions were stopped by the addition of 2 mM phenylmethylsulfonyl fluoride (PMSF; Sigma).
Genistein, PP2, and U0126 (Calbiochem) were resuspended in dimethyl sulfoxide (DMSO) according to the manufacturer's instructions. The immunoglobulin G (IgG) fraction of rabbit antiserum raised against T1L (69) was purified by protein A-Sepharose as described previously (5). Phosphotyrosine 416 (phospho-Y416)-specific Src antibody, phosphotyrosine 527 (phospho-Y527)-specific Src antibody, total Src clone 36D10 antibody (Cell Signaling Technology), JAM-A-specific monoclonal antibody (provided by Charles Parkos, Emory University), human β1 integrin-specific monoclonal antibody MAB2253Z (Millipore), glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific antibody (Sigma), lysosome-associated membrane protein 1 (LAMP1)-specific antibody (Abcam), and actin-specific antibody (Santa Cruz Biotechnology) were used in indirect immunofluorescence and immunoblotting assays. Alexa Fluor-conjugated antibodies (Invitrogen) were used as secondary antibodies.
Src-EGFP plasmid.
A cDNA encoding murine Src was a gift from Carolyn Coyne. Enhanced green fluorescent protein (EGFP) was fused to the carboxy terminus of Src by sublconing Src into pEGFP-N1 (Clontech) by PCR amplification using Platinum Pfx (Invitrogen) and Src-FWD (CGACGAATTCACCATGGGCAGCAACAAGAGCAAGC) and Src-REV (ATCACGAGGGATCCATAGGTTCTCCCCGGGCTGG) oligonucleotide primers. PCR amplification was followed by restriction enzyme digestion and insertion of the Src fragment into the EcoRI and BamHI sites of pEGFP-N1. A linker was inserted between the carboxy terminus of Src and amino terminus of EGFP to preserve native Src localization and access to Y527 (23). The plasmid sequence was confirmed to ensure fidelity of cloning.
Fluorescent focus assay.
HeLa cells plated in 24-well plates (Costar) were either untreated or treated with DMSO or chemical inhibitor at 37°C for 1 h. Cells were adsorbed with reovirus virions or ISVPs in incomplete medium (without serum) at various multiplicities of infection (MOIs) in the presence of DMSO or chemical inhibitor at room temperature for 1 h. The inoculum was removed, cells were washed once with PBS, and complete medium with or without chemical inhibitor was added. Infected cells were incubated at 37°C for 20 h to allow a single cycle of viral replication. Cells were fixed with cold absolute methanol at −20°C for at least 30 min, washed with phosphate-buffered saline (PBS), incubated with PBS-bovine serum albumin (BSA; 5%) for 15 min, and incubated with reovirus-specific polyclonal antiserum (1:1,000) in PBS containing 0.5% Triton X-100 (PBS-TX) at room temperature for 1 h. Cells were washed twice with PBS and incubated with Alexa Fluor 488- or 546-labeled anti-rabbit IgG (1:1,000) in PBS-TX at room temperature for 1 h. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Cells were visualized by indirect immunofluorescence using an Axiovert 200 fluorescence microscope (Carl Zeiss). Infected cells (fluorescent focus units [FFU]) were identified by diffuse cytoplasmic fluorescence staining. Reovirus-infected cells were quantified by scoring either entire wells or random fields in equivalently confluent monolayers for three to five fields of view for triplicate wells (5).
Virus replication.
Monolayers of HeLa CCL2 cells in 24-well plates were treated with DMSO or chemical inhibitor at 37°C for 1 h and adsorbed in triplicate with reovirus at an MOI of 0.01 PFU per cell at room temperature for 1 h in incomplete medium containing DMSO or chemical inhibitor. Cells were washed once with PBS and incubated in medium with serum and DMSO or chemical inhibitor for 0 or 24 h. Cells were frozen and thawed twice prior to determination of viral titer by plaque assay (68).
Cell viability.
HeLa CCL2 cells in 24-well plates were treated with DMSO or chemical inhibitor at 37°C for 24 h, washed once with PBS, lifted from the plate with 0.05% trypsin EDTA (Invitrogen), and resuspended in DMEM supplemented to contain 0.067% trypan blue (Cellgro). Cells were enumerated using a hemocytometer, excluding cells showing blue staining, before and after chemical inhibitor treatment.
Flow cytometry.
HeLa CCL2 cells were treated with DMSO, 100 μM genistein, or 5 μM PP2 at 37°C for 1 h and washed once with PBS. Cells were detached from the plate with Cellstripper (Cellgro) at 37°C, quenched with fluorescence-activated cell sorter (FACS) buffer (PBS with 2% fetal bovine serum), pelleted at 1,000 × g, washed once with PBS, and pelleted a second time at 1,000 × g. Cells were adsorbed with 105 reovirus particles per cell at 4°C for 1 h in the presence of DMSO, 100 μM genistein, or 5 μM PP2. After adsorption, cells were washed once in FACS buffer, pelleted, and stained in FACS buffer with reovirus-specific polyclonal antiserum, β1 integrin-specific antibody (MAB2253Z), human JAM-A-specific antibody (J10.4), or Alexa Fluor-conjugated IgG as a negative control at 4°C for 30 min. Cells were washed twice in FACS buffer, pelleted, stained in FACS buffer containing Alexa Fluor-conjugated antisera at 4°C for 30 min, pelleted, washed twice in FACS buffer, and fixed in FACS fix (PBS with 1% electron microscopy grade paraformaldehyde [Electron Microscopy Sciences]). Cell staining was quantified using FlowJo software.
Confocal microscopy of reovirus internalization.
HeLa cells were plated on no. 1.5 glass coverslips (Thermo Scientific) in 24-well plates and incubated with incomplete medium in the presence of DMSO or chemical inhibitor at 37°C for 1 h, followed by incubation at 4°C for 1 h (39). Virus was adsorbed at 4°C for 1 h, the inoculum was removed, and cells were washed three times with PBS, either fixed with 10% formalin or supplemented with complete medium with DMSO or chemical inhibitor, and incubated at 37°C for various intervals. Cells were washed once with PBS and fixed for 20 min with 10% formalin, quenched with 0.1 M glycine, and washed three times with PBS. Cells were incubated with 1% Triton X-100 for 5 min and PBS-BGT (PBS, 0.5% BSA, 0.1% glycine, 0.05% Tween 20) for 10 min. Cells were incubated with reovirus-specific polyclonal antiserum (1:1,000) in PBS-BGT for 1 h, washed with PBS-BGT, and incubated with Alexa Fluor 488 or Alexa Fluor 546 IgG (1:1,000) or phalloidin conjugated to Alexa Fluor 546 (1:100; Invitrogen) in PBS-BGT for 1 h. Nuclei were visualized by incubating cells with TO-PRO-3 stain (Invitrogen) conjugated to Alexa Fluor 642 (1:1,000) in PBS-BGT for 20 min. Cells were washed with PBS-BGT, and coverslips were removed from wells and placed on slides using Aqua-Poly/Mount mounting medium (Polysciences, Inc.). Images were captured using a Zeiss LSM 510 Meta laser scanning confocal microscope. Internalized particles were quantified by counting the number of green particles within the boundary of a cell, which was established by phalloidin staining of the cell periphery.
Src distribution was visualized by transfecting cells plated on glass coverslips in 24-well plates with pEGFP-N1 or pEGFP-Src using Fugene 6 reagent (Roche), according to the manufacturer's instructions. Following incubation for 48 h, cells were chilled, infected, and prepared for confocal microscopy.
Colocalization analysis was performed using the profile function of Zeiss LSM Image software. Intracellular 20-μm-long sections were selected. Fluorescence peaks with mean fluorescence intensities (MFIs) of ≥50 were scored positive for each fluorescence channel. Areas of overlapping fluorescence peaks with MFIs of ≥50 were interpreted as colocalization.
Immunoblot analysis.
Cells were incubated in incomplete medium at 37°C overnight and either mock infected or adsorbed with reovirus at an MOI of 100 PFU/cell in serum-free medium at 37°C for 1 h. Cells were incubated in incomplete medium at 37°C for 0, 30, or 60 min, immediately chilled, and washed once in ice-cold PBS. Total cell lysates were prepared using RIPA buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% sodium dodecyl sulfate, 0.1% sodium deoxycholate), and protein concentration was determined using the DC protein assay (Bio-Rad). Lysates were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) in 4 to 20% gradient polyacrylamide gels (Bio-Rad) and transferred to 0.2-μm-pore-size Protran nitrocellulose membranes (Millipore). Membranes were incubated for 1 h in blocking buffer (Tris-buffered saline [TBS] with 5% powdered milk), followed by incubation with primary antibodies specific for Y416 Src, Y527 Src, total Src, GAPDH, or actin in TBS-T (TBS with 0.1% Tween 20) at 4°C overnight with gentle agitation. Membranes were washed with TBS-T three times for 5 min each time and incubated for 1 to 2 h with secondary antibodies conjugated to Alexa Fluor dyes diluted 1:1,000 in TBS-T at room temperature. Following three 5-min washes with TBS-T, membranes were rinsed twice with double-distilled water and scanned using an Odyssey imaging system (LI-COR). Blots were quantified by optical densitometry (OD) using Odyssey software (version 3.0; LI-COR).
RNA interference.
Sequence-specific short hairpin RNA (shRNA) constructs complementary to JAM-A (catalog no. 16015), Src (catalog no. 262795), and GAPDH and a nonspecific (scrambled) shRNA were obtained from Open Biosystems (Thermo Scientific). Lentiviruses encoding each shRNA were generated by triple transfection of each shRNA, lentiviral gag/pol (provided by Jim Chappell, Vanderbilt University), and vesicular stomatitis virus G (provided by David Everly, Rosalind Franklin University) into 293T cells using the Fugene 6 reagent. After incubation for 24 h, the medium was replaced and cells were incubated at 32°C for 24 h. Medium containing lentivirus was removed from cells and clarified by centrifugation at 1,000 × g for 5 min.
Cells stably expressing each shRNA construct were produced by transfection with Fugene 6 reagent or transduction with shRNA-encoding lentivirus with 5 μg per ml Polybrene (Millipore) for 24 h. Cells were maintained in medium supplemented to contain 1 μg per ml puromycin (Sigma) and screened for target protein expression by immunoblotting at each passage until maximum knockdown was observed. Cells were adsorbed with reovirus virions or ISVPs and scored for infection by immunofluorescence.
Statistical analysis.
Mean values for at least triplicate samples were compared using paired (normalized data) or unpaired (nonnormalized data) Student's t tests applied in Prism software (GraphPad). P values of <0.05 were considered to be statistically significant.
RESULTS
Reovirus infection is inhibited by genistein and PP2.
To determine whether Src-family kinases are required for reovirus infection, we assessed the effect on reovirus infectivity of genistein, a small-molecule inhibitor of tyrosine-specific protein kinases (1), and PP2, a small-molecule inhibitor of Src-family kinases (33). HeLa CCL2 cells were incubated in the presence of vehicle control (DMSO) or increasing concentrations of genistein (Fig. 1A) or PP2 (Fig. 1B) for 1 h, adsorbed with reovirus, and scored for infectivity by indirect immunofluorescence. Genistein and PP2 demonstrated dose-dependent reductions in the number of infected cells, reaching a threshold at concentrations of 100 μM genistein and 5 μM PP2. These concentrations of genistein and PP2 were used for the remainder of the experiments in the study.
FIG. 1.

Genistein and PP2 inhibit reovirus infectivity. HeLa CCL2 cells were incubated with vehicle control (DMSO) or the indicated concentrations of genistein (A) or PP2 (B) for 1 h prior to adsorption with reovirus at an MOI of 1 PFU/cell. The inoculum was removed, and cells were incubated at 37°C for 20 h, fixed, and stained by indirect immunofluorescence. Infected cells were quantified in four fields of view for triplicate samples. Results are expressed as percent infectivity relative to the level of infectivity for DMSO-treated cells. Error bars indicate standard deviations. *, P < 0.0001 in comparison to DMSO-treated cells. (C) HeLa CCL2 cells were treated with DMSO, 100 μM genistein, 5 μM PP2, or 10 μM U0126 for 1 h and adsorbed with reovirus at an MOI of 0.01 PFU/cell. Viral titers were determined at 0 and 24 h. Results are expressed as viral titers for each chemical inhibitor for triplicate samples. Error bars indicate standard deviations. *, P < 0.0001 in comparison to DMSO-treated cells. (D) HeLa CCL2 cells were incubated in the presence of DMSO or the indicated concentrations of genistein or PP2 at 37°C for 24 h. Cell viability was assessed for four wells of cells per condition by trypan blue staining. Error bars indicate standard deviations.
To determine whether genistein or PP2 inhibits reovirus replication, HeLa CCL2 cells were incubated with DMSO, genistein, PP2, or 10 μM U0126, an extracellular signal-regulated kinase (ERK)-specific small-molecule inhibitor, for 1 h and adsorbed with reovirus. Viral titers were determined at 0 and 24 h by plaque assay (Fig. 1C). Genistein and PP2 inhibited reovirus growth by greater than 10-fold at 24 h postinfection, whereas U0126 did not alter reovirus replication. To confirm that genistein and PP2 do not impair reovirus growth as a consequence of cytotoxicity, HeLa CCL2 cells were incubated in the presence of increasing concentrations of genistein or PP2 for 24 h, stained with trypan blue, and enumerated (Fig. 1D). Cell numbers were equivalent following treatment with DMSO or increasing concentrations of PP2. Treatment with 100 μM or 200 μM genistein inhibited cell proliferation but did not cause significant cell death. These results suggest that tyrosine-specific protein kinases, and perhaps Src kinases in particular, are required for efficient reovirus replication.
Genistein and PP2 inhibit reovirus infection at an early step in viral replication.
To gauge the temporal window in which genistein and PP2 inhibit reovirus replication, HeLa CCL2 cells were incubated in the presence of DMSO, genistein, or PP2 for 1 h prior to adsorption or for various intervals thereafter, adsorbed with reovirus, and scored for infection by indirect immunofluorescence (Fig. 2A and data not shown). As before, addition of genistein or PP2 prior to infection decreased the number of infected cells. In contrast, addition of genistein or PP2 to cells at intervals greater than or equal to 1 h after infection minimally altered reovirus infectivity. These results suggest that genistein and PP2 inhibit reovirus infection at an early step in the infectious cycle coincident with virus entry.
FIG. 2.

Genistein and PP2 inhibit reovirus within the first hour of infection. (A) HeLa CCL2 cells were incubated with DMSO, 100 μM genistein, or 5 μM PP2 for 1 h prior to adsorption (left side of graph) or 1 h following adsorption (right side of graph) with reovirus at an MOI of 0.01 PFU/cell. The inoculum was removed, and cells were incubated at 37°C for 20 h, fixed, and stained by indirect immunofluorescence. Infected cells were quantified in four fields of view for triplicate samples. Results are expressed as percent infectivity relative to the level of infectivity for DMSO-treated cells. Error bars indicate standard deviations. *, P < 0.0001 in comparison to DMSO-treated cells; **, P = 0.0004 in comparison to DMSO-treated cells. (B) Cells were treated with DMSO, 100 μM genistein, or 5 μM PP2 for 1 h prior to adsorption with reovirus virions at an MOI of 0.01 PFU/cell or ISVPs at an MOI of 0.001 PFU/cell. The inoculum was removed, and cells were incubated at 37°C for 20 h, fixed, and stained by indirect immunofluorescence. Infected cells were quantified in four fields of view for triplicate samples. Results are expressed as percent infectivity relative to the level of infectivity for DMSO-treated cells. Error bars indicate standard deviations. *, P < 0.0001 in comparison to DMSO-treated cells; **, P = 0.011 in comparison to DMSO-treated cells.
Like reovirus virions, ISVPs must bind cell surface receptors to infect cells (6), but in contrast to virions, ISVPs likely enter cells by direct penetration at the plasma membrane (9) and do not require proteolytic processing (9, 61). As such, ISVPs do not require sorting into an endocytic compartment for disassembly. To determine whether genistein or PP2 alters infection by ISVPs, HeLa CCL2 cells were treated with DMSO, genistein, or PP2 for 1 h, adsorbed with reovirus virions or ISVPs, and scored for infectivity by indirect immunofluorescence (Fig. 2B). Treatment with genistein or PP2 decreased the number of infected cells following adsorption with virions. In contrast, treatment with genistein or PP2 minimally decreased the number of infected cells following adsorption with ISVPs. These results suggest that genistein and PP2 act to inhibit a step in the infectious cycle that differs between virions and ISVPs, most likely uptake or sorting in the endocytic compartment. Moreover, these results provide additional evidence that genistein and PP2 do not inhibit reovirus infection by virtue of nonspecific cytotoxicity.
Reovirus attachment to cells is not affected by genistein or PP2.
JAM-A serves as a reovirus receptor and is required for infection of a variety of cell types (2, 6). Following viral attachment to JAM-A, β1 integrin mediates internalization of reovirus particles into cells (38, 39). To determine whether genistein and PP2 alter the cell surface expression of JAM-A or β1 integrin, HeLa cells were incubated with DMSO, genistein, or PP2 for 1 h and stained with antibodies specific for JAM-A (Fig. 3A) or β1 integrin (Fig. 3B). Cell surface levels of JAM-A and β1 integrin were not altered by either inhibitor, suggesting that the attachment and internalization mediators are not altered by kinase inhibitor treatment. To determine directly whether genistein and PP2 inhibit reovirus infection by impeding reovirus attachment, HeLa CCL2 cells were incubated with DMSO, genistein, or PP2 for 1 h and tested for reovirus binding using flow cytometry (Fig. 3C). Neither genistein nor PP2 altered the capacity of reovirus to bind to cells. Together, these data suggest that genistein and PP2 do not inhibit reovirus infection by impairing viral attachment to the cell surface.
FIG. 3.

Genistein and PP2 do not alter reovirus attachment to cells. HeLa CCL2 cells were treated with DMSO, 100 μM genistein, or 5 μM PP2 for 1 h, chilled, and adsorbed with 10,000 particles of reovirus per cell or were mock infected at 4°C for 1 h. Cells were stained with JAM-A-specific antibody (A), β1 integrin-specific antibody (B), reovirus-specific antiserum (C), or secondary antibody as a control (white peaks in panels A and B) and analyzed by flow cytometry. Data are expressed as fluorescence intensity.
Internalization of reovirus virions is not inhibited by genistein or PP2.
To determine whether genistein or PP2 blocks internalization of reovirus virions, HeLa CCL2 cells were incubated with DMSO, genistein, or PP2 at 37°C for 1 h, chilled at 4°C, infected with reovirus virions, and warmed to 37°C. After 20 min incubation, cells were imaged for reovirus, actin, or nuclei by confocal microscopy (Fig. 4). Reovirus particles were observed in cells incubated with DMSO (Fig. 4A), genistein (Fig. 4B), and PP2 (Fig. 4C). Quantification of the fluorescence intensity of the confocal micrographic images revealed no differences in the number of particles internalized into cells treated with DMSO or either inhibitor (Fig. 4D). These results suggest that genistein and PP2 do not impair uptake of reovirus particles into cells.
FIG. 4.

Internalization of reovirus particles is not altered by genistein or PP2. HeLa CCL2 cells were incubated with DMSO (A), 100 μM genistein (B), or 5 μM PP2 (C) for 1 h, chilled, and adsorbed with 20,000 reovirus particles per cell at 4°C for 1 h. The inoculum was removed, and cells were incubated at 37°C for 20 min, fixed, stained for actin (red), reovirus (green), and nuclei (blue), and imaged by confocal microscopy. Insets depict enlarged areas of the boxed regions. Representative digital fluorescence images of cells are shown. Scale bars, 10 μm. (D) Quantification of internalized reovirus particles in single planes of view for 8 to 11 cells per condition. Error bars indicate standard deviations.
Genistein and PP2 route reovirus to lysosomes.
Substitution of tyrosine residues with phenylalanine in the NPXY motifs in the β1 integrin cytoplasmic tail results in aberrant targeting of reovirus particles to lysosomes (39). To determine whether genistein or PP2 alters the distribution of reovirus virions in the endocytic pathway, HeLa CCL2 cells were incubated with DMSO, genistein, or PP2 for 1 h, chilled at 4°C, infected with reovirus, and warmed to 37°C. After 60 min incubation, cells were imaged for the lysosomal marker LAMP1, reovirus, or nuclei by confocal microscopy. In cells treated with DMSO (Fig. 5A), the bulk of internalized reovirus particles did not distribute to lysosomes. In contrast, treatment of cells with either genistein (data not shown) or PP2 led to increased distribution of reovirus virions to these organelles (Fig. 5B). The pixel intensity profile of LAMP1-positive areas of cells incubated with DMSO (Fig. 5C) showed little overlap with the profile of reovirus particles (green line) in structures that stained for LAMP1 (red line). In contrast, cells incubated with PP2 (Fig. 5D) showed substantial overlap between the reovirus and LAMP1 spectra, suggesting substantial colocalization. Quantification of the spectral overlap showed increased colocalization of reovirus particles with LAMP1 in genistein- and PP2-treated cells in comparison to those treated with DMSO (Fig. 5E). Thus, genistein and PP2 lead to aberrant transport of reovirus particles to lysosomes, which is a nonproductive pathway of viral entry.
FIG. 5.

Colocalization of reovirus particles with LAMP1 in kinase inhibitor-treated cells. HeLa CCL2 cells were incubated with DMSO (A) or 5 μM PP2 (B) for 1 h, chilled, and adsorbed with 20,000 reovirus particles per cell at 4°C for 1 h. The inoculum was removed, and cells were incubated at 37°C for 60 min, fixed, stained for LAMP1 (red), reovirus (green), and nuclei (blue), and imaged by confocal microscopy. Insets depict enlarged areas of the boxed regions. Representative digital fluorescence images of cells are shown. Scale bars, 10 μm. The white lines in the insets depict the area used to determine the spectral overlap for DMSO (C) or PP2 (D). Spectral overlap is expressed as fluorescence intensity for LAMP1 (red), reovirus (green), and TO-PRO-3 stain (blue). (E) Quantification of spectral overlap of reovirus particles with LAMP1 60 min following infection of cells treated with DMSO, 100 μM genistein, or 5 μM PP2. Spectral overlap is presented as percent colocalization (n = 21 cells). Error bars indicate standard deviations. *, P < 0.0001 in comparison to DMSO-treated cells.
Src is activated following reovirus infection.
Activation of Src kinase is biochemically marked by dephosphorylation of Y527 and subsequent phosphorylation of Y416 (11, 18, 47). To determine whether Src is activated following reovirus infection, HeLa CCL2 cells were incubated in serum-free medium overnight and adsorbed with reovirus. At 0, 30, and 60 min postadsorption, whole-cell lysates were prepared, and levels of phospho-Y416 Src, phospho-Y527 Src, total Src, and actin were assessed by immunoblotting (Fig. 6). In comparison to mock-infected cells, levels of phospho-Y416 Src were increased at 30 and 60 min following infection with reovirus. In these experiments, activated Src levels peaked at 30 min after infection, with levels reaching approximately 3-fold greater than those observed following mock infection. Phospho-Y527 and total Src levels remained largely unchanged throughout the time course. The increased levels of phospho-Y416 without a concomitant decrease in levels of phospho-Y527 suggest that reovirus induces localized activation of Src at early times of infection but does not change the activation state of the entire intracellular Src pool.
FIG. 6.

Src is phosphorylated at Y416 during reovirus cell entry. HeLa CCL2 cells were incubated in serum-free medium overnight and adsorbed with reovirus at an MOI of 100 PFU/cell or mock infected for 1 h. The inoculum was removed, and cells were incubated at 37°C for 0, 30, or 60 min. Total cell lysates were prepared, resolved by SDS-PAGE, and analyzed by immunoblotting using antibodies specific for phosphorotyrosine 416-specific Src (p-Y416, arrow), phosphorotyrosine 527-specific Src (p-Y527, arrow), total Src, and actin.
Reovirus distributes with Src during cell entry.
To define the cellular distribution of Src and reovirus during cell entry, HeLa CCL2 cells were transfected with a plasmid encoding Src-EGFP. At 48 h after transfection, cells were chilled at 4°C, infected with reovirus, and imaged by confocal microscopy after 0, 20, and 60 min incubation (Fig. 7). Inactive Src is maintained in a perinuclear compartment (35, 36), whereas activated Src localizes mostly at the cell periphery (35, 36). At 0 min postinfection, reovirus virions were observed predominantly at the cellular periphery and showed little colocalization with Src. At 20 min postinfection, reovirus particles were detected in the cytoplasm and colocalized with Src at both peripheral and perinuclear sites (Fig. 7A, insets). By 60 min postinfection, most reovirus virions were observed in the cytoplasm and colocalized with Src (Fig. 7A, insets). Quantification of the fluorescence intensity of the confocal micrographic images indicates that the colocalization of reovirus particles with Src increases as the entry process progresses, with the majority of viral particles codistributing with Src by 20 min (Fig. 7B). These data suggest that reovirus and Src distribute to the same intracellular compartment at early times of infection.
FIG. 7.

Reovirus distributes with Src during cell entry. (A) HeLa CCL2 cells were transfected with a plasmid encoding Src-EGFP (green), incubated at 4°C for 1 h, and adsorbed with 20,000 reovirus particles per cell at 4°C for 1 h. The inoculum was removed, and cells were incubated at 37°C for 0, 20, or 60 min. Cells were fixed, stained for reovirus (red), and visualized by confocal microscopy. Insets depict enlarged areas of the boxed regions. Representative digital fluorescence images of cells are shown. Scale bars, 10 μm. (B) Quantification of spectral overlap of reovirus particles with Src. Spectral overlap is presented as percent colocalization (n = 20 cells). Error bars indicate standard deviations.
Src is required for efficient reovirus infection.
To determine whether Src is required for reovirus infection, we used RNA interference to diminish Src expression. Cells stably transfected with scrambled, GAPDH, JAM-A, and Src shRNAs were assessed for Src and actin levels by immunoblotting (Fig. 8A). Cells stably expressing Src shRNA showed about a 60% decrease in Src protein levels. Levels of Fyn and Yes were unaffected in the shRNA-expressing cells (data not shown), suggesting that the knockdown observed in the Src shRNA-expressing cells was specific for Src and not other Src family members. Flow cytometric analysis revealed no effect of the Src shRNA on expression of JAM-A at the cell surface (data not shown), although as anticipated, cells expressing JAM-A shRNA had decreased levels of cell surface JAM-A (data not shown). HeLa CCL2 cells stably expressing scrambled, GAPDH, JAM-A, or Src shRNA were infected with reovirus virions or ISVPs and scored for infectivity by indirect immunofluorescence (Fig. 8B). Expression of JAM-A or Src shRNA yielded 51% and 32% decreases in the number of infected cells following adsorption of virions (P < 0.05). In contrast, expression of JAM-A shRNA, but not Src shRNA, produced a decrease in the number of infected cells following adsorption with ISVPs. These data suggest that Src is required for efficient infection by reovirus virions but not by ISVPs, providing additional evidence that Src promotes an early step in reovirus infection.
FIG. 8.
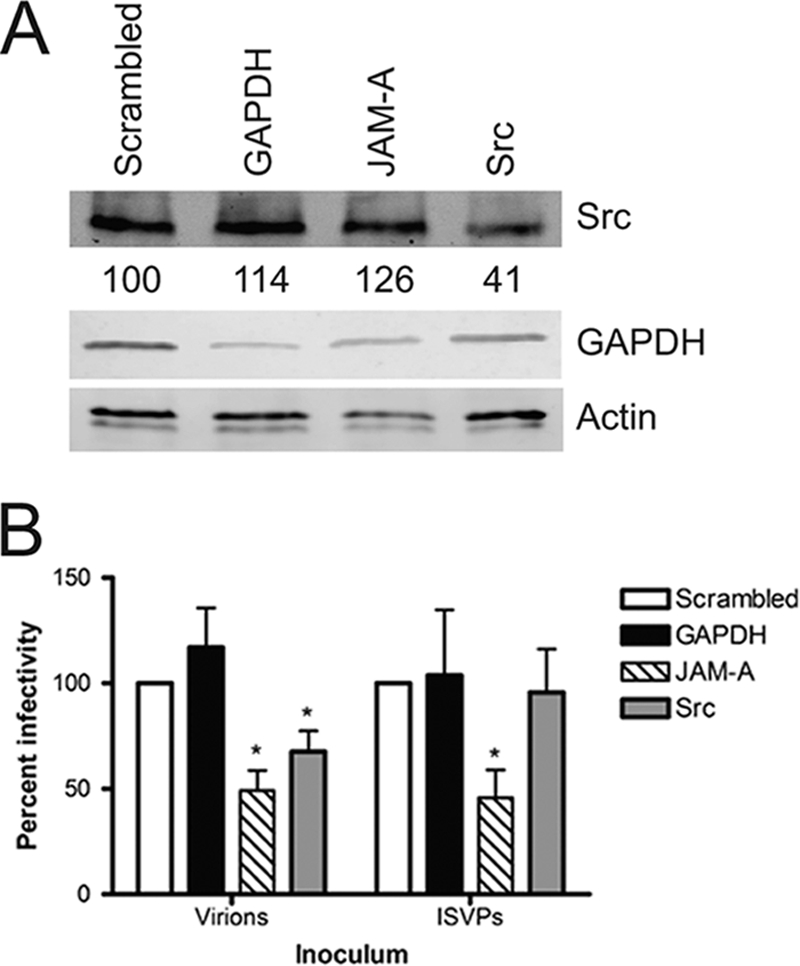
Reovirus infection is diminished in cells expressing Src shRNA. (A) Whole-cell lysates of HeLa CCL2 cells stably expressing scrambled, GAPDH, JAM-A, or Src shRNA were resolved by SDS-PAGE and analyzed by immunoblotting using antibodies specific for total Src, GAPDH, or actin. Src levels are normalized to those in cells with scrambled shRNA. (B) HeLa CCL2 cells stably expressing scrambled, GAPDH, JAM-A, or Src shRNA were adsorbed with reovirus virions at an MOI of 0.02 PFU/cell or ISVPs at an MOI of 0.002 PFU/cell at 37°C for 20 h. The inoculum was removed, and cells were fixed and stained by indirect immunofluorescence. Infected cells were quantified in four fields of view for triplicate samples. Results are expressed as percent infectivity relative to the level of infectivity for cells expressing scrambled shRNA. Error bars indicate standard deviations. *, P < 0.0001 in comparison to cells expressing scrambled shRNA.
DISCUSSION
In this study, we found that genistein and PP2 diminish reovirus infectivity by inhibiting a step in virus entry required for the proper sorting of virions in the endocytic pathway. The key finding is that inhibition of Src-family kinases redirects incoming reovirus virions to lysosomes, which is a nonproductive route of entry. Reovirus induces phosphorylation of Src, and reduction of endogenous Src by RNAi decreases reovirus infectivity. Reovirus colocalizes with Src during entry, suggesting that kinase activity in physical proximity to the virus is necessary for its targeting to an endocytic organelle for disassembly. Together, these data provide strong evidence that Src kinase is required for efficient reovirus entry into host cells.
Reovirus is thought to enter cells via clathrin-dependent endocytosis (26, 39), followed by acid-dependent proteolysis in an endocytic compartment mediated by cathepsin proteases (25, 32). The internalization and disassembly steps culminate in the release of the transcriptionally active core particle into the cytoplasm, which initiates infection (15, 24). However, host factors that guide reovirus virions from attachment at the cell surface to disassembly in an endocytic organelle are poorly understood. Inhibition of functional reovirus entry (i.e., entry that leads to productive infection) by genistein and PP2 provides evidence that reovirus requires tyrosine kinases, and Src kinases in particular, to efficiently enter cells. Interestingly, genistein and PP2 do not diminish internalization of reovirus virions, but, instead, these inhibitors alter intracellular trafficking of reovirus, resulting in targeting of viral particles to lysosomes. These data suggest that in the absence of Src activity, reovirus entry is aberrant, resulting in nonproductive viral internalization.
In previous work, genistein was found to block reovirus protein synthesis (60), but how this occurs was not known. Genistein impairs cell cycle progression, arresting cells at the G2/M checkpoint by inactivating Cdc25 phosphatases (34, 46, 51), and induces apoptosis by an uncharacterized mechanism (51). In this study, we found that genistein produced cytostatic effects on cells (Fig. 1D), although cytopathic effects were not observed until 48 h after treatment (data not shown). Genistein also affects phosphorylation of caveolin (66) and inhibits intracellular signaling that promotes internalization of simian virus 40 into caveolae (16, 20). However, there is no evidence that reovirus enters cells via caveolin-dependent pathways. In fact, we have found that reovirus infection is more efficient in murine embryo fibroblast cells that lack caveolin-1 (B. A. Mainou and T. S. Dermody, unpublished observations). Genistein increases cellular adhesion by increasing the translocation of focal adhesion kinase to focal adhesions (7). Based on the results presented here, we conclude that the effect of genistein on reovirus protein synthesis is mediated by a block at a much earlier step in viral replication. Our data suggest that genistein inhibits a tyrosine kinase function required for proper sorting of incoming virions in the endocytic pathway. Experiments using PP2 and Src-specific RNAi suggest that Src-mediated signaling is required for this function.
The initiating step in the signaling cascade that triggers reovirus entry is likely to occur after JAM-A binding, as reovirus can productively infect cells expressing a truncated form of JAM-A lacking the cytoplasmic tail (JAM-AΔCT) (38). Furthermore, genistein treatment equivalently inhibits reovirus infection of cells expressing either full-length JAM-A or JAM-AΔCT (data not shown). We previously found that the expression of β1 integrin is required for reovirus internalization and endocytic sorting (38, 39), but the underlying mechanism is unknown. Tyrosine residues in the cytoplasmic tail of β1 integrin can be phosphorylated by v-Src (53), which induces the dynamic regulation of focal contacts and extracellular matrix deposition (22, 29, 53). It is therefore possible that reovirus-induced Src activation promotes phosphorylation of β1 integrin, which subsequently recruits cellular adapters that sort reovirus to an endocytic compartment for disassembly. It is also possible that Src activation by reovirus modulates the function of α integrins, although no specific α integrin has been implicated in reovirus infection (38). Src activation could be mediated by the recruitment of signaling molecules to the site of viral entry. Focal adhesion kinase and phosphatidylinositol 3-kinase, which play important roles in cytoskeletal regulation and transduce signals from the plasma membrane, interact directly with Src (17, 27, 57, 62, 70), and these effectors may be involved in reovirus internalization and endocytic sorting.
The activation and utilization of Src-family kinases by coxsackievirus during cell entry are particularly well understood. The coxsackievirus receptor, coxsackievirus and adenovirus receptor (CAR), like JAM-A, distributes to interepithelial tight junctions (8, 19). Coxsackievirus activates Src-family kinases by first engaging the glycosylphosphatidylinositol-anchored protein decay-accelerating factor, and these kinases in turn trigger a signaling cascade that disrupts the tight junction, allowing the virus access to CAR (19). In contrast to coxsackievirus, reovirus does not appear to require Src activity to engage and access JAM-A, although we have not formally tested for a requirement for Src in reovirus infection of polarized cells. Instead, Src activity appears to guide reovirus to an endosomal organelle for disassembly after receptor engagement.
Src kinases localize to distinct intracellular sites. Activated Src distributes to the plasma membrane, whereas inactive Src predominantly localizes to perinuclear regions (35, 36). However, Src also can localize to the endocytic compartment, specifically, to late endosomes (37, 55). Although we did not detect bulk redistribution of Src during reovirus infection in comparison to the findings for mock-infected cells (data not shown), we found that reovirus colocalizes with Src near the plasma membrane and in perinuclear regions during a time course of viral entry (Fig. 7). It is possible that reovirus activates Src near the plasma membrane by recruiting adapter proteins to the site of endocytosis and then is transported with Src to late endosomes, where disassembly is thought to occur. While we observed activation of Src, as evidenced by increased levels of phospho-Y416, at early times of infection, we did not detect reduced levels of inactive Src (phospho-Y527). These findings suggest that reovirus activates localized pools of Src, leaving the bulk of intracellular Src unaffected.
Although both genistein and PP2 impair reovirus infectivity, the effect is by no means absolute. Similarly, decreased Src expression by RNAi does not fully abrogate reovirus infection. These data suggest that although Src is required for maximal reovirus infectivity, it is not essential for viral growth. The increased impairment of infectivity observed with PP2 in comparison to that observed with RNAi points to a putative role for other Src-family members in reovirus entry. Fyn and Yes are ubiquitously expressed and also could facilitate reovirus entry in the absence of Src. In support of this idea, coxsackievirus activates both Fyn and Src during entry, although Fyn activation is more robust (19). It is also possible that reovirus uses multiple pathways to enter cells, some of which do not require Src. For example, JC virus utilizes multiple endocytic sorting pathways to effect cell entry (50).
Like all viruses, reovirus uses endogenous cellular processes to enter host cells. In this study, we identify Src to be an important mediator of reovirus cell entry. Whether Src participates in recruiting other cellular components to guide reovirus through the endocytic pathway to facilitate its orderly disassembly remains to be determined. Understanding how viruses engage Src and other cellular factors to gain access to the cell interior is likely to yield knowledge about how cells internalize macromolecular cargo and may foster development of broadly active antivirals that inhibit key steps in the entry process.
Acknowledgments
We thank Karl Boehme, Jim Chappell, Josh Doyle, and Denise Wetzel for critical reviews of the manuscript and Adrienne Cox (University of North Carolina—Chapel Hill), Carolyn Coyne (University of Pittsburgh), and Marilyn Resh (Memorial Sloan-Kettering Cancer Center) for critical advice. We thank Derek Holmes (Roche) and Natalie Thornburg for technical assistance with the flow cytometry experiments. We are grateful to Anne Kenworthy, Roy Zent, and members of the Dermody lab for useful suggestions during the course of this study.
The flow cytometry experiments were performed in the Vanderbilt Cytometry Shared Resource. The confocal microscopy experiments were conducted in the Vanderbilt Cell Imaging Shared Resource.
This work was supported by Public Health Service awards T32 HL07751 and F32 AI080108 (to B.A.M.) and R01 AI32539 (to T.S.D.) and the Elizabeth B. Lamb Center for Pediatric Research. Additional support was provided by Public Health Service awards P30 CA68485 for the Vanderbilt-Ingram Cancer Center and P60 DK20593 for the Vanderbilt Diabetes Research and Training Center.
Footnotes
Published ahead of print on 19 January 2011.
REFERENCES
- 1.Akiyama, T., et al. 1987. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 262:5592-5595. [PubMed] [Google Scholar]
- 2.Antar, A. A., et al. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baer, G. S., and T. S. Dermody. 1997. Mutations in reovirus outer-capsid protein sigma3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J. Virol. 71:4921-4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baer, G. S., D. H. Ebert, C. J. Chung, A. H. Erickson, and T. S. Dermody. 1999. Mutant cells selected during persistent reovirus infection do not express mature cathepsin L and do not support reovirus disassembly. J. Virol. 73:9532-9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barton, E. S., J. L. Connolly, J. C. Forrest, J. D. Chappell, and T. S. Dermody. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 276:2200-2211. [DOI] [PubMed] [Google Scholar]
- 6.Barton, E. S., et al. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441-451. [DOI] [PubMed] [Google Scholar]
- 7.Bergan, R., et al. 1996. Genistein-stimulated adherence of prostate cancer cells is associated with the binding of focal adhesion kinase to beta-1-integrin. Clin. Exp. Metastasis 14:389-398. [DOI] [PubMed] [Google Scholar]
- 8.Bergelson, J. M., et al. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320-1323. [DOI] [PubMed] [Google Scholar]
- 9.Borsa, J., et al. 1979. Two modes of entry of reovirus particles into L cells. J. Gen. Virol. 45:161-170. [DOI] [PubMed] [Google Scholar]
- 10.Borsa, J., M. D. Sargent, P. A. Lievaart, and T. P. Copps. 1981. Reovirus: evidence for a second step in the intracellular uncoating and transcriptase activation process. Virology 111:191-200. [DOI] [PubMed] [Google Scholar]
- 11.Brown, M. T., and J. A. Cooper. 1996. Regulation, substrates and functions of src. Biochim. Biophys. Acta 1287:121-149. [DOI] [PubMed] [Google Scholar]
- 12.Brugge, J. S., and R. L. Erikson. 1977. Identification of a transformation-specific antigen induced by an avian sarcoma virus. Nature 269:346-348. [DOI] [PubMed] [Google Scholar]
- 13.Campbell, J. A., et al. 2005. Junctional adhesion molecule-A serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J. Virol. 79:7967-7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandran, K., D. L. Farsetta, and M. L. Nibert. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J. Virol. 76:9920-9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandran, K., J. S. Parker, M. Ehrlich, T. Kirchhausen, and M. L. Nibert. 2003. The delta region of outer-capsid protein micro 1 undergoes conformational change and release from reovirus particles during cell entry. J. Virol. 77:13361-13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen, Y., and L. C. Norkin. 1999. Extracellular simian virus 40 transmits a signal that promotes virus enclosure within caveolae. Exp. Cell Res. 246:83-90. [DOI] [PubMed] [Google Scholar]
- 17.Cobb, B. S., M. D. Schaller, T. H. Leu, and J. T. Parsons. 1994. Stable association of pp60src and pp59fyn with the focal adhesion-associated protein tyrosine kinase, pp125FAK. Mol. Cell. Biol. 14:147-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper, J. A., K. L. Gould, C. A. Cartwright, and T. Hunter. 1986. Tyr527 is phosphorylated in pp60c-src: implications for regulation. Science 231:1431-1434. [DOI] [PubMed] [Google Scholar]
- 19.Coyne, C. B., and J. M. Bergelson. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124:119-131. [DOI] [PubMed] [Google Scholar]
- 20.Dangoria, N. S., W. C. Breau, H. A. Anderson, D. M. Cishek, and L. C. Norkin. 1996. Extracellular simian virus 40 induces an ERK/MAP kinase-independent signalling pathway that activates primary response genes and promotes virus entry. J. Gen. Virol. 77(Pt 9):2173-2182. [DOI] [PubMed] [Google Scholar]
- 21.Danthi, P., C. M. Coffey, J. S. Parker, T. W. Abel, and T. S. Dermody. 2008. Independent regulation of reovirus membrane penetration and apoptosis by the μ1 φ domain. PLoS Pathog. 4:e1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Datta, A., Q. Shi, and D. E. Boettiger. 2001. Transformation of chicken embryo fibroblasts by v-src uncouples beta1 integrin-mediated outside-in but not inside-out signaling. Mol. Cell. Biol. 21:7295-7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donepudi, M., and M. D. Resh. 2008. c-Src trafficking and co-localization with the EGF receptor promotes EGF ligand-independent EGF receptor activation and signaling. Cell. Signal. 20:1359-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drayna, D., and B. N. Fields. 1982. Activation and characterization of the reovirus transcriptase: genetic analysis. J. Virol. 41:110-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ebert, D. H., J. Deussing, C. Peters, and T. S. Dermody. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 277:24609-24617. [DOI] [PubMed] [Google Scholar]
- 26.Ehrlich, M., et al. 2004. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 118:591-605. [DOI] [PubMed] [Google Scholar]
- 27.Eide, B. L., C. W. Turck, and J. A. Escobedo. 1995. Identification of Tyr-397 as the primary site of tyrosine phosphorylation and pp60src association in the focal adhesion kinase, pp125FAK. Mol. Cell. Biol. 15:2819-2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forrest, J. C., J. A. Campbell, P. Schelling, T. Stehle, and T. S. Dermody. 2003. Structure-function analysis of reovirus binding to junctional adhesion molecule 1. Implications for the mechanism of reovirus attachment. J. Biol. Chem. 278:48434-48444. [DOI] [PubMed] [Google Scholar]
- 29.Frame, M. C. 2004. Newest findings on the oldest oncogene; how activated src does it. J. Cell Sci. 117:989-998. [DOI] [PubMed] [Google Scholar]
- 30.Furlong, D. B., M. L. Nibert, and B. N. Fields. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J. Virol. 62:246-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gampel, A., et al. 2006. VEGF regulates the mobilization of VEGFR2/KDR from an intracellular endothelial storage compartment. Blood 108:2624-2631. [DOI] [PubMed] [Google Scholar]
- 32.Golden, J. W., J. A. Bahe, W. T. Lucas, M. L. Nibert, and L. A. Schiff. 2004. Cathepsin S supports acid-independent infection by some reoviruses. J. Biol. Chem. 279:8547-8557. [DOI] [PubMed] [Google Scholar]
- 33.Hanke, J. H., et al. 1996. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. 271:695-701. [DOI] [PubMed] [Google Scholar]
- 34.Ismail, I. A., K. S. Kang, H. A. Lee, J. W. Kim, and Y. K. Sohn. 2007. Genistein-induced neuronal apoptosis and G2/M cell cycle arrest is associated with MDC1 up-regulation and PLK1 down-regulation. Eur. J. Pharmacol. 575:12-20. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan, K. B., et al. 1994. Association of the amino-terminal half of c-Src with focal adhesions alters their properties and is regulated by phosphorylation of tyrosine 527. EMBO J. 13:4745-4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaplan, K. B., J. R. Swedlow, H. E. Varmus, and D. O. Morgan. 1992. Association of p60c-src with endosomal membranes in mammalian fibroblasts. J. Cell Biol. 118:321-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasahara, K., et al. 2007. Rapid trafficking of c-Src, a non-palmitoylated Src-family kinase, between the plasma membrane and late endosomes/lysosomes. Exp. Cell Res. 313:2651-2666. [DOI] [PubMed] [Google Scholar]
- 38.Maginnis, M. S., et al. 2006. Beta1 integrin mediates internalization of mammalian reovirus. J. Virol. 80:2760-2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maginnis, M. S., et al. 2008. NPXY motifs in the beta1 integrin cytoplasmic tail are required for functional reovirus entry. J. Virol. 82:3181-3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maratos-Flier, E., M. J. Goodman, A. H. Murray, and C. R. Kahn. 1986. Ammonium inhibits processing and cytotoxicity of reovirus, a nonenveloped virus. J. Clin. Invest. 78:1003-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marcotte, R., L. Zhou, H. Kim, C. D. Roskelly, and W. J. Muller. 2009. c-Src associates with ErbB2 through an interaction between catalytic domains and confers enhanced transforming potential. Mol. Cell. Biol. 29:5858-5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nada, S., et al. 1993. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell 73:1125-1135. [DOI] [PubMed] [Google Scholar]
- 43.Nibert, M. L., A. L. Odegard, M. A. Agosto, K. Chandran, and L. A. Schiff. 2005. Putative autocleavage of reovirus μ1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. J. Mol. Biol. 345:461-474. [DOI] [PubMed] [Google Scholar]
- 44.Odegard, A. L., et al. 2004. Putative autocleavage of outer capsid protein μ1, allowing release of myristoylated peptide μ1N during particle uncoating, is critical for cell entry by reovirus. J. Virol. 78:8732-8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okada, M., and H. Nakagawa. 1989. A protein tyrosine kinase involved in regulation of pp60c-src function. J. Biol. Chem. 264:20886-20893. [PubMed] [Google Scholar]
- 46.Ouyang, G., et al. 2009. Genistein induces G2/M cell cycle arrest and apoptosis of human ovarian cancer cells via activation of DNA damage checkpoint pathways. Cell Biol. Int. 33:1237-1244. [DOI] [PubMed] [Google Scholar]
- 47.Parsons, J. T., and M. J. Weber. 1989. Genetics of src: structure and functional organization of a protein tyrosine kinase. Curr. Top. Microbiol. Immunol. 147:79-127. [DOI] [PubMed] [Google Scholar]
- 48.Ping-Yuan, L., et al. 2006. Avian reovirus activates a novel proapoptotic signal by linking Src to p53. Apoptosis 11:2179-2193. [DOI] [PubMed] [Google Scholar]
- 49.Purchio, A. F., E. Erikson, J. S. Brugge, and R. L. Erikson. 1978. Identification of a polypeptide encoded by the avian sarcoma virus src gene. Proc. Natl. Acad. Sci. U. S. A. 75:1567-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Querbes, W., B. A. O'Hara, G. Williams, and W. J. Atwood. 2006. Invasion of host cells by JC virus identifies a novel role for caveolae in endosomal sorting of noncaveolar ligands. J. Virol. 80:9402-9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramos, S. 2007. Effects of dietary flavonoids on apoptotic pathways related to cancer chemoprevention. J. Nutr. Biochem. 18:427-442. [DOI] [PubMed] [Google Scholar]
- 52.Rubin, D. H., et al. 1992. Receptor utilization by reovirus type 3: distinct binding sites on thymoma and fibroblast cell lines result in differential compartmentalization of virions. Microb. Pathog. 12:351-365. [DOI] [PubMed] [Google Scholar]
- 53.Sakai, T., R. Jove, R. Fassler, and D. F. Mosher. 2001. Role of the cytoplasmic tyrosines of beta 1A integrins in transformation by v-src. Proc. Natl. Acad. Sci. U. S. A. 98:3808-3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sandilands, E., et al. 2007. Src kinase modulates the activation, transport and signalling dynamics of fibroblast growth factor receptors. EMBO Rep. 8:1162-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sandilands, E., et al. 2004. RhoB and actin polymerization coordinate Src activation with endosome-mediated delivery to the membrane. Dev. Cell 7:855-869. [DOI] [PubMed] [Google Scholar]
- 56.Sandilands, E., and M. C. Frame. 2008. Endosomal trafficking of Src tyrosine kinase. Trends Cell Biol. 18:322-329. [DOI] [PubMed] [Google Scholar]
- 57.Schaller, M. D., and J. T. Parsons. 1994. Focal adhesion kinase and associated proteins. Curr. Opin. Cell Biol. 6:705-710. [DOI] [PubMed] [Google Scholar]
- 58.Schiff, L. A., M. L. Nibert, and K. L. Tyler. 2007. Orthoreoviruses and their replication, p. 1853-1915. In D. M. Knipe and P. M. Howley (ed.), Fields virology, vol. 2, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 59.Smith, R. E., H. J. Zweerink, and W. K. Joklik. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791-810. [DOI] [PubMed] [Google Scholar]
- 60.Strong, J. E., and P. W. Lee. 1996. The v-erbB oncogene confers enhanced cellular susceptibility to reovirus infection. J. Virol. 70:612-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sturzenbecker, L. J., M. L. Nibert, D. B. Furlong, and B. N. Fields. 1987. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J. Virol. 61:2351-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas, S. M., and J. S. Brugge. 1997. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13:513-609. [DOI] [PubMed] [Google Scholar]
- 63.Tokunaga, K., et al. 1998. Inhibition of human immunodeficiency virus type 1 virion entry by dominant-negative Hck. J. Virol. 72:6257-6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turk, V., B. Turk, and D. Turk. 2001. Lysosomal cysteine proteases: facts and opportunities. EMBO J. 20:4629-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Veettil, M. V., et al. 2006. RhoA-GTPase facilitates entry of Kaposi's sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J. Virol. 80:11432-11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vepa, S., W. M. Scribner, and V. Natarajan. 1997. Activation of protein phosphorylation by oxidants in vascular endothelial cells: identification of tyrosine phosphorylation of caveolin. Free Radic. Biol. Med. 22:25-35. [DOI] [PubMed] [Google Scholar]
- 67.Virgin, H. W., K. L. Tyler, and T. S. Dermody. 1997. Reovirus, p. 669-699. In N. Nathanson (ed.), Viral pathogenesis. Lippincott-Raven, New York, NY.
- 68.Virgin, H. W., IV, R. Bassel-Duby, B. N. Fields, and K. L. Tyler. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J. Virol. 62:4594-4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wetzel, J. D., J. D. Chappell, A. B. Fogo, and T. S. Dermody. 1997. Efficiency of viral entry determines the capacity of murine erythroleukemia cells to support persistent infections by mammalian reoviruses. J. Virol. 71:299-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xing, Z., et al. 1994. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol. Biol. Cell 5:413-421. [DOI] [PMC free article] [PubMed] [Google Scholar]