

Abstract
We have determined that, in addition to its receptor-destroying activity, the influenza virus neuraminidase is capable of efficiently forming virus-like particles (VLPs) when expressed individually from plasmid DNA. This observation applies to both human subtypes of neuraminidase, N1 and N2. However, it is not found with every strain of influenza virus. Through gain-of-function and loss-of-function analyses, a critical determinant within the neuraminidase ectodomain was identified that contributes to VLP formation but is not sufficient to accomplish release of plasmid-derived VLPs. This sequence lies on the plasma membrane-proximal side of the neuraminidase globular head. Most importantly, we demonstrate that the antiviral restriction factor tetherin plays a role in determining the strain-specific limitations of release competency. If tetherin is counteracted by small interfering RNA knockdown or expression of the HIV anti-tetherin factor vpu, budding and release capability is bestowed upon an otherwise budding-deficient neuraminidase. These data suggest that budding-competent neuraminidase proteins possess an as-yet-unidentified means of counteracting the antiviral restriction factor tetherin and identify a novel way in which the influenza virus neuraminidase can contribute to virus release.
The influenza virus encodes the neuraminidase (NA) which is responsible for cleaving terminal sialic acid residues off glycoconjugates on both the virus particle and the host cell, thereby facilitating virus release (9, 46, 47). While this final stage in the virus life cycle was clearly described many years ago, virus-encoded and cellular factors involved in influenza virus budding have yet to be clearly defined. Early studies identified the matrix protein as the primary budding determinant and suggested participation of the endocytic sorting complexesrequired for transport (ESCRT) machinery (14, 15, 21, 22). While several papers reported this finding, two of them were retracted, adding controversy to the role of M1 in virus assembly (21, 22). Subsequent studies using a plasmid-driven expression system showed that the hemagglutinin (HA) and NA proteins represent the minimal requirements for the formation of virus-like particles (VLPs) in 293T cells (7). In the presence of HA, the enzymatic activity of NA, rather than the protein itself, was sufficient for efficient particle release. Other viral proteins expressed individually or in various combinations were incapable of efficiently forming VLPs. Other findings, however, suggested the existence of additional budding determinants since WSN viruses containing undetectable amounts of HA were present in the medium following infection at the nonpermissive temperature with temperature-sensitive HA plasma membrane transport mutants (48). In addition, VLP production was detected following the coexpression of the viral M1 and M2 proteins (62). Recently, Lai et al. published data demonstrating that sole expression of the NA is also capable of efficiently forming VLPs (29). In their study, they tested the budding competence of the NAs from the novel 2009 H1N1 strain, a seasonal H1N1 (A/Gansu/Chenguan/1129/07) strain, and a highly pathogenic avian influenza virus H5N1 strain (A/Cambodia/JP52a/2005). In addition, they demonstrated that the budding capability of the NA is independent of its enzymatic activity (29). In addition, the most recent publication in the field suggests that the M2 protein alone is capable of substituting for the ESCRT machinery and is actually responsible for the “pinching off” process of budding (52, 53). Taken together, these studies implicate HA, M1, M2, and NA in the morphogenesis of influenza virus.
The budding of influenza virus appears to occur independently of the canonical late domain motif pathways (6, 7). Viral late domain motifs were originally identified in the HIV gag polyprotein and are represented by short peptide regions that recruit members of the ESCRT machinery (3, 17, 19, 26, 39, 41, 50). This machinery is normally involved in the morphogenesis of the multivesicular body, a structure involved in the lysosomal degradation of transmembrane proteins (23). Viral late domains aberrantly recruit this machinery to the plasma membrane to mediate budding (2, 37, 38). Oftentimes, as in the case of many retroviruses, several late domain motifs are present at different locations within the gag polyprotein (64). Since certain motifs tend to vary in importance in a cell-type-dependent manner, this redundancy may allow efficient budding to occur when the primary cellular pathway is absent or inefficient (12, 18, 33, 50). While the ESCRT machinery does not appear to be involved in the budding of influenza virus, cellular factors are still most likely required, since a Rab11-dependent pathway for influenza virus budding was recently identified (5).
While most respiratory viruses encode an enzymatic function to assist the release process, virus release factors are not always enzymatic in nature. In the case of HIV, the small accessory protein vpu enhances virus release without a known enzymatic activity (27, 57, 59). It was found that the enhancement of virus release mediated by vpu was due to the counteraction of an interferon-inducible antiviral host factor, BST-2, now renamed tetherin (45, 49, 61). If left unabated, tetherin inhibits the release of HIV and other viruses, with considerable evidence suggesting that tetherin actually forms a proteinaceous link joining virions together or “tethering” them to the host cell (25, 34, 44, 45, 49, 54). Vpu is able to mediate downregulation of tetherin from the cell surface and in several cases was shown to cause the degradation of tetherin (10, 11, 13, 43, 61). Vpu has also been shown to downregulate and degrade the CD4 cell surface molecule, and its anti-tetherin and CD4 downregulation functions are genetically separable through point mutation (35, 55, 63).
When we discovered that a virus lacking the HA segment completely was able to bud efficiently, we looked into a role for the NA protein in influenza virus morphogenesis. Using a plasmid-driven expression system in 293T cells, we are able to confirm the data presented by Lai et al. and, in addition, extend it to additional strains of the N1 and also the N2 subtype of NA. Furthermore, we identified a residue within the ectodomain of NA that contributes to VLP formation but is not sufficient to mediate the release of NA-driven VLPs. Gain-of-function analysis with an otherwise budding/release-deficient NA suggested that VLP production with this mutant was blocked at the stage of release. Upon further exploration of factors that could inhibit the release of VLPs, we found that the antiviral restriction factor tetherin was contributing to the observed phenotype. Counteraction of tetherin by small interfering RNA (siRNA) knockdown or expression of the HIV-encoded tetherin antagonist vpu rescued the budding and release of otherwise budding-incompetent NA. These data suggest that budding-competent NA proteins possess the ability to counteract the antiviral factor tetherin and therefore possess a redundant function in facilitating virus release.
MATERIALS AND METHODS
Cells, compounds, plasmids, viruses, and antibodies.
MDCK and MDCK-HA cells were maintained in minimum essential medium (Gibco) supplemented with 1× Pen/Strep and 10% fetal bovine serum (FBS). For the MDCK-HA cells, hygromycin (200 μg/ml) was added to the cells occasionally to maintain selection pressure (36). 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Cellgro) supplemented with 1× Pen/Strep and 10% FBS. HeLa cells were generously provided by Dafna Bar-Sagi and maintained in DMEM (Cellgro) supplemented with 1× Pen/Strep and 10% FBS. Cos-1 cells were maintained in DMEM supplemented with 1× Pen/Strep and 10% FBS. They were used for indirect immunofluorescence and thin-section electron microscopy (EM), as they allowed improved visualization. The WSN luciferase virus has been previously described and was grown on MDCK-HA cells (28). Plasmids (pCAGGS) encoding the NA proteins of WSN/33, Udorn/72, and Hong Kong/68 were identical matches to the published GenBank sequences, while the Japan/57 wild-type (WT) NA differed at one amino acid (S367G) from the published GenBank sequence (accession no. CY045806.1). Plasmids (pcDNA) for the expression of tetherin, vpu, and the vpu 2/6 CD4 downregulation mutant were kindly provided by Klaus Strebel. The antibodies used in this study included rabbit anti-hexon (Abcam), which was used for the adenovirus loading control. The rabbit anti-Flag polyclonal antibody (Sigma) and the goat anti-influenza virus Singapore/57 N2 antibody (BEI resources) were used for immunogold labeling and Western blotting. The rabbit polyclonal anti-PR8 virus antibody, mouse anti-WSN HA antibody 2G9, and mouse anti-WSN HA antibody 4B2 were generated in our laboratory (32). 2G9 was used for immunogold labeling of the VLPs, while 4B2 and the rabbit polyclonal antibody were used for Western blot analysis. Fluorescence-activated cell sorter (FACS) analysis was performed with the anti-N2 antibody mentioned above. The E10 anti-M1 and -M2 antibody was generated in our laboratory and was described previously (4). The rabbit polyclonal anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) loading control antibody was purchased from Sigma. The tetherin-related antibodies used included a rabbit anti-BST-2 antibody (kindly provided by Klaus Strebel) and a rabbit anti-vpu antibody (kindly provided by the AIDS Reagent Program). The secondary antibodies used included a donkey anti-goat fluorescein isothiocyanate (FITC)-conjugated antibody for FACS analysis and indirect immunofluorescence. The immunogold secondary antibodies were purchased from Electron Microscopy Sciences and included goat anti-mouse 15-nm gold-conjugated (1:20) and goat anti-rabbit 6-nm gold-conjugated (1:25) antibodies for purified VLP labeling. Anti-goat 12-nm gold-conjugated secondary antibodies were used for thin-section EM analysis. Oseltamivir carboxylate (GS4071) was provided by Gilead Sciences.
VLP generation and purification.
Plasmids (pCAGGs, 3 μg/10-cm dish) for the expression of influenza virus HA and/or NA, as indicated, were transfected using Lipofectamine 2000 (Invitrogen) at a 2:1 Lipofectamine-to-DNA ratio according to the manufacturer's instructions. The tetherin VLP experiments were performed with the following plasmid amounts per 10-cm dish (as indicated): tetherin, 250 ng; WT vpu, 4.5 μg; 2/6 mutant vpu, 3 μg. For Western blot analysis of VLPs, two 10-cm dishes were transfected and VLPs were harvested at 48 h posttransfection. EM analysis was performed with VLPs harvested from the filtered supernatant (0.45 μm, polyvinylidene difluoride; Millipore) of three 10-cm dishes at 48 h posttransfection. For VLP purification, transfected cell supernatants were layered onto a 30% sucrose-NTE (100 mM NaCl, 10 mM Tris [pH 7.4], 1 mM EDTA) cushion and centrifuged at 25,000 rpm in a Beckman SW28 rotor for 2 h at 4°C. The supernatants and cushion were then carefully aspirated, and pellets were resuspended in a 1:1 mixture of radioimmunoprecipitation assay buffer (50 mM Tris pH[8.0], 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) and 2× SDS sample buffer (4% SDS, 150 mM Tris [pH 6.8], 50 mM dithiothreitol, 10% glycerol) for Western blot analysis or NTE buffer in preparation for transmission EM processing. Densitometric analysis was performed using an Alphaimager 3400 (Alpha Innotech).
Immunogold labeling, negative staining, and transmission EM.
Virus/VLPs were allowed to adhere to carbon-coated copper grids (Electron Microscopy Sciences) for 15 min at room temperature (RT). Virus/VLPs were then washed quickly with 1× phosphate-buffered saline (PBS; Gibco), diluted 1:10 in double-distilled H2O, and stained with a 1% solution of phosphotungstic acid (pH 8.0) for 30 s at RT. For immunogold labeling of the VLPs, the grids were blocked with 3% bovine serum albumin (BSA) in PBS for 45 min after VLP adherence. The grid was then incubated with a mixture of both primary antibodies (anti-Flag antibody at 1 μg/ml and anti-HA antibody at 10 μg/ml) for 45 min at RT. Grids were then washed quickly three times with PBS and incubated with both gold-conjugated secondary antibodies (15-nm gold-conjugated anti-mouse antibody and 6-nm gold-conjugated anti-rabbit antibody) for 45 min at RT. After secondary-antibody incubation, grids were washed again three times with PBS, followed by negative staining with 1% phosphotungstic acid. Transmission electron micrographs were captured using a Hitachi 7000 electron microscope.
Thin sectioning and EM.
Cultures grown on Aclar film in 6-cm tissue culture dishes were fixed with 2% glutaraldehyde-1% paraformaldehyde in 0.12 M sodium cacodylate buffer, pH 7.3 to 7.4, for 50 min at RT, postfixed with 1% OsO4, stained en bloc with 1% uranyl acetate in H2O, dehydrated in an ethanol series followed by propylene oxide, and embedded in Epon. Thin sections cut parallel to the growth surface were stained with uranyl acetate and lead citrate and viewed with a JEM-1200EX electron microscope (JEOL USA) equipped with an AMT XR-60 digital camera (Advanced Microscopy Techniques) or a FEI Tecnai12 BioTwinG2 transmission electron microscope equipped with an AMT XR-60 charge-coupled device digital camera system.
Confocal microscopy.
Cells were plated on coverslips and fixed in 4% formaldehyde (Fisher) for 20 min. Permeabilization was done in 0.5% saponin for 15 min. The cells were stained with goat anti-NA antibody to detect NA and a fluorescein isothiocyanate (FITC)-conjugated anti-goat secondary antibody. Hoechst stain was used to detect the nucleus. All images were captured on an inverted fluorescence/differential-interference contrast Zeiss Axiovert 200M deconvoluting fluorescence microscope operated by AxioVision version 4.5 (Zeiss) software. Ten to 20 optimal sections along the z axis were acquired in increments of 0.4 μm. Captured images show the central section. The fluorescence data sets were deconvoluted by using the constrained iterative method (AxioVision).
NA activity assay.
The NA-star assay kit (Applied Biosystems) was used according to the manufacturer's instructions. Tissue culture supernatant containing VLPs was diluted 1:50 in 50 μl of NA-star assay buffer. The transfected cells were washed twice with cold PBS and collected in 1 ml of cold PBS. Cells were then sonicated, and the lysate was diluted 1:50 in NA-star assay buffer. To these samples, an additional 50 μl of assay buffer was added prior to the addition of 10 μl of substrate. The supernatant/lysate was incubated with the detection reagent for 20 min prior to luminescence reading in a 96-well plate reader (Beckman Coulter DTX880). Data were analyzed using the multimode analysis software supplied with the plate reader.
Cell surface expression analysis.
Transfected 293T cells used for VLP generation were processed at 48 h posttransfection. Cells were washed twice and tapped off the plate prior to staining. Roughly 1 × 106 293T cells were then incubated for 1 h with the primary antibody (goat anti-Singapore/57 N2 NA antibody, 1:100 dilution) in standard FACS buffer (3% BSA in PBS). Cells were washed three times and then incubated with the secondary antibody (donkey anti-goat FITC-conjugated antibody; Invitrogen) for 1 h. After secondary-antibody incubation, cells were washed three times with FACS buffer and fixed in 4% formaldehyde in PBS prior to analysis.
Tetherin knockdown experiments.
siRNA against tetherin (validated siRNA against BST-2 [Hs_BST2_5]) and the scrambled custom-designed target sequence (5′-AAGGTAATTGCGCGTGCAACT-3′) were described previously (28) and were purchased from Qiagen. Adherent HeLa cells (3 × 106 cells/dish, three dishes per condition) were cotransfected with the siRNA and the Hong Kong/68 NA (36 μg/10-cm dish, three dishes per condition) expression plasmid with a 2:1 Fugene 6/DNA ratio. The final siRNA concentration was 30 nM, and an additional 15 μl of Fugene 6 (Roche) was added per dish for siRNA transfection. The siRNA-resistant tetherin clone was generated by site-directed mutagenesis to introduce five silent mutations into the targeted sequence of tetherin. The tetherin-resistant clone was transfected concurrently with the other plasmids (1.5 μg/dish) where indicated (2:1 Fugene 6/DNA ratio). One dish was lysed at 40 h posttransfection to determine tetherin knockdown efficiency. VLPs were harvested at 60 h posttransfection and purified as stated above. BSA (0.1%) and sodium bicarbonate (7.5%; 100 μl/dish) were added to the cells at 24 h posttransfection.
RESULTS
The WSN luciferase virus, which does not encode HA, buds with equal efficiency from MDCK-HA and MDCK cells.
WSN luciferase virus (H1N1 background) contains all of the influenza virus segments except that coding for the HA. This segment is replaced with the coding sequence of Renilla luciferase with flanking 5′ and 3′ packaging sequences from the HA segment to allow efficient incorporation into virions (28). This virus is normally grown on an HA-complementing cell line (MDCK-HA), allowing the incorporation of HA into the virion. However, we noted that infection of MDCK-HA and MDCK cells generated particles comparable in size and morphology to influenza virus that could be pelleted through a sucrose cushion. This observation indicated that virus was assembled and released efficiently even though the HA was absent (Fig. 1A and B). To eliminate the possibility that the virus preparation derived from the MDCK cells represented initial input virus or potential contamination with the WT WSN virus, the preparations were examined for viral proteins by Western blotting. As shown in Fig. 1C, both preparations contained NP, M1, and M2; however, only the virus produced from the MDCK-HA cells contained the HA protein, indicating that the virus produced in the MDCK cells does not represent WT contamination or input virus.
FIG. 1.

The WSN luciferase virus buds efficiently in the absence of HA expression. Virus was isolated from MDCK-HA (A) or MDCK (B) cells following infection and examined by EM as described in Materials and Methods. (C) The virus preparations were analyzed by Western blotting with anti-PR8 polyclonal (C1), anti-M1 and -M2 (C2), and anti-HA (C3) antibodies. The MDCK-HA virion preparation was treated with peptide N-glycosidase to demonstrate a deglycosylation shift in the HA protein. The values to the left of panel C1 are molecular sizes in kilodaltons.
NA forms VLPs efficiently in 293T cells.
To further examine the factors that contribute to HA-independent release of VLPs, NA was tested for its release competence when expressed individually from plasmid DNA. Due to their high transfection efficiency and prior characterization for influenza virus budding phenotypes (7), 293T cells were transfected with plasmids encoding Flag-tagged WSN NA only or NA-Flag and HA. Previous studies showed that a C-terminally tagged NA fusion protein is well tolerated (20). VLPs were harvested at 48 h posttransfection and purified over a 30% sucrose-NTE cushion prior to Western blot analysis. As shown in Fig. 2A, comparable amounts of VLPs were detected in the medium of cells expressing WSN NA-Flag in the presence or absence of coexpressed HA. Since Udorn/72 NA has been demonstrated to be incapable of budding efficiently (7), we determined if budding competency is specific to the N1 subtype by testing two N2-containing strains, namely, the 1957 pandemic strain (A/Japan/305/57) and the Udorn strain (A/Udorn/72). An identical experiment performed with untagged N2 NAs demonstrated that while the Udorn/72 NA was inefficient in VLP formation (Fig. 2C), the Japan/57 N2 NA was able to form VLPs efficiently in the absence of coexpressed HA (Fig. 2B). A densitometric analysis (NAVLP/NALysate plus NAVLP) indicated that the VLP release efficiencies of WSN, Japan/57, and Udorn/72 NA were 0.98:1, 0.82:1, and 0.38:1 relative to the respective HANA VLPs. To rule out the possibility that the particles represented membrane blebbing due to NA-induced apoptosis, annexin V staining of transfected cells was performed. The level of staining was equivalent for mock-transfected and transfected cells, indicating that budding-competent NA was not selectively triggering apoptotic blebbing (data not shown). We conclude that certain NAs of both subtypes are capable of forming VLPs independently of additional viral factors.
FIG. 2.
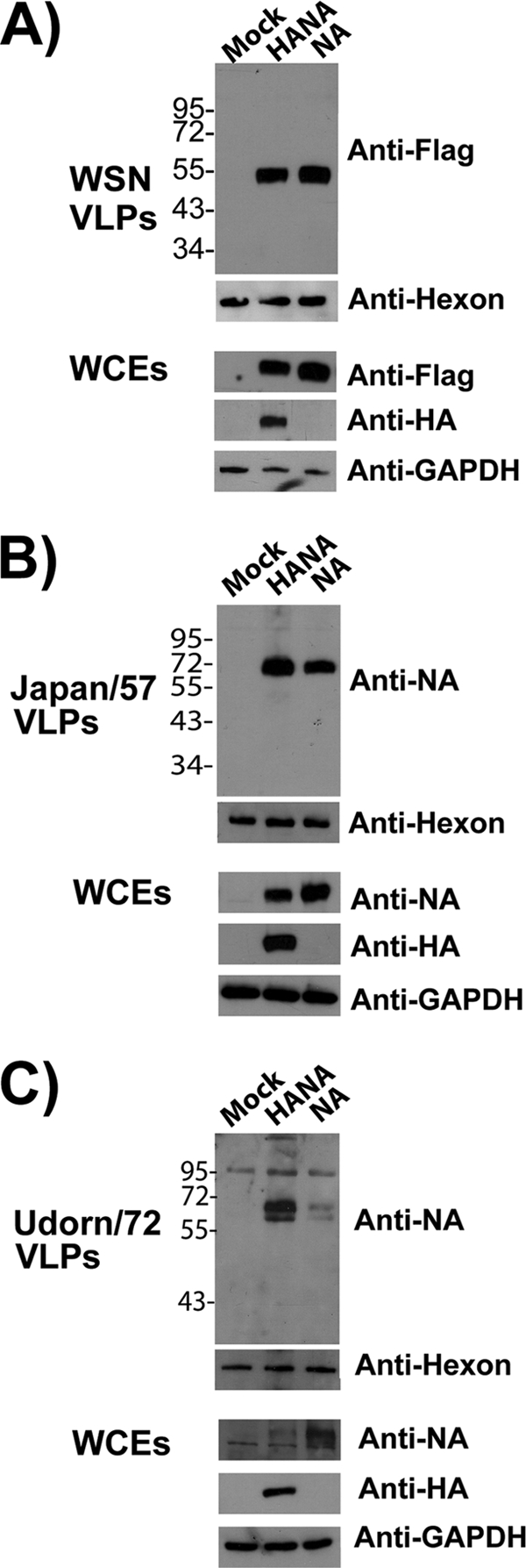
NA forms VLPs efficiently in 293T cells. 293T cells were either left untransfected (mock) or transfected with the indicated plasmids. VLPs were harvested from tissue culture supernatants at 48 h posttransfection and pelleted through a 30% sucrose-NTE cushion as described in Materials and Methods. As a loading control for tissue culture supernatants, equal amounts of WT adenovirus (dl309) were added to each sample prior to centrifugation. The presence of adenovirus was detected by an antibody against the hexon protein component of the capsid. Whole-cell extracts (WCEs) were also prepared and analyzed. Panels: A, WSN virus HA and Flag-tagged NA; B, Japan/57 virus HA and NA; C, Udorn/72 virus HA and NA. GAPDH was used as a loading control for WCEs. The values to the left of the blots are molecular sizes in kilodaltons.
HANA VLPs and NA VLPs appear similar.
VLPs were purified by pelleting through a 30% sucrose cushion and examined by transmission EM. As shown in Fig. 3A (left), VLPs from the Japan/57 H2N2 strain exhibited the morphology of influenza virus whether assembled from HA and NA or from NA alone. In contrast, the Udorn/72 H3N2 strain formed particles that resembled virions only upon HA coexpression with NA (Fig. 3A, right). Similar results were obtained when the WSN NA-Flag construct was coexpressed with WSN HA (Fig. 3B, top). While the average diameter of HANA VLPs and NA-only VLPs was always the same (∼140 nm), it was, on average, 1.5 times the size of the particles in a purified virus preparation. To confirm that the NA-only VLPs contained NA protein, immunogold labeling was performed. The images at the bottom of Fig. 3B represent purified VLPs containing either the HA, the NA, or both viral glycoproteins. These preparations were incubated with anti-HA and anti-Flag primary antibodies conjugated to secondary antibodies tagged with either 6-nm (NA-Flag labeling) or 15-nm (HA labeling) gold beads. As shown in Fig. 3B, while HANA VLPs were labeled with both bead sizes (left), predominantly 15-nm gold beads decorated VLPs from cells expressing HA alone (center; released with soluble NA) (7) and 6-nm beads labeled VLPs assembled using NA-Flag (right). Thus, NA assembles VLPs that resemble authentic influenza virus particles.
FIG. 3.

HANA VLPs and NA VLPs appear similar. (A) Purified VLPs were examined by negative staining and transmission EM. (B) Purified VLPs were incubated with primary and secondary antibodies for the detection of both HA and the Flag tag prior to EM analysis. Secondary antibodies were conjugated with either 6-nm (anti-Flag) or 15-nm (anti-HA) gold beads.
The enzymatic activity of the Japan/57 NA is not required for NA budding capability.
VLPs formed by HA require the enzymatic activity of NA for release. To determine if the enzymatic activity of NA played a role in the observed budding competence, VLP assembly and release by Japan/57 WT NA were examined in the presence or absence of oseltamivir carboxylate. In addition, a catalytically inactive mutant form previously shown to be properly localized and folded, W178L, was also tested (30). As shown in Fig. 4A, Japan/57 WT NA VLPs were released efficiently in the presence or absence of oseltamivir carboxylate at a concentration of 1 μM (Fig. 4A, compare lanes 4 and 5). Control experiments confirmed that the enzymatic activity was completely inhibited. (i) HANA VLPs were not released in the presence of 1 μM oseltamivir carboxylate (compare lanes 2 and 3). (ii) No enzymatic activity was detected in the presence of oseltamivir carboxylate, based on the NA-star assay (Applied Biosystems) (Fig. 4C). Since oseltamivir carboxylate does not penetrate the cell, we also disrupted the NA activity through mutation. As shown in Fig. 4B, VLPs formed from W178L NA were released as efficiently as VLPs assembled from WT NA (compare lanes 3 and 4). We conclude that the enzymatic activity of NA is not required for VLP assembly and release.
FIG. 4.

The enzymatic activity of the Japan/57 NA is not required for NA budding. (A) Transfected cells were incubated in the presence or absence of oseltamivir carboxylate (1 μM). VLPs were isolated at 48 h posttransfection and analyzed by Western blotting. (B) Cells transfected with plasmids for the expression of WT HA and NA, WT NA alone, or W178L NA were isolated and analyzed by Western blotting. It should be noted that the altered mobility of the mutant reflects altered glycosylation due to the lack of NA activity. WCEs, whole-cell extracts. (C) Supernatants and cell lysates from transfected cells were analyzed for total NA activity using the NA-star assay kit for the oseltamivir carboxylate (Oselt) experiment depicted in panel A. RLU, relative light units. The values to the left of panels A and B are molecular sizes in kilodaltons.
Asp286 in the Japan/57 NA contributes to budding competency.
Sequence alignment of the Udorn/72 and Japan/57 NAs indicated differences at 39 positions. Assuming that any budding motif present in the Japan/57 NA would also be present in the WSN/33 NA allowed a narrowing of potential amino acids involved in budding to five. One of these positions lies within a motif, YPX286Φ (where Φ represents a hydrophobic amino acid), demonstrated to be involved in retrovirus budding (51, 56). Specifically, while four out of five of these amino acids did not resemble any late domain sequence, the Japan/57 sequence (284YPDV287) is almost an exact match to a late domain sequence present in the p9 protein of equine infectious anemia virus (YPDL) known to interact with the AIP2 (ALIX) component of the ESCRT machinery (8, 31, 51). Substitution of the Gly286 residue found in the budding-incompetent Udorn/72 NA for the Asp286 residue in the budding-competent Japan/57 NA generated a protein that was incapable of budding when expressed alone, even though it was expressed at WT levels in the cell (Fig. 5A). This mutant NA, however, exhibited WT enzymatic activity, as measured by the NA-star assay (Fig. 5B); rescued release of HA VLPs at the level of efficiency of the WT NA protein (Fig. 5C); and localized to the cell surface appropriately, as indicated by FACS analysis (Fig. 5D). A similar YPDT motif is present in the WSN N1 NA from residues 266 to 269. Mutation of the critical Asp residue in this motif to alanine also eliminated the budding capability of the N1 NA (data not shown). These results indicate that Asp286 in the N2 NA and Asp268 in the N1 NA contribute to the release of NA VLPs.
FIG. 5.

Asp286 in Japan/57 NA contributes to budding competency. 293T cells were either left untransfected (mock) or transfected with plasmids encoding HA and NA, NA alone, or the Asp286Gly mutant NA, and VLPs were isolated at 48 h posttransfection. Prior to centrifugation, the same amount of WT adenovirus (dl309) was added to each sample for use as a loading control. (A) Western analysis. (B) NA activity (NA-star assay) in the supernatant and cell lysate were combined to determine total NA activity. Cells transfected with the indicated plasmids were analyzed at 48 h posttransfection. The lower part of the panel represents Western blot data to demonstrate NA (top) and GAPDH (bottom; loading control) expression levels. RLU, relative light units. (C) Budding assay identical to that in panel A, except that the transfected samples contained WT HA and either WT NA or the D286G NA mutant. (D) Cell surface expression of Japan/57 WT NA and Asp286Gly NA determined by FACS analysis. The percentage of cells staining positive with the anti-NA antibody is indicated. The bottom of the panel demonstrates the expression levels of both proteins as determined by Western analysis. The values to the left of the blots in panels A and C are molecular sizes in kilodaltons.
Replacement of Gly286 with Asp confers partial budding competence on Udorn/72 NA.
To determine whether Asp286 was sufficient to direct NA VLP release, a “gain-of-function” experiment was performed wherein Gly286 was replaced with an Asp residue in budding-incompetent Udorn/72 NA. A budding experiment was performed with this mutant, and no VLPs were detected in the supernatant when either the WT or the G286D mutant NA was expressed in 293T cells (data not shown). However, examination of Cos-1 cells by confocal microscopy indicated a potential gain-of-function phenotype. Cos-1 cells were used for these experiments, since they are larger and provide improved image quality over 293T cells. In addition, a budding experiment was performed with these cells that confirmed the strain-specific budding capabilities of the NA (data not shown). While the WT Udorn/72 NA was primarily localized adjacent to the nucleus with some staining at the cell periphery (Fig. 6A and A1), the G286D mutant NA accumulated largely at the cell periphery (Fig. 6B). This differed from what we observed with the Japan/57 WT and loss-of-function D286G mutant NAs. In the Japan/57 background, the loss-of-function mutation had no effect on the NA cell surface expression by FACS (Fig. 5D) or by confocal microscopy (Fig. 6C and D). Since the confocal data suggested a gain-of-function phenotype, we observed the cells more closely by EM. As expected, no WT Udorn/72 NA VLPs were detected by thin-section EM (Fig. 7C). However, the G286D Udorn/72 mutant NA formed VLPs that accumulated at the cell surface (Fig. 7D and E) but appeared entrapped in large vesicular structures. This differed from results obtained with WT Japan/57 NA (Fig. 7A), where VLPs were detected at the plasma membrane in the process of release or for the release-defective D286G Japan/57 NA mutant, where no particles were detected (Fig. 7B). In order to confirm that the VLP structures trapped at the cell surface for the G286D Udorn/72 mutant contained the NA, immunogold labeling was performed (Fig. 7E). We conclude that D286 conferred the ability to assemble VLPs at the plasma membrane. However, this determinant was not sufficient for particle release, explaining why VLPs were not detected in the medium.
FIG. 6.

Substitution of Asp for Gly286 confers partial budding competence on the Udorn/72 NA. Cos-1 cells transfected with the indicated plasmids were fixed at 48 h posttransfection and analyzed by confocal microscopy (A to D) as described in Materials and Methods. Panels A and B represent the Udorn/72 WT and G286D mutant NAs. Panels C and D represent the Japan/57 WT and D286G mutant NAs. All NA proteins were labeled with FITC-conjugated secondary antibodies.
FIG. 7.

Substitution of Asp for Gly286 confers partial budding competence on the Udorn/72 NA. Cos-1 cells transfected with the indicated plasmids were fixed at 48 h posttransfection, embedded in Durcupan resin, and analyzed by thin-section EM (A to D). In panel E, the VLPs were labeled with goat anti-Singapore/57 primary antibodies and 12-nm gold-conjugated anti-goat secondary antibodies. Open arrows indicate NA protein labeled at the cell surface, while closed arrows indicate NA protein contained within trapped VLPs.
Tetherin inhibits NA VLP release in budding-incompetent NAs, and this inhibition can be alleviated by expression of the HIV tetherin antagonist vpu.
Since the gain-of-function point mutant of Udorn/72 demonstrated a deficiency at the stage of release, we performed an experiment to determine if modulation of the antiviral restriction factor tetherin could alter NA budding capability. At this time, we began using the Hong Kong/68 NA since it bears increased homology to budding-capable Japan/57 yet still remains incapable of budding on its own. Tetherin/vpu expression levels were initially set via an HIV VLP budding experiment conducted prior to the influenza virus experiment (data not shown). Once appropriate levels were established, 293T cells were transfected with plasmids for the expression of HA, NA, and either vpu or tetherin. VLPs present in the supernatant were harvested at 48 h posttransfection as described above. Strikingly, VLP release of the normally budding-incapable Hong Kong 68 WT NA was accomplished by the coexpression of vpu (Fig. 8A). Expression of vpu had a similar effect on the release of the G286D Hong Kong/68 NA (data not shown), while tetherin expression reduced budding below the inefficient baseline (Fig. 8A). This was surprising, since we were unable to detect tetherin expression in the 293T cells by Western blotting. This is consistent with previous reports demonstrating low tetherin expression in 293T cells (49). Since tetherin expression was undetectable in 293T cells, we also performed a competition experiment involving the coexpression of both the tetherin and vpu proteins in addition to the Hong Kong/68 NA. In these experiments, we also tested whether a vpu mutant [vpu (2/6)] could accomplish release of Hong Kong/68 NA VLPs. The vpu (2/6) mutant is deficient in CD4 downregulation and was described previously (55). This mutant retains some ability to downregulate tetherin (55). Once again, the expression of either the WT vpu or the vpu (2/6) mutant rescued the release capabilities of the Hong Kong/68 NA in spite of concurrent tetherin overexpression (Fig. 8C). The increase in VLP yield was not as pronounced in this experiment due to competition resulting from concurrent tetherin overexpression. A densitometric analysis of the Western blots in Fig. 8A revealed a 5-fold increase in VLP yield for the expression of vpu without tetherin coexpression (Fig. 8B). Coexpression of tetherin and WT vpu or the vpu (2/6) mutant form resulted in a 1.6- or 2.5-fold increase in VLP yield, respectively (Fig. 8D). These data suggest that tetherin downregulation is the critical component of vpu-mediated NA budding enhancement since the CD4 downregulation contribution can be eliminated.
FIG. 8.

Expression of vpu can modulate NA-budding capability. (A) 293T cells were either untransfected (mock) or transfected with the Hong Kong/68 NA and either vpu WT or tetherin. VLPs were isolated from the supernatants, and VLP yield in the supernatant and expression levels in the whole-cell extracts (WCEs) were determined by Western blotting. (B) Quantification of the Western blotting results in panel A performed by densitometric quantitation of the signal in panel A using an AlphaImager 3400. Values are presented as percent release, which was calculated by the following formula: % release = [signal supernatant/(signal supernatant + signal cells)] × 100. The percent release of HANA VLPs was set at 100%. (C) 293T cells were either left untransfected (mock) or transfected with the Hong Kong/68 NA and either tetherin alone or tetherin and vpu (WT or 2/6 mutant). VLP yield in the supernatant and expression levels in the whole-cell extracts were determined by Western blotting. (D) Quantification of the Western blotting results in panel C performed by densitometric quantitation of the signal in panel C using an AlphaImager 3400. Values are presented as percent release calculated by the following formula: % release = [signal supernatant/(signal supernatant + signal cells)] × 100. The percent release of HANA VLPs was set at 100%. The values to the left are molecular sizes in kilodaltons.
Knockdown of tetherin by siRNA transfection restores the release phenotype in the Hong Kong/68 NA background.
In order to demonstrate that tetherin was specifically involved in the observed phenotypes, we performed a knockdown experiment with HeLa cells. HeLa cells were used in this experiment because they contain high levels of endogenous tetherin and are able to replicate the budding phenotypes we observed in 293T cells (data not shown). As shown in Fig. 9A, siRNA knockdown of tetherin resulted in an increase in VLP yield at 60 h posttransfection. This increase was not observed when a scrambled siRNA sequence was used (Fig. 9A). In order to demonstrate that this result was a consequence of tetherin modulation, a clone was generated that was resistant to the siRNA by site-directed mutagenesis. Silent mutations were introduced at five locations in the siRNA target sequence. As indicated in Fig. 9A, expression of this tetherin siRNA-resistant clone is able to counteract the increase in VLP yield observed upon tetherin knockdown. The tetherin knockdown efficiency was determined to be 50% (Fig. 9B) based on densitometric quantification of the tetherin signal in whole-cell extracts (Fig. 9A) at 40 h posttransfection. These data demonstrate directly that tetherin is capable of modulating the budding capability of the NA.
FIG. 9.
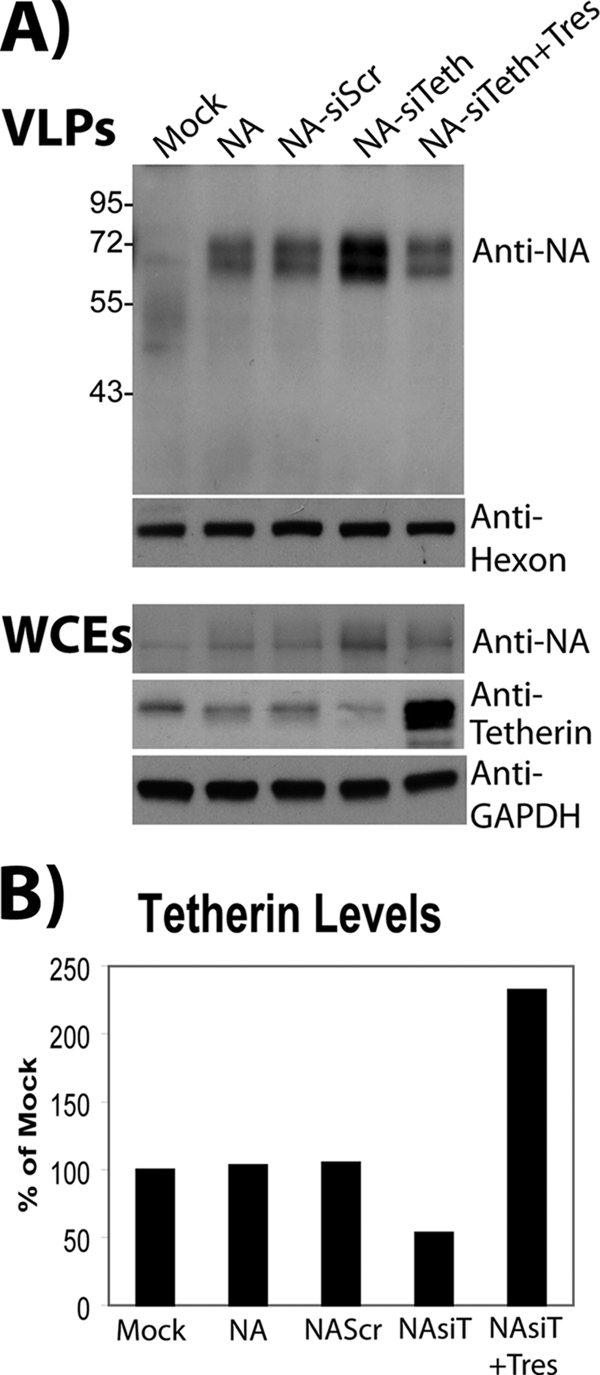
siRNA-mediated knockdown of tetherin enhances NA-dependent budding. (A) HeLa cells were transfected with the Hong Kong/68 NA, the tetherin-resistant clone (as indicated), and 15 nM siRNA representing either a scrambled sequence or the anti-tetherin validated siRNA (as indicated). At 60 h posttransfection, cell lysates and supernatants were harvested and analyzed by Western blotting to determine NA budding efficiency. One of the three plates was harvested at 40 h posttransfection to determine tetherin knockdown efficiency. WCEs, whole-cell extracts. The values to the left are molecular sizes in kilodaltons. (B) Quantification of tetherin knockdown efficiency performed by densitometric quantitation of the tetherin signal in panel A using an AlphaImager 3400. Values are presented as percentages of the value obtained by mock treatment, which represents untransfected HeLa cells.
DISCUSSION
To date, the primary functions of the influenza virus NA have been attributed to its enzymatic activity. These include (i) the cleavage of sialic acid from influenza virus and infected cells to facilitate virus release (46, 47), (ii) the enzymatic cleavage of tracheobronchial airway mucus to allow improved virus penetration of the epithelial cell layer (40), (iii) enhanced infectivity due to enhancement of endosomal traffic by the NA upon entry (58), (iv) WSN-specific enhancement of trypsin-independent cleavage of the HA by recruitment of plasminogen (16), (v) the presence in some N2 NAs of an additional hemadsorption site that has been shown to increase the catalytic activity of the NA toward multimeric substrates (60), and (vi) the fact that NA has also been shown to have an effect on influenza virus virion morphology (24, 42, 65). The studies reported herein and by Lai et al. demonstrate that several NAs possess an additional function in their facilitation of budding (29). This novel function is independent of the protein's enzymatic activity. In addition, we demonstrate that modulation of the antiviral restriction factor tetherin can accomplish the release of otherwise budding-incompetent NA VLPs. Therefore, budding-competent NA proteins should contain an unidentified motif responsible for counteracting tetherin and a novel means of enhancing virus release.
A densitometric comparison (NAVLP/NALysate plus NAVLP) of the budding efficiency of HA alone (released with bacterial NA) and that of HANA VLPs suggests that in strains with budding-competent NA proteins, both the HA and the NA contribute to VLP formation. In the WSN/33 background, the budding efficiency of HA alone was only 56% of the budding efficiency of HANA (data not shown). This discrepancy was not as pronounced in the Udorn background, where the NA is inefficient at the budding process. In this background, the budding efficiency of HA alone was 85% of the budding efficiency of HANA (data not shown). This observation suggests that in the WSN background, both the HA and NA proteins contribute equally to VLP formation. However, we cannot rule out a decrease in the efficiency or specificity of the bacterial NA in these experiments which is required for VLP release in the absence of the influenza virus NA.
In this study, we found that aspartate residue 286 in the N2 NA and aspartate residue 268 in the N1 NA contribute to the budding competency phenotype. However, this residue is not sufficient to confer VLP formation on an otherwise budding-incompetent NA. While the gain-of-function G286D phenotype observed appears to be an intermediate between budding and release of the VLPs, no signal is detectable in the supernatants of transfected cells in the Udorn background. Several attempts were made to accomplish a complete gain of function through Japan/1957-Udorn/72 chimeric NA proteins but have proved unsuccessful to date. Furthermore, it is unlikely that the relatively low enzymatic activity of the Udorn NA (data not shown) explains its budding incapability for two reasons. First, we are able to demonstrate that the budding capability is independent of the enzymatic activity. In addition, neither the Udorn WT, the G286D mutant, nor the chimeric NAs generated demonstrated enhanced budding in the presence of exogenous bacterial NA (data not shown). It is likely that mutation of several residues throughout the NA protein is required to generate a gain-of-function mutant protein. Thin-section analysis by EM reveals a sequestration of the NA VLPs into large vesicular structures. We demonstrated by immunogold labeling that the VLPs contained within these vesicles do indeed contain the NA; however, they do not result in a known tetherin phenotype. These vesicular structures may represent an internalization of the tetherin-restricted VLPs, and experiments are under way to characterize these structures and determine if tetherin plays a role in their formation. Endosomal internalization of tetherin-inhibited HIV particles has been observed previously (45).
It is quite peculiar that the critical sequence contributing to NA budding contains a consensus late domain sequence. The sequence, YPDV in Japan/57 NA and YPDT in WSN NA, mimics a motif known to interact with the cellular proteins ALIX and AP2 (51, 56). A key factor differentiating the NA peptide sequence from all known late domain sequences is its location in the extracellular matrix. This sequence lies on the membrane-proximal side of the globular head domain of the NA, and this localization would allow interaction with the extracellular domains of other transmembrane proteins. In spite of this difference, we are currently exploring a potential role for the ALIX protein and other ESCRT family members in NA-dependent budding. As mentioned previously, several groups have demonstrated that influenza virus budding occurs independently of the ESCRT machinery; however, the complexities of functional redundancy suggest that the ESCRT pathway requires more detailed exploration.
ADDENDUM IN PROOF
Whi1e this article was in press, Tisoncik et al. (J. R. Tisoncik, Y. Guo, K. Cordero, J. Yu, J. Wang, Y. Cao, L. Rong, Virology 8:14, 2011) reported that a residue two amino acids C-terminal to the YPD motif contributed to enhanced release of pseudotype particles (containing the A/Goose/Qinghai/59/2005 [H5N1] HA and either the A/PR/8/34 strain NA or the A/chicken/Henan/12/2004 [H5N1] strain NA).
Acknowledgments
We acknowledge Rong Hai, Taia Wang, Domenico Tortorella, Adolfo García-Sastre, and Zsuzsanna Varga for helpful discussions. In addition, we thank Lily Ngai, Min Chen, Bill Janssen, and Susan VanHorn (Stony Brook University Central Microscopy Imaging EM core facility) for technical support. We thank Patrick Hearing for providing numerous reagents and dl309 adenovirus type V. We thank Klaus Strebel for providing numerous tetherin reagents. We thank Michael J. M. Hitchcock from Gilead Sciences for generously providing oseltamivir carboxylate (GS4071).
This study was supported by RC1 award AI086061-01 (P.P.) and UO1 award AI1074539-02 (P.P.). This study was also supported by award T32 AI07647-06 (M.A.Y.). In addition, this study was partially supported by Targeted Research Opportunity funding for Pandemic Influenza Virus Research from the Stony Brook University School of Medicine (1) and R01 award AI068463 (C.C.).
Footnotes
Published ahead of print on 5 January 2011.
REFERENCES
- 1.Abdul Jabbar, M., and D. P. Nayak. 1987. Signal processing, glycosylation, and secretion of mutant hemagglutinins of a human influenza virus by Saccharomyces cerevisiae. Mol. Cell. Biol. 7:1476-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bieniasz, P. D. 2009. The cell biology of HIV-1 virion genesis. Cell Host Microbe 5:550-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bieniasz, P. D. 2006. Late budding domains and host proteins in enveloped virus release. Virology 344:55-63. [DOI] [PubMed] [Google Scholar]
- 4.Bourmakina, S. V., and A. Garcia-Sastre. 2005. The morphology and composition of influenza A virus particles are not affected by low levels of M1 and M2 proteins in infected cells. J. Virol. 79:7926-7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruce, E. A., P. Digard, and A. D. Stuart. 2010. The Rab11 pathway is required for influenza a virus budding and filament formation. J. Virol. 84:5848-5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruce, E. A., et al. 2009. Budding of filamentous and non-filamentous influenza A virus occurs via a VPS4 and VPS28-independent pathway. Virology 390:268-278. [DOI] [PubMed] [Google Scholar]
- 7.Chen, B. J., G. P. Leser, E. Morita, and R. A. Lamb. 2007. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 81:7111-7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, C., F. Li, and R. C. Montelaro. 2001. Functional roles of equine infectious anemia virus Gag p9 in viral budding and infection. J. Virol. 75:9762-9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Compans, R. W., N. J. Dimmock, and H. Meier-Ewert. 1969. Effect of antibody to neuraminidase on the maturation and hemagglutinating activity of an influenza A2 virus. J. Virol. 4:528-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Douglas, J. L., et al. 2010. The great escape: viral strategies to counter BST-2/tetherin. PLoS Pathog. 6:e1000913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douglas, J. L., et al. 2009. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a {beta}TrCP-dependent mechanism. J. Virol. 83:7931-7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujii, K., J. H. Hurley, and E. O. Freed. 2007. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat. Rev. Microbiol. 5:912-916. [DOI] [PubMed] [Google Scholar]
- 13.Goffinet, C., et al. 2009. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5:285-297. [DOI] [PubMed] [Google Scholar]
- 14.Gómez-Puertas, P., C. Albo, E. Perez-Pastrana, A. Vivo, and A. Portela. 2000. Influenza virus matrix protein is the major driving force in virus budding. J. Virol. 74:11538-11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gómez-Puertas, P., et al. 1999. Efficient formation of influenza virus-like particles: dependence on the expression levels of viral proteins. J. Gen. Virol. 80(Pt. 7):1635-1645. [DOI] [PubMed] [Google Scholar]
- 16.Goto, H., and Y. Kawaoka. 1998. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 95:10224-10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Göttlinger, H. G., T. Dorfman, J. G. Sodroski, and W. A. Haseltine. 1991. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. U. S. A. 88:3195-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gottwein, E., et al. 2003. The Mason-Pfizer monkey virus PPPY and PSAP motifs both contribute to virus release. J. Virol. 77:9474-9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gruenberg, J., and H. Stenmark. 2004. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 5:317-323. [DOI] [PubMed] [Google Scholar]
- 20.Huang, I. C., et al. 2008. Influenza A virus neuraminidase limits viral superinfection. J. Virol. 82:4834-4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hui, E. K., S. Barman, D. H. Tang, B. France, and D. P. Nayak. 2006. YRKL sequence of influenza virus M1 functions as the L domain motif and interacts with VPS28 and Cdc42. J. Virol. 80:2291-2308. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Hui, E. K., S. Barman, T. Y. Yang, and D. P. Nayak. 2003. Basic residues of the helix six domain of influenza virus M1 involved in nuclear translocation of M1 can be replaced by PTAP and YPDL late assembly domain motifs. J. Virol. 77:7078-7092. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Hurley, J. H., and S. D. Emr. 2006. The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu. Rev. Biophys. Biomol. Struct. 35:277-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin, H., G. P. Leser, J. Zhang, and R. A. Lamb. 1997. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 16:1236-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaletsky, R. L., J. R. Francica, C. Agrawal-Gamse, and P. Bates. 2009. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 106:2886-2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kikonyogo, A., et al. 2001. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl. Acad. Sci. U. S. A. 98:11199-11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klimkait, T., K. Strebel, M. D. Hoggan, M. A. Martin, and J. M. Orenstein. 1990. The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 64:621-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.König, R., et al. 2010. Human host factors required for influenza virus replication. Nature 463:813-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai, J. C., et al. 2010. Formation of virus-like particles from human cell lines exclusively expressing influenza neuraminidase. J. Gen. Virol. 91(Pt. 9):2322-2330. [DOI] [PubMed] [Google Scholar]
- 30.Lentz, M. R., R. G. Webster, and G. M. Air. 1987. Site-directed mutation of the active site of influenza neuraminidase and implications for the catalytic mechanism. Biochemistry 26:5351-5358. [DOI] [PubMed] [Google Scholar]
- 31.Li, F., C. Chen, B. A. Puffer, and R. C. Montelaro. 2002. Functional replacement and positional dependence of homologous and heterologous L domains in equine infectious anemia virus replication. J. Virol. 76:1569-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li, S., et al. 1993. Chimeric influenza virus induces neutralizing antibodies and cytotoxic T cells against human immunodeficiency virus type 1. J. Virol. 67:6659-6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Licata, J. M., et al. 2003. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 77:1812-1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mansouri, M., et al. 2009. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi's sarcoma-associated herpesvirus. J. Virol. 83:9672-9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Margottin, F., et al. 1998. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1:565-574. [DOI] [PubMed] [Google Scholar]
- 36.Marsh, G. A., R. Hatami, and P. Palese. 2007. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J. Virol. 81:9727-9736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin-Serrano, J. 2007. The role of ubiquitin in retroviral egress. Traffic 8:1297-1303. [DOI] [PubMed] [Google Scholar]
- 38.Martin-Serrano, J., and M. Marsh. 2007. ALIX catches HIV. Cell Host Microbe 1:5-7. [DOI] [PubMed] [Google Scholar]
- 39.Martin-Serrano, J., T. Zang, and P. D. Bieniasz. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7:1313-1319. [DOI] [PubMed] [Google Scholar]
- 40.Matrosovich, M. N., T. Y. Matrosovich, T. Gray, N. A. Roberts, and H. D. Klenk. 2004. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J. Virol. 78:12665-12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medina, G., et al. 2005. The functionally exchangeable L domains in RSV and HIV-1 Gag direct particle release through pathways linked by Tsg101. Traffic 6:880-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mitnaul, L. J., M. R. Castrucci, K. G. Murti, and Y. Kawaoka. 1996. The cytoplasmic tail of influenza A virus neuraminidase (NA) affects NA incorporation into virions, virion morphology, and virulence in mice but is not essential for virus replication. J. Virol. 70:873-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyagi, E., A. J. Andrew, S. Kao, and K. Strebel. 2009. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. U. S. A. 106:2868-2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neil, S. J., V. Sandrin, W. I. Sundquist, and P. D. Bieniasz. 2007. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe 2:193-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neil, S. J., T. Zang, and P. D. Bieniasz. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425-430. [DOI] [PubMed] [Google Scholar]
- 46.Palese, P., and R. W. Compans. 1976. Inhibition of influenza virus replication in tissue culture by 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid (FANA): mechanism of action. J. Gen. Virol. 33:159-163. [DOI] [PubMed] [Google Scholar]
- 47.Palese, P., K. Tobita, M. Ueda, and R. W. Compans. 1974. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 61:397-410. [DOI] [PubMed] [Google Scholar]
- 48.Pattnaik, A. K., D. J. Brown, and D. P. Nayak. 1986. Formation of influenza virus particles lacking hemagglutinin on the viral envelope. J. Virol. 60:994-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perez-Caballero, D., et al. 2009. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139:499-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pincetic, A., G. Medina, C. Carter, and J. Leis. 2008. Avian sarcoma virus and human immunodeficiency virus, type 1 use different subsets of ESCRT proteins to facilitate the budding process. J. Biol. Chem. 283:29822-29830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Puffer, B. A., L. J. Parent, J. W. Wills, and R. C. Montelaro. 1997. Equine infectious anemia virus utilizes a YXXL motif within the late assembly domain of the Gag p9 protein. J. Virol. 71:6541-6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rossman, J. S., et al. 2010. Influenza virus m2 ion channel protein is necessary for filamentous virion formation. J. Virol. 84:5078-5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rossman, J. S., X. Jing, G. P. Leser, and R. A. Lamb. 2010. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142:902-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakuma, T., T. Noda, S. Urata, Y. Kawaoka, and J. Yasuda. 2009. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 83:2382-2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schubert, U., and K. Strebel. 1994. Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J. Virol. 68:2260-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strack, B., A. Calistri, S. Craig, E. Popova, and H. G. Göttlinger. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689-699. [DOI] [PubMed] [Google Scholar]
- 57.Strebel, K., T. Klimkait, and M. A. Martin. 1988. A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science 241:1221-1223. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki, T., et al. 2005. Sialidase activity of influenza A virus in an endocytic pathway enhances viral replication. J. Virol. 79:11705-11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Terwilliger, E. F., E. A. Cohen, Y. C. Lu, J. G. Sodroski, and W. A. Haseltine. 1989. Functional role of human immunodeficiency virus type 1 vpu. Proc. Natl. Acad. Sci. U. S. A. 86:5163-5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uhlendorff, J., T. Matrosovich, H. D. Klenk, and M. Matrosovich. 2009. Functional significance of the hemadsorption activity of influenza virus neuraminidase and its alteration in pandemic viruses. Arch. Virol. 154:945-957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Damme, N., et al. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang, D., et al. 2010. The lack of an inherent membrane targeting signal is responsible for the failure of the matrix (M1) protein of influenza A virus to bud into virus-like particles. J. Virol. 84:4673-4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Willey, R. L., F. Maldarelli, M. A. Martin, and K. Strebel. 1992. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 66:7193-7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuan, B., X. Li, and S. P. Goff. 1999. Mutations altering the Moloney murine leukemia virus p12 Gag protein affect virion production and early events of the virus life cycle. EMBO J. 18:4700-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang, J., A. Pekosz, and R. A. Lamb. 2000. Influenza virus assembly and lipid raft microdomains: a role for the cytoplasmic tails of the spike glycoproteins. J. Virol. 74:4634-4644. [DOI] [PMC free article] [PubMed] [Google Scholar]