

Abstract
Hox genes encode transcription factors that regulate morphogenesis in all animals with bilateral symmetry. Although Hox genes have been extensively studied, their molecular function is not clear in vertebrates, and only a limited number of genes regulated by Hox transcription factors have been identified. Hoxa2 is required for correct development of the second branchial arch, its major domain of expression. We now show that Meox1 is genetically downstream from Hoxa2 and is a direct target. Meox1 expression is downregulated in the second arch of Hoxa2 mouse mutant embryos. In chromatin immunoprecipitation (ChIP), Hoxa2 binds to the Meox1 proximal promoter. Two highly conserved binding sites contained in this sequence are required for Hoxa2-dependent activation of the Meox1 promoter. Remarkably, in the absence of Meox1 and its close homolog Meox2, the second branchial arch develops abnormally and two of the three skeletal elements patterned by Hoxa2 are malformed. Finally, we show that Meox1 can specifically bind the DNA sequences recognized by Hoxa2 on its functional target genes. These results provide new insight into the Hoxa2 regulatory network that controls branchial arch identity.
In vertebrates, development of the face and neck starts with formation of the frontonasal mass and the branchial arches. The branchial arches are transient, repetitive structures in which cells of the cranial neural crest (CNC) and mesoderm are encapsulated by epithelia. Upon differentiation, each of the branchial arches contributes to head- and neck-specific skeletal elements, their associated muscles, blood supply, and nerves. Morphogenesis of the branchial arches depends on a number of transcription factors and signaling molecules. Transcription factors of the Dlx and Hox families have a most prominent role, with the first regulating proximodistal patterning within each branchial arch (8) and the latter controlling branchial arch identity (3, 11, 17, 25).
The second branchial arch (IIBA) contributes to the outer and middle ear and to part of the neck. Development of the IIBA is controlled by the transcription factor Hoxa2. In Hoxa2 mutant embryos, IIBA skeletal derivatives are replaced by typical first branchial arch (IBA) skeletal elements in mirror image configuration (3, 11, 25). Hoxa2 belongs to the large family of Hox transcription factors, whose members specify the body axis of a bilaterian organism and whether a segment of the embryo will form the head, thorax, or abdomen (7). The identification of Hox downstream effectors in vertebrates is complicated by the high redundancy of Hox genes, represented by multiple paralogs in vertebrate genomes. Although Hox genes have been the subject of extensive genetic analysis, few target genes have been identified (22, 29). The IIBA is one of the few embryonic districts in which inactivation of a single Hox gene has an unambiguous effect in mouse and an effective model system to define Hox molecular function in vertebrate embryogenesis.
How does Hoxa2 specify IIBA identity? Hoxa2 control of IIBA development appears to involve the repression of few transcription factors. At embryonic day 10.0 (E10.0), before overt differentiation has begun in the IIBA, Hoxa2 negatively regulates transcription of Ptx1 and Lhx6; their characterization has suggested that Hoxa2 patterns the IIBA by changing the competence of the CNC to respond to skeletogenic signals (5). At the same developmental stage, Hoxa2 directly represses Six2 expression, and this partially mediates Hoxa2 control over the insulin-like growth factor (IGF) system (13, 14). Genetic experiments to dissect the role of the genes downstream of Hoxa2 have shown that correcting the expression levels of Six2 and Ptx1 ameliorates the Hoxa2 mutant phenotype (5, 13, 14), indicating that repression of Six2 and Ptx1 is indeed required for IIBA-specific morphogenesis. However, the limited extent of the rescue by inactivation of individual targets and its incomplete penetrance suggest the existence of strong redundancies among these genes in the morphogenesis of the IIBA.
A number of observations indicate that Hoxa2 controls IIBA identity by blocking a first arch default fate. Ptx1, Lhx6, and Six2, and the few additional genes identified as regulated by Hoxa2 (6, 28), are normally expressed in the IBA, but not in the IIBA, suggesting that Hoxa2 functions as a transcriptional repressor to prevent the expression of IBA-specific genes in the IIBA. Moreover, the IIBA gives rise to duplicates of IBA skeletal elements in the absence of Hoxa2. Is IIBA identity achieved simply by blocking a default IBA state or is there more to it? The finding that Hoxa2 is functionally relevant at E9.5 (28), which is earlier than the stages at which the expression levels of most identified Hoxa2 targets become apparent in Hoxa2 mutant IIBAs, indicates that the gene regulatory network (GRN) controlled by Hoxa2 in the IIBA must include additional target genes expressed at earlier developmental stages. Their identification is essential to clarify the structure and organization of the GRN that governs IIBA identity downstream of Hoxa2.
Here we show that the earliest event controlled by Hoxa2 in the IIBA is the activation of the gene encoding the transcription factor Meox1. The Meox1 homeobox gene is strongly expressed in the somites during embryogenesis and controls formation and differentiation of the somites and their derivatives (16). We find that, in the developing IIBA, Hoxa2 is associated with a highly conserved region of Meox1 chromatin. We further show that the interaction of Hoxa2 with the Meox1 promoter is sequence specific and required for Meox1 activation. Our results show that Meox1 is genetically downstream of Hoxa2 and is a direct target. In addition, we find that Meox1 can specifically bind the DNA sequences recognized by Hoxa2 on its functional target genes, suggesting that these transcription factors may share the control of downstream targets in the IIBA. Finally, the analysis of Meox1 Meox2 combined mutants reveals that IIBA derivatives develop abnormally in the absence of Meox genes. These findings link two coexpressed transcription factors in a novel molecular pathway in mouse embryonic development. In addition, they uncover a previously unidentified role for Meox genes in the morphogenesis of the branchial arches.
MATERIALS AND METHODS
Mutant and transgenic animals and embryos.
Hoxa2 Meox1 Meox2 null mice were described previously (11, 16, 16a). Meox2+/tm1(lacZ)Mnko contains a internal ribosome entry site-nuclear localization signal-lacZ knocked into Meox2's first exon. Animal experiments were carried out under Animals (Scientific Procedures) Act 1986.
Molecular and phenotypic analyses.
Whole-mount and section in situ hybridization (ISH) was performed as described previously (12a) using Hoxa2 (15), Crabp1 (a gift from Rudolf Grosschedl), and Meox1 probes, amplified from IIBA cDNA using primers Meox1F (5′-CACAGGAGCAGGACCGAGAGG-3′) and Meox1R (5′-CCGGAACACGCAGGATAGGTCC-3′). Skeletal phenotypes were analyzed by Alcian blue-Alizarin red staining (15a).
RT-PCR.
Reverse transcription (RT)-PCR was performed as described previously (13). RNA was extracted from 106 cells dissociated from the IBA and IIBA of E10.5 embryos using 1% trypsin-0.1% EDTA, filtered through a cell strainer (BD), and cultured in Dulbecco modified Eagle medium (DMEM)-10% fetal calf serum (FCS) for 3 days. cDNA was subjected to 24 cycles of amplification using the following primers: Lhx6F (5′-GGAGATCTACTGCAAGATGGACTAC-3), Lhx6R (5′-CCGTCATGTCCGCTAGCTTCTG-3), Pitx1F (5′-AATCGTCCGACGCTGATCTGCC-3), Pitx1R (5′-CCTTGCACAGGTCCAACTGCTG-3′), and Six2 and Hoxa2 (14).
ChIP and EMSA.
Chromatin immunoprecipitation (ChIP) was performed according to Kutejova et al. (14). Meox1 mouse cDNA was amplified from E10.0 IIBA cDNA and cloned into pcDNA3 (Invitrogen). pcDNA3-Meox1-HA contains a hemagglutinin (HA) tag before the stop codon; the pcDNA3-Hoxa2 construct has been described previously (13). An electrophoretic mobility shift assay (EMSA) was performed as described previously (13). The Meox1 promoter and its mutant versions were labeled using [γ-32P]dATP. For the supershift experiment, 40 ng of anti-HA antibodies (rat monoclonal antibody 3F10; Roche) was added to the reaction.
Cell transfection.
IBA mandibular components were isolated from 10 mouse embryos collected at E10.5. Cells were dissociated and resuspended in DMEM-10% FCS. A total of 500 ng DNA (250 ng Meox1-lacZ or mutMeox1-lacZ construct and 250 ng pCDNA3-Hoxa2 or pCDNA3) and 3 μl FuGENE (Roche) were added to identical aliquots of the resuspended cells. Each aliquot was plated onto a well of a 24-well plate. After 24 h of incubation at 37°C, cells were fixed and stained for β-galactosidase activity.
RESULTS
Hoxa2 regulates Meox1 expression in the IIBA.
To identify early targets of Hoxa2, we compared the expression profiles of wild-type and Hoxa2 mutant IIBAs in mouse at E9.5, when migration of the Hoxa2-positive CNC into the IIBA has just been completed (15, 24). The gene encoding the transcription factor Meox1 was among the few genes differentially expressed, and its signal was decreased 2-fold in the absence of Hoxa2.
Meox1 signal was not detected in the Hoxa2-positive CNC prior to or during migration to the IIBA (not shown). The Meox1 signal was first evident in the branchial area at E9.0 and predominantly in the IIBA (Fig. 1 A). At E9.5, strong expression of Meox1 was detected in the proximal area of the IIBA, and expression was excluded from the distal domain of the arch; strong expression was also found in the third arch (IIIBA) (Fig. 1B). Around E10.0, only a small area of the IIBA and the IIIBA remained positive for Meox1 (Fig. 1C), and after E10.5, no Meox1 signal could be detected in the IIBA (not shown). At all the stages examined, Meox1 expression was excluded from the IBA.
FIG. 1.

Meox1 expression in the branchial arches. Whole-mount and section ISH on wild-type (wt) (A, B, C, D, F, G, H, and I) and Hoxa2 mutant (E) embryos using Meox1 (A, B, C, D, E, and H), Hoxa2 (F and G), and Crabp1 (I) probes. (A) Meox1 expression is first detected at E9.0 in the IIBA. At E9.5, Meox1-positive cells occupy most of the proximal area of the IIBA (B); they are still detected at E10.0 (C) and are no longer visible after E10.5 (not shown). (D) Meox1 is expressed at E9.5 in both IIBA and IIIBA. (E) Meox1 expression is specifically lost in the IIBA of the Hoxa2 mutant. (F) Hoxa2-positive cells are detected in the entire IIBA and in rhombomere 4 (asterisk). The dotted line delineates the proximal Meox1-positive area of the IIBA. (G, H, and I) Adjacent parasagittal sections of the E9.5 embryo. Hoxa2 (G) and Crabp1 (I) expression patterns demarcate cranial neural crest cells. (H) Meox1 is transcribed in a subpopulation of the cranial neural crest, anterior to the second arch artery (asterisk). In panels A to E, arrowheads and arrows point at IIBA and IIIBA, respectively; I, first branchial arch.
To confirm that Hoxa2 regulates Meox1 expression, we performed ISH on E9.5 Hoxa2 mutant embryos. Meox1 expression was specifically lost in the IIBA of Hoxa2 mutant embryos (Fig. 1E).
At E9.5, when migration of Hoxa2-positive cells has been completed, Hoxa2 was expressed in most of the IIBA (Fig. 1F), while Meox1 was transcribed only in the proximal area of the IIBA (Fig. 1D). Meox1 is expressed in a subpopulation of the Hoxa2-positive and Crabp1-positive cranial neural crest of the IIBA (Fig. 1G to I).
These results indicate that Hoxa2 positively controls Meox1 expression in the IIBA. They also show that the temporal and spatial expression patterns of Meox1 are more restricted than those of Hoxa2. Therefore, Hoxa2 appears to be necessary but not sufficient to activate Meox1 expression.
Hoxa2 binds a conserved sequence within the Meox1 proximal promoter.
Hoxa2 contains a DNA binding domain, which interacts in a sequence-specific fashion with the chromatin of target genes to control their transcription. The association of Hoxa2 with Meox1 chromatin in vivo is therefore an essential requisite for Meox1 direct regulation by Hoxa2. Loss of Hoxa2 function generates identical IIBA phenotypes in mouse, frog, and fish (2, 3, 11, 12, 25), suggesting that Hoxa2-responsive elements should be conserved across vertebrates. The CORG database (9) identified the Meox1 proximal promoter (corresponding to positions −236 to −33 in mouse) as perfectly conserved from human to fish (Fig. 2 A). IIBA-extracted Hoxa2-immunoprecipitated chromatin showed a substantial enrichment for the most proximal Meox1 promoter region, while no enrichment was detected for an unrelated control promoter (Fig. 2B). These results demonstrate that at E10.0, when Hoxa2 is strongly expressed (Fig. 2C) and still required for activation of Meox1 transcription in the IIBA, Hoxa2 is bound to Meox1 chromatin in vivo.
FIG. 2.

Hoxa2 binds the Meox1 proximal promoter. (A) Clustal alignment of Meox1 proximal promoter sequences from different vertebrate species. Numbers indicate nucleotide positions relative to position +1 (transcriptional start site) in mouse. Red shading highlights Hoxa2 binding sites. The chromatin sequence amplified in ChIP is enclosed in a red rectangle. (B) The conserved Meox1 proximal promoter (red rectangle in panel A) is enriched in ChIP assays performed on E10.0 IIBAs in the presence of Hoxa2 antibody (α-a2, Hoxa2 antibody; IgG, nonspecific antibody; EB, elution buffer). The input was diluted 1:300 prior to amplification. Specific enrichment is also detected for Hoxa2 direct target Six2, while no enrichment is observed for Intein, an unrelated control promoter. ChIP was performed on three independent pools of samples. PCRs were performed in duplicate on each pool. The results shown are from a representative set. (C) ISH of E10.5 embryo shows strong Hoxa2 expression in the IIBA (surrounded by red dots). (D) A labeled Meox1 proximal promoter (red rectangle in panel A), incubated in the presence of Hoxa2-programmed reticulocytes, gives rise to a retarded complex (arrowhead) supershifted by the addition of anti-Hoxa2 antibody (Ab) (arrow). Nucleotide substitutions in single mutated (m) Hoxa2 binding sites (changes are shown in panel A) do not abolish complex formation, while no complex formation is observed when the probe contains nucleotide substitutions in both binding sites. (E) The formation of the complexes (arrowhead) is competed by the addition of cold double-stranded oligonucleotides containing wild-type Hoxa2 binding sites but not of oligonucleotides with the mutated sites. Cold oligonucleotides were added at 200-fold (lanes 3, 4, 8, and 9) and 400-fold (lanes 5, 6, 10, and 11) molar excesses.
Binding of Hoxa2 to its in vivo target Six2 is mediated by two conserved TAAT consensus sites (13). The Meox1 promoter region enriched in ChIP assays contains an ATTA motif, embedded in a perfectly conserved stretch of flanking nucleotides, and a second one located on the opposite strand (Fig. 2A). Incubation of the conserved Meox1 proximal promoter with in vitro-translated Hoxa2 in the electrophoretic mobility shift assay (EMSA) resulted in the formation of a retarded complex, which was supershifted by the addition of an anti-Hoxa2 antibody (Fig. 2D). In contrast, incubation of the probe in the presence of unprogrammed reticulocytes did not result in any retarded complex, nor did addition of the antibody have any effect (Fig. 2D). Mutating both TAAT consensus sites completely abolished binding of Hoxa2 to the probe (Fig. 2D), while nucleotide substitutions introduced in each of the single TAAT sites partially reduced Hoxa2 binding (Fig. 2D). Finally, Hoxa2 binding to the probe was competed by increasing the molar concentrations of wild-type oligonucleotides reproducing the TAAT motif and flanking nucleotides but not mutant oligonucleotides (Fig. 2E). The oligonucleotide reproducing binding site 1 (BS1) was more efficient in competing Hoxa2 binding to the probe, indicating a higher affinity of Hoxa2 for binding site 1 (Fig. 2E).
We used IBA-derived cells to investigate if binding of Hoxa2 to the Meox1 promoter may control gene transcription. Experimental evidence indicates that IBA and IIBA cells share similar ground states, which are modified in the IIBA by the presence of Hoxa2 (3, 11, 21, 25); IBA cells therefore appear to be capable of providing an environment equivalent to that of IIBA cells (without Hoxa2). When isolated from E10.0 branchial arches and grown in monolayers, these cells maintain the molecular identity of the area of origin, as estimated by the expression of Hoxa2 and a few of its target genes in IBA- and IIBA-derived cells after 3 days in culture (Fig. 3 A and B).
FIG. 3.
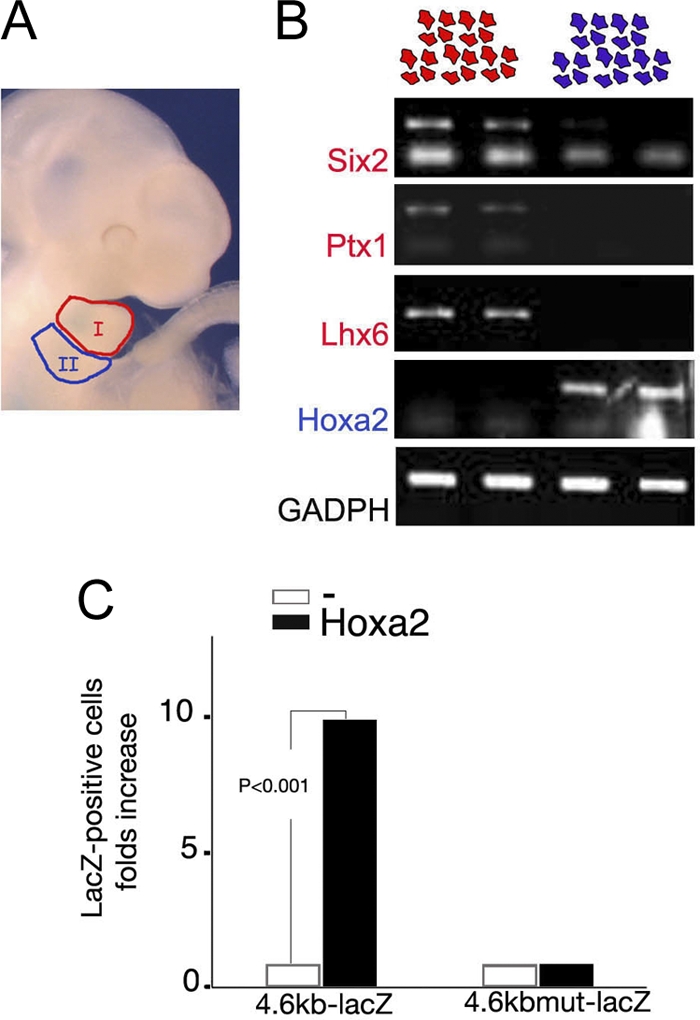
Hoxa2 activates the Meox1 promoter. (A) Craniofacial area of E10.5 mouse embryo showing first (I) and second (II) arches (red and blue, respectively). (B) Semiquantitative RT-PCR on RNA extracted from duplicates of IBA- and IIBA-derived mesenchymal cells cultured for 3 days (red and blue cells on top, respectively). The expression of IBA-specific genes (Six2, Ptx1, and Lhx6) is maintained in IBA-derived cultures. IIBA-derived cultures do not express these genes. Hoxa2 expression is still detected in IIBA-derived cultures and is absent from IBA-derived cell cultures. (C) The number of β-galactosidase-stained IBA cells is significantly increased when 4.6-kb Meox1-lacZ is cotransfected with Hoxa2 (P < 0.001 for three independent experiments). Hoxa2 fails to activate LacZ expression driven by the same promoter containing point mutation in BS1 and BS2 (4.6-kb mutMeox1; mutations are as shown in Fig. 2A). White bars show the basal activity of lacZ constructs; black bars show lacZ construct activity in the presence of Hoxa2. The results shown are the averages of three independent experiments, each performed in duplicate.
Cells dissociated from E10.0 IBAs were transfected with a 4.6-kb Meox1-lacZ construct, containing the Meox1 promoter (positions −4642 to +22) fused to a lacZ reporter gene. Meox1 promoter activity, assessed by counting the number of cells that turned blue after transfection, was very low in these cells (fewer than 20 blue cells were counted in each individual experiment). A significant increase in the number of cells expressing lacZ (>200; i.e., 10-fold increase) was observed when Hoxa2 was cotransfected together with 4.6-kb Meox1-lacZ (Fig. 3C). Introducing the nucleotide substitutions shown to abolish Hoxa2 binding (4.6-kb mutMeox1-lacZ) (Fig. 2D) did not affect the basal activity of the promoter. However, unlike its wild-type version, addition of Hoxa2 failed to activate the reporter gene (Fig. 3C). The lack of effect described above shows that the expression of Hoxa2 in these cells did not affect cell number; this observation was also confirmed by using an unrelated promoter (not shown).
In conclusion, these results show that Hoxa2 interacts with the proximal region of the Meox1 promoter in vivo and that this interaction, which is sequence specific and mediated by two TAAT sites, is required for Meox1 activation.
Meox homeodomain transcription factors control formation of IIBA-specific cartilages.
We analyzed Meox1 mutant mice to understand whether Meox1 activation contributes to IIBA morphogenesis. Skeletal analysis of Meox1 mutant mice showed no evident phenotype in CNC derivatives of the IIBA, but when the Meox1 mutation was combined with mutation in the homolog Meox2 (also not showing an IIBA phenotype), two of the three elements that are controlled by Hoxa2 were abnormal. As sporadically observed in Hoxa2 heterozygous mice (20), the styloid process was split in two fragments (Fig. 4 B). Its distal part failed to extend distally and formed close to IBA cartilages, resembling an intermediate situation toward the Hoxa2 mutant phenotype, in which the IIBA cartilages are formed much closer to their IBA mirror-image counterparts with respect to the wild type (Fig. 4A to C). The lesser horn of the hyoid bone also developed abnormally (not shown). In addition, similar to the Hoxa2 mutants, whose basioccipital bone has an abnormal shape, Meox1 single mutants showed defects in this structure. These abnormalities were enhanced in Meox1−/− Meox2−/− mutants, resulting in a hypoplastic basioccipital bone (Fig. 4E).
FIG. 4.

Middle ear skeletal phenotype of Meox1−/− Meox2−/− mouse mutants. Skeletal phenotype of wild-type (A and D), Meox1−/− Meox2−/− (B and E), and Hoxa2−/− (C and F) E18.5 fetuses. (A to C) Dissected otic capsules. In the absence of Hoxa2 stapes (s), the styloid process (st) (highlighted in green in panel A) is replaced by mirror-image copies of IBA skeletal elements (highlighted in green in panel C); wild-type IBA cartilages, incus (i) and malleus (M), are highlighted in orange in panels A to C. In Meox1−/− Meox2−/− mutants, the styloid process is truncated and is formed much closer to IBA skeletal derivatives than in the wild type (compare double arrows in panels A and B; dissection of six otic capsules revealed identical phenotypes). (D to F) Ventral view of the posterior cranial base, showing an hypomorphic basioccipital bone (bo) in Meox1−/− Meox2−/− and Hoxa2−/− mutants compared to that in the wild type.
The data above indicate that Meox1 functions redundantly with its homolog Meox2 during the development of the IIBA, consistent with the experimental evidence that Meox1 and Meox2 act redundantly to control somite development (16). Meox2 expression in the IIBA is not equivalent to that of Meox1 and appears later in the development of the IIBA; although we could detect Meox2 transcripts in the developing somites by ISH, we were unable to detect Meox2 expression in the branchial arches at stages earlier than E12.5. However, LacZ staining of Meox2+/tm1(lacZ)Mnko embryos indicated that the Meox2 allele is expressed in the posterior-distal domain of the IIBA already at E10.75 (Fig. 5).
FIG. 5.

Meox2 expression in the branchial arches. Whole-mount LacZ staining of E10.75 Meox2+/tm1(lacZ)Mnko embryos reveals lacZ-positive cells in the posterior region of the IIBA (arrow). I, first branchial arch.
Meox1 binds Hoxa2 target sequences.
The identification of the genes regulated by Meox1 in the IIBA is complicated by the apparent redundant function of Meox1 and Meox2. Meox1 regulates Bapx1 expression in developing somites (26), and Bapx1 is under Hoxa2 regulation in the IIBA (28), suggesting that Meox1 may mediate Hoxa2 regulation of Bapx1 in the IIBA.
The Meox1 binding site on its functional target Bapx1 (26) is very similar to the binding sites recognized by Hoxa2 on its direct targets, Six2 (13) and Meox1 itself. High-resolution analysis of sequence preferences attributes identical DNA binding specificities to Meox1 and Hoxa2 (4). We ran a BLAST search (1) using the Meox1 homeodomain and identified the Hoxa2 homeodomain as a hit, with 68% residue identity and up to 75% positive residues. These observations predict that Meox1 and Hoxa2 may bind the same sequences in vitro. Hoxa2 directly regulates transcription of the Six2 gene by binding two closely spaced sites on the Six2 proximal promoter (13). In band shift assays, the Six2 proximal promoter interacted similarly with Meox1- and Hoxa2-programmed reticulocytes (Fig. 6 A). Double-stranded oligonucleotides, reproducing the Hoxa2 binding sites identified in the Six2 gene, specifically competed Meox1 and Hoxa2 binding to the Six2 promoter, and both Hoxa2 and Meox1 showed a higher affinity for binding site 1 (BS1). Conversely, double-stranded oligonucleotides containing mutations in the TAAT core did not disturb complex formation (Fig. 6A). Meox1, like Hoxa2, binds the Meox1 promoter; incubation of a probe containing the Meox1 proximal promoter gave rise to a strong complex in the presence of Meox1-HA-programmed reticulocytes. The specificity of the complex was confirmed by the addition of an anti-HA antibody (Fig. 6B).
FIG. 6.

Meox1 and Hoxa2 display very similar binding activities in vitro. (A) A labeled mouse Six2 promoter (nucleotide positions −181 to −48), incubated in the presence of Meox1- or Hoxa2-programmed reticulocytes, gives rise to a retarded complex (arrowhead and arrow, respectively). Both complexes are similarly competed by the addition of cold double-stranded oligonucleotides containing the wild-type Hoxa2 binding sites identified on the Six2 promoter but not of oligonucleotides containing mutated Hoxa2 binding sites. Cold oligonucleotides (sequences are shown) were added at 200- and 400-fold molar excesses. (B) A labeled Meox1 promoter (nucleotide positions −235 to −102), incubated in the presence of Meox1-HA- or Hoxa2-programmed reticulocytes, gives rise to retarded complexes (arrows) supershifted by the addition of specific antibodies (arrowheads).
The finding that Meox1 and Hoxa2 bind the same sequences in vitro suggests that these transcription factors could, at least in part, share the control of downstream targets in the IIBA.
DISCUSSION
Activation of Meox1 by Hoxa2.
Hoxa2 controls IIBA identity. Diverse experimental evidence indicates that Hoxa2 controls the production of IIBA structures by blocking a first arch default fate. First, morphogenesis in the IIBA follows patterns typical of the proximal region of the IBA in the absence of Hoxa2 (3, 11, 25). Second, Hoxa2 appears to act as a repressor and mainly negatively regulates the expression of IBA-specific developmental regulators in the IIBA (5, 6, 13, 28).
Here we identified a new downstream target of Hoxa2, the transcription factor Meox1. The addition of Meox1 to the Hoxa2 GRN uncovers a novel aspect of Hoxa2 activity in the IIBA, which does not directly function to prevent the execution of the molecular program that imposes a first arch fate. Unlike all the previously identified Hoxa2 target genes, Meox1 expression is never detected in the IBA. Activation of Meox1 by Hoxa2 is mediated by the direct interaction of Hoxa2 with a phylogenetically conserved region in the Meox1 promoter. Meox1 expression is detected as early as E9.0, when Hoxa2-positive cells settle in the IIBA. Meox1 is transiently expressed, and by the time the other known Hoxa2 targets appear in the Hoxa2 mutant IIBA (i.e., around E10.5), its expression has ceased.
Few similarities can be found between the regulation of Meox1 and that of another bona fide Hoxa2-regulated promoter in the IIBA, Six2 (13, 14). Hoxa2 binding sites are closely spaced and located in the proximity of the transcription start site in both Six2 and Meox1 promoters. Also, in both cases the activity of Hoxa2 appears to require additional factors, namely, Pax and Eya, to regulate Six2 (31). The spatial and temporal expression patterns of Meox1 in the IIBA are more restricted than the expression of Hoxa2 (15, 24), indicating that Hoxa2 is necessary but not sufficient to activate Meox1 expression. However, Hoxa2 activates Meox1 and represses Six2 transcription; Hoxa2 repressor activity has been mapped to the protein region N terminal to the homeodomain (27, 31). The molecular basis of this regulatory switch, also common to other Hox proteins (23, 27), is unknown. The molecular composition of the IIBAs at different developmental stages (the temporal dynamics of Meox1 activation and Six2 repression are clearly different, almost complementary) or the presence of additional cis-regulatory modules in Six2 and Meox1 promoters, able to recruit specific coactivators or corepressors, may affect Hoxa2 function.
Hoxa2 GRN: a highly redundant network?
Hoxa2 controls morphogenesis of the IIBA and defines the shape and position of IIBA cartilages. Hoxa2 regulates Meox1 expression, and Meox1 and Meox2 control morphogenesis of IIBA skeletal elements. Taken together, it is highly likely that Meox1 partially mediates Hoxa2 function in the IIBA. The finding that Meox1 null mutants do not display an IIBA phenotype but Meox1 Meox2 null mutants have defects in the skeletal elements that are controlled by Hoxa2 suggests that Meox1 function in the IIBA can be compensated by Meox2. This is similar to what was observed in the developing somites, where Meox2 and Meox1 act redundantly (16). However, unlike what was observed in the somites, Meox1 and Meox2 spatiotemporal expression patterns in the IIBA are almost complementary. Meox2 is expressed in distal areas of the IIBA, in contrast to the anteroproximal expression of Meox1. Expression profiling of wild-type and mutant IIBAs, collected from E10.0 to E11.5, indicates that in the absence of Hoxa2, Meox1 transcript levels are downregulated at all stages examined, while changes in Meox2 expression are detected only at the latest stage examined (E11.5), suggesting that they are likely to be indirect effects of Hoxa2 absence (N. Bobola, unpublished results). The combined Meox1 Meox2 null phenotype in the IIBA indicates that Meox1 and Meox2 can compensate for each other's loss. Their differences in expression, however, raise questions about the effective capacity of Meox2 to compensate for the loss of Meox1 in the cells where Meox1 is normally expressed. Could other factors, in addition to Meox2, compensate for Meox1's absence?
One of the most intriguing findings resulting from the analysis of Hoxa2 activity during branchial arch development is that, while Hoxa2 loss of function generates strong phenotypes, the network downstream of Hoxa2 appears to be highly resistant to perturbations. Correcting Six2, Ptx1, and Gbx2 upregulation in Hoxa2 mutant embryos has a mild or no effect on the development of the IIBA (5, 14; M. Carapuco and M. Mallo, unpublished results). Similarly, we show here that Meox1 null mice do not have a phenotype, while Meox1 Meox2 null mutants have IIBA defects. The simplest interpretation is that we observe mild defects because the genes analyzed do not include the crucial Hoxa2 functional targets, which still need to be identified.
An alternative possibility, which takes into account that two of the four genes analyzed (Meox1 and Six2) are bona fide direct targets of Hoxa2, is that the GRN downstream of Hoxa2 is highly robust and is able to cope with modifications in the activity of its members. Indeed, the variability observed in the rescue of the phenotype in Hoxa2 Six2 null mutants (5, 14) indicates a high degree of redundancy, with other genes able to compensate Six2 function. Although we currently lack systematic evidence to substantiate this hypothesis, it is interesting to speculate further. Hoxa2 GRN robustness may derive from recruiting genes with considerable redundant roles, e.g., genes that belong to families, and whose members are also present in the IIBA (Meox1 and Meox2 and Six2, Six1, and Six4).
It is often found that changing the level of a transcription factor alters the expression level of a small subset of its predicted target genes; one of the possible explanations for this finding, other than a lack of function, is that related family members might bind to the same sites and have the same function (10). The structure of the Hoxa2 transcriptional network, where most of the Hoxa2 downstream targets identified so far encode homeodomain transcription factors (5, 13, 14), may also contribute to the functional stability of the network. Homeodomain proteins regulate transcription of their target genes by binding to specific nucleotide sequences. A survey of the binding preferences of Hoxa2 targets, according to the interactive prediction tool developed by Noyes et al. (19) and high-throughput binding site selection (4), shows that Meox1, Lhx6 (5), Ptx1 (5), Msx1 (28), Gbx2 (6), and Hoxa2 itself interact with very similar, if not identical, nucleotides sequences (Fig. 7). We showed in this paper that Meox1 specifically interacts with the sequences recognized by Hoxa2, and its binding abilities are comparable to those of Hoxa2.
FIG. 7.
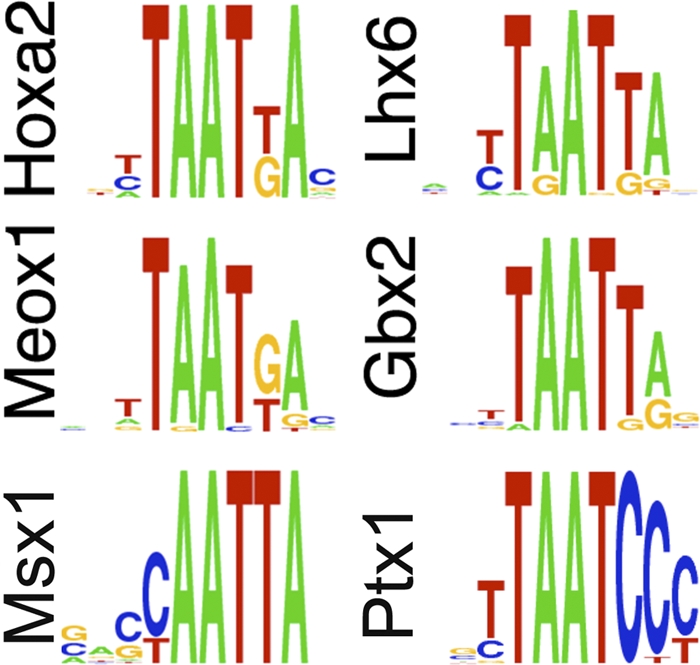
Members of the Hoxa2 GRN display similar binding preferences. The sequence logo indicates the DNA recognition sequence determined by the prediction tool at http://ural.wustl.edu/flyhd. The same recognition sequences were independently identified by high-throughput binding site selection (4).
Recent genomewide profiling of site-specific transcription factors has discovered that transcription factors bind thousands of binding sites in the genome (10), pointing at functional redundancy as a built-in safeguard for maintaining accurate regulation of the genome. The observed enrichment in transcription factors with similar binding affinities could provide a quantitative backup to Hoxa2 GRN function. For instance, elimination of one GRN member could allow a higher level of binding of another GRN member. Homeodomain proteins recognize short sequences that are widespread throughout the genome, and it is believed that only a small percentage of all occurrences of a motif are actually bound by these proteins (18). A global map of the binding sites of Hoxa2 GRN members in vivo could discover potential overlaps on target promoters. These data would provide a molecular basis for the network redundancy and explain the recurring finding that loss of function of Hoxa2 targets has only a very partial effect, if any, on the phenotype.
The developing somites represent the main domain of Meox1 expression and the area of the embryo most affected in the absence of Meox1 (16). Hox genes are expressed in somites and control morphogenesis of the axial skeleton (30). These observations raise the intriguing possibility that Meox1 might be a target of other Hox proteins, in addition to Hoxa2. The use of common target genes to control diverse developmental processes has been documented only for Six2 (13, 14, 31), but it could represent a more widespread aspect of Hox function in vertebrates.
Acknowledgments
We thank members of the Piper-Hanley and Hentges labs for help with sectioning and Rudolf Grosschedl for the Crabp1 probe.
This work was supported by BBSRC grant BB/E017355/1 to N.B. The Bobola group is supported by the Manchester Academic Health Science Centre and the Manchester NIHR Biomedical Research Centre. M.M. was supported by grant PTDC/BIA-BCM/71619/2006 and by Centro de Biologia do Desenvolvimento POCTI-ISFL-4-664.
Footnotes
Published ahead of print on 18 January 2011.
REFERENCES
- 1.Altschul, S. F., et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baltzinger, M., M. Ori, M. Pasqualetti, I. Nardi, and F. M. Rijli. 2005. Hoxa2 knockdown in Xenopus results in hyoid to mandibular homeosis. Dev. Dyn. 234:858-867. [DOI] [PubMed] [Google Scholar]
- 3.Barrow, J. R., and M. R. Capecchi. 1999. Compensatory defects associated with mutations in Hoxa1 restore normal palatogenesis to Hoxa2 mutants. Development 126:5011-5026. [DOI] [PubMed] [Google Scholar]
- 4.Berger, M. F., et al. 2008. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133:1266-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobola, N., et al. 2003. Mesenchymal patterning by Hoxa2 requires blocking Fgf-dependent activation of Ptx1. Development 130:3403-3414. [DOI] [PubMed] [Google Scholar]
- 6.Carapuco, M., A. Novoa, N. Bobola, and M. Mallo. 2005. Hox genes specify vertebral types in the presomitic mesoderm. Genes Dev. 19:2116-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carroll, S. B. 1995. Homeotic genes and the evolution of arthropods and chordates. Nature 376:479-485. [DOI] [PubMed] [Google Scholar]
- 8.Depew, M. J., T. Lufkin, and J. L. Rubenstein. 2002. Specification of jaw subdivisions by Dlx genes. Science 298:381-385. [DOI] [PubMed] [Google Scholar]
- 9.Dieterich, C., H. Wang, K. Rateitschak, H. Luz, and M. Vingron. 2003. CORG: a database for comparative regulatory genomics. Nucleic Acids Res. 31:55-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farnham, P. J. 2009. Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 10:605-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gendron-Maguire, M., M. Mallo, M. Zhang, and T. Gridley. 1993. Hoxa-2 mutant mice exhibit homeotic transformation of skeletal elements derived from cranial neural crest. Cell 75:1317-1331. [DOI] [PubMed] [Google Scholar]
- 12.Hunter, M. P., and V. E. Prince. 2002. Zebrafish hox paralogue group 2 genes function redundantly as selector genes to pattern the second pharyngeal arch. Dev. Biol. 247:367-389. [DOI] [PubMed] [Google Scholar]
- 12a.Kanzler, B., S. J. Kuschert, Y. H. Liu, and M. Mallo. 1998. Hoxa-2 restricts the chondrogenic domain and inhibits bone formation during development of the branchial area. Development 125:2587-2597. [DOI] [PubMed] [Google Scholar]
- 13.Kutejova, E., B. Engist, M. Mallo, B. Kanzler, and N. Bobola. 2005. Hoxa2 downregulates Six2 in the neural crest-derived mesenchyme. Development 132:469-478. [DOI] [PubMed] [Google Scholar]
- 14.Kutejova, E., et al. 2008. Six2 functions redundantly immediately downstream of Hoxa2. Development 135:1463-1470. [DOI] [PubMed] [Google Scholar]
- 15.Mallo, M. 1997. Retinoic acid disturbs mouse middle ear development in a stage-dependent fashion. Dev. Biol. 184:175-186. [DOI] [PubMed] [Google Scholar]
- 15a.Mallo, M., and I. Brändlin. 1997. Segmental identity can change independently in the hindbrain and rhombencephalic neural crest. Dev. Dyn. 210:146-156. [DOI] [PubMed] [Google Scholar]
- 16.Mankoo, B. S., et al. 2003. The concerted action of Meox homeobox genes is required upstream of genetic pathways essential for the formation, patterning and differentiation of somites. Development 130:4655-4664. [DOI] [PubMed] [Google Scholar]
- 16a.Mankoo, B. S., et al. 1999. Max2 is a component of the genetic hierarchy controlling limb muscle development. Nature 400:69-73. [DOI] [PubMed] [Google Scholar]
- 17.Manley, N. R., and M. R. Capecchi. 1997. Hox group 3 paralogous genes act synergistically in the formation of somitic and neural crest-derived structures. Dev. Biol. 192:274-288. [DOI] [PubMed] [Google Scholar]
- 18.Moens, C. B., and L. Selleri. 2006. Hox cofactors in vertebrate development. Dev. Biol. 291:193-206. [DOI] [PubMed] [Google Scholar]
- 19.Noyes, M. B., et al. 2008. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133:1277-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohnemus, S., N. Bobola, B. Kanzler, and M. Mallo. 2001. Different levels of Hoxa2 are required for particular developmental processes. Mech. Dev. 108:135-147. [DOI] [PubMed] [Google Scholar]
- 21.Pasqualetti, M., M. Ori, I. Nardi, and F. M. Rijli. 2000. Ectopic Hoxa2 induction after neural crest migration results in homeosis of jaw elements in Xenopus. Development 127:5367-5378. [DOI] [PubMed] [Google Scholar]
- 22.Pearson, J. C., D. Lemons, and W. McGinnis. 2005. Modulating Hox gene functions during animal body patterning. Nat. Rev. Genet. 6:893-904. [DOI] [PubMed] [Google Scholar]
- 23.Pinsonneault, J., B. Florence, H. Vaessin, and W. McGinnis. 1997. A model for extradenticle function as a switch that changes HOX proteins from repressors to activators. EMBO J. 16:2032-2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prince, V., and A. Lumsden. 1994. Hoxa-2 expression in normal and transposed rhombomeres: independent regulation in the neural tube and neural crest. Development 120:911-923. [DOI] [PubMed] [Google Scholar]
- 25.Rijli, F. M., et al. 1993. A homeotic transformation is generated in the rostral branchial region of the head by disruption of Hoxa-2, which acts as a selector gene. Cell 75:1333-1349. [DOI] [PubMed] [Google Scholar]
- 26.Rodrigo, I., P. Bovolenta, B. S. Mankoo, and K. Imai. 2004. Meox homeodomain proteins are required for Bapx1 expression in the sclerotome and activate its transcription by direct binding to its promoter. Mol. Cell. Biol. 24:2757-2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saleh, M., I. Rambaldi, X. J. Yang, and M. S. Featherstone. 2000. Cell signaling switches HOX-PBX complexes from repressors to activators of transcription mediated by histone deacetylases and histone acetyltransferases. Mol. Cell. Biol. 20:8623-8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santagati, F., M. Minoux, S. Y. Ren, and F. M. Rijli. 2005. Temporal requirement of Hoxa2 in cranial neural crest skeletal morphogenesis. Development 132:4927-4936. [DOI] [PubMed] [Google Scholar]
- 29.Svingen, T., and K. F. Tonissen. 2006. Hox transcription factors and their elusive mammalian gene targets. Heredity 97:88-96. [DOI] [PubMed] [Google Scholar]
- 30.Wellik, D. M. 2007. Hox patterning of the vertebrate axial skeleton. Dev. Dyn. 236:2454-2463. [DOI] [PubMed] [Google Scholar]
- 31.Yallowitz, A. R., K. Q. Gong, I. T. Swinehart, L. T. Nelson, and D. M. Wellik. 2009. Non-homeodomain regions of Hox proteins mediate activation versus repression of Six2 via a single enhancer site in vivo. Dev. Biol. 335:156-165. [DOI] [PMC free article] [PubMed] [Google Scholar]