

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) lytic infection increases the expression of viral and human interleukin-6 (vIL-6 and hIL-6, respectively), an important factor for cell growth and pathogenesis. Here, we report genome-wide analysis of viral RNA targets of KSHV ORF57 by a novel UV-cross-linking and immunoprecipitation (CLIP) assay. We identified 11 viral transcripts as putative ORF57 targets and demonstrate that vIL-6 mRNA is an authentic target of ORF57. Disrupting the ORF57 gene in the KSHV genome leads to inefficient expression of vIL-6. With transient transfection, the expression of vIL-6 could be enhanced greatly in the presence of ORF57 in a dose-dependent manner. We found that the open reading frame (ORF) region of vIL-6 RNA contains an MRE (MTA [ORF57]-responsive element) composed of two motifs, MRE-A and MRE-B, and binding of ORF57 to these two motifs stabilizes vIL-6 RNA and promotes vIL-6 translation. We demonstrate that vIL-6 MRE-B bears an miR-1293 binding site and that, mechanistically, ORF57 competes with miR-1293 for the same binding site to interact with vIL-6 RNA, thereby preventing vIL-6 RNA from association with the miR-1293-specified RNA-induced silencing complex (RISC). Consistent with this, ORF57 also interacts with an miR-608 binding site in the hIL-6 ORF and prevents miR-608 repression of hIL-6. Collectively, our results identify a novel function of ORF57 in being responsible for stabilization of viral and human IL-6 RNAs and the corresponding enhancement of RNA translation. In addition, our data provide the first evidence that a tumor virus may use a viral protein to interfere with microRNA (miRNA)-mediated repression of an miRNA target to induce cell proliferation and tumorigenesis during virus infection.
Kaposi's sarcoma-associated herpesvirus (KSHV) or human herpesvirus 8 (HHV-8) is a lymphotropic DNA tumor virus, and when it infects B cells, it leads to development of body cavity-based B cell lymphoma and multicentric Castleman's disease (11, 32). A hallmark indication of the infection is the increased expression of both viral interleukin-6 (vIL-6) and human interleukin-6 (hIL-6), which are used for clinical diagnosis and therapeutic targeting (2, 16, 31, 33, 36, 37). Although the causes of increased expression of vIL-6 and hIL-6 during KSHV infection are not fully understood, increased IL-6 appears to be important to maintain cancer cell proliferation (6) and has been ascribed to transcriptional activation (6, 8, 9). Other studies indicate that various RNA elements in the 3′ untranslated region (UTR) of hIL-6 mRNA contribute to rapid decay of hIL-6 mRNA and regulate hIL-6 expression primarily at the posttranscriptional level. These include an AU-rich element (ARE) interacting with AUF1 (38) and tristetraprolin (40), a non-ARE element interacting with Zc3h12a endonuclease (30), and microRNA (miRNA) seed matches binding let-7 (15) and miR-26 (17).
An miRNA is a noncoding RNA that finely regulates the expression of target genes at the posttranscriptional level. In general, miRNAs are 18 to 25 nucleotides in length and function in an RNA-induced silencing complex (RISC) by base pairing with complementary nucleotide sequences (seed matching) of target mRNAs (4) to inhibit translation (partial matching) or to induce mRNA decay (complete matching) (3, 41). Although miRNA binding sites are common in the 5′ and 3′ UTRs of target RNAs, the open reading frame (ORF) regions of vIL-6 and hIL-6 contain a binding site for miR-1293 and miR-608, respectively, and are subject to miRNA-mediated regulation (J.-G. Kang et al., submitted for publication).
During lytic infection, the KSHV genome encodes a multifunctional protein of 455 amino acid (aa) residues from its open reading frame 57 (ORF57) that is essential for virus production (13, 23, 27). ORF57 functions primarily at the posttranscriptional level and enhances mRNA transcript accumulation (thus giving ORF57 the name MTA) of intronless viral RNAs (12, 18) and RNA splicing of intron-containing viral RNAs (24). Previous studies, including those from our lab, had demonstrated that KSHV ORF57 promotes the RNA accumulation of intronless viral RNA transcripts of ORF8 (glycoprotein B), ORF47 (glycoprotein L), ORF59 (DNA polymerase processivity factor), mCP (minor capsid protein), and PAN (polyadenylated nuclear RNA) (5, 13, 18, 23, 25, 35) and stimulates the expression of intron-containing viral RNA transcripts of ORF50, ORF56, K8, and K8.1 (24). Through preferential interaction with low-abundance viral pre-mRNAs and several components of cellular splicing machinery, ORF57 promotes viral RNA splicing (22, 24). Several studies have suggested that ORF57 accumulates intronless viral RNAs by enhancing RNA export through a CRM1-independent pathway (28) or, more recently, by interacting with other components in the TREX complex (transcription-export complex) (5). However, the data from our lab (21, 25) and others (35) indicate that the interaction between ORF57 and Aly/REF, an essential component of TREX complex, is not necessary for ORF57-mediated accumulation of ORF59 transcripts. Thus, it remains to be understood how ORF57 accumulates intronless viral RNA transcripts.
To better understand the function of ORF57 in the promotion of viral gene expression, we searched for ORF57 targets by conducting genome-wide UV-cross-linking and immunoprecipitation (CLIP) assays using an anti-ORF57 antibody to identify viral RNA transcripts associated with ORF57 protein in B cells with KSHV lytic infection. The principle supporting this approach is that when a protein intimately binds to RNA, an intermolecular cross-link can be generated by UV irradiation, without chemical modification of either the RNA or the protein. The cross-linking is performed by irradiating cells containing native RNA-protein complexes with short-wavelength UV light. Covalent cross-links form when the nucleotides or amino acids in the RNA and protein are photochemically converted to reactive species. In combination with immunoprecipitation and cDNA clone screening, we were able to specifically pull down RNA-protein complexes cross-linked with ORF57 and, thereby, identify unique RNA motifs that interact with ORF57 protein. In this study, we report the viral RNA targets of ORF57 and demonstrate that ORF57 interacts with an miRNA binding site on the vIL-6 RNA and prevents miRNA-mediated RNA degradation and translational repression of vIL-6. Thus, we provide functional evidence as to how ORF57 regulates the expression of vIL-6 during KSHV lytic infections of B cells.
MATERIALS AND METHODS
Cells.
HEK293 and HeLa cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (FBS). HCT116 cells were cultured in McCoy's 5A medium with 10% FBS. JSC-1 cells (KSHV+/Epstein-Barr virus positive [EBV+]) and BCBL-1 cells (KSHV+) were cultured in RPMI 1640 containing 10% FBS and were induced with sodium butyrate (3 mM) for lytic infection. HEK293 cell lines stably harboring the KSHV Bac36 wild-type (wt) or Bac36 Δ57 null genome (23) were induced with 1 mM valproate (VA) or sodium butyrate (3 mM) for lytic infection. Doxycycline (DOX)-inducible TREx BCBL-1 RTA and vector control cell lines were cultured as described previously (34).
Cross-linking immunoprecipitation (CLIP) assay.
Approximately 1 × 107 JSC-1 cells were induced with 3 mM butyrate for 24 h and were washed twice with 1× phosphate-buffered saline (PBS) before UV-cross-linking at 480,000 μJ/cm2 as described previously (25, 44). Briefly, a cell lysate containing protein-RNA complexes was prepared from the cell pellet by direct lysis in 1 ml of 1× radioimmunoprecipitation assay (RIPA) buffer. Protein A agarose beads (Upstate Biotechnology, Lake Placid, NY) were washed with 1× immunoprecipitation (IP) buffer (25 mM HEPES [pH 7.5], 150 mM NaCl, 0.5 mM EDTA, 1 mM EGTA, 10% glycerol, 0.1% NP-40, 1 mM NaF, 1 mM 2-glycerophosphate, 1 mM Na3VO4, 1× Complete Mini EDTA-free protease inhibitor cocktail [Roche, Indianapolis, IN]) and were coated with an anti-ORF57 antibody (25) or control IgG. The cell lysates were precleaned 3 times with the control IgG-coated beads and immunoprecipitated with anti-ORF57 antibody-coated beads at 4°C overnight. The protein-RNA complexes on the beads after IP were washed 3 times with 1× IP buffer and were briefly (for a few seconds) digested with RNase T1 (0.005 U; Ambion, Austin, TX), followed by treatment with proteinase K (0.2 mg/ml; Roche). RNA was extracted with a phenol-chloroform mixture and precipitated in ethanol. RNA was dephosphorylated with Antarctic phosphatase (NEB, Ipswich, MA) and ligated to a 3′ linker (oNP21; 5′-rUrUrUAACCGCGAATTCCAG/3AmMC6/-3′; IDT, Coralville, IA) which was labeled with [γ-32P]ATP using T4 polynucleotide kinase (Invitrogen, Carlsbad, CA). The linker-ligated RNA was resolved in a 15% denaturing polyacrylamide gel (PAGE) and eluted from the gel in probe elution buffer (Ambion). Smart reverse transcription (RT) was performed using BD Power Script reverse transcriptase (Clontech, Mountain View, CA) and a random primer (Applied Biosystems) in the presence of Smart primer (5′-AAGCAGTGGTATCAACGCAGAGTACGCGGG-3′) as instructed by Clontech's protocol. The cDNAs were amplified by PCR, cloned into pCR II-Topo vectors (Invitrogen), and sequenced by Agencourt Bioscience Corporation (Beverly, MA). The inserted sequences were extracted and aligned against the KSHV genome sequence (39).
CLIP and RT-PCR.
CLIP assays were conducted for butyrate-treated (3 mM for 24 h) JSC-1 cells with or without UV irradiation as described above in the CLIP assay, with omission of RNase T1 digestion in order to recover the targeted RNAs as full-length RNAs. Total RNA used as an input control was isolated from the corresponding cell lysates used for IP after cross-linking. Reverse transcription-PCR (RT-PCR) was performed by using a vIL-6-specific primer pair (oNP35, 5′-TACTCAGGATCC/CTTATCGTGGACGTCAGGAGTCA-3′, and oNP29, 5′-CTGGTCGGTTCACTGCTGGT-3′) or an hIL-6-specific primer pair (oJGK24, 5′-ATACGACGCGGCCGCACC/ATGAACTCCTTCTCCACAAG-3′, and oJGK27, 5′-TCAGAAGCTC/CACAGCTCTGGCTTGTTCCT-3′) on DNase-treated RNAs which were isolated from the input cell lysates or from the CLIP complexes obtained with an anti-ORF57 antibody or rabbit IgG (a negative control).
Plasmids.
pEGFP-N1 (Clontech) was used for green fluorescent protein (GFP) expression. ORF57-FLAG (pVM7) was used for ORF57 expression (25). vIL-6-3×FLAG (pNP4), miR-re vIL-6-3×FLAG (pJGK10), and hIL-6-3×FLAG (pJGK6) plasmids were used for vIL-6, mutant vIL-6, and hIL-6 expression, respectively (Kang et al., submitted). The various vIL-6 deletion mutants were constructed from pNP4 by overlapping PCR and expressed as 3×FLAG-tagged proteins from the p3×FLAG-CMV-14 vector: plasmid pJGK1 for ΔB, pJGK2 for ΔC, pJGK3 for ΔD, and pJGK5 for ΔE. The hemagglutinin (HA)/FLAG-human Ago2 (hAgo2) plasmid was purchased from Addgene (Cambridge, MA). All plasmids were confirmed by sequencing.
Western blot analysis.
Protein samples were prepared by lysis of cells in 1× RIPA buffer and the same volume of 2× SDS-protein sample buffer containing freshly added 2-mercaptoethanol. The following antibodies were used in the Western blot analysis: rabbit polyclonal or mouse monoclonal anti-ORF57 antibodies against synthetic peptide (amino acids 119 to 132 of ORF57), monoclonal anti-vIL-6 antibody kindly provided by Giovanna Tosato (NCI/NIH), polyclonal anti-vIL-6 antibody from John Nicholas (Johns Hopkins University), polyclonal anti-Ago2 (Upstate Biotechnology), monoclonal anti-β-tubulin (Sigma), and monoclonal anti-FLAG (M2; Sigma), together with corresponding horseradish peroxidase-conjugated secondary antibodies (Sigma). The signal on blots was detected with a West Pico or Femto chemiluminescence substrate (Thermo Scientific, Rockford, IL).
Northern blotting.
Total RNA was isolated from butyrate-induced JSC-1 and BCBL-1 cells and VA-induced Bac36 stable cells with a wild-type (wt) or ORF57-null (Δ57) KSHV genome. Each sample, containing 5 μg of total RNA, was mixed with NorthernMax formaldehyde load dye (Ambion), denatured at 75°C for 10 min, separated on a 1% agarose gel, and transferred onto a nylon membrane. After 1 h of prehybridization, hybridization was carried out for 24 h at 42°C in Super-Hyb hybridization buffer (Sigma). The antisense oligonucleotides oNP28 (5′-TGGGTGGACTGTAGTGCGTC-3′) for vIL-6 and oZMZ270 (5′-TGAGTCCTTCCACGATACCAAA-3′) for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RNA were labeled with [γ-32P]ATP and were used for the hybridizations. After hybridization, the membrane was washed once with a 2× SSPE (1× SSPE is 0.18 M NaCl, 10 mM NaH2PO4, and 1 mM EDTA [pH 7.7])-0.5% SDS solution for 5 min at room temperature and twice with 0.2× SSPE-0.1% SDS for 20 min at 42°C and exposed to a phosphorimager screen.
Transient cotransfection and protein purification.
Cells were cotransfected with the indicated combinations of plasmids in each figure using Lipofectamine 2000 (Invitrogen) for Western blot analysis 24 h after transfection. Pre-miR-1293 was transfected using siPORT-NeoFX (Ambion). To purify human Ago2 (hAgo2), the plasmid expressing HA/FLAG-tagged hAgo2 (Addgene) was transfected into HEK293 cells, and a cell lysate was prepared 48 h after transfection in 1× RIPA buffer. HA/FLAG-hAgo2 was immunopurified first with an anti-HA affinity gel and then by elution with HA peptide (Sigma).
RNA-protein pulldown assay.
RNA oligomers labeled with biotin at the 5′ end (synthesized by IDT) were immobilized on NeutrAvidin beads (Thermo Scientific). The sequences of the RNA oligomers are presented in Fig. 3A. oJGK50 (5′-biotin-AAGAAUCUAGAUGCAAUAACCACCCCUGA-3′) is an RNA oligomer harboring a putative MRE of hIL-6. The cell lysates indicated in each figure were applied to oligomer-immobilized beads in 1× binding buffer (20 mM Tris [pH 7.5], 200 mM NaCl, 6 mM EDTA, 5 mM potassium fluoride, 5 mM 2-glycerophosphate, and 1 tablet of protease inhibitors [Roche]/10 ml). Pulldown was conducted at 4°C overnight with rotation. Samples were washed 3 times with 1× binding buffer. SDS sample buffer was directly applied to the beads.
IP.
For immunoprecipitation (IP) using anti-FLAG or anti-HA affinity gel (Sigma), a lysate of HEK293 cells ectopically expressing vIL-6-FLAG, ORF57-FLAG, and HA/FLAG-hAgo2 was prepared in 1× RIPA buffer. Other IP conditions were the same as described for the CLIP assay.
In vitro transcription.
DNA templates for in vitro transcription of full-length vIL-6 were amplified from pNP4 by PCR using a primer pair of T7 chimeric oJGK46 (5′-TAATACGACTCACTATAGGG/AACAGCCACC/ATGTGCTGGTTCAAGTTG-3′) and oJGK47 oligomer dT (T30/2 stop codons/vector sequence, 5′-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTT/TCACTA/CTTGTCATCGTCATCCTT-3′), which provides a poly(A) tail in mRNA. The 3′ halves of wt and deletion mutant vIL-6 were prepared for in vitro transcription with the Riboprobe system (Promega, Madison, WI) from individual plasmids by PCR using a T7 chimeric primer pair, oJGK48 (5′-TAATACGACTCACTATAGGG/ AACAGCCACCATG/TCAGTGATAAACGTGGACG-3′) and oJGK47.
In vitro translation assay.
In vitro translation in the presence of an miRNA duplex was performed using a modification of previously published protocols (29, 45). The 5′ ends of the miR-1293 duplex and the nonspecific control small interfering RNA (siRNA; Dharmacon, Lafayette, CO) were phosphorylated with T4 kinase (Invitrogen). Equal amounts (12.5 nM each) of vIL-6 mRNA and firefly luciferase mRNA were mixed with 12.5 nM miRNA duplex, denatured at 70°C for 3 min, and cooled down to room temperature to anneal the miRNA to the target mRNA. Firefly luciferase mRNA served as an internal control. Translation mix (nuclease-treated rabbit reticulocyte lysate [Promega], RNase inhibitor, amino acids [−Met] mix, and [35S]Met [Perkin-Elmer, Shelton, CT]) was added in accordance with the manufacturer's instructions. The total reaction volume was 10 μl. Translation was performed at 30°C for 20 min and stopped on ice by the addition of SDS sample buffer. Translated proteins were resolved in a 4%-12% SDS-PAGE gel (Invitrogen) and transferred onto a membrane for capturing images on a phosphorimager screen or X-ray film.
Quantitative RT-PCR (qRT-PCR).
The primer pair for vIL-6 amplification was previously described (47). The primer pair oZMZ269 and oZMZ270 (24) was used for GAPDH amplification. First-strand cDNA was synthesized from 100 ng of total RNA using random hexamers and SuperScript II reverse transcriptase (Invitrogen). The quantitative PCR (qPCR) was carried out using Platinum SYBR green qPCR SuperMix-UDG (Invitrogen) and a Cepheid Smart Cycler (Sunnyvale, CA).
For detection of endogenous miR-1293, a TaqMan microRNA assay (Applied Biosystems) was used in accordance with the manufacturer's protocol. The threshold cycle (CT) values of qRT-PCR data from 3 repeats were analyzed by the 2−ΔΔCT method (20) and are presented as bar graphs with means ± standard deviations (SD).
Confocal microscopy.
Cells grown on glass coverslips were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 in PBS. After being washed in PBS, cells were blocked with 2% bovine serum albumin (BSA) in PBST (PBS containing 0.05% Tween 20). Indicated antibodies were applied in blocking solution. Alexa Fluor dye-conjugated secondary antibodies were used. Fluorescence images were collected using a fluorescence microscope or a confocal microscope. Confocal fluorescence optical slices, 2.0 μm in thickness, were acquired using a Zeiss LSM510 Meta (Carl Zeiss MicroImaging, Inc., Thornwood, NY) microscope equipped with a 20× plan-apochromat (numerical aperture [NA], 0.8) object lens.
Protein transfection.
Protein transfection was carried out using Chariot reagent (Active Motif, Carlsbad, CA) according to the manufacturer's instructions. Briefly, cells at 5 × 105 cells were transfected with 2 μg of protein. Samples were collected 24 h after transfection. After being washed twice in PBS buffer, the number of cells was determined. Protein and RNA were isolated from the same number of cells (4 × 105) using the Paris kit (Ambion) and examined by Western blotting and by real-time RT-PCR, respectively.
Enzyme-linked immunosorbent assay (ELISA).
Before the assay, the medium from KSHV-infected cells was cleared by centrifugation. The levels of individual cytokines in the supernatant were determined by single- or multianalyte ELISArray kits (SABiosciences, Frederick, MD).
RESULTS
Identification of genome-wide viral RNA targets of KSHV ORF57 by CLIP assays.
To search for genome-wide viral RNA targets of ORF57 in KSHV-infected cells, we established a very reliable and efficient CLIP assay modified from our previous publication where we reported that ORF57 protein associates with a viral ORF59 RNA and can be cross-linked to the associated ORF59 RNA in cells by UV irradiation (25). In this study, the RNA-protein complexes pulled down with anti-ORF57 antibody in the modified CLIP assay were briefly digested with RNase T1 to remove the protein-free parts of the RNAs within the complexes and then thoroughly digested with proteinase K to remove ORF57 protein from the remaining RNA fragments. The resultant ORF57-bound RNA fragments protected from RNase digestion were extracted and ligated to a 32P-labeled 3′ adaptor. Following gel purification, the ligated RNA fragments were reverse transcribed, amplified, and cloned for sequence analysis (Fig. 1). After screening nearly 9,500 bacterial clones, we found a cDNA insert in 5,791 clones, and 199 of them came from 30 viral transcripts (Table 1; see also Table S1 in the supplemental material). We chose viral transcripts with the same sequence cloned twice or more as putative viral targets of ORF57. With this cutoff, we were able to identify 11 viral transcripts as putative ORF57 targets. Among them, ORF56, ORF59, and PAN RNA were authentic ORF57 targets previously reported in other studies (18, 25, 26).
FIG. 1.

Isolation of RNAs in ORF57-RNA complexes obtained from butyrate-activated JSC-1 cells. (A) Key steps of the ORF57 CLIP assay. Chemically induced JSC-1 cells were UV irradiated. The protein-RNA complexes cross-linked in the cell lysates were pulled down by an anti-ORF57 antibody and partially (for a few seconds) digested with RNase T1, followed by proteinase K digestion. ORF57-bound RNA remaining from the RNase digestion was extracted, dephosphorylated, ligated to a 32P-labeled 3′ RNA adaptor, gel purified, amplified, cloned, and sequenced. (B) ORF57 in the pulldown assay. One percent of the input and 3% of the immunoprecipitate were separated in the gel. The lower band indicates a caspase-7-cleaved ORF57 (22). (C) RNAs in the ORF57 pulldowns performed by 3′ RNA ligation assay (indicated by a vertical line on the gel) that were used to create a cDNA library.
TABLE 1.
Genome-wide viral RNA targets of ORF57 identified from CLIP assaysa
| Gene name | No. of clones |
|---|---|
| ORF4 | 1 |
| ORF6 | 1 |
| ORF8 | 2b |
| ORF9 | 1 |
| K2 (vIL-6) | 18 |
| K4 | 9 |
| K6 | 2b |
| PAN | 91 |
| ORF17 | 6 |
| ORF25 | 2b |
| ORF29a | 1 |
| ORF35 | 1 |
| ORF37 | 1 |
| ORF38 | 1 |
| ORF43 | 1 |
| ORF44 | 1 |
| ORF46 | 1 |
| ORF50 | 2 |
| ORF52 | 6 |
| ORF56/57 | 4 |
| K9 | 4 |
| K10 | 2b |
| vIRF-2 | 1 |
| ORF58/59 | 9 |
| ORF61 | 2b |
| ORF62 | 1 |
| ORF63 | 1 |
| Kaposin | 21 |
| ORF73 | 5 |
| K14 | 1 |
| Total | 199 |
The viral RNA sequences associated with ORF57 in CLIP assays were cloned, sequenced, and aligned against the KSHV genome. Results in the table are from 5 CLIP assays.
Two different sequences from different regions of the same transcript (see Table S1 in the supplemental material).
Identification of viral IL-6 RNA as a major target of ORF57.
Interestingly, we found that one of the major ORF57 targets was viral IL-6 (vIL-6 or K2) (Table 1), the viral homolog of hIL-6. This result is intriguing because vIL-6 is a critical factor for maintaining tumor cell proliferation during KSHV infection (2, 16, 31). The interaction between vIL-6 mRNA and ORF57 was verified using CLIP/RT-PCR (Fig. 2A), where vIL-6 RNA was specifically pulled down by using an anti-ORF57 antibody under UV-cross-linking conditions. In contrast, an abundant cellular RNA, coding for GAPDH, was not associated with ORF57. Similarly to results reported previously (36, 42), vIL-6 expression at both RNA and protein levels was increased in lymphoma-derived B cell lines JSC-1 and BCBL-1 (Fig. 2B) with lytic KSHV induction. Bac36 wt cells with stable transfection of a wt KSHV genome also responded to virus induction for vIL-6 expression, whereas Bac36 Δ57 cells stably transfected with an ORF57-null KSHV genome (23) did not (Fig. 2B). These results were confirmed further by vIL-6 immunofluorescence assays (Fig. 2C), showing a remarkable number of cells being activated for vIL-6 expression in Bac36 wt cells but only a few in Bac36 Δ57 cells (Fig. 2C, bar graph). We found that cells expressing vIL-6 in the cytoplasm always coexpressed ORF57 in the nucleus (Fig. 2D). These data indicate that vIL-6 expression in viral lytic infection is associated with ORF57.
FIG. 2.

vIL-6 expression depends on viral ORF57. (A) ORF57 is associated with vIL-6 RNA during lytic KSHV infection. RT-PCR was performed by using a vIL-6- or GAPDH-specific primer pair on RNA isolated from input lysates or from the CLIP complexes obtained with an affinity-purified anti-ORF57 antibody from butyrate-treated JSC-1 cells. (B to D) Expression of vIL-6 mRNA and protein depends on ORF57 during lytic KSHV infection. Total RNA obtained from BCBL-1 and JSC-1 cells induced with butyrate (Bu, 3 mM) or from Bac36 cells with a wt KSHV genome (Bac36 wt) and Bac36 Δ57 cells with an ORF57-null KSHV genome (23) induced with valproate (VA, 1 mM) was examined by Northern blotting (NB) for vIL-6 mRNA (B, top). GAPDH mRNA served as a loading control. Total cell extracts from the corresponding cells were examined by Western blotting (WB) with a polyclonal anti-vIL-6 antibody (B, bottom). Bac36 wt and Bac36 Δ57 cells (C) with or without 24 h of butyrate (3 mM) induction were stained for vIL-6 expression using a rabbit polyclonal anti-vIL-6 antibody and then anti-rabbit IgG Alexa Fluor-546 and examined by confocal microscopy. As a Bac genome has a GFP gene insertion, all stable cells expressed GFP independently of induction. Bar graphs below the confocal images indicate numbers of vIL-6+ cells per 1,000 Bac36 cells with or without butyrate induction. Bars represent means ± SD (n = 5). Bac36 wt cells after butyrate induction were also stained with a mouse monoclonal anti-ORF57 antibody in combination with anti-mouse IgG Alexa Fluor-647 and examined by confocal microscopy for ORF57 and vIL-6 coexpression (D). ORF57 in panel D was converted to green during the imaging process. Bars (D), 10 μm. (E and F) Induction of vIL-6 expression by ORF57 in HEK293 (E) and HeLa and HCT116 (F) cells. Protein samples were immunoblotted 24 h after cotransfection of a control GFP plasmid or a vIL-6 plasmid in the presence or absence of an ORF57 expression vector. (G) ORF57 promotes vIL-6 expression in a dose-dependent manner. HEK293 cells were cotransfected with 100 ng of a vIL-6-expressing vector and an indicated amount of an ORF57-FLAG-expressing vector and immunoblotted with anti-FLAG 24 h posttransfection. One representative gel of 3 repeats is presented. The bar graph shows relative levels of vIL-6 in each sample found by Western blotting after being normalized to β-tubulin. Bars represent means ± SD (n = 3).
Next, we demonstrated that ORF57 promotes vIL-6 expression. The ORF57-mediated enhancement of vIL-6 expression is independent of other viral factors as indicated by cotransfection of several human epithelial cell lines. From our CLIP assays, we found that ORF57 interacts with the coding region of vIL-6; therefore, when designing expression vectors for vIL-6, we included only the coding region of vIL-6 mRNA. When cotransfecting these expression vectors with ORF57, we found that ORF57 by itself was capable of efficiently promoting vIL-6 production in HEK293 (Fig. 2E) and HeLa and HCT116 (Fig. 2F) cells but not the production of the GFP control (Fig. 2E). We also found, in HEK293 cells, that the increase in vIL-6 production was dependent on the amount of ORF57 present (Fig. 2G). The direct effects of ORF57 protein on vIL-6 expression in vivo were also examined by transfection of recombinant ORF57 protein into BCBL-1 and JSC-1 cells. Similarly, direct transfection of ORF57 protein into these cells stabilized vIL-6 RNA and increased vIL-6 protein expression (see Fig. S1A and B in the supplemental material), whereas the control (transfection of β-galactosidase protein) had no effect on vIL-6 expression. Although the transfected ORF57 protein appeared toxic to the cells as indicated by tubulin level (Fig. S1A), we observed a 2-fold increase in both vIL-6 RNA and protein levels due to the presence of ORF57 protein compared to the β-galactosidase controls. Together, these data indicate that the enhanced expression of vIL-6 depends on viral ORF57.
vIL-6 RNA has an ORF57-responsive element that contains two ORF57 binding sites, one of them overlapping an miR-1293 seed match.
We then set out to determine the vIL-6 sequence that ORF57 specifically interacts with. Our ORF57 CLIP assay allowed us to identify an RNA sequence protected by bound ORF57 protein from partial RNase digestion, and therefore, we could determine the specific vIL-6 region that interacts with ORF57 (Fig. 3A). We named this sequence of the vIL-6 RNA an MTA-responsive element (MRE). Subsequent RNA folding analysis indicated that the vIL-6 MRE is composed of two separate stem-loop structures, MRE-A and MRE-B (Fig. 3B). Therefore, we designed a series of RNA oligomers (oNP41 to oNP44) covering the majority of the MRE and examined each RNA oligomer for its binding affinity to ORF57 protein. When ORF57 was expressed in either HEK293 or JSC-1 cells, it was observed to interact with oNP42, oNP43, and oNP44 but not with oNP41 (Fig. 3C). As oNP43 covers a partial sequence of oNP44 and showed only a weak affinity for ORF57, we designated the vIL-6 RNA sequences covered by oNP42 and oNP44 active MRE-A (indicated in blue) and MRE-B (indicated in red) motifs, respectively. Interestingly, the MRE-B motif was also identified as an miR-1293 binding site that regulates vIL-6 expression and shows no similarity to any other cellular miRNA seed matches (J.-G. Kang and Z.-M. Zheng, unpublished observation). Together, these data suggest that vIL-6 RNA has at least two distinct sites for ORF57 binding and that the MRE-B motif is a dual binding site for both miR-1293 and ORF57.
FIG. 3.

Sequence motif of vIL-6 RNA in association with ORF57 protein. (A) Sequences of 18 vIL-6 cDNA clones from CLIP assays. Numbers above the sequences are the nucleotide positions in the KSHV genome (39). Numbers in parentheses indicate the same sequences in multiple clones. Lines immediately below the sequences, along with dotted vertical lines, are where biotin-labeled RNA oligomers oNP41 to oNP44 were derived for RNA pulldown assays. (B) Secondary structure of the identified vIL-6 MRE. Colored lines on stem-loops of the MRE are the most active regions (labeled as MRE-A and MRE-B) in ORF57 binding and functions. (C) The mapped regions of the MRE in vIL-6 RNA interact with KSHV ORF57. Cell lysates from HEK293 cells transfected with an ORF57-FLAG expression vector or from JSC-1 cells induced with 3 mM butyrate for 24 h were used for the RNA pulldown assays with each biotinylated RNA oligomer in panel A. ORF57 associated with RNA oligomers in the pulldowns was immunoblotted using an anti-ORF57 antibody. RNA oligomers oNP42 and oNP44 marked with colors shown in their corresponding MRE regions (B) are the two most active RNA regions mapped for ORF57 binding and functions, which were designated an active MRE-A region and an active MRE-B region in this study, respectively.
The vIL-6 MRE is crucial for RNA stability and translation.
To further analyze the function of the newly discovered vIL-6 MRE motifs, various deletion mutants of vIL-6 (Fig. 4A) were compared with a wt vIL-6-expressing vector in response to the presence of ORF57 in HEK293 cells. As shown in Fig. 4B to D, deletion of the entire MRE (ΔB) exhibited vIL-6 RNA instability and reduced vIL-6 expression in the presence of ORF57. This instability of vIL-6 RNA in the ΔB mutant and unresponsiveness to ORF57 were gradually reversed when the deletion was narrowed down to only MRE-B (Fig. 4B to D). The data indicate that the MRE functionally mediates ORF57-enhanced vIL-6 expression, with MRE-A functioning specifically to stabilize vIL-6 RNA. Unexpectedly, we noticed that deletions 5′ to 3′ of the MRE increased vIL-6 protein expression, regardless of whether ORF57 was present in cells. This was most noticeable in regard to the ΔD mutant, which carries a deletion of the MRE-B sequence. Even in the absence of ORF57, the ΔD mutant exhibited a remarkable amount of vIL-6 protein that was almost comparable to the level observed in the presence of ORF57 (Fig. 4B and C). This result indicates that MRE-B is mainly involved in translational repression of vIL-6, in contrast to the role of MRE-A in vIL-6 RNA instability.
FIG. 4.

The MRE in vIL-6 RNA is responsible for vIL-6 RNA stability and translation regulation. (A) Diagrams of wt vIL-6 and its mutants with the nucleotide positions in the KSHV genome (39) of the deletions indicated. MRE-A (striped box) and MRE-B (black box) are depicted. (B) Expression of wt vIL-6 and its deletion mutants in response to ORF57. Total protein and RNA from HEK293 cells transfected with a wt or a mutant vIL-6 expression vector were analyzed for vIL-6 expression by Western (WB) and Northern (NB) blotting, respectively. (C and D) Densitometry data for Fig. 3B were collected by using Image J software (http://rsb.info.nih.gov/ij/) for Western blots (C) and PhosphorImager for Northern blots (D). vIL-6 protein or RNA level in each sample was normalized to β-tubulin (C) or rRNA (D) for sample loading and is shown in the corresponding bar graph, with the wt vIL-6 expression level set as 1 in the absence of ORF57.
An miR-1293 seed match in the vIL-6 MRE-B sequence is required for efficient vIL-6 expression in response to ORF57.
To determine the importance of the vIL-6 MRE-B interaction with ORF57 in modulating vIL-6 translation, we transiently cotransfected HEK293 cells with ORF57 and either vIL-6 or miR-re vIL-6 expression vectors. The miR-re vIL-6 expression vector used in this assay contains a disrupted miR-1293 seed match (Fig. 5A). As shown in Fig. 5B and C, disruption of the miR-1293 seed match in vIL-6 RNA remarkably increased, in the absence of ORF57, the miR-re vIL-6 protein expression compared to that of wt vIL-6 when the same levels of mRNAs were assayed. Despite the fact that ORF57 was capable of promoting the expression of both wt vIL-6 and miR-re vIL-6 RNAs, it appeared more potent at increasing the production of wt vIL-6 protein. Our results indicate the importance of miR-1293 in modulating the translational repression of vIL-6. Given that ORF57 stimulates IL-6 expression and interacts directly with the vIL-6 MRE-B containing an miR-1293 binding site, we find that ORF57 functions to disrupt the miR-1293-mediated repression of vIL-6 translation.
FIG. 5.

An miR-1293 seed match in vIL-6 MRE-B is responsible for repression of vIL-6 expression and association with Ago2 and for ORF57 to function. (A) The sequences of the miR-1293 seed match in MRE-B of wt vIL-6 mRNA and its point mutations in the miR-re vIL-6. (B and C) Disruption of the miR-1293 seed match in the vIL-6 mRNA increases vIL-6 expression and decreases ORF57 regulation of vIL-6 production. The expression levels of vIL-6 protein (B) and RNA (C) from a wt or miR-re vIL-6 expression vector are compared in HEK293 cells in the absence and in the presence of ORF57. The bar graph in panel C shows relative levels of vIL-6 RNA in panel B after being normalized to GAPDH RNA (a sample loading control). Bars represent mean ± SD (n = 3). (D and E) ORF57 prevents the association of Ago2 with vIL-6 mRNA in vivo. HEK293 cells were cotransfected with HA-Ago2 plus the indicated expression vectors and UV irradiated 48 h after cotransfection. The cell lysates were used as total RNA to detect relative levels of vIL-6 mRNA (D) in transfected cells or used for anti-HA pulldown assays to detect Ago2-associated vIL-6 mRNA (E) in the IP complexes by qRT-PCR. Bars represent mean ± SD (n = 3). (F and G) ORF57 affects the miR-1293-mediated Ago2 binding to the MRE-B RNA. RNA-protein pulldown assays were conducted with a biotinylated RNA oligomer, oNP42 (MRE-A), which contains no miRNA seed match (F), or oNP44 (MRE-B), which harbors a vIL-6 miR-1293 binding site (G). The cell lysates for the RNA oligomer pulldown were prepared from HEK293 cells transfected with miR-NC (a negative control) or miR-1293. An increased amount of ORF57 protein was added to the cell lysate before the RNA pulldown. ORF57 and endogenous Ago2 in the pulldowns were blotted with an anti-ORF57 or anti-Ago2 antibody.
Additional experiments further supported our findings: first, we found that ORF57 enhanced vIL-6 expression in wt RKO cells but not in RKOdicer− cells (see Fig. S2A in the supplemental material); second, ORF57 disrupts miR-1293-mediated repression of vIL-6 translation in the cytoplasm (Fig. S2B to D). By using a cytoplasmic version of the N-terminal half (aa 1 to 251) of ORF57, containing point mutations in all three of its nuclear localization signals (25), we observed that this mutant ORF57 stabilized vIL-6 RNA less efficiently than did the N-terminal half of wt ORF57. However, the mutant ORF57 functioned similarly to wt N-terminal ORF57 in terms of vIL-6 protein expression in relief of miR-1293-mediated repression of vIL-6 translation when the protein/RNA ratio was taken into account (Fig. S2B and C). In addition, the mutant ORF57 also increased vIL-6 translation in a dose-dependent manner (Fig. S2D).
ORF57 prevents Ago2/miR-1293 binding to vIL-6 RNA in vivo and in vitro.
To understand the molecular mechanism by which ORF57 disrupts miR-1293-mediated translational repression of vIL-6, we performed a CLIP assay in cotransfected HEK293 cells expressing HA-Ago2. In the assay an anti-HA antibody was used to measure the vIL-6 RNA in miR-1293-mediated association with Ago2, a major component of RISC. The assay was performed either in the presence or in the absence of ORF57. miR-re vIL-6, which lacks an miR-1293 binding site, served as a negative control. As expected, cotransfection with ORF57 promoted the accumulation of both vIL-6 and miR-re IL-6 RNA, but cotransfection with a luciferase vector did not (Fig. 5D). Strikingly, we found that vIL-6 mRNA was associated with Ago2 efficiently in the presence of luciferase but much less efficiently in the presence of ORF57 (Fig. 5E; also compare with the corresponding level of total vIL-6 RNA in Fig. 5D). In contrast, miR-re vIL-6 lacking an miR-1293 binding site has no association with Ago2 either in the presence of luciferase or in the presence of ORF57. As miRNAs are tightly associated with RISC in vivo (43), our data indicate that Ago2 association with vIL-6 is mediated specifically by cellular miR-1293 in HEK293 cells and that this association is disrupted in the presence of ORF57.
To verify biochemically that the association of Ago2 with vIL-6 RNA is miRNA specific and that ORF57 functionally modulates this association, the influences of the MRE-A (Fig. 5F) and MRE-B (Fig. 5G) RNA sequences of vIL-6 on Ago2 binding were compared in RNA-protein pulldown assays by using HEK293 cell lysates with ectopically expressed miR-NC or miR-1293. ORF57 protein was added to the cell lysates before the RNA pulldown. The vIL-6 MRE-A motif, which does not contain an miRNA binding site, exhibited a minimal background level of binding to Ago2, independently of a specific miRNA and ORF57 (Fig. 5F). In contrast, the MRE-B motif, which has an miR-1293 binding site, exhibited strong binding of Ago2 only in the presence of miR-1293. Not surprisingly, ORF57, which interacts with the MRE-B RNA, could block this binding in a dose-dependent manner (Fig. 5G).
We further examined the specific interaction of miR-1293 with vIL-6 seed match region by a RISC assembly and RNase protection assay using in vitro-transcribed vIL-6 RNAs. The protein-RNA complexes mediated by miR-1293 in HEK293 cell lysates containing HA-Ago2 were UV cross-linked, immunoprecipitated with an anti-HA antibody, digested by RNase T1, and finally incubated with proteinase K. We found, by gel electrophoresis, that the vIL-6 RNA with the MRE-B region was partially protected by association with HA-Ago2 only in the presence of miR-1293 (Fig. 6A and B, compare RNA ΔD to RNA ΔE), indicating that miR-1293 mediated specific association of Ago2 with MRE-B. However, this specific association could be inhibited by ORF57 (Fig. 6C). Collectively, these data provide evidence that ORF57 prevents association of miR-1293-specific RISC with vIL-6 RNA and, therefore, disrupts miRNA-mediated translational repression of the target and leads to increased expression of vIL-6.
FIG. 6.
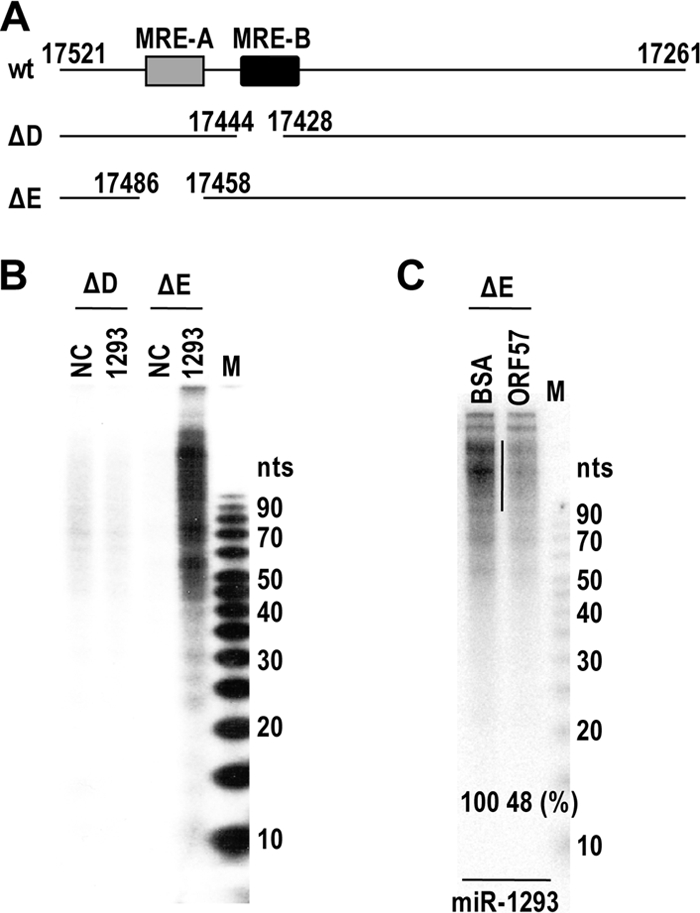
miR-1293-mediated specific association of Ago2 with MRE-B in the context of vIL-6 mRNA is preventable in the presence of ORF57. (A) The 3′ halves of vIL-6 RNAs with deletion of MRE-B (ΔD) or MRE-A (ΔE). (B) miR-1293-mediated Ago2 association with MRE-B of vIL-6 RNA. The indicated RNA ΔD and ΔE were transcribed in vitro and were mixed at 106 cpm with 300 nM miR-1293 or a negative-control miRNA (NC) in the same volume of HEK293 lysates containing HA-Ago2. The mixture was incubated at 30°C for 2 h to allow miR-1293-associated RISC formation on its targeted vIL-6 RNA. After UV cross-linking, the protein-RNA complexes were immunoprecipitated with an anti-HA antibody and digested with RNase T1 and then with proteinase K. The digested RNA was extracted and resolved in a 15% denatured PAGE gel. (C) ORF57 inhibits the specific association of Ago2 with MRE-B of vIL-6 RNA. The experimental conditions and procedures are identical to those in panel B except for the addition of BSA or ORF57 (10 μg) to the indicated reaction mixtures and the use of in vitro-transcribed 3′ halves of vIL-6 RNA (ΔE). A vertical line on the gel indicates a region used to quantify the protected RNA fragments. M, 5- to 10-bp ladders.
Ago2/miR-1293 competes with ORF57 for the same binding site in MRE-B of vIL-6 RNA.
We next used a reciprocal approach to confirm how ORF57 disrupts the miR-1293-mediated translational repression of vIL-6 by competition with miR-1293/Ago2 for the same binding site. We observed that the endogenous miR-1293-mediated Ago2 association with MRE-B of vIL-6 RNA prevents ORF57 from binding to the MRE-B region. This was accomplished by using an anti-Ago2 antibody to deplete Ago2 from HEK293 cell lysates, and then the binding of ORF57 to the MRE-B RNA was compared to that with non-Ago2-depleted lysates in RNA pulldown assays. By Western blotting (Fig. 7A), we observed that Ago2 binding to the MRE-B RNA was reduced by 50% in the presence of ORF57. Conversely, depletion of Ago2 (RISC) from the lysates increased ORF57 binding to the MRE-B RNA by 70%, indicating a competition between ORF57 and Ago2 (RISC) for MRE-B RNA binding. The binding of miR-1293/Ago2 to the MRE-B RNA of vIL-6 was also examined in RNA-protein pulldown assays using HEK293 cell lysates with ectopically expressed miR-1293 in the presence of BSA or ORF57. The MRE-B RNA was found to be strongly bound by miR-1293-associated endogenous Ago2, and this binding, as expected (Fig. 5G), was reduced by 30% in the presence of ORF57 (Fig. 7B). Consistent with this result, miR-1293 binding to the MRE-B RNA was also decreased by 30% in the presence of ORF57 compared to the control BSA (Fig. 7C). Altogether, these data provide further evidence that ORF57 competes with the miR-1293-specific RISC for binding to vIL-6 RNA.
FIG. 7.

ORF57 competes with Ago2 and miR-1293 for binding to vIL-6 MRE-B RNA. (A) Depletion of endogenous Ago2 from cell lysates increases ORF57 binding to the miR-1293 binding site. Cell lysates of HEK293 cells were prepared in 1× RIPA buffer containing protease inhibitors and RNase inhibitors. After depletion of Ago2 by three rounds of immunoprecipitation with an anti-Ago2 antibody, the cell lysates with or without Ago2 depletion were compared for Ago2 and ORF57 binding to biotinylated oNP44 RNA, which contains an miR-1293 binding site, in RNA pulldown assays in the presence or absence of directly added ORF57. About 10% input and 40% pulldown samples were resolved in a 4 to 12% SDS-PAGE gel and immunoblotted with anti-Ago2 and ORF57 antibodies. (B and C) ORF57 reduces the interaction of Ago2/miR-1293 with MRE-B. Biotinylated oNP44 (MRE-B) RNA pulldown assays were performed with 25 nM miR-1293-transfected HEK293 cell lysates prepared 48 h after transfection. About 5 μg of BSA or recombinant ORF57 was mixed with the cell lysates immediately before the pulldown. The relative levels of Ago2 bound to oNP44 RNA oligomer in the presence of BSA or ORF57 are quantified from 3 separate experiments and shown below a representative Ago2 blot. Relative amount of miRNA-1293 detected by qRT-PCR (C) in the corresponding pulldowns (B) after being normalized to U6. Bars represent means ± SD (n = 3). **, P < 0.01 (t test).
ORF57 relieves translational repression of vIL-6 by miR-1293 in an in vitro translation assay.
To better separate ORF57-mediated translational enhancement from ORF57-mediated RNA accumulation, we examined whether ORF57 directly interferes with the miR-1293-mediated repression of vIL-6 translation, using an in vitro translation assay in the presence of purified human HA-Ago2. A firefly luciferase RNA that has no miR-1293 binding site served as an internal control in the study. We found that the translation of vIL-6 RNA in vitro, but not that of firefly luciferase RNA, could be inhibited by 32% in the presence of miR-1293 (Fig. 8A and B). When supplemented in the assays, ORF57 was found to relieve, in a dose-dependent manner, the miR-1293-mediated translational repression of a fixed amount of in vitro-transcribed vIL-6 mRNA (Fig. 8A and B). Together, these data confirm that ORF57 does exercise a major function in promoting vIL-6 translation by disrupting miR-1293-mediated translational repression, which is separate from its function in promoting vIL-6 RNA accumulation.
FIG. 8.
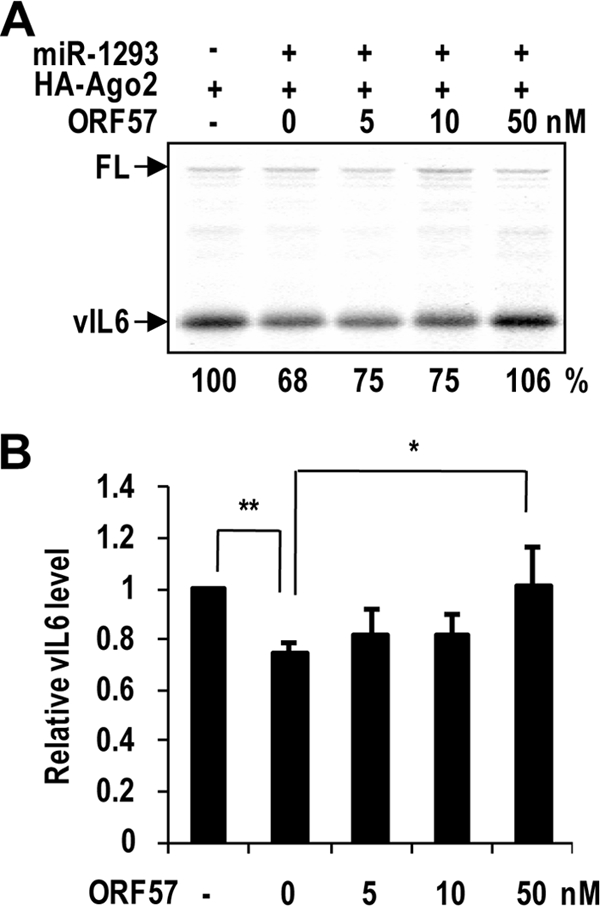
ORF57 prevents the miRNA-mediated translational repression of vIL-6 in vitro. (A) A representative of multiple gels in in vitro translation assays. The mRNA and miRNA duplex were denatured at 70°C for 3 min and quenched on ice. hAgo2 (25 nM) with or without ORF57 was then added and incubated at 30°C for 10 min. Each reaction mixture was then mixed with translation mixture and incubated at 30°C for 20 min. The reaction was stopped on ice by the addition of SDS sample buffer. Translated proteins were resolved in a 4% to 12% SDS-PAGE gel and transferred onto a membrane for capturing images on a phosphorimager screen or X-ray film. The relative translation efficiencies in each reaction are shown below the gel. Firefly luciferase RNA (FL) was included as an internal translation control. (B) The bar graph shows relative levels of vIL-6 protein after being normalized to firefly luciferase. Bars represent means ± SD (n = 3). **, P < 0.01; *, P < 0.05 (t test).
ORF57 enhances hIL-6 expression via a mechanism similar to that for vIL-6 expression.
Lytic KSHV infection also induces hIL-6 expression in B cells (1, 10) and in TREx BCBL-1 RTA cells (Fig. 9A and B). The interaction between hIL-6 RNA and ORF57 in JSC-1 cells with lytic KSHV induction was detected by CLIP/RT-PCR (data not shown). Consistent with this, ORF57 itself promoted hIL-6 expression in cotransfection of HEK293 cells (Fig. 9C). ORF57 protein transfection into JSC-1 cells could also enhance hIL-6 expression (Fig. 9D). These data suggest that, during KSHV infection, enhanced hIL-6 expression is dependent on viral ORF57.
FIG. 9.

ORF57 regulates hIL-6 expression by a mechanism similar to that for vIL-6. (A and B) KSHV lytic infection increases the expression of both vIL-6 and hIL-6. TREx BCBL-1 RTA cells carrying an episomal KSHV genome and a tetracycline-inducible RTA (ORF50) expression vector were cultivated in the presence of doxycycline (DOX; 1 μg/ml) to induce KSHV lytic infection. After 12 h of induction, the culture medium and cells were separated by centrifugation. The pelleted cell lysates were immunoblotted for vIL-6, ORF57, and β-tubulin (A). The levels of secreted hIL-6 and hIL-12 in the culture medium were determined by using the multianalyte ELISArray kit (B). (C) Induction of hIL-6 expression by ORF57 in HEK293 cells. Protein samples were immunoblotted 24 h after cotransfection of an hIL-6 expression vector in the absence or presence of an ORF57 expression vector as described for Fig. 2E. (D) ORF57 protein transfection enhances hIL-6 expression in JSC-1 cells. JSC-1 cells were transfected with 2 μg of β-galactosidase (β-Gal, negative control) or ORF57-FLAG protein. Samples were collected 24 h after transfection. The level of secreted hIL-6 in the culture medium was determined by using an hIL-6 analyte ELISArray kit as described for panel B. Bars represent means ± SD (n = 3). (E) hIL-6 has an MRE responsive to ORF57. RNA-protein pulldown assays were conducted with a biotinylated RNA oligomer (oJGK50) which harbors an hIL-6 miR-608 binding site. The cell lysates for the RNA pulldown assays were prepared from HEK293 cells transfected with miR-NC (a negative control) or miR-608. Increasing amounts of ORF57 protein were added to the cell lysate before the pulldown. ORF57 and endogenous Ago2 in the pulldowns were blotted with an anti-ORF57 or anti-Ago2 antibody as described for Fig. 5F and G.
The hIL-6 ORF contains an miR-608 seed match in the corresponding region of vIL-6 MRE-B, and its expression could be greatly increased in RKOdicer− cells (Kang and Zheng, unpublished); therefore, it seems plausible that hIL-6, like vIL-6, could be an additional target of ORF57. To determine whether this seemed reasonable, a biotinylated RNA oligomer (oJGK50) was synthesized based on the hIL-6 MRE region containing an miR-608 seed match and was used for RNA pulldown assays. The cell lysates for the pulldown assays, either with or without ORF57, were prepared from HEK293 cells transfected with miR-NC (a negative control) or miR-608, and the proteins in the pulldowns were examined by Western blotting. As shown in Fig. 9E, ORF57 binds to the MRE RNA (oJGK50) of hIL-6 in a dose-dependent manner, regardless of whether an HEK293 cell lysate with ectopically expressed miR-NC or miR-608 was used in the RNA pulldown assays. However, a high-affinity binding of Ago2 to the MRE RNA of hIL-6 could be conferred only when HEK293 cell lysates with ectopically expressed miR-608 were used in the assays. The binding could be gradually diminished by increased amounts of ORF57. In contrast, when HEK293 cell lysates containing miR-NC were used, we observed a very-low-affinity interaction (background) of Ago2 with the hIL-6 MRE. Overall, these results indicate that ORF57 promotes hIL-6 expression by modulating the interaction of miR-608-specific RISC with the hIL-6 MRE.
DISCUSSION
A clearly demonstrated function of KSHV ORF57 is to accumulate several intronless viral RNA targets in cells, with some accumulating more in the nucleus and others accumulating more in the cytoplasm (18, 23, 25). This accumulation function of ORF57 appears to be independent of RNA export since ORF57, in the context of the virus genome, promotes accumulation of a noncoding nuclear viral RNA, PAN RNA (23). In addition, this function is distinct from ORF57 association with Aly/REF, because ORF57 functions well in the cells with a deficiency of Aly/REF (25). Our findings that ORF57 interacts with an MRE region in specific viral transcripts of KSHV-infected cells and that this interaction functions to stabilize target RNAs and to disrupt miRNA-mediated translational repression of the targeted RNAs reveal a novel function of ORF57.
In this report, we have characterized KSHV ORF57 as a suppressor of miRNA-mediated repression of vIL-6 expression, highlighting an important function of ORF57 in promotion of intronless vIL-6 production during KSHV lytic infection. By covering an miRNA binding site in both vIL-6 and hIL-6 ORF regions, ORF57 competes for the same binding site with miR-1293 or miR-608 exhibiting a constant level in KSHV-infected B cells from latent to lytic infection (data not shown) and, consequently, relieves the repressive action by miR-1293 on vIL-6 and by miR-608 on hIL-6 (Fig. 10). Undoubtedly, in the presence of ORF57, vIL-6 mRNA became more stable and translatable. Observations of functional miRNA-mRNA interactions that extend from RNA 3′ UTRs to coding regions of targeted RNAs have been made recently (7, 19). Moreover, a cellular protein has also been identified as an RNA stabilizer against miRNA-mediated degradation (7). Thus, our data provide the first evidence for a KSHV viral protein responsible for modulating the function of cellular miRNAs.
FIG. 10.
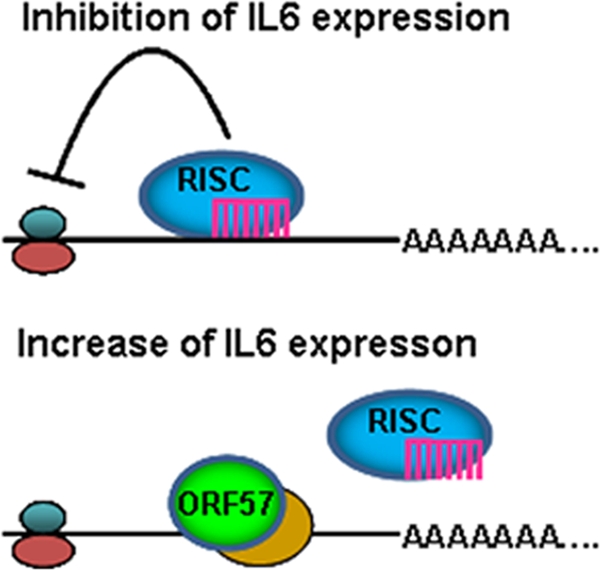
A simplified model of how ORF57 may prevent miRNA-mediated inhibition of IL-6 expression. ORF57 prevents miRNA binding to IL-6 RNA (bottom) by competing with miR-1293 or miR-608 for the same RNA binding site. ORF57 in living cells is always associated with another RNA binding protein(s) in yellow.
The CLIP assay used in this study included an affinity-purified anti-ORF57 antibody that allowed us to determine a specific RNA motif(s) interacting with ORF57 protein in living cells. In these CLIP assays, we did not detect any KSHV miRNAs in association with ORF57. The ORF57-interacting MRE identified in the vIL-6 ORF region was examined for its functional regulation of vIL-6 expression. The identified MRE contains two ORF57 binding sites, MRE-A and MRE-B. Although both MRE-A and MRE-B bind Ago2 and ORF57, MRE-A has much less affinity for Ago2 and the association of Ago2 and related protein(s) can be strengthened by other factors bound to MRE-B (data not shown). In addition, MRE-A does not have a seed match to any known miRNAs. Although the exact function of MRE-A remains to be determined, the MRE-A-ORF57 interaction appears responsible for stabilization of vIL-6 RNA, as shown by vIL-6 retaining an MRE-A region being relatively stable in the presence of ORF57 (Fig. 4). Compared to MRE-A, the function of vIL-6 MRE-B was carefully defined in this study. In addition to Ago2 and ORF57 binding, MRE-B also contains an miR-1293 binding site and the interaction with miR-1293 results in translational repression of vIL-6 RNA. Interestingly, we found that the strong interaction of Ago2 with MRE-B is specifically mediated by miR-1293, which is attributed to the tight association of miRNAs with RISC (43) exemplified by Ago2 being a major component of RISC (14). Importantly, the miR-1293-mediated interaction of MRE-B with Ago2 is disrupted by ORF57, thereby freeing vIL-6 from association with Ago2/miR-1293 in KSHV lytic infection and relieving miR-1293-mediated translational repression. Consistent with this, vIL-6 RNA with MRE-B deletion confers an efficient translation in the absence of ORF57.
The importance of ORF57 functioning in the suppression of the miR-1293 interaction with vIL-6, and in promotion of vIL-6 RNA stability and translation, was highlighted further by discovery of a similar mechanism for ORF57 in regulation of hIL-6 expression. KSHV infection also leads to an increase of hIL-6 expression (1), which is attributed to viral RTA (ORF50) transactivation of the hIL-6 promoter (8). The discovery and characterization of an miR-608 seed match in hIL-6 ORF, rather than in the 3′ UTR, indicate an important role of miRNA-mediated posttranscriptional regulation in control of hIL-6 expression. By binding to the miR-608 seed match sequence, ORF57 prevents miR-608 interaction with hIL-6 RNA and promotes hIL-6 expression during KSHV infection. It is possible that regulation by ORF57 applies to other miRNA-targeted viral or cellular RNAs, but further studies will be required to confirm whether this is a common regulatory function of ORF57. Our preliminary study showed that ORF57 in cotransfection with miR-34a in HeLa cells had no effect on miR-34a-mediated suppression of CDK4 expression or p18Ink4c (46), suggesting that the miR-34a binding site of CDK4 or p18Ink4c is not a binding site for ORF57 to function. Based on all results from this study, we conclude that KSHV employs ORF57 to disrupt the miRNA-mediated suppression of vIL-6 and hIL-6 expression during lytic infection of target cells. Our findings shed light on how an oncogenic virus utilizes an essential viral protein to evade the miRNA pathway to promote viral and human IL-6 production and induce B cell malignancies.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the NCI, Center for Cancer Research, National Institutes of Health.
We thank Giovanna Tosato and John Nicholas for providing anti-vIL-6 antibodies, Bert Vogelstein for providing RKO Dicer knockout cells, Jae Jung for providing doxycycline-inducible TREx BCBL-1 RTA and vector cell lines, and other members of the Zheng laboratory for their assistance and critical comments.
Footnotes
Published ahead of print on 5 January 2011.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Aoki, Y., R. Yarchoan, J. Braun, A. Iwamoto, and G. Tosato. 2000. Viral and cellular cytokines in AIDS-related malignant lymphomatous effusions. Blood 96:1599-1601. [PubMed] [Google Scholar]
- 2.Aoki, Y., et al. 2001. Detection of viral interleukin-6 in Kaposi sarcoma-associated herpesvirus-linked disorders. Blood 97:2173-2176. [DOI] [PubMed] [Google Scholar]
- 3.Baek, D., et al. 2008. The impact of microRNAs on protein output. Nature 455:64-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel, D. P. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyne, J. R., K. J. Colgan, and A. Whitehouse. 2008. Recruitment of the complete hTREX complex is required for Kaposi's sarcoma-associated herpesvirus intronless mRNA nuclear export and virus replication. PLoS Pathog. 4:e1000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chatterjee, M., J. Osborne, G. Bestetti, Y. Chang, and P. S. Moore. 2002. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 298:1432-1435. [DOI] [PubMed] [Google Scholar]
- 7.Chi, S. W., J. B. Zang, A. Mele, and R. B. Darnell. 2009. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460:479-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng, H., J. T. Chu, M. B. Rettig, O. Martinez-Maza, and R. Sun. 2002. Rta of the human herpesvirus 8/Kaposi sarcoma-associated herpesvirus up-regulates human interleukin-6 gene expression. Blood 100:1919-1921. [DOI] [PubMed] [Google Scholar]
- 9.Deng, H., M. J. Song, J. T. Chu, and R. Sun. 2002. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 76:8252-8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foussat, A., et al. 1999. Human interleukin-6 is in vivo an autocrine growth factor for human herpesvirus-8-infected malignant B lymphocytes. Eur. Cytokine Netw. 10:501-508. [PubMed] [Google Scholar]
- 11.Ganem, D. 2006. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu. Rev. Pathol. 1:273-296. [DOI] [PubMed] [Google Scholar]
- 12.Gupta, A. K., V. Ruvolo, C. Patterson, and S. Swaminathan. 2000. The human herpesvirus 8 homolog of Epstein-Barr virus SM protein (KS-SM) is a posttranscriptional activator of gene expression. J. Virol. 74:1038-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han, Z., and S. Swaminathan. 2006. Kaposi's sarcoma-associated herpesvirus lytic gene ORF57 is essential for infectious virion production. J. Virol. 80:5251-5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hutvagner, G., and M. J. Simard. 2008. Argonaute proteins: key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 9:22-32. [DOI] [PubMed] [Google Scholar]
- 15.Iliopoulos, D., H. A. Hirsch, and K. Struhl. 2009. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139:693-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones, K. D., et al. 1999. Involvement of interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi's sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood 94:2871-2879. [PubMed] [Google Scholar]
- 17.Jones, M. R., et al. 2009. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat. Cell Biol. 11:1157-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirshner, J. R., D. M. Lukac, J. Chang, and D. Ganem. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 57 encodes a posttranscriptional regulator with multiple distinct activities. J. Virol. 74:3586-3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lecellier, C. H., et al. 2005. A cellular microRNA mediates antiviral defense in human cells. Science 308:557-560. [DOI] [PubMed] [Google Scholar]
- 20.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 21.Majerciak, V., M. Deng, and Z. M. Zheng. 2010. Requirement of UAP56, URH49, RBM15, and OTT3 in the expression of Kaposi sarcoma-associated herpesvirus ORF57. Virology. 407:206-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majerciak, V., M. Kruhlak, P. K. Dagur, J. P. McCoy, Jr., and Z. M. Zheng. 2010. Caspase-7 cleavage of Kaposi sarcoma-associated herpesvirus ORF57 confers a cellular function against viral lytic gene expression. J. Biol. Chem. 285:11297-11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majerciak, V., N. Pripuzova, J. P. McCoy, S. J. Gao, and Z. M. Zheng. 2007. Targeted disruption of Kaposi's sarcoma-associated herpesvirus ORF57 in the viral genome is detrimental for the expression of ORF59, K8alpha, and K8.1 and the production of infectious virus. J. Virol. 81:1062-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Majerciak, V., et al. 2008. Kaposi sarcoma-associated herpesvirus ORF57 functions as a viral splicing factor and promotes the expression of intron-containing viral lytic genes in spliceosome-mediated RNA splicing. J. Virol. 82:2792-2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majerciak, V., K. Yamanegi, S. H. Nie, and Z. M. Zheng. 2006. Structural and functional analyses of Kaposi sarcoma-associated herpesvirus ORF57 nuclear localization signals in living cells. J. Biol. Chem. 281:28365-28378. [DOI] [PubMed] [Google Scholar]
- 26.Majerciak, V., K. Yamanegi, and Z. M. Zheng. 2006. Gene structure and expression of Kaposi's sarcoma-associated herpesvirus ORF56, ORF57, ORF58, and ORF59. J. Virol. 80:11968-11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Majerciak, V., and Z. M. Zheng. 2009. Kaposi's sarcoma-associated herpesvirus ORF57 in viral RNA processing. Front. Biosci. 14:1516-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malik, P., D. J. Blackbourn, and J. B. Clements. 2004. The evolutionarily conserved Kaposi's sarcoma-associated herpesvirus ORF57 protein interacts with REF protein and acts as an RNA export factor. J. Biol. Chem. 279:33001-33011. [DOI] [PubMed] [Google Scholar]
- 29.Mathonnet, G., et al. 2007. MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science 317:1764-1767. [DOI] [PubMed] [Google Scholar]
- 30.Matsushita, K., et al. 2009. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 458:1185-1190. [DOI] [PubMed] [Google Scholar]
- 31.Moore, P. S., C. Boshoff, R. A. Weiss, and Y. Chang. 1996. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274:1739-1744. [DOI] [PubMed] [Google Scholar]
- 32.Moore, P. S., and Y. Chang. 2003. Kaposi's sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu. Rev. Microbiol. 57:609-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nador, R. G., et al. 1996. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood 88:645-656. [PubMed] [Google Scholar]
- 34.Nakamura, H., et al. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 77:4205-4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nekorchuk, M., Z. Han, T. T. Hsieh, and S. Swaminathan. 2007. Kaposi's sarcoma-associated herpesvirus ORF57 protein enhances mRNA accumulation independently of effects on nuclear RNA export. J. Virol. 81:9990-9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicholas, J., et al. 1997. Kaposi's sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 3:287-292. [DOI] [PubMed] [Google Scholar]
- 37.Parravicini, C., et al. 1997. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman's disease. Am. J. Pathol. 151:1517-1522. [PMC free article] [PubMed] [Google Scholar]
- 38.Paschoud, S., et al. 2006. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol. Cell. Biol. 26:8228-8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russo, J. J., et al. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862-14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sauer, I., et al. 2006. Interferons limit inflammatory responses by induction of tristetraprolin. Blood 107:4790-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selbach, M., et al. 2008. Widespread changes in protein synthesis induced by microRNAs. Nature 455:58-63. [DOI] [PubMed] [Google Scholar]
- 42.Staskus, K. A., et al. 1999. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. J. Virol. 73:4181-4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang, F., et al. 2008. MicroRNAs are tightly associated with RNA-induced gene silencing complexes in vivo. Biochem. Biophys. Res. Commun. 372:24-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ule, J., et al. 2003. CLIP identifies Nova-regulated RNA networks in the brain. Science 302:1212-1215. [DOI] [PubMed] [Google Scholar]
- 45.Wang, B., T. M. Love, M. E. Call, J. G. Doench, and C. D. Novina. 2006. Recapitulation of short RNA-directed translational gene silencing in vitro. Mol. Cell 22:553-560. [DOI] [PubMed] [Google Scholar]
- 46.Wang, X., C. Meyers, M. Guo, and Z. M. Zheng. Up-regulation of p18Ink4c expression by oncogenic HPV E6 via p53-miR-34a pathway. Int. J. Cancer, in press. [DOI] [PMC free article] [PubMed]
- 47.Zhao, J., et al. 2007. K13 blocks KSHV lytic replication and deregulates vIL6 and hIL6 expression: a model of lytic replication induced clonal selection in viral oncogenesis. PLoS One. 2:e1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.