

Abstract
Morphogenesis of vaccinia virus begins with the appearance of crescent-shaped membrane precursors of immature virions in cytoplasmic factories. During the initial characterization of the product of the L2R reading frame, we discovered that it plays an important role in crescent formation. The L2 protein was expressed early in infection and was associated with the detergent-soluble membrane fraction of mature virions, consistent with two potential membrane-spanning domains. All chordopoxviruses have L2 homologs, suggesting an important function. Indeed, we were unable to isolate an infectious L2R deletion mutant. Consequently, we constructed an inducible mutant with a conditional lethal phenotype. When L2 expression was repressed, proteolytic processing of the major core proteins and the A17 protein, which is an essential component of the immature virion membrane, failed to occur, suggesting an early block in viral morphogenesis. At 8 h after infection in the presence of inducer, immature and mature virions were abundantly seen by electron microscopy. In contrast, those structures were rare in the absence of inducer and were replaced by large, dense aggregates of viroplasm. A minority of these aggregates had short spicule-coated membranes, which resembled the beginnings of crescent formation, at their periphery. These short membrane segments at the edge of the dense viroplasm increased in number at later times, and some immature virions were seen. Although the L2 protein was not detected under nonpermissive conditions, minute amounts could account for stunted and delayed viral membrane formation. These findings suggested that L2 is required for the formation or elongation of crescent membranes.
Poxviruses are a family of double-stranded DNA viruses that replicate in the cytoplasm of their host cells (24). Vaccinia virus (VACV), the most intensively characterized member of the family, has a genome of approximately 190 kbp that encodes about 200 proteins. Few of the nearly 100 genes that are conserved in all chordopoxviruses remain uncharacterized. One of the latter, the L2R gene (VACWR089), was found by genome-wide analyses to be transcribed early during infection (3, 46), but no additional information regarding it was available. Poxvirus transcription is regulated in a cascade fashion, with early genes expressed prior to viral genome replication and intermediate- and late-stage genes expressed after replication. Most early genes are involved in synthesis of mRNA or DNA or in host defense, whereas the postreplicative genes mainly encode virion-associated proteins. In the present study, we were surprised to find that the L2 protein was involved in virion morphogenesis despite being an early protein. (Note that the letters L for “left” and R for “right,” which are used to designate the direction of transcription of a gene, are omitted when referring to the protein product.)
The first recognizable structure that appears during VACV morphogenesis is the crescent-shaped membrane (11), with an external lattice constituted of trimers of the D13 protein (39). The crescents engulf electron-dense viroplasm containing core proteins or their precursors, and condensed DNA nucleoids are inserted prior to membrane closure (23). The resulting spherical immature virion (IV) undergoes further maturation involving the removal of the scaffold (6) and the proteolytic processing of several major proteins (25) to form the infectious brick-shape mature virion (MV). Some MVs are wrapped with a double membrane modified from the trans-Golgi network or endosomal cisternae (17, 34, 41), and the wrapped virions (WVs) are transported via microtubules to the plasma membrane and undergo exocytosis, during which the outermost membrane is lost (36). The extracellular enveloped virions (EVs) are largely responsible for cell-to-cell transmission.
Neither the origin, mode of formation, nor composition of the crescent viral membrane is understood. An initial hypothesis, based on the apparent spatial separation of the crescents from other cellular membranes, is that they are assembled de novo (10). An alternative origin from the intermediate compartment between the endoplasmic reticulum and Golgi apparatus was proposed based on ultrastructural evidence for an association of some viral proteins with this compartment (30, 37). Further studies, however, demonstrated that endoplasmic reticulum to Golgi membrane transport is not required for the formation of IVs and that viral proteins can traffic to IVs from the endoplasmic reticulum (18, 20). Nevertheless, no coherent model for the formation of the IV membrane has been established.
In order to understand the initial steps of morphogenesis, it is necessary to identify the components involved in the process. Studies with conditional lethal mutants suggested roles for the products of several VACV genes in the assembly of the viral membrane. An inability to form the viral membrane is associated with conditional lethal mutants of several nonmembrane proteins, namely, A11 (VACWR130) (29), F10 (VACWR049) (40, 42), G5 (VACWR082) (9), H5 (VACWR103) (13), and H7 (VACVWR105) (33). The G5 and H5 proteins have other roles (8, 12, 21, 35), however, and the phenotypes in which viral membranes fail to form have been demonstrated only with elevated-temperature-sensitive alanine substitution mutants, raising the possibility of indirect effects. In cells infected with inducible mutants of two late genes that encode the membrane proteins A14 (VACWR133) (32, 43) and A17 (VACWR137) (31, 45), vesicular or tubular structures accumulate at the boundaries of dense viroplasm under nonpermissive conditions. Repressed expression of the scaffold protein D13 (VACVWR118) produces a phenotype in which irregular membranes surround electron-dense viroplasm (49), similar to that seen with the drug rifampin (26, 27). F10 is a protein kinase that phosphorylates some cellular and viral proteins, including the two transmembrane proteins A14 (5, 43) and A17 (14). In addition to viral components, the cellular coatomer and KDEL receptor proteins have been suggested to have a role in viral membrane formation (48).
In the present study, we investigated the product of the previously uncharacterized L2R gene and demonstrated that it is expressed early in infection and is associated with the virion membrane. A conditionally lethal, inducible L2R mutant was constructed, and an assembly block occurring prior to the formation of viral crescents was found.
MATERIALS AND METHODS
Cells, virus, and antibodies.
BS-C-1 (ATCC CCL-26) and HeLa cells were grown in minimum essential medium with Earle's salt (E-MEM) and Dulbecco's minimum essential medium (DMEM), respectively, supplemented with heat-inactivated 10% fetal bovine serum (FBS), 100 U of penicillin, and 100 μg of streptomycin per ml (Quality Biologicals, Gaithersburg, MD). The VACV Western Reserve (WR) strain and the recombinant virus vT7lacOI (1) were propagated as described previously (15). The newly constructed recombinant vL2Ri was grown in the presence of 50 μM isopropyl-β-d-thiogalactopyranoside (IPTG). MVs were purified by sedimentation twice through 36% sucrose cushions followed by banding on a 25 to 40% sucrose gradient as described previously (16). Antiserum to a peptide corresponding to the N-terminal 15 amino acids of the L2R open reading frame (ORF) followed by a cysteine residue (MEVITDRLDDIVKQNC) was produced in rabbits (Covance, Denver, PA). The viral proteins were detected with rabbit antisera to A17-N (4), A3 (unpublished data), F13 (19), D13 (38), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Covance, Emeryville, CA).
Plasmid and recombinant VACV construction.
To construct pVOTE-L2R, the L2R open reading frame (ORF) was amplified by PCR from genomic DNA with the oligonucleotides 5′-GCGCATGTCATATGGAAGTTATCACTGATCGTCT-3′ and 5′-TAGGGATCCTTATAGTATAAAGTAATAAAAAATAGTTAATGTG-3′. The PCR product was inserted into the NdeI and BamHI sites of pVOTE2 (44). Lipofectamine 2000 (Invitrogen) was used for transfections. The repressible copy of L2R was introduced into the A56R locus of vT7lacOI by homologous recombination with pVOTE-L2R, and vL2Ri was isolated by xanthine-guanine phosphoribosyltransferase selection (1). The inducible L2R copy was verified by PCR and sequencing. Subsequently, DNA containing the enhanced green fluorescent protein (EGFP) ORF flanked by VACV sequences replaced the endogenous L2R ORF.
Plaque assay and virus yield determination.
For plaque assays, BS-C-1 cell monolayers in 6-well tissue culture plates were infected with 10-fold dilutions of virus. After 1 h of absorption, the virus inoculum was removed, the cells were washed with E-MEM plus 2.5% FBS, and medium containing 0.5% methylcellulose with or without 50 μM IPTG was added. The infection was allowed to proceed for 48 h at 37°C, the monolayer was then stained with crystal violet, and the plaques were counted.
For virus yield determination, BS-C-1 monolayers in 12-well tissue culture plates were infected with 3 PFU of vL2Ri or vT7lacOI virus per cell. After 1 h of absorption, the inoculum was removed and the cells were washed with E-MEM plus 2.5% FBS. The infected cells were incubated with medium in the presence or absence of 50 μM IPTG. The cells were lysed by three freeze-thaw cycles and sonicated. Virus titers were determined by plaque assay in the presence of 50 μM IPTG.
Labeling of proteins with [35S]methionine-cysteine.
BS-C-1 cells in 24-well plates were infected with 5 PFU of either vL2Ri or vT7LacOI per cell for 1 h at 37°C. The inocula were removed, and the cells were incubated with complete E-MEM plus 2.5% FBS in the presence or absence of 50 μM IPTG for vL2Ri and with or without 100 μg of rifampin per ml for vT7LacOI. After 8 h, the cells were incubated with cysteine- and methionine-free medium for 30 min at 37°C, labeled with 100 μCi of [35S]methionine-cysteine (PerkinElmer, Waltham, MA) for 30 min, and then harvested. Cells were washed once with cold phosphate-buffered saline (PBS), lysed in 1× NuPAGE lithium dodecyl sulfate sample loading buffer containing 1× reducing agent (Invitrogen), and heated at 70°C for 10 min. The proteins were resolved by electrophoresis in 4 to 12% Novex NuPAGE acrylamide gels (Invitrogen), dried, and detected by autoradiography.
For the pulse and chase, the cells were infected as described above. After 6 h, the cells were incubated with cysteine- and methionine-free medium for 30 min at 37°C and pulse-labeled for 30 min with 100 μCi of [35S]methionine-cysteine. The labeling medium was replaced with complete E-MEM plus 2.5% FBS, the cells were incubated for 12 h, and the proteins were analyzed as described above.
Western blotting.
Proteins were resolved by electrophoresis on 4 to 12% Novex NuPAGE acrylamide gels and transferred to nitrocellulose membranes by using Mini iBlot gel transfer stacks (Invitrogen). The membranes were blocked for 1 h at room temperature (RT) with 5% nonfat dried milk in PBS containing 0.05% Tween 20 and incubated with the primary antibody for 1 h at RT. The membranes were washed four times with PBS with 0.05% Tween and then incubated with the secondary antibody for 1 h at RT. The secondary antibodies were species specific and conjugated with horseradish peroxidase (Pierce, Rockford, IL). The membranes were washed and developed with Dura or Femto chemiluminescent substrate (Pierce).
Detergent extraction of purified virions.
Purified virus particles were incubated in 50 mM Tris-HCl (pH 7.5) containing 150 mM NaCl and 1% Nonidet P-40 detergent in the presence and absence of 50 mM dithiothreitol for 1 h at 37°C. The soluble and insoluble materials were separated by centrifugation at 20,000 × g for 30 min at 4°C. Proteins in the pellet and the supernatant were analyzed by electrophoresis on 4 to 12% Novex NuPAGE acrylamide gels in the absence of reducing agent followed by Western blotting.
Confocal microscopy.
HeLa cells grown on coverslips were infected as described above. The cells were fixed after 16 h with 4% paraformaldehyde in PBS for 15 min at RT and later washed four times with PBS. The cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min at RT and washed four times. The cells were blocked with 10% FBS for 30 min and then incubated with the primary antibody in PBS containing 10% FBS for 1 h at RT. Cells were washed and incubated with the secondary antibody conjugated to dye (Molecular Probes, Eugene, OR) for 1 h. The coverslips were washed and mounted on a glass slide by using Prolong Gold (Invitrogen).
Electron microscopy.
For transmission electron microscopy, infected BS-C-1 cells in 60-mm-diameter plates were fixed in 2% glutaraldehyde-0.1 M sodium cacodylate buffer, washed in 0.1 M sodium cacodylate buffer, postfixed with reduced osmium tetroxide, and washed in buffer. Cells were dehydrated with 50%, 70%, and 100% ethyl alcohol and propylene oxide in series. The samples were embedded in EMbed 812, and sections were cut on an EM UC7 ultramicrotome (Leica Microsystems, Wetzlar, Germany). Thin sections were stained with 7% uranyl acetate in 50% ethanol and then 0.01% lead citrate. Sections were reviewed and photographed on the Tecnai G2 Spirit transmission electron microscope fitted with a Gatan charge-coupled device (CCD) camera (FEI, Hillsboro, OR). Chemicals were purchased from Electron Microscopy Sciences (Hatfield, PA).
RESULTS
L2 homologs are present in all chordopoxviruses.
The L2R ORF is predicted to encode an 87-amino-acid, 10.2-kDa protein with two hydrophobic domains in the C-terminal half (Fig. 1 A). The protein has no recognizable signal peptide, coiled-coil segment, glycosylation site, or other motif. Homologs of the L2R ORF are present in all chordopoxviruses for which sequences have been obtained, but no nonpoxvirus homologs were discerned. The length of the putative L2-related proteins ranges from 87 to 99 amino acids in representatives of the different chordopoxvirus genera (Fig. 1B), and each homolog has N-terminal hydrophilic and C-terminal hydrophobic regions. L2 has 96 to 100% identity to the other orthopoxvirus homologs (data not shown) and 31 to 39% identity to those of other genera of the chordopoxvirus subfamily (Fig. 1B).
FIG. 1.

L2R is predicted to encode a highly conserved transmembrane protein. (A) Kyte-Doolittle hydrophilicity plot of L2R amino acid sequence analyzed using MacVector version 11.1.2 software for hydrophilicity prediction; (B) CLUSTAL alignment of L2-related open reading frames from representative species of each genus of the Chordopoxvirinae subfamily. VACV, vaccinia virus strain Western Reserve; SWPV, swinepox virus strain Nebraska 17077-99; RFV, rabbit fibroma virus strain Kasza; DPV, deerpox virus strain W-848-83; LSDV, lumpy skin disease virus strain Neethling 2490; YMTV, yaba monkey tumor virus strain Amano; ORFV, orf virus strain Orf-11; ZWE, crocodilepox virus strain Zimbabwe. Amino acids conserved in all sequences are marked by asterisks; colons denote conserved amino acids in all but one species, and periods indicate similar amino acids.
L2 is expressed early in infection and is associated with MVs.
Inspection of the DNA sequence upstream of the L2R ORF suggested the presence of an early promoter (not shown), which is consistent with recent genome-wide transcriptome analyses (3, 46). In order to determine the kinetics of L2 protein synthesis, BS-C-1 cells were infected with the Western Reserve strain of VACV and harvested at successive times. Total cell lysates were solubilized with SDS plus reducing agent, and the proteins were resolved on polyacrylamide gels and analyzed by Western blotting with antibody made in rabbits immunized with a L2 N-terminal peptide. L2 was detected at 2 h as a 6-kDa band relative to standard markers, although the predicted mass is 10.2 kDa (Fig. 2 A). Additional higher-molecular-weight cross-reacting protein bands were detected, but these are unrelated to L2 as they were still present when L2 expression was repressed in later experiments (data not shown). L2 increased greatly in amount by 6 h and then only slightly more during the next 18 h. The synthesis of L2 in the presence of cytosine arabinoside, an inhibitor of DNA replication, confirmed the classification of L2 as an early protein (Fig. 2A). As controls for late protein synthesis, A3 and L1 were detected at 6 h and neither was made in the presence of cytosine arabinoside (Fig. 2A). The trace amounts of viral proteins present at zero time are due to virion association. L2 has a single cysteine, and approximately 23% of the protein migrated as a 12-kDa species in the absence of reducing agent (Fig. 2B). The significance of the latter is unknown, and the possibility that disulfide bond formation occurred after cell lysis was not excluded (Fig. 2B).
FIG. 2.
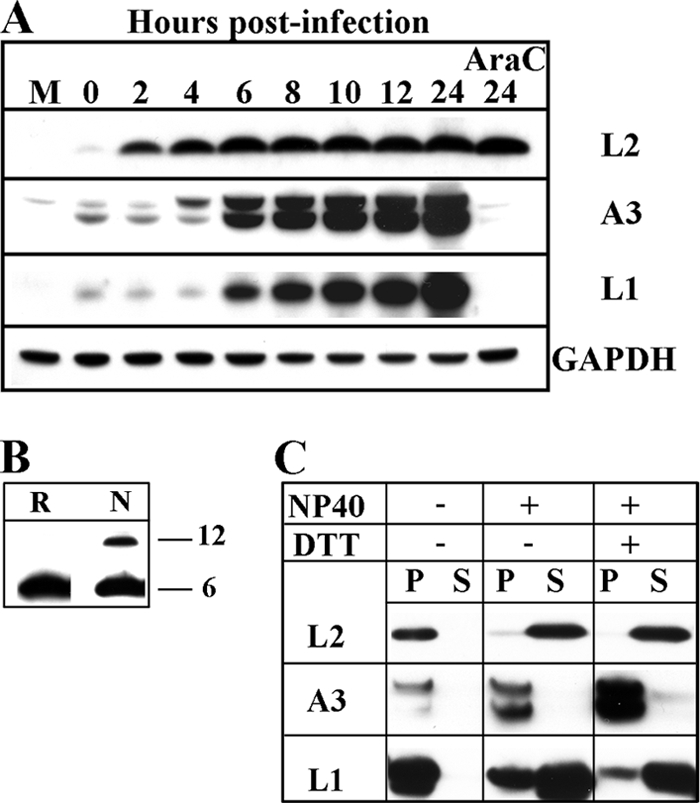
L2 was expressed early during VACV infection, incorporated into MVs, and released by NP-40 detergent. (A) Temporal synthesis of L2. BS-C-1 cells were mock infected (M) or infected with VACV at a multiplicity of 10 PFU per cell in the absence or presence of cytosine arabinoside (AraC) and harvested at the indicated times. The proteins from whole-cell extracts were extracted with lithium dodecyl sulfate buffer containing reducing agent, resolved by SDS-PAGE, and analyzed by Western blotting with antibodies to the indicated viral proteins or the cellular protein GAPDH as a loading control. (B) Unreduced L2 migrated as two species on SDS-PAGE. Cells were infected as in panel A for 18 h, and whole-cell lysates were solubilized with (R) or without (N) reducing agent, resolved by SDS-PAGE, and analyzed by Western blotting with L2 antibody. The masses of the two proteins indicated on the right were estimated from the mobilities of markers. (C) Extraction of L2 from purified virions. Purified MVs were incubated at 37°C in Tris buffer with (+) or without (−) 1% NP-40 and 50 mM dithiothreitol (DTT) and centrifuged to separate the soluble (S) and insoluble (P) fractions. The insoluble fraction was resuspended in a volume equal to the soluble fraction, and aliquots of each were resolved by SDS-PAGE. The proteins were analyzed by Western blotting using antibodies to the indicated viral proteins.
L2 was detected by Western blotting of virions purified by sedimentation through two sucrose cushions and a gradient and was extracted with NP-40 alone or with dithiothreitol, similar to the L1 membrane protein and distinct from the A3 core protein (Fig. 2C). A putative dimer was detected by Western blotting in the absence of reducing agent (not shown), similar to that in cell lysates (Fig. 2B). Our analyses suggested that L2 is synthesized early after infection, remains stable during the remainder of the replication cycle, and is a membrane component of the MV.
L2R is an essential gene.
Attempts to replace the L2R gene with DNA encoding EGFP were unsuccessful, suggesting that L2 has an essential role in VACV replication. To determine the role of L2, we constructed a recombinant virus in which the expression of L2 was regulated by the inducer IPTG. The inducible virus was generated in two steps, starting with vT7lacOI, which expresses the lac repressor regulated by an early/late promoter and the T7 RNA polymerase under the control of the lac operator and a late promoter (44). First, the L2R gene containing a bacteriophage T7 promoter, the Escherichia coli lac operator, and the untranslated leader sequence of encephalomyocarditis virus RNA was inserted into the A56R locus of vT7LacOI. The intermediate virus vL2R/L2Ri contained the endogenous L2R gene and the inducible one. In the second step, the endogenous L2R gene was replaced with EGFP by homologous recombination. The final recombinant virus, vL2Ri (Fig. 3 A), was isolated in the presence of 100 μM IPTG, and the genotype was confirmed by sequencing.
FIG. 3.

Construction and plaque phenotype of the recombinant VACV with an inducible L2R ORF (vL2Ri). (A) Schematic representation of the genome structure of vL2Ri. P11, a VACV late promoter; lacO, lac operator; T7pol, bacteriophage T7 RNA polymerase ORF; P7.5, a VACV early/late promoter; lacI, E. coli lac repressor ORF; EGFP, enhanced green fluorescent protein ORF; gpt, E. coli guanine phosphoribosyltransferase ORF; PT7, bacteriophage T7 promoter; EMC, encephalomyocarditis virus cap-independent translation enhancer element. (B) Plaque phenotype of vL2Ri. BS-C-1 cells were infected with the recombinant virus vL2Ri in the presence (+) or absence (−) of 50 μM IPTG. After 48 h, the cells were stained with crystal violet.
vL2Ri did not form visible plaques in the absence of IPTG, indicating that L2 expression is essential for replication or virus spread (Fig. 3B). In order to further determine the effect of IPTG on virus replication, BS-C-1 cells were infected with vL2Ri at a range of IPTG concentrations. Increased replication occurred at 5 μM IPTG and reached a plateau between 50 and 100 μM IPTG, correlating with the induction of L2 protein synthesis shown by Western blotting (Fig. 4 A). In contrast, replication of the parental vT7lacOI was independent of IPTG.
FIG. 4.

Dependence of vL2Ri replication on IPTG. (A). Effects of IPTG concentration. BS-C-1 cells were mock infected (M) or infected with the recombinant virus vL2Ri or vT7lacOI at a multiplicity of 3 PFU per cell in the presence of 0, 5, 10, 25, 50, 100, or 200 μM IPTG. After 24 h, the cells were harvested and the virus yields were determined by plaque assay in the presence of 50 μM IPTG. L2 synthesized under these conditions was determined by Western blotting. M, mock infected. GAPDH was detected as a loading control. (B) One-step growth curve of vL2Ri. BS-C-1 cells were infected at a multiplicity of 3 PFU per cell with vT7lacOI in the absence of IPTG or vL2Ri in presence or absence of 50 μM IPTG. After 0, 6, 9, 12, 24, 36, and 48 h, the infected cells were harvested and the virus titers were determined by plaque assay in the presence of 50 μM IPTG.
The kinetics of vL2Ri replication in the presence of IPTG indicated a delay of several hours compared to the parental vT7LacOI strain (Fig. 4B). This may be due to the difference in the regulation of L2 synthesis. In vT7LacOI and wild-type VACV, the L2R ORF is under the control of an early promoter and is expressed before viral DNA replication. However, in vL2Ri the L2 ORF is under the control of the T7 promoter and must be expressed after viral DNA replication. Thus, these data suggest that early expression of L2 is advantageous but not essential.
Synthesis and processing of late proteins.
To determine whether L2 is required for the synthesis of viral proteins, BS-C-1 cells were infected with vT7lacOI or vL2Ri in the presence and absence of IPTG. Infected cells were labeled with radioactive amino acids for 30 min at several times postinfection and harvested. The proteins were resolved by SDS-PAGE and analyzed by autoradiography. The synthesis of cellular proteins decreased by 6 h postinfection, at which time the synthesis of viral late proteins was evident. The proteins synthesized during infections with vL2Ri in the presence or absence of IPTG and the parental vT7LacOI were similar, except for the 85-kDa product of the A56R gene, which was disrupted during insertion of the L2R ORF to make vL2Ri (Fig. 5 A).
FIG. 5.

Synthesis and processing of viral proteins. (A) Pulse-labeling of proteins during infection. BS-C-1 cells were mock infected (M) or infected at a multiplicity of 3 PFU per cell with vT7lacOI (vT7) or with vL2Ri in the presence (+) or absence (−) of 50 μM IPTG. Infected cells were labeled with 100 μCi of [35S]methionine-cysteine for 30 min at the indicated times postinfection. The whole-cell lysates were analyzed by SDS-PAGE, followed by autoradiography. The positions and masses of marker proteins in kDa are indicated on the left. The arrow points to the 85-kDa A56 protein made only in cells infected with vT7lacOI. (B) Proteolytic processing of viral proteins. BS-C-1 cells were mock infected (M) or infected at a multiplicity of infection of 5 PFU cell with vT7lacOI (vT7) in the presence (+) or absence (−) of 100 μg/ml of rifampin (RIF) or with vL2Ri in the presence or absence of 50 μM IPTG. After 6 h, the cells were pulse-labeled with 100 μCi of [35S]methionine-cysteine for 30 min. Cells were either harvested immediately (Pulse) or incubated with excess unlabeled methionine and cysteine for an additional 12 h (Chase). The proteins were resolved by SDS-PAGE and detected by autoradiography. The major core protein precursors (P4a and P4b) and their processed forms (4a and 4b) are indicated on the right. The positions and masses in kDa of the marker proteins are shown on the left. (C) Proteolytic processing of membrane and core proteins analyzed by Western blotting. BS-C-1 cells were infected as in panel B. After 18 h, the cells were lysed and the proteins were resolved by SDS-PAGE and analyzed by Western blotting with antibodies to the viral MV membrane protein A17, the MV core protein A3 (P4b), or the EV membrane protein F13, as indicated on the right.
Proteolytic processing of several core proteins by the I7 protease occurs during VACV infection but not when morphogenesis is prevented either by the drug rifampin or by certain mutant viruses. To ascertain whether L2 expression was required for protein processing, BS-C-1 cells were infected with vL2Ri in presence or absence of IPTG and with vT7lacOI in presence or absence of rifampin as a control. At 6 h after infection, the cells were labeled with radioactive amino acids for 30 min and either harvested or chased with unlabeled amino acids for 12 h to allow morphogenesis and then harvested. The labeled proteins were analyzed by SDS-PAGE and visualized by autoradiography. The protein patterns after the 30-min pulse were similar in each sample, except for the additional 85-kDa A56 band in vT7LacOI (Fig. 5B). After the chase, the protein patterns from cells infected with vT7LacOI in the presence of rifampin and vL2Ri in the absence of IPTG were unchanged from the pulse, indicating the absence of processing of the major core proteins. In contrast, the P4a (A10R) and P4b (A3R) core proteins were cleaved in the absence of rifampin and presence of IPTG.
To confirm the block in the processing of core proteins, lysates from cells infected for 18 h with vT7LacOI in the presence or absence of rifampin and with vL2Ri in the presence or absence of IPTG were analyzed by Western blotting and probing with antibodies to several viral proteins. L2 was detected in lysates of cells infected with vT7LacOI but not from cells infected with vL2Ri in the absence of IPTG, as expected (Fig. 5C). The core protein P4B (A3) was not cleaved under the latter conditions nor in the cells infected with vT7LacOI in the presence of rifampin (Fig. 5C). Proteolytic processing of the A17 membrane protein, like that of the core proteins, depends on the I7 protease but occurs earlier and is not inhibited by rifampin (2), as confirmed in the Western blot (Fig. 5C). Processing of A17 was clearly reduced in the absence of L2 expression but not completely abrogated (Fig. 5C), suggesting that the block occurred prior to formation of the IV. F13, an EV membrane protein, was used as a control because it does not undergo processing.
Effects of L2 repression on VACV morphogenesis.
The localizations of the core protein A3, the membrane protein A17, and the IV scaffold protein D13 in cells infected with vL2Ri in the presence and absence of IPTG were compared by confocal microscopy. 4′,6-Diamidino-2-phenylindole (DAPI) staining was used to visualize nuclei and DNA-containing virus factories, which usually have a juxtanuclear localization. In the presence of IPTG, antibodies to L2, A3, A17, and D13 stained factory regions in particulate patterns (Fig. 6). In the absence of IPTG, antibody staining of L2 was very faint, as expected due to repression of expression. Interestingly, the A3 antibody staining appeared to surround dark holes, which were also not stained by DAPI, sometimes giving a ring-like appearance. In contrast, antibodies to A17 and D13 had more particulate patterns than A3 in the absence of IPTG (Fig. 6). Ring-like staining had previously been seen in cells infected with inducible A11 and H7 mutants and was due to the presence of large, dense masses of viroplasm that excluded antibody (33).
FIG. 6.

Confocal microscopy of cells infected with vL2Ri in the presence or absence of IPTG. HeLa cells were infected with vL2Ri in the presence or absence of 50 μM IPTG at a multiplicity of 5 PFU per cell. After 16 h, the infected cells were fixed, permeabilized, and stained with primary antibodies for L2, A3, A17, and D13, followed by goat anti-rabbit IgG coupled to Alexa Fluor 594 and DAPI.
Transmission electron microscopy was used to visualize VACV morphogenesis at high magnification. Cells infected with vL2Ri at 1 PFU per cell, with or without IPTG, were fixed at 8 h, and thin sections were prepared. In the presence of IPTG, all stages of VACV morphogenesis were seen, including crescents, IVs, MVs, and WVs (Fig. 7 A). We did not detect IVs, MVs, or WVs, and only rare cells contained IVs in the absence of IPTG. Under the latter conditions, the majority of infected cells had large, circular, electron-dense masses of viroplasm, likely corresponding to the confocal images of holes that excluded DNA and antibody, and separate lower-density inclusions (Fig. 7B). At 8 h, two-thirds of the dense masses (26/39) had no visible membranes at their periphery (Fig. 7C), whereas the remainder had short “spicule”-coated membrane structures resembling crescent segments (Fig. 7B and D). At 19 h, however, short crescent segments were associated with the majority of the dense masses, and IVs and more mature forms were present in some cells (not shown). At higher multiplicities of infection, the membrane structures appeared earlier. In cells infected with the inducible H7 mutant under nonpermissive conditions, similar dense masses and lower-density inclusions were seen; immunogold labeling indicated that the dense masses contained core protein and the less-dense inclusions contained D13 and membranes with A17 (33).
FIG. 7.

Transmission electron microscopy of cells infected with vL2Ri in the presence or absence of IPTG. BS-C-1 cells were infected with vL2Ri at a multiplicity of 1 PFU per cell in the presence (A) or absence (B, C, and D) of 50 μM IPTG. At 8 h, the infected cells were fixed and prepared for transmission electron microscopy. Short crescent membranes are on the periphery of dense aggregates of viroplasm in panels B and D but are absent in panel C. Abbreviations: C, crescents; IV, immature virion; nu, IV that contains a nucleoid; WV, wrapped virion; EV, enveloped virion; SC, short crescent-like structures; V, dense masses of viroplasm; and *, putative D13 accumulation sites. Magnifications are indicates by scale bars at the bottom of each panel.
DISCUSSION
L2 was one of the few remaining proteins conserved in all chordopoxviruses that had not yet been characterized. Inspection of the ORF revealed two hydrophobic domains comprising the C-terminal half of the putative protein, suggesting that it might be membrane associated, but there were no other motifs or clues to its function. The presence of a consensus early promoter and the failure to detect L2 by mass spectrometry of purified MVs (7, 28, 47) initially led us to think that L2 might be involved in an important host interaction and possibly nonessential in tissue culture, despite its conservation. We confirmed that L2 is expressed early in infection, but determined that it is an essential membrane-associated component of MVs and is required for an early step in morphogenesis. The failure to detect L2 by mass spectrometry may be related to the long hydrophobic domains and possibly low abundance (22).
Because our attempts to obtain a viable L2R knockout virus failed, we constructed an inducible mutant and demonstrated that it had a conditional lethal phenotype. The mode of construction of the mutant requires additional comment. The lac operator system has not been used with VACV early promoters because of the consideration that the lac repressor would have to be packaged in virions for stringent regulation. We avoided this potential problem in the following way. The lac repressor gene was regulated by the P7.5 early/late promoter, the T7 RNA polymerase gene was regulated by the stringent P11 late promoter (and lac operator), and the L2 gene was regulated by a T7 promoter (and lac operator). Therefore, repressor was made immediately after infection in the absence or presence of IPTG, whereas L2 was not expressed until after DNA replication and late synthesis of the T7 RNA polymerase in the presence of IPTG. As with authentic late proteins, there should be no need to package repressor. However, this system could only work if de novo synthesis of L2 was not required at early times, which was apparently the case. We did note a delay in the formation of infectious virus that might be due to altered timing of L2 expression.
When expression of L2 was repressed, the synthesis of viral proteins and the usual decline in host protein synthesis appeared normal. However, proteolytic processing of the A17 membrane protein was reduced, and processing of the A3 and A10 core proteins was prevented, indicating an early block in virus assembly. The latter was confirmed by confocal and electron microscopy of infected cells. Neither crescents nor IVs were seen. Instead, there was an accumulation of electron-dense masses of viroplasm, similar in appearance to the interior of IVs but much larger. In some cells, there were short membrane-like structures with a “spicule” coat resembling stunted crescents on the exterior of the dense masses. The overall appearance was indistinguishable from that occurring in cells infected with an H7-inducible mutant (33) and similar to that of cells infected with an A11-inducible mutant (29). In those studies, we showed that the dense masses contained core proteins and the less-dense inclusions had D13 and membranes with A17.
The formation of the short membrane segments on the exterior of some of the dense masses might be due to minute amounts of L2 made in the absence of IPTG, although this was undetectable by Western blotting. An analogous explanation was proposed for the phenotype of an H7-inducible mutant (33). Alternatively, L2 and H7 may be important for efficient elongation of the short membrane arcs, which have delayed appearance and fail to progress to IVs in their absence. The construction of complementing cell lines and knockout mutants may provide a way to distinguish between these possibilities, although this procedure has rarely been achieved with VACV—possibly because of a need to express proteins within the cytoplasmic factory.
Despite the very similar null phenotypes of L2, A11, and H7 mutants, the latter two proteins do not have putative transmembrane regions and were not detected in purified virions. Although it seems reasonable to think that these proteins have interrelated roles, we have been unable to detect a stable physical interaction between them by immunoprecipitation. A14 and A17 are MV membrane proteins, but their null phenotypes are clearly distinguishable from that of L2. A prominent feature of the A17 and A14 mutants is the accumulation of small vesicles, which was not seen with L2, A11, or H7 mutants.
The formation of the viral membrane is the most enigmatic process in the biology of poxviruses. The identification of an additional viral protein involved in this process represents an important step toward elucidating the mechanism. When all of the proteins are known, it may be possible to investigate membrane formation in a virus-free system.
Acknowledgments
We thank Catherine Cotter for preparation of cells and Owen Schwartz for help with confocal microscopy.
The research was supported by the Division of Intramural Research, NIAID, NIH.
Footnotes
Published ahead of print on 12 January 2011.
REFERENCES
- 1.Alexander, W. A., B. Moss, and T. R. Fuerst. 1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA polymerase and the Escherichia coli lac repressor. J. Virol. 66:2934-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ansarah-Sobrinho, C., and B. Moss. 2004. Role of the I7 protein in proteolytic processing of vaccinia virus membrane and core components. J. Virol. 78:6335-6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Assarsson, E., et al. 2008. Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc. Natl. Acad. Sci. U. S. A. 105:2140-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Betakova, T., E. J. Wolffe, and B. Moss. 1999. Membrane topology of the vaccinia virus A17L envelope protein. Virology 261:347-356. [DOI] [PubMed] [Google Scholar]
- 5.Betakova, T., E. J. Wolffe, and B. Moss. 1999. Regulation of vaccinia virus morphogenesis: phosphorylation of the A14L and A17L membrane proteins and C-terminal truncation of the A17L protein are dependent on the F10L protein kinase. J. Virol. 73:3534-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bisht, H., A. S. Weisberg, P. Szajner, and B. Moss. 2009. Assembly and disassembly of the capsid-like external scaffold of immature virions during vaccinia virus morphogenesis. J. Virol. 83:9140-9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung, C. S., et al. 2006. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 80:2127-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cresawn, S. G., and R. C. Condit. 2007. A targeted approach to identification of vaccinia virus postreplicative transcription elongation factors: genetic evidence for a role of the H5R gene in vaccinia transcription. Virology 363:333-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.da Fonseca, F. G., A. S. Weisberg, M. F. Caeiro, and B. Moss. 2004. Vaccinia virus mutants with alanine sbstitutions in the conserved G5R gene fail to initiate morphogenesis at the nonpermissive temperature. J. Virol. 78:10238-10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dales, S., and E. H. Mosbach. 1968. Vaccinia as a model for membrane biogenesis. Virology 35:564-583. [DOI] [PubMed] [Google Scholar]
- 11.Dales, S., and L. Siminovitch. 1961. The development of vaccinia virus in Earle's L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 10:475-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D'Costa, S. M., et al. 2010. Vaccinia H5 is a multifunctional protein involved in viral DNA replication, postreplicative gene transcription, and virion morphogenesis. Virology 401:49-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeMasi, J., and P. Traktman. 2000. Clustered charge-to-alanine mutagenesis of the vaccinia virus H5 gene: isolation of a dominant, temperature-sensitive mutant with a profound defect in morphogenesis. J. Virol. 74:2393-2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derrien, M., A. Punjabi, R. Khanna, O. Grubisha, and P. Traktman. 1999. Tyrosine phosphorylation of A17 during vaccinia virus infection: involvement of the H1 phosphatase and the F10 kinase. J. Virol. 73:7287-7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Earl, P. L., N. Cooper, L. S. Wyatt, B. Moss, and M. W. Carroll. 1998. Preparation of cell cultures and vaccinia virus stocks, p. 16.16.1-16.16.3. In F. M. Ausubel et al. (ed.), Current protocols in molecular biology, vol. 2. John Wiley and Sons, New York, NY. [DOI] [PubMed] [Google Scholar]
- 16.Earl, P. L., B. Moss, L. S. Wyatt, and M. W. Carroll. 1998. Generation of recombinant vaccinia viruses, p. 16.17.1-16.17.19. In F. M. Ausubel et al. (ed.), Current protocols in molecular biology, vol. 2. Greene Publishing Associates & Wiley Interscience, New York, NY. [Google Scholar]
- 17.Hiller, G., and K. Weber. 1985. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol. 55:651-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Husain, M., and B. Moss. 2003. Evidence against an essential role of COPII-mediated cargo transport to the endoplasmic reticulum-Golgi intermediate compartment in the formation of the primary membrane of vaccinia virus. J. Virol. 77:11754-11766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Husain, M., and B. Moss. 2005. Role of receptor-mediated endocytosis in the formation of vaccinia virus extracellular enveloped particles. J. Virol. 79:4080-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Husain, M., A. S. Weisberg, and B. Moss. 2006. Existence of an operative pathway from the endoplasmic reticulum to the immature poxvirus membrane. Proc. Natl. Acad. Sci. U. S. A. 103:19506-19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovacs, G. R., and B. Moss. 1996. The vaccinia virus H5R gene encodes late gene transcription factor 4: purification, cloning and overexpression. J. Virol. 70:6796-6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu, B., D. B. McClatchy, J. Y. Kim, and J. R. Yates III. 2008. Strategies for shotgun identification of integral membrane proteins by tandem mass spectrometry. Proteomics 8:3947-3955. [DOI] [PubMed] [Google Scholar]
- 23.Morgan, C. 1976. The insertion of DNA into vaccinia virus. Science 193:591-592. [DOI] [PubMed] [Google Scholar]
- 24.Moss, B. 2007. Poxviridae: the viruses and their replication, p. 2905-2946. In D. M. Knipe and P. M. Howley (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 25.Moss, B., and E. N. Rosenblum. 1973. Protein cleavage and poxvirus morphogenesis: tryptic peptide analysis of core precursors accumulated by blocking assembly with rifampicin. J. Mol. Biol. 81:267-269. [DOI] [PubMed] [Google Scholar]
- 26.Moss, B., E. N. Rosenblum, E. Katz, and P. M. Grimley. 1969. Rifampicin: a specific inhibitor of vaccinia virus assembly. Nature 224:1280-1284. [DOI] [PubMed] [Google Scholar]
- 27.Nagayama, A., B. G. T. Pogo, and S. Dales. 1970. Biogenesis of vaccinia: separation of early stages from maturation by means of rifampicin. Virology 40:1039-1051. [DOI] [PubMed] [Google Scholar]
- 28.Resch, W., K. K. Hixson, R. J. Moore, M. S. Lipton, and B. Moss. 2007. Protein composition of the vaccinia virus mature virion. Virology 358:233-247. [DOI] [PubMed] [Google Scholar]
- 29.Resch, W., A. S. Weisberg, and B. Moss. 2005. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J. Virol. 79:6598-6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Risco, C., et al. 2002. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 76:1839-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez, J. R., C. Risco, J. L. Carrascosa, M. Esteban, and D. Rodriguez. 1997. Characterization of early stages in vaccinia virus membrane biogenesis: implications of the 21-kilodalton protein and a newly identified 15-kilodalton envelope protein. J. Virol. 71:1821-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez, J. R., C. Risco, J. L. Carrascosa, M. Esteban, and D. Rodriguez. 1998. Vaccinia virus 15-kilodalton (A14L) protein is essential for assembly and attachment of viral crescents to virosomes. J. Virol. 72:1287-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satheshkumar, P. S., A. Weisberg, and B. Moss. 2009. Vaccinia virus H7 protein contributes to the formation of crescent membrane precursors of immature virions. J. Virol. 83:8439-8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmelz, M., et al. 1994. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans Golgi network. J. Virol. 68:130-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Senkevich, T. G., E. V. Koonin, and B. Moss. 2009. Predicted poxvirus FEN1-like nuclease required for homologous recombination, double-strand break repair and full-size genome formation. Proc. Natl. Acad. Sci. U. S. A. 106:17921-17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith, G. L., and M. Law. 2004. The exit of vaccinia virus from infected cells. Virus Res. 106:189-197. [DOI] [PubMed] [Google Scholar]
- 37.Sodeik, B., et al. 1993. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J. Cell Biol. 121:521-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sodeik, B., G. Griffiths, M. Ericsson, B. Moss, and R. W. Doms. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J. Virol. 68:1103-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szajner, P., A. S. Weisberg, J. Lebowitz, J. Heuser, and B. Moss. 2005. External scaffold of spherical immature poxvirus particles is made of protein trimers, forming a honeycomb lattice. J. Cell Biol. 170:971-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szajner, P., A. S. Weisberg, and B. Moss. 2004. Evidence for an essential catalytic role of the F10 protein kinase in vaccinia virus morphogenesis. J. Virol. 78:257-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tooze, J., M. Hollinshead, B. Reis, K. Radsak, and H. Kern. 1993. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur. J. Cell Biol. 60:163-178. [PubMed] [Google Scholar]
- 42.Traktman, P., A. Caligiuri, S. A. Jesty, and U. Sankar. 1995. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of morphogenesis. J. Virol. 69:6581-6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Traktman, P., et al. 2000. Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: construction and characterization of a tetracycline-inducible recombinant. J. Virol. 74:3682-3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ward, G. A., C. K. Stover, B. Moss, and T. R. Fuerst. 1995. Stringent chemical and thermal regulation of recombinant gene expression by vaccinia virus vectors in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 92:6773-6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolffe, E. J., D. M. Moore, P. J. Peters, and B. Moss. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang, Z., D. P. Bruno, C. A. Martens, S. F. Porcella, and B. Moss. 2010. Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:11513-11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoder, J. D., et al. 2006. Pox proteomics: mass spectrometry analysis and identification of vaccinia virion proteins. Virol. J. 3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang, L. L., et al. 2009. A role for the host coatomer and KDEL receptor in early vaccinia biogenesis. Proc. Natl. Acad. Sci. U. S. A. 106:163-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang, Y., and B. Moss. 1992. Immature viral envelope formation is interrupted at the same stage by lac operator-mediated repression of the vaccinia virus D13L gene and by the drug rifampicin. Virology 187:643-653. [DOI] [PubMed] [Google Scholar]