

Abstract
Infections with certain human papillomaviruses (HPV), such as type 16 (HPV16), 18, or 31, are a necessary risk factor for the development of cervical cancer. Transcript analyses of several HPV revealed that the viral E2 gene encodes both the E2 regulator protein and the E8∧E2C protein, which differ in their amino termini. Up to now, functional studies have focused on HPV31 E8∧E2C and demonstrated that it is a potent repressor of viral transcription and replication. However, recent analyses of HPV16 genomes have suggested that E8∧E2C proteins may differ in their activities. Therefore, we performed a comparative analysis of E8∧E2C proteins of HPV16, -18, and -31. All E8∧E2C proteins potently inhibited HPV E6/E7 oncogene promoters, and also displayed long-distance transcriptional-repression activities. Furthermore, the expression of all E8∧E2C proteins inhibited the growth of HeLa cells. Expression of E8∧E2C proteins rapidly increased the protein levels of the E6 and E7 targets p53 and p21, consistent with the repression of the endogenous HPV18 E6/E7 promoter. All E8∧E2C proteins induced G1 arrest more efficiently than E2 proteins and activated senescence markers. Furthermore, we demonstrate that the 31E8 domain can be functionally replaced by the KRAB repression domain derived from KOX1. The KRAB-E2C fusion protein possesses long-distance transcriptional-repression activity and inhibits the growth of HeLa cells comparably to E8∧E2C. Taken together, our results suggest that the E8∧E2C proteins of HPV16, -18, and -31 are highly conserved transcriptional repressors that inhibit the growth of HeLa cells by repression of E6/E7 transcription but do not have proapoptotic activities.
Persistent infections with human papillomaviruses (HPV), such as HPV type 16 (HPV16), -18, or -31, are a necessary risk factor for the development of invasive cervical cancer (4, 42, 47). HPV16 accounts for ∼55%, HPV18 for ∼16%, and HPV31 for only ∼4% of cervical cancers worldwide (3), but the underlying differences accounting for these behaviors are not well understood. The viral E2 gene expresses crucial regulatory proteins involved in replication, transcription, and maintenance of viral genomes (19, 40). The E2 protein is a sequence-specific DNA binding protein that recognizes four E2 binding sites (E2BS) upstream of the HPV E6/E7 promoter through its C-terminal domain (E2C) (26). The amino-terminal domain of E2 (E2TA) is responsible for the activation of transcription, the activation of viral replication, and attachment to mitotic chromosomes (19, 40). In addition to E2, several HPV express a spliced RNA that expresses a fusion protein consisting of the small E8 domain fused to E2C (9, 29, 33, 36, 37). The functions of the E8∧E2C protein have been mainly investigated with HPV31. It was shown that E8∧E2C knockout HPV31 genomes displayed a strong overreplication of viral genomes in short-term analyses (37). Despite this, in stable cell lines, HPV31 E8∧E2C (31E8∧E2C) knockout genomes were not maintained as episomes but only found integrated into the host chromosomes, suggesting that E8∧E2C is required for the long-term extrachromosomal maintenance of viral genomes (37). Genetic and biochemical analyses of the 31E8∧E2C protein have demonstrated that the 31E8 domain is required for transcriptional repression. This is due to the recruitment of cellular corepressors, such as the histone deacetylase 3 (HDAC3)/N-CoR complex, by the 31E8 domain (1, 31). The analysis of E8∧E2C functions in the context of HPV16, the most potent cancer-inducing HPV, has revealed differences from HPV31. While HPV16 E8∧E2C knockout genomes also display a short-term overreplication phenotype, 16E8∧E2C is not required for stable maintenance of HPV16 episomes (21). This suggested that E8∧E2C activities may vary among different papillomaviruses (PV).
The expression of E2 proteins in HeLa cells leads to growth arrest. This is mainly due to the transcriptional repression of the endogenous HPV18 upstream regulatory region (URR) promoter, which drives the expression of the E6 and E7 oncoproteins. Shutdown of E6 and E7 expression by E2 reactivates key cellular E6 and E7 target proteins, such as p53, p21, and the Rb family (6, 10, 13, 17, 44). This causes permanent growth arrest and coincides with the appearance of markers for replicative senescence, such as senescence-associated beta-galactosidase (SA-β-Gal) (8, 15, 44). Interestingly, in some studies, the E2 proteins derived from the most prevalent carcinogenic HPV types, 16 and 18, have been shown to induce apoptosis (6, 7, 43). In the case of HPV18, E2 apoptosis induction has been linked to an interaction of the E2TA domain with caspase 8 (2, 41). Taken together, the E2 proteins from HPV16 and -18 may inhibit the growth of HeLa cells by repression of E6/E7 transcription, leading to senescence and the induction of apoptosis independently of other HPV gene products.
Interestingly, fusion proteins of viral or cellular transcription activation domains and the E2C DNA binding domain have failed to inhibit URR promoter activity or to induce growth arrest in HeLa cells (10, 14, 28). In contrast, the 31E8∧E2C protein inhibits the growth of HeLa cells as well as 31E2 (31, 38). Growth inhibition by 31E8∧E2C was correlated with a reduction of E6/E7 transcripts and the induction of p53, p21, and pRb proteins (38). This suggested that 31E8∧E2C mainly induces growth inhibition by transcriptional repression of the integrated HPV18 URR promoter.
Due to the reported additional activities of 16E2 and 18E2 proteins compared to other PV E2 proteins and the differences between HPV16 and HPV31 E8∧E2C knockout genomes, we performed comparative analyses of E8∧E2C proteins from HPV16, -18, and -31. We found that the different E8∧E2C proteins share similar transcription repression activities on several E2BS-dependent reporter plasmids. All E8∧E2C proteins are able to prevent the growth of HeLa cells, and this coincides with the rapid increase of p53 and p21 protein levels. We also provide evidence that short-term expression of the different E8∧E2C proteins and the respective E2 proteins induces a cell cycle arrest in the G1 phase and that long-term expression induces markers for senescence. In addition, we demonstrate that the E8 domain can be functionally replaced by the KRAB (Krüppel-associated box) domain, derived from the cellular KOX1 protein. Together, our results suggest that the E8∧E2C proteins of the carcinogenic HPV types 16, 18, and 31 are potent transcriptional repressors that inhibit growth of HeLa cells by inhibition of E6/E7 transcription but do not have proapoptotic activities.
MATERIALS AND METHODS
Plasmid construction.
The luciferase reporter plasmids pC18-SP1-luc, pGL 31URR-luc, pGL 31URR-BS2,3,4 mt-luc, and pGL 18URR-luc and the expression plasmids for HPV31 E8∧E2C (pSG 31E8∧E2C) hemagglutinin (HA)-tagged 31E8∧E2C-HA, HPV31 E2, and HPV31 E2-HA have been previously described (38, 39). To generate the luciferase reporter plasmid pGL 16URR-luc, a fragment (HPV16 nucleotides [nt] 7141 to 7173) was amplified by PCR using the cloned HPV16 114b genome (kindly provided by M. Dürst) as a template and primers 16URR XhoI-F (5′-ATATATCTCGAGTATTGTATGTATGT-3′) and 16URR NcoI-R (5′-ATATATCCATGGTGCAGTTCTCTTTTG-3′). The amplicon was digested with XhoI and NcoI and cloned into XhoI/NcoI-digested pGL3-basic (Promega, Mannheim, Germany). Reverse transcription (RT)-PCR products were generated with RNA isolated from HPV18-positive cells and primers HPV18 1288 forward (5′-AAAAAGGCGGCTGTTTACAA-3′) and HPV18 3996 reverse (5′-GACATGGCAGCACACATAC-3′) and then cloned into pDrive, giving rise to pDrive-18E8∧E2C. The fragment was sequenced and then excised by EcoRI digestion and inserted into pSG5 (Stratagene, La Jolla, CA). The HPV16 E8∧E2C cDNA (HPV16 nt 1258 to 3913) was amplified by RT-PCR using RNA of HPV16-positive W12 cells as a template and primers HPV16 1258 forward (5′-AGCGGGTATGGCAATACTGA-3′) and HPV16 3913 reverse (5′-GCACACAAAAGCAAAGCAAA-3′). The amplicon was used as a template for PCR with the primers 16E8 RI F (5′-ATCGAGAATTCATGGCAATACTGAA -3′) and 16E2 Bam R (5′-ACTGACGGATCCTCATATAGACATAA3′), digested with EcoRI and BamHI, and cloned into EcoRI/BamHI-digested pSG5. The internal HA tag was inserted into the HPV18 E8∧E2C sequence between nt 3509 and 3510 by PCR (pSG 18E8∧E2C-HA). To generate pSG 16E8∧E2C-HA, a double-stranded oligonucleotide encoding the HA epitope sequence was inserted into the MscI site of pSG 16E8∧E2C. The coding sequences for HPV16 and -18 E2 were amplified by PCR using the cloned HPV16 114b genome or the HPV18 genome and then inserted between the EcoRI and BamHI sites of plasmid pSG5. Plasmids pSG 16E2-HA and pSG 18E2-HA, encoding internally HA-tagged proteins, were generated by overlap extension PCR. Plasmid pSG KRAB-E2C was generated by PCR using pKRAB-Gal4 (1) as a template and primers KRAB/E2C F 5′-CTCTGAGAATTCCATGGATGCTAAGTCACTA-3′ and KRAB/E2C R 5′-ATATCTGAATTCCCTGCAGTCTCTGAATCAGG-3′. The amplicon was digested with EcoRI and used to replace the small EcoRI fragment in pSG 31E8∧E2C. To generate pSG KRAB-E2C-HA, an HA epitope was inserted by PCR between the KRAB and the E2C coding sequences. pSG KRAB-HA was generated by inserting the EcoRI fragment encoding KRAB-HA into EcoRI-digested pSG5.
Cell culture.
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (no. 41665-062; Invitrogen, Karlsruhe, Germany) supplemented with gentamicin and 10% fetal bovine serum (Seromed Biochrom, Berlin, Germany). Colony reduction assays were essentially performed as previously described (38). Briefly, 3 × 105 HeLa cells were transfected with Fugene HD (Roche) in 60-mm dishes with 0.8 μg of the respective expression vectors and 0.2 μg of pPur. Cells were selected with 0.4 μg/ml puromycin for 10 to 12 days. Colonies were then fixed with acetone-methanol (1:1 [vol/vol]), stained with eosin solution, and then counted. Human keratinocytes harboring replicating HPV18 genomes (20) or replicating HPV16 (W12) genomes (kindly provided by L. A. Laimins) were maintained in the presence of NIH 3T3 feeder layers as previously described (37). The HPV-negative keratinocyte RTS3b cell line was maintained in epithelial cell medium containing 5% fetal bovine serum (32).
HeLa cells were transfected with control small interfering RNA (siRNA) (siAllstar; Qiagen, Hilden, Germany) or siRNA18E7 (22) using HiPerfect reagent (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
Luciferase assays.
Approximately 3 × 104 HeLa or RTS3b cells were seeded into 24-well dishes the day before transfection. The cells were cotransfected with 50 ng of luciferase reporters and 10 ng of the respective expression vector DNA or empty vector pSG5, as indicated in the figure legends. Transfections were carried out using Fugene HD (Roche) and Opti-MEM (Invitrogen). Luciferase assays were carried out 48 h after transfection, as previously described (39).
Immunoblot analyses.
Transfected cells were lysed 48 h posttransfection in 30 μl of 4× SDS gel loading buffer (Carl Roth, Karlsruhe, Germany), heated to 95 C°, and separated in a 12% SDS-PAGE. The proteins were transferred in 10 mM N-cyclohexyl-3-amino propanesulfonic acid (pH 10.3) onto a nitrocellulose membrane (Protran; Whatman, Dassel, Germany). The membranes were blocked by incubation in Tris-buffered saline (TBS)-0.1% Tween 20-50 mg/ml nonfat dry milk for 30 min and then incubated with diluted primary antibodies (HA-probe [Covance MMS-101P], 1:1,000; HSP90 [Santa Cruz sc-69703], 1:1,500; CDKN1A/p21 [BD Pharmingen 556430], 1:1,500; and p53 [DO-1; Santa Cruz sc-126], 1:1,000). Bound antibodies were detected with anti-rabbit (polyclonal swine anti-rabbit immunoglobulin-horseradish peroxidase [HRP]; Dako, Hamburg, Germany; 1:3,000) or anti-mouse (polyclonal rabbit anti-mouse immunoglobulin-HRP; Dako; 1:3,000) antibodies conjugated to horseradish peroxidase and Super-Signal West Dura reagent (Perbio Science, Bonn, Germany). Chemiluminescent signals were recorded with a FluorSMax imaging system (Bio-Rad, Munich, Germany).
Senescence associated beta-galactosidase assay.
HeLa cells (3 × 105) were treated as described for colony-forming assays, and senescence associated β-galactosidase staining was conducted as described previously (8). In brief, 5 to 7 days after transfection, cells were washed with phosphate-buffered saline (PBS), fixed for 5 min in 4% formaldehyde at room temperature, and then incubated at 37°C (without CO2) with fresh staining solution (1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside [X-Gal] per ml [stock, 20 mg of dimethylformamide per ml], 40 mM citric acid-sodium phosphate, pH 6.0, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, 2 mM MgCl2) for 48 h. Pictures were recorded with a Zeiss Axiovert 400 M microscope using a bright field and a 10× objective.
Flow cytometry analysis.
Transfected HeLa cells were harvested 48 h posttransfection. Both adherent and detached cells were harvested in culture medium by scraping and concentrated by centrifugation. The cell pellets were then lysed in a hypotonic lysis buffer containing 1% sodium citrate, 0.1% Triton X-100, and 50 μg/ml propidium iodide and subsequently analyzed by flow cytometry. Signals left of the peak diploid DNA content were considered apoptotic. The data were registered in logarithmic scale. Flow cytometric analyses were performed on a FACSCalibur (Beckton Dickinson, Heidelberg, Germany) using CellQuest Pro analysis software.
RESULTS
The long-distance repression activity of E8∧E2C is conserved among high-risk HPV.
We first investigated whether HPV18, which is the second most prevalent HPV in cervical cancer and belongs to alphapapillomavirus 7 species, expresses an E8∧E2C transcript. Sequence analysis revealed that the HPV18 genome has a putative E8 open reading frame (ORF) from nt 1269 to 1361 with an ATG at 1323 to 1325 and a potential splice donor site at 1357, which is similar to the alpha 9 viruses HPV16, -31, and -33 (9, 36, 37). RT-PCR was carried out with primers within 18E8 (5′ end at nt 1288) and at the end of the E2 gene using RNA from human keratinocyte cell lines harboring extrachromosomally replicating HPV18 genomes (20). In parallel, the HPV16 E8∧E2C transcript was amplified by RT-PCR using RNA from human keratinocyte cell lines harboring extrachromosomally replicating HPV16 genomes. For both virus types, a major band of ∼ 600 nt was obtained that was cloned and sequenced. This revealed that the HPV18 primer pair detects a spliced RNA in which an exon from nt 1288 to 1357 is spliced to an exon from nt 3434 to 3996 (Fig. 1). Conceptual translation of the RNA revealed a fusion protein consisting of 11 residues of the 18E8 ORF fused to residues 206 to 365 of 18E2. The overall structure of this protein is very similar to that of 16E8∧E2C and 31E8∧E2C, and E8 residues 1, 2, 4 to 7, and 10 are conserved, including the K5W6K7 motif, which is important for the repression activity of 31E8∧E2C (39) (Fig. 1). Therefore, we suggest calling this protein 18E8∧E2C.
FIG. 1.

Schematic structure of E8∧E2C. E8∧E2C consists of the small E8 part and the E2C domain, which is comprised of a less conserved “hinge” (H) region and the highly conserved DNA binding domain (DBD). The nucleotide positions for the E8 ATG and the splice donor sites and the splice acceptor and the stop codon for the E2 gene are indicated. Below are the amino acid sequences of the respective E8 parts. Conserved residues are shown in gray. The conserved KWK motif that is responsible for repression activity of the 31 E8∧E2C protein is underlined.
To comparatively analyze the functions of 16E8∧E2C, 18E8∧E2C, and 31E8∧E2C, the respective cDNAs were inserted into the eukaryotic expression plasmid pSG5. Cotransfection experiments were carried out in HeLa cells with plasmids pGL 16URR-luc, pGL 18URR-luc, and pGL 31URR-luc, in which the HPV16, HPV18, or HPV31 E6/E7 promoter, respectively, drives luciferase expression and the different E8∧E2C expression constructs. 31E8∧E2C repressed the homologous promoter to 11.6%, the 16URR promoter to 6.4%, and the 18URR promoter to 3.9% of the basal activity (Fig. 2a). 16E8∧E2C repressed the homologous promoter to 1.8% and both the 18URR and the 31URR to 0.7% of the basal activity (Fig. 2a). 18E8∧E2C inhibited the homologous promoter to 0.9%, the 16URR promoter to 3.4%, and the 31URR promoter to 2.1% of the basal activity (Fig. 2a). This indicated that all E8∧E2C proteins are potent repressors of URR promoter activity but that 31E8∧E2C appears to be slightly less active than 16- and 18E8∧E2C. In contrast to 31E2, the 31E8∧E2C protein exerts a long-distance repression activity that can be determined using either a 31URR reporter construct in which only the promoter-distal E2BS1 is intact and the promoter-proximal E2BS 2, 3, and 4 are mutated (pGL31 URR-BS2,3,4mt-luc) or a synthetic reporter construct with multimerized E2BS located upstream of a minimal promoter driving luciferase (pC18-Sp1-luc) (39). Reporter assays revealed that all E8∧E2C proteins displayed long-distance repression activity (Fig. 2b). 16- and 31E8∧E2C equally repressed the pGL31 URR-BS2,3,4mt-luc plasmid activity to 8% that of the control, whereas 18E8∧E2C inhibited activity to 18.6% that of the control (Fig. 2b). The basal activity of pC18-Sp1-luc was repressed nearly identically by 16E8∧E2C to 18%, by 18E8∧E2c to 23%, and by 31E8∧E2C to 27% of the basal activity (Fig. 2c). Similar repression efficiencies of the pGL18 URR-luc (3 to 9% of the control) and the pC18-Sp1-luc (9 to 22% of the control) reporter plasmids by E8∧E2C proteins were also observed in HPV-negative RTS3b keratinocytes (Fig. 2d). Taken together, these results suggested that the E8∧E2C proteins of the high-risk types 16, 18, and 31 display similar inhibitory effects on HPV E6/E7 promoters in HPV-positive and -negative cells.
FIG. 2.

Transcriptional-repression activity is conserved among the E8∧E2C proteins of HPV16, -18, and -31. (a to c) HeLa cells were transfected with 50 ng each of pGL 16URR-luc (HPV16 URR), pGL 18URR-luc (HPV18 URR), or pGL 31URR-luc (HPV31 URR) (a); 50 ng of pGL 31URR-luc BS2,3,4 mt (HPV31 URR BS 2,3,4 mt) (b); or 50 ng of pC18-SP1-luc (pC18 SP1) (c) and 10 ng of expression plasmids for 16E8∧E2C (bar 16), 18E8∧E2C (bar 18), 31E8∧E2C (bar 31), or the empty vector pSG5 (vector). (d) HPV-negative RTS3b keratinocytes were cotransfected with 50 ng of pGL 18URR-luc (HPV 18 URR) or pC18-SP1 luc (pC18 SP1) and 10 ng of expression plasmids for 16E8∧E2C (bar 16), 18E8∧E2C (bar 18), 31E8∧E2C (bar 31), or the empty vector pSG5 (vector). Luciferase activities were determined 48 h after transfection. The data represent the averages of at least 3 independent transfections performed in duplicate. Luciferase activities are relative to the activity of the indicated luciferase reporter plasmid in the presence of the empty vector, which was set to 1. The error bars indicate the standard deviations.
The E8∧E2C proteins of HPV16, -18, and -31 inhibit the growth of HeLa cells via induction of cell cycle arrest and senescence.
We recently demonstrated that 31E8∧E2C is able to inhibit the growth of HeLa cells by binding to HPV promoter sequences and repressing the expression of the viral oncogenes E6 and E7 (38). To investigate whether the E8∧E2C proteins of HPV16 and -18 also lead to growth arrest, HeLa cells were transfected with the pPur plasmid, providing puromycin resistance, and the eukaryotic expression vector pSG5 or derivatives expressing E8∧E2C of HPV16, -18, or -31 in a 4:1 ratio. The cells were selected with puromycin, and resistant colonies were stained and counted after 12 to 14 days. Figure 3 shows the average colony numbers derived from 3 independent experiments. The E8∧E2C proteins of HPV16, -18, and -31 all showed decreased colony numbers compared to the empty vector. Even fewer colonies could be observed with 16E8∧E2C and 18E8∧E2C than with 31E8∧E2C. These data indicate that the E8∧E2C proteins of high-risk HPV types 16, 18, and 31 are equally able to inhibit the growth of HeLa cells.
FIG. 3.
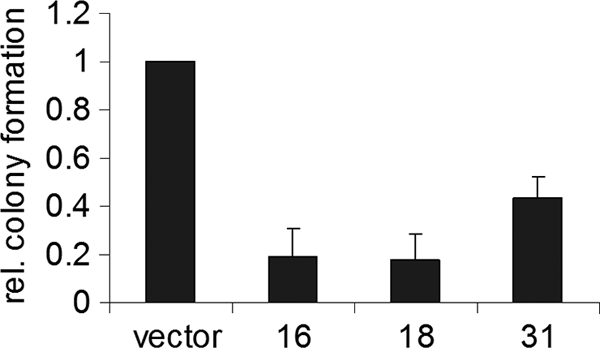
Expression of E8∧E2C of HPV16, -18, and -31 reduces colony formation of HeLa cells. HeLa cells were transfected with a puromycin resistance plasmid, pPur, and expression plasmids for 16-, 18-, or 31E8∧E2C (bars 16, 18, and 31) or the empty vector pSG5 (vector). After selection with puromycin for 12 to 14 days, the resistant colonies were counted. The graph represents the average of the puromycin-resistant colonies derived from three independent experiments relative (rel.) to the colonies formed by the empty vector, which was set to 1. Standard deviations are indicated by error bars.
To address the growth-inhibitory activities in more detail and to evaluate whether differences between E8∧E2C and E2 proteins exist, the protein levels of p53 and p21 were analyzed, since p53 and the p53 target gene CDKN1A/p21 are inhibited by the HPV18 E6 and E7 proteins. To be able to monitor the expression levels of the different E8∧E2C and E2 proteins, HA epitopes were inserted internally in HPV16 and HPV18 E2 and E8∧E2C genes as previously described for 31E2 and 31E8∧E2C (38). HeLa cells were transfected with the empty vector or the respective expression vectors encoding the different E8∧E2C or E2 proteins. Cell lysates were prepared 48 h posttransfection and then analyzed by Western blotting. As demonstrated in Fig. 4a, the expression of 16-, 18-, or 31E2-HA or -E8∧E2C-HA increased the p53 and p21 protein levels compared to the empty vector. However, all E8∧E2C proteins induced slightly higher p53 protein levels than the E2 proteins. This was not due to different protein amounts, as all E2 and E8∧E2C proteins were expressed at similar levels (Fig. 4a). In parallel, the DNA content of transfected cells was subjected to flow cytometry analysis to determine the cell cycle profile and the rates of apoptosis (Fig. 4b). The expression of 16-, 18-, or 31E8∧E2C proteins increased the average percentages of cells in the G1 population from 52.1% to 63.4%, 65.5%, and 66.1% and decreased the number of cells in G2 phase from 19.3% to 10.2%, 10.9%, and 9.5%, respectively (Fig. 4b). The corresponding E2 proteins also increased the number of G1 phase cells and decreased the number of G2 phase cells but were slightly less efficient than E8∧E2C proteins (Fig. 4b). This indicated that the different E2 and E8∧E2C proteins induce a cell cycle arrest consistent with the loss of E7, reactivation of Rb proteins, and induction of p21. Furthermore, we observed for 16-, 18-, or 31E2 a very moderate increase in the number of apoptotic cells (DNA content < 2n) from 10.2% to 12.5%, 13.8%, and 15.9%, respectively (Fig. 4b). The effects induced by 16-, 18-, and 31E8∧E2C proteins were similar or even smaller, resulting in 13.3%, 11.6%, and 12.1%, respectively (Fig. 4b). To address if these weak proapoptotic activities are E6/E7-independent effects of E2 and E8∧E2C proteins, as reported for HPV16 and -18 E2 (6, 43), we inhibited E6/E7 expression by siRNA targeting the E6/E7 region (si18E7) (16, 22). Transfection of the siRNA increased p53 levels comparably to the transfection of E2 or E8∧E2C expression plasmids (Fig. 4c). Flow cytometry analyses of siRNA-transfected cells revealed an increase from 56.0% to 65.2% in G1 phase cells, a reduction in G2 phase cells from 22.1% to 9.5%, and an increase in apoptotic cells from 7.0 to 10.7% (Fig. 4c). As the effects of the 18E7 siRNA on the cell cycle and apoptosis rates are very similar to the expression of E2 and E8∧E2C proteins, this strongly suggests that the major mechanism of growth inhibition of HeLa cells by E8∧E2C proteins from HPV16, -18, and -31 is the induction of cell cycle arrest via E6/E7 repression, and not apoptosis.
FIG. 4.

Expression of E8∧E2C or E2 proteins of HPV16, -18, and -31 in HeLa cells leads to stabilization of p53 and CDKN1A/p21 and to cell cycle arrest. (a) HeLa cells were transfected with 1 μg of empty vector pSG5 (V); expression vectors for the respective HA-tagged E8∧E2C or E2 proteins of HPV16, HPV18, or HPV31; or the KRAB-E2C fusion protein. Whole-cell lysates were prepared and analyzed by immunoblotting for the presence of p53, p21, and HSP90 as a loading control. E8∧E2C-HA, E2-HA, and KRAB-E2C-HA proteins were detected using an anti-HA antibody. (b) For flow cytometry analysis, HeLa cells were transfected with 1 μg of pSG5 (vector) or expression vectors for the respective E8∧E2C or E2 proteins. DNA was stained with propidium iodide, and the DNA content was determined using CellQuest Pro analysis software and a FACSCalibur. The averaged percentages and standard errors of cell numbers in G1 phase (G1), G2 phase (G2), or sub-G1 phase (apoptosis) derived from five independent transfection experiments are shown. (c) HeLa cells were transfected with an HPV18E7 siRNA or a control siRNA (siControl). Forty-eight hours posttransfection, p53 and HSP90 levels were determined by immunoblotting. A molecular mass marker in kDa is shown on the right of the gel. Cell cycle distribution and apoptotic cells were measured as described above and represent averages derived from three independent transfection experiments.
To investigate the effects of long-term E8∧E2C expression, HeLa cells were transfected with E8∧E2C or E2 expression plasmids and a puromycin resistance plasmid and drug selected as described for the colony formation assays. Approximately 5 days posttransfection, E8∧E2C- and E2-transfected cells changed their morphology. In contrast to the vector-transfected control, E8∧E2C- and E2-transfected cells appeared flat and enlarged and grew as single cells (Fig. 5). To determine whether this was due to the induction of cellular senescence, the cells were fixed and stained for SA-β-Gal activity (8). This showed that the majority of the single, flat cells in E8∧E2C and E2 transfectants were SA-β-Gal positive, whereas no staining was observed in the control (Fig. 5). This strongly suggested that E8∧E2C proteins induce a cell cycle arrest that leads to cellular senescence.
FIG. 5.
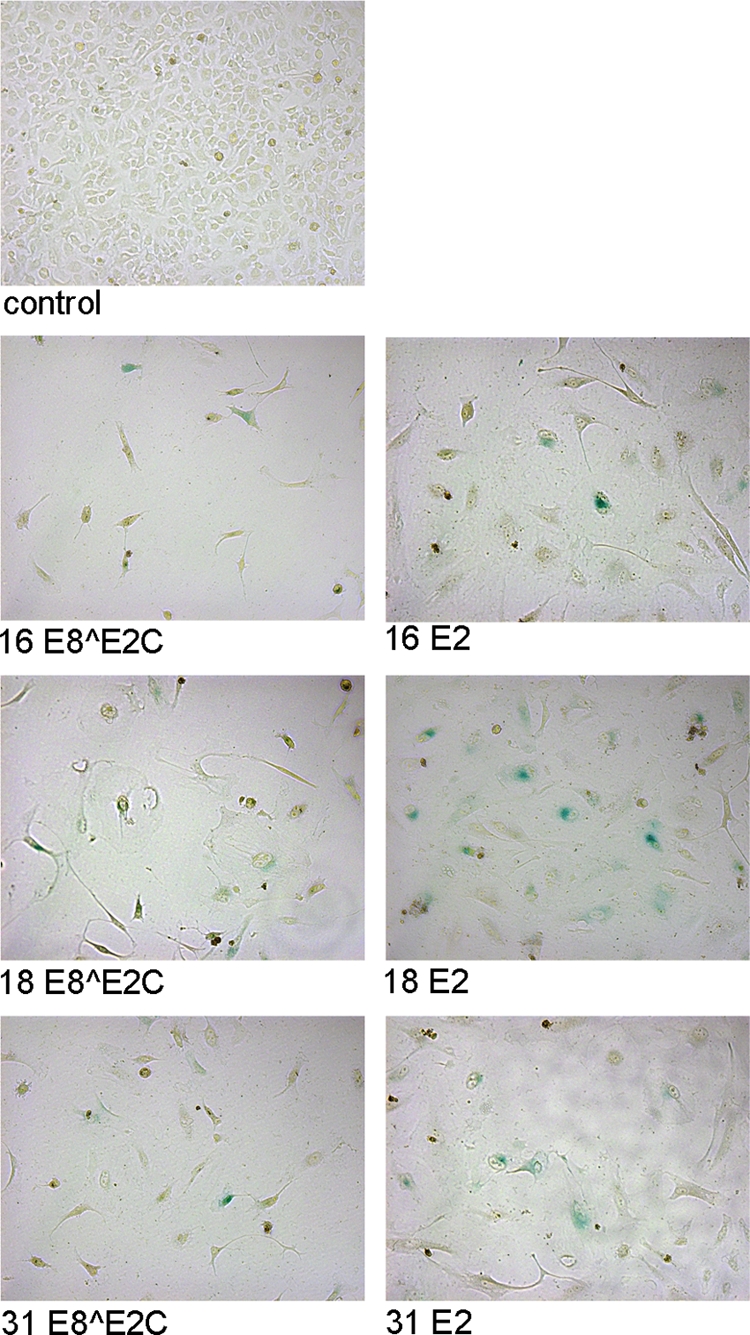
E8∧E2C and E2 expression in HeLa cells results in morphological changes and senescence markers. HeLa cells were transfected with a puromycin resistance plasmid and plasmids expressing 16E8∧E2C, 18E8∧E2C, 31E8∧E2C, 16E2, 18E2, 31E2, or the empty vector pSG5 (control). After drug selection, the cells were fixed and stained for senescence-associated β-galactosidase activity (blue cells). The pictures were recorded with a Zeiss Axiovert 400 M microscope using a bright field and a 10× objective.
An artificial KRAB-E2C fusion protein acts as a transcriptional repressor that inhibits HeLa cell growth.
In contrast to viral E2 and E8∧E2C proteins, fusion proteins consisting of the papillomavirus E2C DNA binding domain and activation domains from different transcription factors have not acted as transcriptional repressors of the HPV URR promoter or inhibited the growth of HeLa cells (10, 14, 28). Thus, the viral amino-terminal E2 and E8 domains may have acquired unique functions to repress the HPV URR. To test this, we created a fusion between the well-characterized KRAB domain derived from KOX1 (27) and the HPV31 E2C domain (KRAB-E2C) (Fig. 6a). The repression activity of KRAB-E2C was compared with that of 31E8∧E2C using the reporter constructs for the HPV18 URR and HPV31 URR, as well as reporter constructs specific for the long-distance repression activity of E8∧E2C proteins (pGL 31URR BS2,3,4 mt and pC18-SP1-luc). This revealed that KRAB-E2C inhibited transcription from all constructs as well as or even better than 31E8∧E2C in both HPV18-positive HeLa and HPV-negative RTS3b keratinocyte cells (Fig. 6b). To test whether the HPV18 E6/E7 promoter integrated in HeLa cells also responds to KRAB-E2C, colony reduction assays were performed. The expression of KRAB-E2C resulted in a reduction of colonies similar to that of the expression of 31E8∧E2C (Fig. 6c). In contrast, the expression of the KRAB domain alone gave rise to colony numbers comparable to those of the control (Fig. 6c), which suggests that growth inhibition is mediated by targeting the fusion protein to the HPV18 URR and is not due to nonspecific effects of the KRAB domain. The transient expression of an HA-tagged version of KRAB-E2C-HA revealed that it is present in amounts similar to those of the E2 and E8∧E2C proteins (Fig. 4a). Consistent with the repression of E6/E7 transcripts by KRAB-E2C, p53 and p21 protein levels were increased in KRAB-E2C-transfected cells (Fig. 4a). Similar to the effects of the different E8∧E2C and E2 proteins, expression of KRAB-E2C induced a change in the cell cycle profile, a weak increase in apoptotic cells, morphological changes, and the appearance of SA-β-Gal-positive cells (Fig. 6c and d). Taken together, these data suggest that the viral E2TA and E8 domains of E2 and E8∧E2C, respectively, can be replaced by the cellular KOX1 KRAB domain in order to generate a transcriptional repressor specific for the HPV URR.
FIG. 6.

(a) Structures of E2, E8∧E2C, and the artificial repressor KRAB-E2C. E2 consists of the amino-terminal E2TA and the E2C domain, which comprises the hinge and the DNA binding domain. In E8∧E2C, the E2TA domain is replaced by the short E8 domain. To generate KRAB-E2C, the KRAB domain, which is derived from the KOX1 protein (residues 1 to 90), was fused to the E2C domain. (b) HeLa cells were transfected with 50 ng of pGL 18URR-luc (HPV18 URR), pGL 31URR-luc (HPV31 URR), pGL 31URR-luc BS2,3,4 mt (HPV31 URR BS 2,3,4 mt), or 50 ng of pC18-SP1-luc (pC18 SP1) and 10 ng of expression plasmids for 31E8∧E2C, KRAB-E2C, or the empty vector pSG5 (vector). HPV-negative RTS3b keratinocytes were cotransfected with 50 ng of pGL 18URR-luc or pC18-SP1 luc and 10 ng of expression plasmids for KRAB-E2C, 31E8∧E2C (bars 31), or the empty vector pSG5 (vector). Luciferase activities were determined 48 h after transfection. The data represent the averages of at least 3 independent transfections performed in duplicate. Luciferase activities are relative to the activity of the indicated luciferase reporter plasmid in the presence of the empty vector, which was set to 1. The error bars indicate the standard deviations. (c) HeLa cells were transfected with a puromycin resistance plasmid and plasmids expressing KRAB-E2C, the KRAB domain alone (KRAB), 31E8∧E2C, or the empty vector pSG5 (vector) in a 1:4 ratio. After selection with puromycin for 12 to 14 days, the resistant colonies were counted. The graph represents the averages of the puromycin-resistant colonies derived from 3 independent experiments relative to the colonies formed by the empty vector, which was set to 1. Standard deviations are indicated by error bars. (Right) KRAB-E2C or control cells were fixed and stained for senescence-associated β-galactosidase activity. (d) Flow cytometry analysis of HeLa cells transiently transfected with the KRAB-E2C expression plasmid or the empty vector (vector) was performed as described in the legend to Fig. 4b. The graph displays the averages and standard deviations of five independent experiments.
DISCUSSION
In contrast to other papillomavirus proteins, no comparative analyses have been performed for the E8∧E2C protein. Currently, transcript analyses suggest that HPV1, -11, -16, -31, and -33 express E8∧E2C transcripts (9, 29, 33, 36, 37). Genetic analyses of HPV E8∧E2C in the context of complete genomes have been carried out for HPV16 and -31. This has revealed that the loss of E8∧E2C leads in both viruses to genome overreplication, but only 31E8∧E2C is required for long-term extrachromosomal maintenance (21, 37). Also taking into account that the E2 proteins of HPV16 and -18 have been reported to display proapoptotic activities that appear to be different from those of other PV E2 proteins, we performed the first comparative study with different E8∧E2C proteins. In addition to HPV16 and -31, we demonstrated for the first time that replicating HPV18 genomes express an E8∧E2C transcript that is very similar to other HPV E8∧E2C transcripts. This suggests that many, if not all, alphapapillomaviruses may express an E8∧E2C protein. Consistent with this idea, sequence analyses of 33 α-HPV genomes revealed that putative E8 ORFs followed by downstream consensus splice donor sites (AGGT) are highly conserved (see Fig. S1 in the supplemental material). Furthermore, the KWK motif, essential for repression by 31E8∧E2C, is highly conserved in 27 out of 33 α-HPV types analyzed (see Fig. S1 in the supplemental material). Interestingly, the remaining 6 α-HPV types have a conserved central WK motif (see Fig. S1 in the supplemental material), which has been demonstrated by single-amino-acid mutations (W6A and K7A) to be sufficient for repressing the replication of HPV31 genomes (39, 46). This also suggests that the repression activities of all α-HPV E8∧E2C proteins might be due to a conserved interaction of the E8 domain with an HDAC3/N-CoR complex, as demonstrated for 31E8∧E2C, and also to interaction with the CHD6 protein via the E2C domain (11, 31).
Reporter assays revealed that all E8∧E2C proteins are potent repressors of the homologous URR, as well as heterologous URRs (Fig. 2a). However, the repression of URRs by 31E8∧E2C seemed to be slightly less efficient than repression by 16- or 18E8∧E2C. Consistent with this, 31E8∧E2C gave rise to more colonies than 16- or 18E8∧E2C in HeLa colony reduction assays (Fig. 3). Since all proteins were expressed to similar levels (Fig. 4a), this is most likely not due to different protein stabilities. Interestingly, when reporter constructs that measure E8-dependent repression activity were used (39), no differences between the three proteins were observed (Fig. 2b and c). The most likely explanation for the differences in URR repression is that the DNA binding affinities of 16- and 18E8∧E2C for E2BS2, -3, or -4 are slightly higher than those of 31E8∧E2C, and therefore, URR repression is more efficient.
Growth inhibition of HeLa cells by all E2 proteins tested so far has been ascribed to the repression of the URR promoter driving E6/E7 oncogene expression (6, 10, 13, 17, 18, 44). In addition, the 16- and 18E2 proteins have been shown to induce apoptosis in HPV-positive and -negative cells (5-7, 30, 43). This has led to the model that growth inhibition of HPV-positive cells by 16- and 18E2 is due to both URR repression and an independent induction of apoptosis. In our hands, the 16-, 18-, and 31E8∧E2C proteins were as able as the respective E2 proteins to inhibit the growth of HeLa cells. This coincides with the stabilization of p53 and p21 proteins, which are targets of E6 and E7 proteins, an increase in G1 cells, and a decrease in G2 cells 48 h after transfection of E8∧E2C expression vectors (Fig. 4). Consistent with permanent cell cycle arrest being the major cause of the growth inhibition, approximately 5 days posttransfection, morphological changes and senescence markers became apparent. We also observed a slight increase in apoptotic cells 48 h after transfection of E8∧E2C and E2 expression plasmids. However, knocking down E6/E7 transcription via siRNA recapitulated the effects of E8∧E2C and E2 proteins on cell cycle and apoptosis induction in HeLa cells, strongly suggesting that the weak apoptosis induction is a consequence of E6/E7 inhibition and not an independent function of E8∧E2C proteins.
Our studies have not revealed a significant difference between the repression activities of the HPV16 and -31 E8∧E2C proteins. Thus, the differences in the HPV16 and -31 E8∧E2C knockout genomes in long-term assays might be due to additional activities of E8∧E2C proteins, which require further studies (21, 37).
Up to now, only the viral E2 and E8∧E2C proteins have been shown to inhibit HPV URRs and inhibit the growth of HPV-positive cells, and this required, in addition to the E2C domain, either the E2TA or the E8 domain, respectively (6, 10, 17, 38). Attempts to create fusion proteins consisting of the E2C domain and transcription modulatory domains from viral or cellular transcription factors have failed so far to generate a transcriptional repressor that represses HPV URR promoter activity and inhibits the growth of HeLa cells (10, 15, 28). We have now demonstrated for the first time that the KRAB domain derived from the cellular KOX1 transcriptional repressor is able to functionally replace the E2 and the E8 domains to inhibit URR activity and also to inhibit the growth of HeLa cells. Molecular markers for E6/E7 oncogene inhibition in KRAB-E2C-expressing cells are identical to those in E8∧E2C-expressing cells, suggesting that KRAB-E2C acts as a bona fide transcriptional repressor of the URR.
Recent studies have indicated that transcriptional repression of the HPV URR by the E2 and 31E8∧E2C proteins is due to interaction with different sets of cellular proteins via the amino termini and to a conserved interaction of the E2C domain with CHD6 (11). The transcriptional-repression activity of E2 has been linked to the interaction with the cellular Brd4, SMCX, and EP400 proteins (35, 45). In contrast, the transcriptional-repression activity of 31E8∧E2C has been linked to the interaction of the E8 domain with cellular corepressors, such as HDAC3 and the N-CoR protein (1, 31). The KRAB domain of KOX1 represses transcription through the recruitment of the corepressor TRIM28/KAP1/TIF1β/KRIP1, heterochromatin protein 1 (HP1) alpha, HP1 gamma, and the histone methyltransferase SETDB1 (12, 23, 25, 34). While 31E8∧E2C was also found to interact with TRIM28 and SETDB1 (1), siRNA knockdown experiments indicated that these proteins do not contribute to the repression of the HPV URR by 31E8∧E2C (31). Furthermore, while HDAC inhibitors relieved repression by 31E8∧E2C, this was not the case for the KOX1 KRAB domain (1, 24). These data suggest that the repression of the URR by E8∧E2C and KRAB-E2C is achieved by the recruitment of different transcription factors and possibly by different repression pathways. Interestingly, KRAB-E2C also displayed a long-distance repression similar to that of E8∧E2C, which has not been observed for E2 (39). It appears that the failure to reconstitute HPV URR repressors was due to the choice of activation instead of repression domains that were fused to the E2C domain and not by the fact that the E2TA domain has unique transcriptional-repression properties for the HPV URR.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 773 C2) to F.S.
Footnotes
Published ahead of print on 29 December 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Ammermann, I., M. Bruckner, F. Matthes, T. Iftner, and F. Stubenrauch. 2008. Inhibition of transcription and DNA replication by the papillomavirus E8-E2C protein is mediated by interaction with corepressor molecules. J. Virol. 82:5127-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blachon, S., S. Bellanger, C. Demeret, and F. Thierry. 2005. Nucleo-cytoplasmic shuttling of high risk human Papillomavirus E2 proteins induces apoptosis. J. Biol. Chem. 280:36088-36098. [DOI] [PubMed] [Google Scholar]
- 3.Castellsague, X., et al. 2007. HPV and cervical cancer in the world 2007 report. Vaccine 25:C1-C26. [DOI] [PubMed] [Google Scholar]
- 4.Cogliano, V., et al. 2005. Carcinogenicity of human papillomaviruses. Lancet Oncol. 6:204. [DOI] [PubMed] [Google Scholar]
- 5.Demeret, C., A. Garcia-Carranca, and F. Thierry. 2003. Transcription-independent triggering of the extrinsic pathway of apoptosis by human papillomavirus 18 E2 protein. Oncogene 22:168-175. [DOI] [PubMed] [Google Scholar]
- 6.Desaintes, C., C. Demeret, S. Goyat, M. Yaniv, and F. Thierry. 1997. Expression of the papillomavirus E2 protein in HeLa cells leads to apoptosis. EMBO J. 16:504-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desaintes, C., S. Goyat, S. Garbay, M. Yaniv, and F. Thierry. 1999. Papillomavirus E2 induces p53-independent apoptosis in HeLa cells. Oncogene 18:4538-4545. [DOI] [PubMed] [Google Scholar]
- 8.Dimri, G. P., et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U. S. A. 92:9363-9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doorbar, J., et al. 1990. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology 178:254-262. [DOI] [PubMed] [Google Scholar]
- 10.Dowhanick, J. J., A. A. McBride, and P. M. Howley. 1995. Suppression of cellular proliferation by the papillomavirus E2 protein. J. Virol. 69:7791-7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fertey, J., et al. 2010. Interaction of the papillomavirus E8-E2C protein with the cellular CHD6 protein contributes to transcriptional repression. J. Virol. 84:9505-9515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman, J. R., et al. 1996. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 10:2067-2078. [DOI] [PubMed] [Google Scholar]
- 13.Goodwin, E. C., and D. DiMaio. 2000. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. U. S. A. 97:12513-12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodwin, E. C., L. K. Naeger, D. E. Breiding, E. J. Androphy, and D. DiMaio. 1998. Transactivation-competent bovine papillomavirus E2 protein is specifically required for efficient repression of human papillomavirus oncogene expression and for acute growth inhibition of cervical carcinoma cell lines. J. Virol. 72:3925-3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodwin, E. C., et al. 2000. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl. Acad. Sci. U. S. A. 97:10978-10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall, A. H., and K. A. Alexander. 2003. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells. J. Virol. 77:6066-6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang, E. S., L. K. Naeger, and D. DiMaio. 1996. Activation of the endogenous p53 growth inhibitory pathway in HeLa cervical carcinoma cells by expression of the bovine papillomavirus E2 gene. Oncogene 12:795-803. [PubMed] [Google Scholar]
- 18.Hwang, E. S., et al. 1993. Inhibition of cervical carcinoma cell line proliferation by the introduction of a bovine papillomavirus regulatory gene. J. Virol. 67:3720-3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadaja, M., T. Silla, E. Ustav, and M. Ustav. 2009. Papillomavirus DNA replication—from initiation to genomic instability. Virology 384:360-368. [DOI] [PubMed] [Google Scholar]
- 20.Karstensen, B., et al. 2006. Gene expression profiles reveal an upregulation of E2F and downregulation of interferon targets by HPV18 but no changes between keratinocytes with integrated or episomal viral genomes. Virology 353:200-209. [DOI] [PubMed] [Google Scholar]
- 21.Lace, M. J., J. R. Anson, G. S. Thomas, L. P. Turek, and T. H. Haugen. 2008. The E8-E2 gene product of human papillomavirus type 16 represses early transcription and replication but is dispensable for viral plasmid persistence in keratinocytes. J. Virol. 82:10841-10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lea, J. S., et al. 2007. Silencing of HPV 18 oncoproteins With RNA interference causes growth inhibition of cervical cancer cells. Reprod. Sci. 14:20-28. [DOI] [PubMed] [Google Scholar]
- 23.Lechner, M. S., G. E. Begg, D. W. Speicher, and F. J. Rauscher III. 2000. Molecular determinants for targeting heterochromatin protein 1-mediated gene silencing: direct chromoshadow domain-KAP-1 corepressor interaction is essential. Mol. Cell. Biol. 20:6449-6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenz, P., D. Koczan, and H. J. Thiesen. 2001. Transcriptional repression mediated by the KRAB domain of the human C2H2 zinc finger protein Kox1/ZNF10 does not require histone deacetylation. Biol. Chem. 382:637-644. [DOI] [PubMed] [Google Scholar]
- 25.Margolin, J. F., et al. 1994. Kruppel-associated boxes are potent transcriptional repression domains. Proc. Natl. Acad. Sci. U. S. A. 91:4509-4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McBride, A. A., H. Romanczuk, and P. M. Howley. 1991. The papillomavirus E2 regulatory proteins. J. Biol. Chem. 266:18411-18414. [PubMed] [Google Scholar]
- 27.Moosmann, P., O. Georgiev, H. J. Thiesen, M. Hagmann, and W. Schaffner. 1997. Silencing of RNA polymerases II and III-dependent transcription by the KRAB protein domain of KOX1, a Kruppel-type zinc finger factor. Biol. Chem. 378:669-677. [DOI] [PubMed] [Google Scholar]
- 28.Nishimura, A., et al. 2000. Mechanisms of human papillomavirus E2-mediated repression of viral oncogene expression and cervical cancer cell growth inhibition. J. Virol. 74:3752-3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palermo-Dilts, D. A., T. R. Broker, and L. T. Chow. 1990. Human papillomavirus type 1 produces redundant as well as polycistronic mRNAs in plantar warts. J. Virol. 64:3144-3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parish, J. L., et al. 2006. E2 proteins from high- and low-risk human papillomavirus types differ in their ability to bind p53 and induce apoptotic cell death. J. Virol. 80:4580-4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Powell, M. L. C., et al. 2010. NCoR1 mediates papillomavirus E8∧E2C transcriptional repression. J. Virol. 84:4451-4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Purdie, K. J., et al. 1993. Malignant transformation of cutaneous lesions in renal allograft patients: a role for human papillomavirus. Cancer Res. 53:5328-5333. [PubMed] [Google Scholar]
- 33.Rotenberg, M. O., C. M. Chiang, M. L. Ho, T. R. Broker, and L. T. Chow. 1989. Characterization of cDNAs of spliced HPV-11 E2 mRNA and other HPV mRNAs recovered via retrovirus-mediated gene transfer. Virology 172:468-477. [DOI] [PubMed] [Google Scholar]
- 34.Schultz, D. C., K. Ayyanathan, D. Negorev, G. G. Maul, and F. J. Rauscher III. 2002. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16:919-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith, J. A., et al. 2010. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. U. S. A. 107:3752-3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snijders, P. J., et al. 1992. Human papillomavirus type 33 in a tonsillar carcinoma generates its putative E7 mRNA via two E6* transcript species which are terminated at different early region poly(A) sites. J. Virol. 66:3172-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stubenrauch, F., M. Hummel, T. Iftner, and L. A. Laimins. 2000. The E8E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J. Virol. 74:1178-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stubenrauch, F., E. Straub, J. Fertey, and T. Iftner. 2007. The E8 repression domain can replace the E2 transactivation domain for growth inhibition of HeLa cells by papillomavirus E2 proteins. Int. J. Cancer 121:2284-2292. [DOI] [PubMed] [Google Scholar]
- 39.Stubenrauch, F., T. Zobel, and T. Iftner. 2001. The E8 domain confers a novel long-distance transcriptional repression activity on the E8E2C protein of high-risk human papillomavirus type 31. J. Virol. 75:4139-4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thierry, F. 2009. Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma. Virology 384:375-379. [DOI] [PubMed] [Google Scholar]
- 41.Thierry, F., and C. Demeret. 2008. Direct activation of caspase 8 by the proapoptotic E2 protein of HPV18 independent of adaptor proteins. Cell Death Differ. 15:1356-1363. [DOI] [PubMed] [Google Scholar]
- 42.Walboomers, J. M., et al. 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 189:12-19. [DOI] [PubMed] [Google Scholar]
- 43.Webster, K., et al. 2000. The human papillomavirus (HPV) 16 E2 protein induces apoptosis in the absence of other HPV proteins and via a p53-dependent pathway. J. Biol. Chem. 275:87-94. [DOI] [PubMed] [Google Scholar]
- 44.Wells, S. I., et al. 2000. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21(CIP)-dependent pathways. EMBO J. 19:5762-5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu, S. Y., et al. 2006. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 20:2383-2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zobel, T., T. Iftner, and F. Stubenrauch. 2003. The papillomavirus E8-E2C protein represses DNA replication from extrachromosomal origins. Mol. Cell. Biol. 23:8352-8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.zur Hausen, H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat. Rev. Cancer. 2:342-350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.