

Abstract
We performed analyses of the molecular mechanisms involved in the regulation of phospholipase Cγ2 (PLCγ2). We identified several regions in the PLCγ-specific array, γSA, that contribute to autoinhibition in the basal state by occlusion of the catalytic domain. While the activation of PLCγ2 by Rac2 requires stable translocation to the membrane, the removal of the domains required for membrane translocation in the context of an enzyme with impaired autoinhibition generated constitutive, highly active PLC in cells. We further tested the possibility that the interaction of PLCγ2 with its activator protein Rac2 was sufficient for activation through the release of autoinhibition. However, we found that Rac2 binding in the absence of lipid surfaces was not able to activate PLCγ2. Together with other observations, these data suggest that an important consequence of Rac2 binding and translocation to the membrane is that membrane proximity, on its own or together with Rac2, has a role in the release of autoinhibition, resulting in interfacial activation.
Phosphoinositide-specific phospholipase C (PLC)-catalyzed formation of the second messengers inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) from its substrate phosphatidylinositol 4,5-bisphosphate (PIP2) constitutes one of the major cell signaling responses (3, 36). There are six families of PLC enzymes (PLCβ, γ, δ, ɛ, η, and ζ) consisting of 13 isoforms in humans. Enzymes from each PLC family are uniquely integrated into complex signaling networks through diverse regulatory mechanisms and contribute to the regulation of a variety of biological functions. Despite this diversity, some common principles of their regulation at the molecular level have been proposed for several PLC families (16). However, such mechanistic concepts need to be tested further for each of the families and, in particular, for PLCγ enzymes, which represent a branch separate from all other PLC families (22).
One of the two members of the PLCγ family, PLCγ2, is most highly expressed in cells of the hematopoietic system and plays a key role in the regulation of the immune response. The regulatory interactions that control PLCγ2 in B cells have been well characterized and involve phosphorylation on critical tyrosine residues by Src and Tec family kinases; similar types of regulatory interactions have been described for the regulation of PLCγ1 in T-cell responses (2). Both PLCγ enzymes can be activated in response to growth factor stimulation (26, 37). A number of studies of the ubiquitously expressed PLCγ1 in different cell types support the critical importance of tyrosine phosphorylation for PLCγ activation. In addition to the regulatory mechanisms that are shared by the two PLCγ enzymes, the Rac GTPases Rac1, Rac2, and Rac3 have been specifically implicated in PLCγ2 regulation that is not dependent on tyrosine phosphorylation of this enzyme (29). However, like stimulation via tyrosine kinase-linked receptors, Rac2 also leads to membrane translocation (29). Several studies suggest signaling connectivity between PLCγ2 and Rac GTPases in B cells, dendritic cells, and platelets (7, 14, 30). Notably, it has been shown that Rac1 is essential for the activation of PLCγ2 in platelets and for subsequent aggregation and thrombus formation in vivo (14, 30). Despite a large number of studies covering signaling links, the exact molecular mechanisms of enzyme regulation have not been fully defined for either PLCγ1 or PLCγ2.
With respect to domain organization, PLCγ enzymes have the unique insertion of a highly structured region (the PLCγ-specific array [γSA]) between the two halves of the catalytic triosephosphate isomerase (TIM) barrel (5, 22, 36). This region in all other isoforms is shorter and largely unstructured (10, 21). The γSA is critical for the activation process and comprises a split pleckstrin homology (spPH) domain flanking two tandem Src homology 2 domains (SH2n and SH2c [N-terminal and C-terminal SH2 domains]) and an SH3 domain (23). Data obtained for both PLCγ isoforms suggest that for this family, the γSA, in addition to its role as the main site of regulatory interactions, also incorporates elements of autoinhibition (3). However, these findings require further, more comprehensive support. Specifically, for PLCγ2 regulation, the extent of conformational change required for activation and the relative contributions of interacting molecules, tyrosine phosphorylation, and membrane proximity to this process remain poorly understood.
Here, we first analyzed mechanisms that keep PLCγ2 activity in a basal state and then examined further how PLC activation can occur following stimulation by Rac2. We found that autoinhibitory constraints in PLCγ2 could involve elements from different regions of the γSA. Furthermore, we show that Rac is not able to release autoinhibition in PLCγ2 through the formation of PLCγ2/Rac2 complexes alone. Instead, activation is likely to result from the release of intramolecular inhibitory constraints in the proximity of the membrane following stable, Rac-mediated membrane association.
MATERIALS AND METHODS
Construction of vectors.
Plasmids for the expression of full-length human PLCγ2 (pMT2-, pTriEx4-, and pEGFPC1-PLCγ2) in mammalian cells and in Sf9 cells (pVL1393-PLCγ2) have been described previously (11, 26, 32, 40). The deletion of amino acids 471 to 913 encoding the γSA generated variant PLCγ2ΔSA, also referred to as PLCγ2-core; this variant was made in pMT2, pTriEx4, and pEGFPC1 plasmids. For protein expression in bacteria, PLCγ2-core was expressed from a pTriEx4 plasmid backbone.
Most PLCγ2 variants incorporating deletions and point mutations were made in pMT2-PLCγ2, and the changes were introduced by site-directed mutagenesis (QuikChange PCR mutagenesis; Stratagene). A similar strategy was used to generate PLCγ2 variants incorporating fluorescent tags. Site-directed mutagenesis was used to create N-terminally green fluorescent protein (GFP)-tagged PLCγ2-core (Δ471-913) and PLCγ2ΔPCI (726-733) and a variant in which PLC is inactive (H327A) from pEGFPC1-PLCγ2. PLCγ2 variant PLCγ2ΔSH3(mCherry)GFP was generated as follows: PLCγ2 in pTriEx4 was truncated (amino acids [aa] 1 to 1,192) and tagged with GFP at its C terminus. Site-directed mutagenesis was used to delete the SH3 domain and introduce an NruI site in its place. In-Fusion PCR (Clontech) was then used to insert mCherry into the NruI site. These pTriEx4 vectors were used to produce baculoviruses using a BacVector-3000 kit (Novagen).
For the construction of the PLCγ2/PLCɛ chimera, plasmids pCMV-PLCɛ1a (described in reference 34) and pTriEx4-PLCγ2 were used. The PLCɛ linker was inserted into pTriEx4-PLCγ2-core (Δ471-913) by In-Fusion PCR cloning (Clontech) following the introduction of an SfoI site in place of the γSA by site-directed mutagenesis. The PLCɛ linker was deleted from pCMV PLCɛ1a by site-directed mutagenesis.
In some experiments, cDNAs encoding either wild-type (1,265 aa, GenBank accession number NP_002652) or mutant human PLCγ2 endowed with a carboxyl-terminal c-myc epitope tag were used in pcDNA3.1(+) (12). The cDNAs encoding the PLCγ2/PLCβ2 chimera were constructed using the PCR overlap extension method to join three pieces of DNA encoding the N-terminal portion of PLCγ2 (aa 1 to 457), the human PLCβ2 (1,181 aa, GenBank accession number Q00722) X-Y linker (aa 465 to 536 for PLCγ2-core/β2linker or aa 465 to 515 and 536 for PLCγ2-core/β2linkerΔ20), and the C-terminal portion of PLCγ2 (aa 925 to 1,265). The replacement of the sequence encoding the outermost 21 carboxyl-terminal residues (516 to 536) of the PLCβ2-derived X-Y linker of PLCγ2-core/β2 linker by the sequence encoding aa 904 to 924 of PLCγ2 was performed using standard PCR-mediated mutagenesis (QuikChange XL site-directed mutagenesis kit; Stratagene), resulting in DNA encoding PLCγ2-core/β2 linkerΔ21+21. The same technique was used to create constructs encoding the mutant PLCγ2Δ16, lacking aa 908 to 923 of PLCγ2, the mutants PLCγ2Δ16+β2 and PLCγ2Δ16+γ1, carrying the corresponding regions of human PLCβ2 (EEIKKMQSDEGTAGLE) and human PLCγ1 (GenBank accession number ABB84466; QTADARLTEGKIMERR), respectively, and the mutant PLCγ2Y919A/N922A.
Plasmids encoding Rac2 and Rac2V12 for expression in mammalian cells [pcDNA3.1(+)-Rac 2 and -Rac2G12V] and the vectors used for expression and protein purification were described previously (40).
Preparation of recombinant proteins.
For the expression and purification of different PLCγ2 full-length variants (with and without fluorescent tags), 400 ml of Sf9 cells at 1.8 × 106 cells/ml in a 2-liter roller bottle were infected with baculovirus at a multiplicity of infection (MOI) of two. Following incubation for 72 h at 28°C with shaking, the cells were pelleted by centrifugation at 2,000 × g for 15 min. The pellets were snap-frozen in liquid nitrogen and then stored at −80°C. Lysis was performed using Sf9 lysis buffer with a reduced salt concentration [50 mM Tris-Cl, pH 8.0, 50 mM NaCl, 1 mM MgCl2, 1% Triton X-100, 1 mM Tris(2-carboxyethyl) phosphine (TCEP), 1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF), 1× Complete EDTA-free protease inhibitor cocktail, 8 units/ml DNase I]. Following clearing of the lysate by centrifugation, a 5-ml HiTrap Q HP column (GE Healthcare) was used to obtain a fraction enhanced for the full-length protein. A 1-ml HisTrap column was used to further purify the protein. His buffers A and B were used as described below. After dilution of the NaCl concentration to 100 mM, a 1-ml HiTrap heparin HP column (GE Healthcare) was used to concentrate the protein and remove imidazole. Fractions from the heparin column were pooled, and the protein concentration was calculated using calculated extinction coefficients and the absorbance at 280 nm.
For expression and purification of a truncated PLCγ2 construct, PLCγ2-core, 5 liters of 2× YT microbial medium (sigma) was inoculated with the bacterial strain C41(DE3) harboring the vector pTriEx4-PLCγ2ΔSA. Bacteria were cultured at 37°C and 250 rpm until the optical density at 600 nm (OD600) reached 0.4. The cultures were cooled to 18°C for 1.5 h, and then expression was induced with 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG). Expression continued for 16 h at 18°C and 250 rpm. Bacteria were harvested, and pellets frozen for 30 min at −20°C. Each pellet (derived from 1 liter of bacterial culture) was lysed with 25 ml of lysis buffer (25 mM Tris-HCl, 250 mM NaCl, 40 mM imidazole, 20 mM benzamidine, 50 μg/ml lysozyme, pH 8.0) for 30 min on an orbital shaker at 100 rpm at 4°C. Subsequently, 5 ml of a 10% (vol/vol) solution of Triton X-100 and 8 units/ml DNase I were added and lysis continued for 1 h. Lysates were clarified through centrifugation in an SS34 rotor at 15,000 rpm for 1 h at 4°C. The supernatants were applied to 5-ml HisTrap columns (GE Healthcare) and subsequently washed with buffer A (25 mM Tris-Cl, 500 mM NaCl, 40 mM imidazole, 1 mM TCEP, pH 8.0). Proteins were eluted with buffer B (25 mM Tris-Cl, 500 mM NaCl, 500 mM imidazole, 1 mM TCEP, pH 8.0). Eluted protein was dialyzed overnight with tobacco etch virus (TEV) protease (10 μg per 1 mg of protein) in dialysis buffer (25 mM Tris-Cl, 100 mM NaCl, 1 mM TCEP, pH 8.0) at 4°C. PLCγ2ΔSA with the His tag removed was collected directly by flowing the dialyzed protein over a 5-ml HisTrap column (GE Healthcare). A 1-ml HiTrap heparin HP column (GE Healthcare) was used to concentrate the protein and remove imidazole. Fractions from the heparin column were pooled, and the protein concentration was calculated using calculated extinction coefficients and the absorbance at 280 nm.
Purification of Rac2 proteins was as previously described (40).
In vitro reconstitution assay.
In vitro reconstitution of purified Rac2 and purified PLCγ2 and determination of PLC activity were done as previously described (29, 40). The concentrations of functionally competent Rac2 GTPases were measured in purified preparations by quantitative 35S-GTPγS binding as detailed before (29). To examine the functional role of C-terminal geranylgeranylation of Rac2, purified soluble wild-type Rac2 was first incubated for 15 min at 30°C in a volume of 20 μl containing 8 μg purified type I geranylgeranyltransferase, 60 μM geranylgeranylpyrophosphate, 65 mM Tris-maleate, pH 7.3, 0.7 mM EDTA, 70 mM NaCl, 0.7 mM MgCl2, 0.7 mM dithiothreitol (DTT), 70 μM phenylmethylsulfonyl fluoride (PMSF), 0.7 μM GDP, and 10 μM ZnCl2. At the end of the isoprenylation reaction, the samples were reconstituted with purified PLCγ2 and incubated in a final volume of 60 μl for 60 min at 30°C with phospholipid vesicles containing PIP2. The incubation was in the presence of 30 nM free Ca2+, 1 mM sodium deoxycholate, 1.9 mM sodium cholate, and either 100 μM GDP or 100 μM GTPγS.
PLC activity in mixed micelles.
The assay was based on the method described in Ellis et al. (9). Briefly, the reaction mixture contained 20 mM Tris-Cl, pH 6.8, 0.4 mg/ml bovine serum albumin (BSA), 5 mM 2-mercaptoethanol, 4 mM EGTA, 2 mM CaCl2, 100 mM NaCl, 0.4% sodium cholate, 200 μM PIP2, and 25 ng protein in 50 μl. The reaction mixtures were incubated at 37°C for 20 min. The results of these experiments are shown as the means ± standard deviations (SDs) of duplicate samples and are representative of three or more independent experiments.
Cell culture, transfection, and fractionation.
COS-7 and HEK293 cells were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (Invitrogen) and 2.5 mM glutamine. Prior to transfection, cells were seeded into 6-well plates at a density of 2.5 × 105 cells/well and grown for 16 h in 2 ml/well of the same medium. For transfection of COS-7, 1.0 μg of PLCγ DNA was mixed with 1 μl PlusReagent and 7 μl Lipofectamine (Invitrogen) and the mixture added to the cells in 0.8 ml DMEM without serum. The cells were incubated for 3.5 h at 37°C, 5% CO2 before the transfection mixture was removed and replaced with DMEM containing serum.
Analysis of PLCγ2 distribution in fractions from HEK293 and COS7 cells was as previously described (29). Cells were transfected with PLCγ2 together with empty vector or Rac2 by the calcium phosphate method. Twenty-four hours later, the cells were scraped in 100 μl fractionation buffer (20 mM Tris-HCl, pH 7.5, 2 mM EDTA, 2× Complete EDTA-free protease inhibitor cocktail, 0.1 M AEBSF). The lysate was then passed 10 times through a 0.45- by 23-mm needle and centrifuged at 200 × g for 10 min at 4°C. The supernatant was centrifuged again at 12,000 × g for 15 min at 4°C to produce the soluble cytosolic fraction and the particulate membrane fraction, which was resuspended in 50 μl fractionation buffer.
Analysis of inositol phosphate formation in intact COS-7 cells.
This analysis was essentially as described previously (11, 40). Briefly, 24 h posttransfection, cells were labeled with 1.5 μCi/ml myo-[2-3H]inositol. After a further 24 h, the cells were incubated in 1.2 ml inositol-free DMEM, without serum, containing 20 mM LiCl with or without stimulation with 100 ng/ml epidermal growth factor (EGF; Calbiochem) for 1 h. The cells were lysed by the addition of 1.2 ml 4.5% perchloric acid, and supernatants and pellets were separated. Inositol phosphates were collected using AG1-X8 200-400 columns (Bio-Rad). The levels of inositol phosphates were quantified by liquid scintillation counting using Ultima-Flo scintillation fluid (PerkinElmer). The PLC activity analyzed only by this standard measurement is given as “PLC activity (cpm).” Data shown are the means ± SDs of triplicate samples and are representative of three or more independent experiments.
In a number of experiments, we also analyzed the lipid fraction. The pellets from the lysates were treated with chloroform-methanol-HCl to separate aqueous and lipid phases. An aliquot of the lipid phase was used for scintillation counting with Ultima-Flo scintillation fluid. This value was used for normalization of PLC activity so that PLC activity is expressed as the total inositol phosphates formed relative to the amount of [3H]inositol in the phospholipid pool. Because the differences in steady-state labeling of inositol lipids are small (within 20%), this normalized PLC activity corresponds closely to PLC values expressed as total inositol phosphates; however, the standard errors between the results for the duplicates are generally smaller. The PLC activity derived from these measurements is given as “normalized PLC activity.” The data shown are the means ± SDs of duplicate samples and are representative of three or more independent experiments.
FRET.
Fluorescence resonance energy transfer (FRET) measurements were performed in solution using a multidimensional spectrofluorometer capable of resolving fluorescence with respect to wavelength, lifetime, and polarization (25).
The fluorescent PLCγ2 was thawed on ice, and 300 μl of 0.5 μM protein was added to a glass cuvette and placed in the spectrofluorometer. Donor (GFP) emission was measured at 510 nm upon excitation at 488 nm. Acceptor (mCherry) emission was detected at 600 nm upon either direct excitation at 570 nm or following donor excitation at 488 nm. Fluorescence lifetime (FLIM) measurements were undertaken, with the emission collected after passing a polarizer set at the magic angle (54.7°). Time-resolved measurements of the fluorescence anisotropy were also undertaken, for which the emission was measured with the polarizer set parallel and perpendicular relative to the excitation. Data were collected for fluorescent PLCγ2 alone and after adding increasing concentrations (0.1 to 25 μM) of Rac2G12V protein, in the presence of GTPγS. The fluorescence intensity and anisotropy decay profiles were analyzed using TRFA software (Scientific Software Technologies Center, Minsk, Belarus), utilizing its global analysis capability. For the construct undergoing FRET, the GFP fluorescence intensity decay was analyzed as the sum of two exponential functions and the mean decay time was calculated according to the following formula (24):
 |
(1) |
The FRET efficiency, E, was determined from the mean lifetime of GFP according to the formula
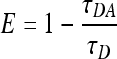 |
(2) |
These E values were used to estimate the donor-acceptor distance as follows:
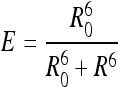 |
(3) |
The time-resolved anisotropy decay profiles of the acceptor (mCherry) by donor (GFP) excitation were analyzed as the sum of two exponential functions,
 |
(4) |
where φ1 is the rotational correlation time due to the rotation of the whole PLCγ2 molecule and φ2 is the rotational correlation time due to FRET.
The contribution of the long rotational correlation time (β1) of mCherry can be used to estimate the average angle, θ, between the dipoles of GFP and mCherry as follows (2):
 |
(5) |
where r0 is the initial anisotropy.
For FRET analysis in transfected cells, FLIM images were acquired using a pulsed Ti:Sapphire laser (Maitai, Spectra Physics), frequency doubled to deliver the 470-nm excitation wavelength. Images were taken with a Leica SP5 microscope equipped with a Becker and Hickl acquisition card. Emission was collected with a band pass filter (500 to 550 nm) and detected with an external hybrid detector (Becker and Hickl, Germany). Each image was acquired for 2 min. Analysis was performed with a home-made program using a monoexponential decay model, and convolution with an instrument response function obtained from a solution of erythrosine B in water. The lifetime maps were merged with the fluorescence intensity image. In this case, the color corresponds to the lifetime value, and the brightness is proportional to the number of photons accumulated in each pixel. In this way, the pixels with a low number of photon counts (where the error in analyzing the lifetime value is higher) contribute less to the total image.
SPR.
All surface plasmon resonance (SPR) measurements were carried out as previously described (11, 35). Measurements were carried out at 23°C using a lipid-coated L1 chip in the BIACORE X system as described previously. The 400-μg/ml solutions of vesicles were prepared in running buffer (50 mM Tris-HCl, pH 7.4, 0.16 M KCl). After the sensor chip surface was washed with the running buffer, POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine)/POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine)/POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-l-serine])/PI (phosphatidylinositol)/cholesterol (12:33:22:8:22%) vesicles with either 3% PI(4,5)P2 or PI(3,4,5)P3 or an extra 3% POPC and POPC (100%) vesicles were injected at 5 μl/min into the active surface and the control surface, respectively, to give the same resonance unit values. The flow rate was maintained at 15 μl/min for both association and dissociation phases.
RESULTS
Critical role of the γSA in the regulation of basal activity and activation of PLCγ2.
As shown in Fig. 1 A, the domains within the γSA and their linkers comprise a large insertion (∼470 amino acids) placed between the two halves (TIM-X and TIM-Y) of the catalytic domain. By deletion of the γSA (residues 471 to 913), we generated the common core unit to compare the PLC activity of this PLCγ2 variant (PLCγ2ΔSA/PLCγ2-core) and that of the full-length protein in transfected cells (Fig. 1B and C). When PLCγ2-core was expressed in COS-7 cells, the basal activity of this variant was greatly elevated and was higher than the PLC activity of the full-length protein following stimulation by either EGF or Rac (Rac2G12V) (Fig. 1B and C). Thus, deletion of the γSA removed intramolecular inhibitory constraints, resulting in a constitutively active PLC enzyme.
FIG. 1.

Critical role of γSA in regulation of basal activity and activation of PLCγ2. (A) Schematic representation of the domain organization of PLCγ2 highlighting the components of the “core” and the γSA. The domains are scaled according to the number of amino acid residues. Numbering of the amino acids is for human PLCγ2. (B) Activity measurements for full-length PLCγ2 and PLCγ2-core [also known as PLCγ2ΔSA (Δ471-913)] were performed in COS-7 cells transfected with full-length PLCγ2 or PLCγ2-core with and without EGF stimulation. Western blotting was used to show equal expression (inset). (C) PLC activity was measured in COS-7 cells cotransfected with full-length PLCγ2 or PLCγ2-core and Rac2G12V. Western blotting was used to show equal expression (inset). (D) The effects of point mutations on PLCγ2 activity were measured in COS-7 cells transfected with wild-type PLCγ2 and constructs containing point mutations (R564A, R672/674A, and W899A), with and without EGF stimulation. (E) PLC activity was measured in COS-7 cells cotransfected with wild-type PLCγ2 or constructs containing point mutations (R564A, R672/674A, and W899A) and Rac2G12V. Data are representative of at least three independent experiments.
The ability of the core to be fully active even when the domains required for membrane translocation (through EGF receptor or Rac2 binding) were not present was unexpected; notably, translocation has been shown to be a requirement for activation of intact PLCγ enzymes (reviewed in reference 23). Indeed, PLCγ2 activation of the full-length variants with nonfunctional SH2 domains (SH2n and SH2c [N-terminal and C-terminal SH2] and R564A and R672/674A, respectively [26]) was greatly reduced in response to EGF (similar to the levels in control mock-transfected cells), while the activation by Rac2 was less affected (Fig. 1D and E). Notably, the point mutation W899A in the spPH domain that greatly reduced Rac2 activation had no effect on stimulation by EGF (Fig. 1D and E), consistent with our previous findings of different structural requirements for activation resulting from interaction with the membrane-bound EGF receptor and Rac2 (4). In contrast to the deletion of the γSA, point mutations that specifically affect the function of the SH2 domains or the spPH domain were not able to increase basal PLC activity (Fig. 1D and E).
X-Y linkers from other PLC enzymes inhibit the PLC activity of PLCγ2-core.
We further tested the possibility that the γSA could be involved in autoinhibition by occluding the catalytic domain and, in particular, the active site; this role has been suggested for X-Y linkers from PLCβ, PLCδ, and PLCɛ (17). More specifically, based on structural information (17), the PLCβ2 X-Y linker region, highlighted in Fig. 2 A, was identified as interacting directly with the PLCβ2 core. We therefore tested the possibility that the X-Y region from PLCβ2 can impose autoinhibition on PLCγ2-core. As shown in Fig. 2B and C, the variant combining PLCγ2-core and the X-Y linker from PLCβ2 (PLCγ2-core+β2 linker) had low basal activity and could not be stimulated by Rac2 (Fig. 2B). Furthermore, the deletion of 20 amino acid residues that are directly implicated in the interaction with the core (17) generated a variant (PLCγ2-core+β2Δ20 linker) with high basal activity (Fig. 2C), as initially observed for PLCγ2-core (Fig. 1B).
FIG. 2.

Effect of X-Y linker from PLCβ2 on activity of PLCγ2-core. (A) Diagram of the core PLC enzyme (core) with the linker from PLCβ2 (β2), showing its amino acid sequence. The 20 residues shown in gray contain amino acids that interact directly with the PLCβ2 core, resulting in autoinhibition of PLCβ2. (B) COS-7 cells were cotransfected as indicated at the abscissa with 500 ng each per well of either empty vector (Mock) or vector encoding wild-type PLCγ2 (PLCγ2) or variant comprising PLCγ2-core and X-Y linker from β2 (PLCγ2-core+β2 linker) together with 25 ng each per well of either empty vector (None) or vector encoding wild-type Rac2 (Rac2) or constitutively active Rac2G12V (Rac2G12V). (C) COS-7 cells were transfected with increasing amounts per well of vector encoding a PLCγ2 variant comprising PLCγ2-core and X-Y linker from PLCβ2 with the deletion of 20 amino acid residues indicated in panel A (PLCγ2-core+β2 linkerΔ20) or a variant replacing the 21 amino acids (the indicated 20 aa in gray plus a Val residue) with a potentially similar region of 21 residues from PLCγ2 (aa 904 to 924) (PLCγ2-core+β2 linkerΔ21 PLCγ2-21). COS-7 cells were also transfected with a single amount (500 ng) of vector encoding wild-type PLCγ2 (PLCγ2) or the variant PLCγ2-core+β2 linker, as described for panel B. (D) Cells from one well were lysed in 100 μl of SDS-PAGE sample preparation buffer, and immunoblotting was performed using antibodies against c-myc epitope tag. For the experiments whose results are shown in panels B and C, the total amount of DNA was maintained constant at 525 ng in each transfection by adding empty vector. Twenty-four hours after transfection, the cells were incubated for 20 h in the presence of myo-[2-3H]inositol (2.5 μCi/ml) and 10 mM LiCl, and the levels of inositol phosphates were then determined as described in Materials and Methods. The results are presented as the means ± standard deviations of triplicate determinations.
Based on these findings, we explored the possibility that a region in γSA similar to the 20-amino-acid stretch in the PLCβ2 X-Y linker could be the critical component of PLCγ2 autoinhibition. Amino acid sequence comparison followed by secondary structure prediction (6) identified aa residues 904 to 924 as a candidate region for such a role. However, insertion of this region into the PLCβ2 linker (β2-linkerΔ21+γ2-21) failed to confer autoinhibition (Fig. 2C). Similarly, neither the deletion of this region within native PLCγ2 (sparing residues contributing to the structure of the spPH) nor replacement by the corresponding regions of PLCβ2 or PLCγ1 resulted in high basal activity (see Fig. S1B in the supplemental material). Interestingly, the latter PLCγ2 variants were also resistant to activation by Rac2 in intact cells and in a cell-free, reconstituted system (see Fig. S1B and C). This finding indicated that the resistance of the mutant to activated Rac2 was not due to an unspecific disarray of the enzyme's catalytic core, since the deletion mutant PLCγ2Δ16 was indistinguishable from wild-type PLCγ2 in the presence of 10 μM free Ca2+ (see Fig. S1C, left, in the supplemental material). Collectively, these data suggest that different PLC enzymes could have different surfaces and interactions that occlude the common core and suppress PLC activity in a basal state. Consistent with this possibility, the 110-residue-long X-Y linker from PLCɛ that contributes to the maintenance of low basal activity of this PLC enzyme (see Fig. S2B in the supplemental material) significantly inhibited PLCγ2-core when inserted in place of the γSA (see Fig. S2C).
Analysis of domains and interdomain linkers in the γSA for their contribution to autoinhibition.
To identify domains and interdomain linkers in the γSA that contribute to autoinhibition, we measured the PLC activity of a panel of deletion variants (Fig. 3). The PLCγ2 variants with elevated basal activity resulted from deletions in the spPH domain (Fig. 3B) and the deletion of the SH2c domain (Fig. 3C). In contrast to the results for the deletion of the entire γSA (Fig. 1B), it was possible to further activate several of these variants (ΔspPHn, ΔspPHc, and ΔSH2c) by EGF stimulation (Fig. 3B and C). In a previous study, a similar deletion of the spPH domain in PLCγ1 resulted in higher phosphorylation in the basal state that was further enhanced above the wild-type levels upon stimulation of the B-cell receptor (8). Deletion of the SH2n domain did not affect basal activity but, like the point mutation in this domain, reduced the activation of PLCγ2 by EGF (Fig. 3D). The deletion of the SH3 domain had no effect on basal activity or PLCγ2 activation (Fig. 3C). In contrast to activation by EGF, activation by Rac2 required an intact spPH, as described in detail in our previous studies (4, 40); however, activation of ΔSH2c and ΔSH3 variants by Rac2 was observed (data not shown), similar to that seen for EGF in Fig. 3C.
FIG. 3.

Analysis of domains and interdomain linkers in γSA. (A) Diagram showing the catalytic (TIM-X and TIM-Y) and γSA domains of PLCγ. Numbering refers to the position in the human PLCγ2 protein sequence. The gray lines numbered 1 through 5 indicate the approximate positions of the linker deletions. The sequences of these linker deletions and secondary structure predictions for these regions (using published structures and PSIPRED) are shown below the diagram. (B to F) PLC activity was measured in COS-7 cells transfected with wild-type PLCγ2 and constructs containing deletions. Activity was measured with or without EGF stimulation, as indicated. (B) Wild-type PLCγ2, ΔspPH (Δ471-513-841-913), ΔspPHn (Δ471-513), and ΔspPHc (Δ841-913). (C) Wild-type PLCγ2, ΔSH2n (Δ530-634), ΔSH2c (Δ642-738), and ΔSH3 (Δ767-831). (D) Wild-type PLCγ2, ΔspPHn-SH2 (Δ523-534), ΔSH2n-SH2c (ΔSH-SH) (Δ626-633), ΔSH2c-SH3 (ΔSH2-SH3) (Δ748-755), ΔSH3-spPHc (ΔSH3-spPH) (Δ834-842), and ΔspPHc-Y (ΔspPH-Y) (Δ914-921). (E) Wild-type PLCγ2 and PLCγ2ΔPCI (726-733). (F) Wild-type PLCγ2, PLCγ2ΔPCI (726-733), and ΔSH2c (Δ642-738). nspPH/spPHn, N-terminal spPH; cspPH/spPHc, C-terminal spPH; nSH2/SH2n, N-terminal SH2; cSH2/SH2c, C-terminal SH2.
Comparison of deletion variants in the interdomain linkers (the precise boundaries of these deleted regions are listed in Fig. 3A, bottom) showed that none of five deleted regions greatly enhanced basal activity (Fig. 3D). These regions were relatively short (8 to 12 aa), to minimize the chance of significantly disrupting the overall conformation of the γSA, and include sequences conserved between PLCγ1 and PLCγ2. All these deletions affected basal PLC activity only slightly. However, a small deletion within the long SH2c-SH3 linker (residues 748 to 755) and the deletions in the linkers connecting the spPH domain to the SH3 or the SH2n domain considerably enhanced the level of activation in stimulated cells. In contrast, a deletion within the linker connecting the spPH domain to the catalytic TIM barrel did not enhance PLC activation (spPH-TIM-Y linker), as was the case for the PLCγ2 variant with the deletion in the relatively short SH2n-SH2c linker. Interestingly, the region within spPH-TIM-Y linker that was identified as similar to the autoinhibitory region in PLCβ2 had reduced activation by EGF (Fig. 3D), in addition to the reduced sensitivity to activated Rac2 noted above and shown by the results in Fig. S1 in the supplemental material. For all other variants with deletions in the interdomain linkers, essentially the same data were obtained for activation with Rac2G12V (data not shown) as for EGF stimulation. The finding that some of the small deletions in interdomain linkers increase the basal level of activity only slightly, while considerably enhancing activation, could be due to their effect of destabilizing but not overcoming autoinhibition; recently described gain-of-function single point mutations in PLCγ2 show similar effects (11).
The deletion of the SH2c domain included eight amino acids from the C-terminal end of this domain, previously referred to as the phospholipase C inhibitor (PCI) peptide. The PCI peptide was reported to be inhibitory when added to PLC enzymes in vitro and in cells (18, 19). Therefore, we deleted this region (residues 726 to 733) and found that its integrity was important for autoinhibition (Fig. 3E). As observed for the deletion of the SH2c domain, the enhanced basal activity of the ΔPCI variant could be stimulated further by EGF (Fig. 3E), and the basal PLC activities of the two variants, ΔPCI and ΔSH2c, were similar (Fig. 3F).
Subsequent experiments using purified PLCγ2-core and the γSA showed absence of binding in a micromolar range and, similarly, no inhibition of PLC activity by isolated γSA. Only a small peptide corresponding to the 8-aa PCI from the SH2c domain, used here at the same, high concentrations as in previous publications (18, 19), inhibited PLC (data not shown).
Together, these data suggest that autoinhibitory constraints in PLCγ2 could involve elements from different regions that have an effect on PLCγ2-core through a limited direct interaction, i.e., mainly by the relative intramolecular positioning of the γSA and the core. The type of deletion analysis performed here implicates the spPH domain, SH2c domain, and to some degree also, the internal linkers in the central part of the γSA in maintaining the low basal activity of PLCγ2.
Analysis of PLCγ2 variants with constitutively elevated PLC activity and their effects in transfected cells.
The finding that PLCγ2-core is highly active in cells even though it lacks the domains required for membrane translocation (ether by Rac or EGFR) was unexpected. One possibility is that the removal of the γSA exposes surfaces involved in membrane interactions, leading to association of the core enzyme with the membrane in unstimulated cells. To test this possibility, we analyzed the localization of GFP-tagged versions of full-length PLCγ2 and core in COS-7 cells. When COS-7 cells were transfected with PLCγ2-core, their viability and cell-cycle profile were not affected (data not shown). However, these cells appeared to round up and adhere less strongly (Fig. 4). For this reason, we also analyzed a PLC-inactive variant of PLCγ2-core that incorporated a single point mutation of the critical active site residue (H327Q). This variant had no effect on cell morphology following transfection. Analysis of transfected cells did not reveal any clear differences in the cellular localization of PLCγ2-core proteins compared to the localization of the full-length protein (Fig. 4A). The full-length (Fig. 4A, top) and PLCγ2-core proteins (Fig. 4A, middle) did not appear to localize to the plasma membrane in unstimulated cells. The same conclusion was reached from the analysis of a ΔPCI construct and its inactive-PLC variant (Fig. 4A, bottom).
FIG. 4.

Analysis of PLCγ2 variants with constitutively elevated PLC activity. (A) COS-7 cells were transfected with N-terminally GFP-tagged PLCγ2 variants. Top, full-length PLCγ2 (FL) and its inactive-PLC variant (H327A); middle, PLCγ2-core (Δ471-913) and its inactive-PLC variant (H327A); and bottom, PLCγ2ΔPCI (726-733) and its inactive-PLC variant (H327A). At 24 h posttransfection, the cells were fixed in 4% paraformaldehyde. Images were captured using a Bio-Rad confocal microscope with a 60× lens objective. (B) HEK293 cells were transfected with full-length PLCγ2 or PLCγ2-core with or without Rac2G12V. The cells were fractionated to separate membrane (M) and cytosolic (C) fractions. Proteins were visualized by Western blotting. Gβ1 was used as a marker of the membrane fraction. Essentially the same data were obtained using COS-7 cells.
The cellular distribution of the full-length PLCγ2 and PLCγ2-core was also analyzed by cell fractionation and subsequent Western blotting, as this allowed better assessment of the relative amounts of PLCγ2 in different compartments. It has been shown previously that, under basal conditions, PLCγ2 is present almost exclusively in the soluble fraction; however, following coexpression with Rac2G12V, it is distributed between the soluble and membrane fractions (29) (Fig. 4B, top). In contrast, PLCγ2-core was present in the soluble fraction under all conditions (Fig. 4B, middle). Thus, it seems that stable translocation to the membrane is not required for PLCγ2 activation and substrate access when autoinhibitory constraints are removed.
Insights into mechanisms of PLCγ2 activation from analyses in vitro.
Our analysis of PLCγ2 in a cellular setting included two different mechanisms of regulation, activation following cell stimulation by EGF and activation by Rac2 after coexpression with the Rac2G12V variant (Fig. 1). However, the molecular mechanisms of activation through either EGF receptor or Rac2 have not been elucidated. In the case of activation of PLCγ2 by Rac GTPases, this process can be analyzed not only in a cellular setting but also in a reconstitution system in vitro. Furthermore, our previous studies in this system using purified PLCγ2 and Rac2 proteins and lipid vesicles demonstrated that no additional components or tyrosine phosphorylation are required to achieve stimulation of PLC activity (40). We therefore used this system to further analyze PLCγ2 activation. As shown in Fig. 5 A, we first compared the full-length PLCγ2 with PLCγ2-core. While the full-length protein required the presence of the GTPγS-bound form of Rac2 for activation (Fig. 5A, left), PLCγ2-core was not stimulated by activated Rac2 and was constitutively active (Fig. 5A, right); the basal activity of the core was estimated to be at least about 100-fold higher than full-length PLCγ2. Although these observations are consistent with the data obtained in COS-7 cells (Fig. 1B and C), they are unexpected. As stated earlier, there is extensive experimental evidence that stable translocation is required for the high PLC activity. As shown in Fig. 5B, activation of PLCγ2 required that Rac2 was lipid modified, suggesting that its ability to associate with the membrane and to mediate PLCγ2 translocation to lipid vesicles was critical for the activation process. This is illustrated by direct comparison of Rac2 with and without lipid modification in the in vitro reconstitution system (Fig. 5B). The results suggest that interaction between PLCγ2 and Rac2 in solution is not sufficient for the activation. These findings again raised the question of whether PLCγ2-core had an enhanced ability to interact with lipid vesicles. Using surface plasmon resonance analysis, we could assess the binding of PLCγ2-core to lipid vesicles and found that it was similar to that observed for the full-length PLCγ2; in fact, PLCγ2-core had a somewhat higher dissociation rate (Fig. 5C). This was observed under more physiological, low-binding conditions (lines 3 and 4), as well as under conditions of enhanced binding due to incorporation of PI(3,4,5)P3 (lines 1 and 2). This further reinforced our findings in cells that the PLC enzyme lacking autoinhibitory constraints can access its substrate without stable association with the membrane.
FIG. 5.

Analysis of PLCγ2 regulation by Rac in vitro. (A) In vitro reconstitution of purified Rac2 with purified full-length PLCγ2 (left) was compared with the activity of purified PLCγ2ΔSA (PLCγ2-core) (right) in the presence and absence of Rac2 (loaded with GDP or GTPγS). (B) To examine the functional role of C-terminal geranylgeranylation of Rac2, purified soluble wild-type Rac2 was reconstituted without modification or was first incubated for 15 min with purified type I geranylgeranyltransferase (GGTase I). Two hundred nanograms of purified full-length PLCγ2 was used. (C) SPR was used to measure the interaction of purified full-length PLCγ2 (black lines) and PLCγ2-core (Δ471-913) (gray lines) with plasma membrane mimetic (lines 3 and 4) or further addition of PI(3,4,5)P3 (lines 1 and 2). Binding to 100% phosphocholine was used to obtain control readings. (D) Activity assays using mixed PIP2-cholate micelles were carried out using full-length PLCγ2 and PLCγ2ΔSA (PLCγ2-core). DPM, disintegrations per minute.
We further analyzed PLCγ2 in a detergent-PIP2 mixed-micelle assay containing only PIP2 substrate and sodium cholate. This assay assesses more directly the accessibility of the active site to substrate and the ability of the enzyme to bind and hydrolyze PIP2. Interactions with the lipid surface by PLCγ2 surfaces other than the active site, which contribute to PLC activity in assays using lipid vesicles (such as those in reconstitution experiments), typically affect the PLC activity much less in this detergent-PIP2 mixed-micelle assay (9, 11). Comparison of the full-length PLCγ2 and PLCγ2-core proteins in the mixed-micelle assay showed that PLCγ2-core had much greater basal activity (about 10-fold) (Fig. 5D), suggesting that the active site is more exposed in this construct; this is consistent with the possibility of the active site occlusion. Interestingly, neither Rac2 without lipid modification nor the Rac2 that was lipid modified by geranylgeranylation at the C terminus was able to stimulate the full-length PLCγ2 in this assay lacking more extensive lipid surfaces (see Fig. S3 in the supplemental material). As further discussed below, one possibility is that regulation mediated by Rac2 requires interactions between membrane surfaces and regions in the PLCγ2 protein that are distinct from the PLC active site, leading to interfacial activation.
Analysis of conformational changes using FRET.
Although the results of reconstitution experiments did not support the possibility that interaction between Rac2 and PLCγ2 alone was sufficient for the activation, we performed in vitro fluorescent resonance energy transfer (FRET) experiments to directly test whether the Rac2 interaction with PLCγ2 causes observable conformational changes in PLCγ2 protein that could contribute to activation. In order to use FRET, a construct was created which contained two fluorophores selected to provide appropriate spectral overlap: GFP (donor) and mCherry (acceptor) (41). As shown in Fig. 6 A, the SH3 domain was deleted from PLCγ2 and mCherry was inserted in its place, since the deletion of the SH3 domain had no significant effect on PLCγ2 activity (Fig. 3D). GFP was added to the C-terminal end of the protein. As a control, a construct containing only the GFP tag at the C terminus was also made. As illustrated for PLCγ2ΔSH3(mCherry)GFP, when expressed in COS-7 cells, these constructs retained the ability to be activated by coexpression with Rac2G12V (Fig. 6B, left) and by stimulation with EGF (data not shown). Similarly, purified proteins incorporating these fluorophores, as shown for the wild-type PLCγ2 (Fig. 5A), were stimulated by lipid-modified Rac2-GTP (Fig. 6B, right). These purified proteins were subsequently used in solution in the absence or presence of purified Rac2 for in vitro FRET measurements using a multidimensional spectrofluorometer (25).
FIG. 6.

Analysis of PLCγ2 regulation by Rac using FRET. (A) Diagram showing the domains of PLCγ2 and the positions of the fluorophores. The red fluorophore, mCherry, has been inserted in place of the SH3 domain, and GFP has been added after the C2 domain with the deletion of the C-terminal portion of PLCγ2. Numbering refers to the position in the wild-type human PLCγ2 sequence. (B) Inositol phosphates were measured in COS-7 cells transfected with PLCγ2ΔSH3(mCherry)GFP and Rac2G12V (left). Protein expression was analyzed by Western blotting (inset). Purified PLCγ2 and PLCγ2ΔSH3(mCherry)GFP variant was used in a reconstitution assay (right) as described for Fig. 5A. (C) Experiments were carried out using purified PLCγ2GFP and PLCγ2ΔSH3(mCherry)GFP proteins in vitro. Fluorescence decay was measured with no Rac2G12V present and with a 100-fold excess of Rac2G12V. (D) Variation of the GFP average fluorescence decay time within the double-tagged PLCγ2 protein (top) and variation of the distance R between GFP and mCherry in the double-tagged PLCγ2 protein (bottom) were expressed as a function of Rac concentration. The distance was calculated from equations 2 and 3, assuming a Förster distance (R0) of 5.2 nm for the GFP-mCherry pair from the data previously published in the literature (1, 27, 33, 38) and a nonquenched GFP lifetime of τD = 2.6 ns from our measurements of the single-tagged PLCγ2 construct. (E) Variation of the time-resolved anisotropy values for the short and long rotational correlation times in the mCherry anisotropy decay upon GFP excitation as a function of Rac concentration (top). The shorter rotational correlation time component is due to FRET, while the longer component is due to the rotation of the whole PLCγ2 molecule. Variation of the angle between the mCherry and GFP dipoles is shown as a function of Rac concentration (bottom). All error bars were calculated by the analysis software based on the photon statistics within the measured decay curves. (F) Representative FLIM images for cells transfected with different constructs, as indicated, all presented on the same scale bar (1.2 to 3 ns). Images are corrected for intensity to eliminate the effect of the lifetime in different parts of the cell.
First, PLCγ2 proteins containing fluorophores were analyzed and the donor and acceptor lifetimes were measured. A reduced value of the GFP fluorescence lifetime (2.15 ns) compared to that of the PLCγ2 labeled only with GFP (2.6 ns) suggested that FRET occurs in this construct even in the absence of Rac2 (Fig. 6C). When Rac2 and GTPγS were added so that the concentration of Rac2 was in excess (100-fold), there was no difference in lifetime for PLCγ2GFP; similarly, there was no obvious change for PLCγ2ΔSH3(mCherry)GFP. This was analyzed further using PLCγ2ΔSH3(mCherry)GFP and a range of increasing concentrations of Rac2, where a further, marginal reduction in the average decay time of GFP was observed, indicating a very small decrease in the distance between the two fluorophores (GFP and mCherry), from 6.75 nm in the absence of Rac2 to 6.60 nm at 25 μM Rac2 (Fig. 6D).
The multidimensional spectrofluorometer was used to measure the time-resolved anisotropy decay profiles and, thus, to calculate the rotational correlation time of each fluorophore and the average angle between GFP and mCherry (Fig. 6E; also see Fig. S4 in the supplemental material). The rotational correlation time of GFP (488 nm excitation, 510 nm emission) was estimated to be ∼30 ns, which is longer than the value reported in the literature for free GFP (∼16 ns) (39) (see Fig. S4B, left, in the supplemental material). Direct excitation of mCherry at 570 nm yielded a much longer rotational correlation time, estimated to be 200 ns (see Fig. S4B, middle). This suggests that the GFP bound to the C terminus of PLCγ2 retains some rotational freedom, while mCherry appears to be rigidly attached to PLCγ2 and moves only as part of the whole molecule. The average angle between the GFP and mCherry dipoles was calculated (according to equation 5) to be about 40° and was not affected by the addition of Rac2 (Fig. 6E, bottom).
These results in vitro show that there are no substantial conformational changes in PLCγ2 upon Rac2 binding alone. Although some small, localized changes, not detectable by this FRET system, could occur due to the Rac2 binding, based on the experimental results described in the previous section (Fig. 5B; also see Fig. S3 in the supplemental material), these changes would be insufficient for the activation. Our further experiments in transfected cells where the lifetimes of PLCγ2GFP and PLCγ2ΔSH3(mCherry)GFP were measured (Fig. 6F) showed a clear difference between the two PLCγ2 variants, consistent with the in vitro data (Fig. 6C). At this scale, we could not detect any significant overall changes due to the presence of Rac2G12V, and any further changes for the GFP-mCherry PLCγ2 variant in Rac2G12V cells were only indicated in some areas of the cell.
DISCUSSION
It is generally accepted that studies of signaling events that occur in the proximity of the membrane should take into account the influence of the membrane surface environments (13, 28). For PLC enzymes, including PLCγ, it has been assumed that the main consequence of stable translocation to the membrane, critical for PLC regulation, is to increase the relative concentrations of the PLC and PIP2 substrate. However, these assumptions are now considered simplistic. For example, they did not take into account that PLC complexes at the membrane could in fact decrease PLC mobility compared to a free diffusion in the cytoplasm. As PLCs do not require membrane penetration to access a scissile bond of PIP2, membrane complexes could restrict the pool of accessible substrate. Furthermore, it is becoming increasingly apparent that there is yet another level of complexity; namely, the activation of PLCs requires a release of intramolecular inhibitory constraints (3). Our data suggest that the membrane environment, rather than being a simple source enriched in the PLC substrate, contributes to the activation process. These observations are consistent with a number of studies that illustrate effects of the membrane environment on protein function and signaling (13, 28).
We first demonstrate here that PLCγ2 is an autoinhibited enzyme and that the removal of these constraints results in constitutively active PLC in cells. We found that PLCγ2 activity is increased after deletion of the γSA both in cells and in vitro (Fig. 1B and C and 5A and D). Our in vitro data are consistent with the results of a previous analysis of a PLCγ1 variant lacking this region (12, 20). Importantly, we further show that the most likely mechanism of autoinhibition of PLCγ2 activity is by occlusion of the active site (Fig. 2, 3, and 5D; also see Fig. S1 and S2 in the supplemental material). Based on mutational analysis in PLCγ2 (Fig. 3), the autoinhibitory surface of γSA involves elements from the SH2c domain, spPH domain, and several interdomain linkers (Fig. 3). However, sites of direct interaction between the γSA and the core are likely to be limited. One such site is the region at the C terminus of the SH2c domain that contains the PCI peptide previously shown to inhibit PLC activity (19). The PLCγ2ΔPCI variant had higher basal activity, which further facilitates activation independent of the nature of the stimulus.
Another new finding from our study (Fig. 1B and C) is that the deletion of the γSA generates constitutively and highly active PLC in cells, with basal activity that is higher than the activity of wild-type PLCγ2 stimulated by either EGF or Rac2. The activity of PLCγ2ΔSA (also designated PLCγ2-core) is not increased after cell stimulation (Fig. 1B and C). This contrasts with studies of PLCβ2, where the deletion of a smaller, largely disordered X-Y linker resulted in an increase of basal activity that was activated further by different regulatory molecules (17). Furthermore, while the enhanced basal activity of PLCβ2 could be attributed to interaction between the PLCβ2 variant with compromised autoinhibition and a fraction of regulatory molecules that are active even in nonstimulated cells, this does not apply to PLCγ2ΔSA. This PLCγ2 variant is lacking regions that are required for interaction with both EGF receptor and Rac GTPases. Additional experiments in cells and in vitro further demonstrate that PLCγ2ΔSA has no enhanced capacity to bind lipid membranes (Fig. 4 and 5C). While there is firm evidence in the literature that membrane translocation is a required step for the activation process of the wild-type enzyme, our data show that for the PLCγ2 enzyme in the absence of autoinhibitory constraints, stable accumulation at the cellular membrane is no longer an important step. This suggests that such stable translocation of the PLCγ2 enzyme to the cellular membrane, leading solely to an increase in the relative concentration of the substrate available to PLCγ2, could not explain the activation mechanism.
We further analyzed the activation of PLCγ2 by RacG12V, since this activation could be demonstrated in cells and in a reconstitution system in vitro. As PLCγ2ΔSA is constitutively active without the requirement for stabile translocation to the membrane, we tested the possibility that the key regulatory step of PLCγ2 activation requires only binding of Rac2, resulting in the release of intramolecular inhibitory constraints. We previously determined the strength of Rac binding to the wild-type PLCγ2 (Kd [dissociation constant], ∼4 μM) (12), structurally defined the interaction surface between Rac2 and the spPH domain, and demonstrated that the critical residues required for Rac2 binding (present in the β5-strand and α-helix of the spPH and switch I and switch II regions of Rac2) are also critical for PLCγ2 activation (4). One possible model for PLCγ2 activation by Rac2, therefore, could be that the PLCγ2 binding to Rac2 causes substantial conformational changes that result in unmasking of the active site, resulting in an active complex with properties similar to those of the constitutively active PLCγ2 variant (PLCγ2ΔSA/PLCγ2-core). However, our findings that the activation of PLCγ2 in a reconstitution system requires lipid-modified Rac (Fig. 5B), that Rac2 with or without lipid modification is not able to activate PLCγ2 in a simple mixed-micelle assay (see Fig. S3 in the supplemental material), and that Rac2 binding in vitro does not result in substantial conformational changes of PLCγ2 as assessed using FRET (Fig. 6) do not support this possibility. These data suggest the importance of membrane surfaces for PLCγ2 activation and the possibility that a consequence of Rac2 binding and translocation to the membrane could be that membrane proximity, on its own or together with Rac2, has a role in the release of autoinhibition and subsequent activation (Fig. 7).
FIG. 7.

Model for PLCγ2 regulation by Rac. PLCγ2 is represented by the core domains (EF-hands [EF], catalytic TIM barrel [TIM], and C2 domain [C2]) and the γSA. Under basal conditions (inactive), autoinhibitory constraints in PLCγ2 could involve elements from different regions of γSA (represented as a black lid) that have an effect on the PLCγ2 active site. Upon Rac binding and translocation to the membrane proximity, changes in γSA result in the removal of the inhibitory region and exposure of the active site.
The above-described findings on the regulation of PLCγ2 are consistent with a model proposed for the activation of PLCβ2, also an effector of Rac GTPases (16). Comparison of the crystal structure of PLCβ2 on its own and in a complex with Rac1 has demonstrated that Rac binding surfaces do not include an autoinhibitory region and that there is no conformational change in PLCβ2 following Rac1 binding (17, 21). Regions implicated in autoinhibition within X-Y linkers of several PLC enzymes are characterized by clusters of charged amino acid residues, and according to the model, these regions are shifted by membrane proximity (due to steric and electrostatic factors) to expose the active site (16, 17). More generally, the findings supporting the possibility that PLC activation is restricted to sites of membrane interaction are consistent with the requirements for their stringent regulation. Recent studies of PLCβ2, for example, suggest that, depending on the type of activating protein, PLCβ2 can be transiently recruited either to confined regions or to undergo fast, surfing-like diffusion along a larger area of the plasma membrane, thus tightly controlling PIP2 hydrolysis in time and space (15).
Previous studies of molecular mechanisms of the activation of PLCγ family members mainly focused on the PLCγ1 isozyme following stimulation of tyrosine kinase-linked receptors. A model for activation by platelet-derived growth factor, mediated by phosphorylation of tyrosine-783, proposed that an intramolecular interaction between the SH2c domain and tyrosine-783 results in conformational changes and subsequent activation (31); further in vitro characterization of PLCγ1 is consistent with this model (12). Although a contribution of the membrane was not analyzed directly, the demonstration of higher PLC activity of phosphorylated PLCγ1 was dependent on substrate presentation and could not be observed in a standard PI-deoxycholate micelle assay (31). While having some elements in common, it is possible that interaction with tyrosine kinase receptors and subsequent phosphorylation of PLCγ1 overcomes the autoinhibition through different changes in the γSA than the interaction between Rac GTPases and PLCγ2 in the proximity of the membrane. In the case of another, more extensively studied signaling component that is regulated by tyrosine kinase receptors and small GTPases, PI3-kinase α, analysis of gain-of-function mutations and structural studies suggest that these two input signals regulate activation through changes in different regions of this enzyme (42, 43). Nevertheless, despite the diversity of regulatory molecules converging on PLCγ and PLC enzymes in general, the findings presented here support some common themes for autoinhibition and, in the case of PLCγ2 regulation by Rac GTPases, for similar activation mechanisms in different PLC families.
Supplementary Material
Acknowledgments
We are grateful to Youngdae Yoon and Wohnwa Cho for SPR measurements and to Banafshe Larijani for helpful comments.
K.L.E. was supported by MRC and Cancer Research UK. Work in M.K.'s laboratory was funded by Cancer Research UK and BBSRC, and support to P.G.'s laboratory was by the DFG (grant SFB 497). The work from P.M.W.F.'s laboratory is supported by BBSRC.
Footnotes
Published ahead of print on 18 January 2011.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Albertazzi, L., D. Arosio, L. Marchetti, F. Ricci, and F. Beltram. 2009. Quantitative FRET analysis with the EGFP-mCherry fluorescent protein pair. Photochem. Photobiol. 85:287-297. [DOI] [PubMed] [Google Scholar]
- 2.Borst, J. W., et al. 2008. Structural changes of yellow Cameleon domains observed by quantitative FRET analysis and polarized fluorescence correlation spectroscopy. Biophys. J. 95:5399-5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bunney, T. D., and M. Katan. 24 September 2010. PLC regulation: emerging pictures for molecular mechanisms. Trends Biochem. Sci. [Epub ahead of print.] doi: 10.1016/j.tibs.2010.08.003. [DOI] [PubMed]
- 4.Bunney, T. D., et al. 2009. Structural insights into formation of an active signaling complex between Rac and phospholipase C gamma 2. Mol. Cell 34:223-233. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter, G., and Q. Ji. 1999. Phospholipase C-gamma as a signal-transducing element. Exp. Cell Res. 253:15-24. [DOI] [PubMed] [Google Scholar]
- 6.Cole, C., J. D. Barber, and G. J. Barton. 2008. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36:W197-W201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cremasco, V., et al. 2010. Phospholipase C gamma 2 is critical for development of a murine model of inflammatory arthritis by affecting actin dynamics in dendritic cells. PLoS One 5:e8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeBell, K., et al. 2007. Intramolecular regulation of phospholipase C-gamma1 by its C-terminal Src homology 2 domain. Mol. Cell. Biol. 27:854-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellis, M. V., et al. 1998. Catalytic domain of phosphoinositide-specific phospholipase C (PLC). Mutational analysis of residues within the active site and hydrophobic ridge of plcdelta1. J. Biol. Chem. 273:11650-11659. [DOI] [PubMed] [Google Scholar]
- 10.Essen, L. O., O. Perisic, R. Cheung, M. Katan, and R. L. Williams. 1996. Crystal structure of a mammalian phosphoinositide-specific phospholipase C delta. Nature 380:595-602. [DOI] [PubMed] [Google Scholar]
- 11.Everett, K. L., et al. 2009. Characterization of phospholipase C gamma enzymes with gain-of-function mutations. J. Biol. Chem. 284:23083-23093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gresset, A., S. N. Hicks, T. K. Harden, and J. Sondek. 2010. Mechanism of phosphorylation-induced activation of phospholipase C-{gamma} isozymes. J. Biol. Chem. 285:35836-35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groves, J. T., and J. Kuriyan. 2010. Molecular mechanisms in signal transduction at the membrane. Nat. Struct. Mol. Biol. 17:659-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guidetti, G. F., et al. 2009. Integrin alpha2beta1 induces phosphorylation-dependent and phosphorylation-independent activation of phospholipase Cgamma2 in platelets: role of Src kinase and Rac GTPase. J. Thromb. Haemost. 7:1200-1206. [DOI] [PubMed] [Google Scholar]
- 15.Gutman, O., C. Walliser, T. Piechulek, P. Gierschik, and Y. I. Henis. 2010. Differential regulation of phospholipase C-beta2 activity and membrane interaction by Galphaq, Gbeta1gamma2, and Rac2. J. Biol. Chem. 285:3905-3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harden, T. K., S. N. Hicks, and J. Sondek. 2009. Phospholipase C isozymes as effectors of Ras superfamily GTPases. J. Lipid Res. 50(Suppl.):S243-S248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hicks, S. N., et al. 2008. General and versatile autoinhibition of PLC isozymes. Mol. Cell 31:383-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homma, M. K., M. Yamasaki, S. Ohmi, and Y. Homma. 1997. Inhibition of phosphoinositide hydrolysis and cell growth of Swiss 3T3 cells by myristoylated phospholipase C inhibitor peptides. J. Biochem. 122:738-742. [DOI] [PubMed] [Google Scholar]
- 19.Homma, Y., and T. Takenawa. 1992. Inhibitory effect of src homology (SH) 2/SH3 fragments of phospholipase C-gamma on the catalytic activity of phospholipase C isoforms. Identification of a novel phospholipase C inhibitor region. J. Biol. Chem. 267:21844-21849. [PubMed] [Google Scholar]
- 20.Horstman, D. A., K. DeStefano, and G. Carpenter. 1996. Enhanced phospholipase C-gamma1 activity produced by association of independently expressed X and Y domain polypeptides. Proc. Natl. Acad. Sci. U. S. A. 93:7518-7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jezyk, M. R., et al. 2006. Crystal structure of Rac1 bound to its effector phospholipase C-beta2. Nat. Struct. Mol. Biol. 13:1135-1140. [DOI] [PubMed] [Google Scholar]
- 22.Katan, M. 2005. New insights into the families of PLC enzymes: looking back and going forward. Biochem. J. 391:e7-e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katan, M., R. Rodriguez, M. Matsuda, Y. M. Newbatt, and G. W. Aherne. 2003. Structural and mechanistic aspects of phospholipase Cgamma regulation. Adv. Enzyme Regul. 43:77-85. [DOI] [PubMed] [Google Scholar]
- 24.Lakowicz, J. R. 2006. Principles of fluorescence spectroscopy, 3rd ed. Springer, New York, NY.
- 25.Manning, H. B., et al. 2008. A compact, multidimensional spectrofluorometer exploiting supercontinuum generation. J. Biophotonics 1:494-505. [DOI] [PubMed] [Google Scholar]
- 26.Matsuda, M., et al. 2001. Real time fluorescence imaging of PLC gamma translocation and its interaction with the epidermal growth factor receptor. J. Cell Biol. 153:599-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merzlyak, E. M., et al. 2007. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods 4:555-557. [DOI] [PubMed] [Google Scholar]
- 28.Phillips, R., T. Ursell, P. Wiggins, and P. Sens. 2009. Emerging roles for lipids in shaping membrane-protein function. Nature 459:379-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piechulek, T., et al. 2005. Isozyme-specific stimulation of phospholipase C-gamma2 by Rac GTPases. J. Biol. Chem. 280:38923-38931. [DOI] [PubMed] [Google Scholar]
- 30.Pleines, I., et al. 2009. Rac1 is essential for phospholipase C-gamma2 activation in platelets. Pflugers Arch. 457:1173-1185. [DOI] [PubMed] [Google Scholar]
- 31.Poulin, B., F. Sekiya, and S. G. Rhee. 2005. Intramolecular interaction between phosphorylated tyrosine-783 and the C-terminal Src homology 2 domain activates phospholipase C-gamma1. Proc. Natl. Acad. Sci. U. S. A. 102:4276-4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez, R., et al. 2001. Tyrosine residues in phospholipase Cgamma 2 essential for the enzyme function in B-cell signaling. J. Biol. Chem. 276:47982-47992. [DOI] [PubMed] [Google Scholar]
- 33.Shcherbo, D., et al. 2009. Practical and reliable FRET/FLIM pair of fluorescent proteins. BMC Biotechnol. 9:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sorli, S. C., T. D. Bunney, P. H. Sugden, H. F. Paterson, and M. Katan. 2005. Signaling properties and expression in normal and tumor tissues of two phospholipase C epsilon splice variants. Oncogene 24:90-100. [DOI] [PubMed] [Google Scholar]
- 35.Stahelin, R. V., and W. Cho. 2001. Differential roles of ionic, aliphatic, and aromatic residues in membrane-protein interactions: a surface plasmon resonance study on phospholipases A2. Biochemistry 40:4672-4678. [DOI] [PubMed] [Google Scholar]
- 36.Suh, P. G., et al. 2008. Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep. 41:415-434. [DOI] [PubMed] [Google Scholar]
- 37.Sultzman, L., C. Ellis, L. L. Lin, T. Pawson, and J. Knopf. 1991. Platelet-derived growth factor increases the in vivo activity of phospholipase C-gamma 1 and phospholipase C-gamma 2. Mol. Cell. Biol. 11:2018-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Visser, A. J., et al. 2010. Time-resolved FRET fluorescence spectroscopy of visible fluorescent protein pairs. Eur. Biophys. J. 39:241-253. [DOI] [PubMed] [Google Scholar]
- 39.Volkmer, A., V. Subramaniam, D. J. Birch, and T. M. Jovin. 2000. One- and two-photon excited fluorescence lifetimes and anisotropy decays of green fluorescent proteins. Biophys. J. 78:1589-1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walliser, C., et al. 2008. rac regulates its effector phospholipase Cgamma2 through interaction with a split pleckstrin homology domain. J. Biol. Chem. 283:30351-30362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu, B., Y. Chen, and J. D. Muller. 2009. Fluorescence fluctuation spectroscopy of mCherry in living cells. Biophys. J. 96:2391-2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao, L., and P. K. Vogt. 2008. Class I PI3K in oncogenic cellular transformation. Oncogene 27:5486-5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao, L., and P. K. Vogt. 2008. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. U. S. A. 105:2652-2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.