

Abstract
Converging lines of evidence indicate dysregulation of the key immunoregulatory molecule CD45 (also known as leukocyte common antigen) in Alzheimer's disease (AD). We report that transgenic mice overproducing amyloid-β peptide (Aβ) but deficient in CD45 (PSAPP/CD45−/− mice) faithfully recapitulate AD neuropathology. Specifically, we find increased abundance of cerebral intracellular and extracellular soluble oligomeric and insoluble Aβ, decreased plasma soluble Aβ, increased abundance of microglial neurotoxic cytokines tumor necrosis factor-α and interleukin-1β, and neuronal loss in PSAPP/CD45−/− mice compared with CD45-sufficient PSAPP littermates (bearing mutant human amyloid precursor protein and mutant human presenilin-1 transgenes). After CD45 ablation, in vitro and in vivo studies demonstrate an anti-Aβ phagocytic but proinflammatory microglial phenotype. This form of microglial activation occurs with elevated Aβ oligomers and neural injury and loss as determined by decreased ratio of anti-apoptotic Bcl-xL to proapoptotic Bax, increased activated caspase-3, mitochondrial dysfunction, and loss of cortical neurons in PSAPP/CD45−/− mice. These data show that deficiency in CD45 activity leads to brain accumulation of neurotoxic Aβ oligomers and validate CD45-mediated microglial clearance of oligomeric Aβ as a novel AD therapeutic target.
Introduction
Deposition of amyloid-β peptide (Aβ) as β-amyloid plaques is a defining pathological hallmark of Alzheimer's disease (AD) and occurs with increased abundance of soluble Aβ and activation of microglia-mediated inflammatory responses (Sedgwick et al., 1991). However, reactive microglia ultimately fail to clear Aβ in brains of AD patients and in mouse models of the disease (McGeer et al., 1987; Benzing et al., 1999). It has even been suggested that chronic microglial immune responses contribute to AD pathogenesis by promoting Aβ plaque formation (Frackowiak et al., 1992; Wisniewski and Frackowiak, 1998; Townsend et al., 2005), but the molecular mechanisms underlying this deleterious response have remained elusive.
CD45 (also known as leukocyte common antigen), the most abundant transmembrane protein tyrosine phosphatase, is expressed on all nucleated hematopoietic cells and plays an important role in regulating immune responses (Thomas and Brown, 1999; Penninger et al., 2001). In the periphery, CD45 promotes antigen-specific B- and T-cell responses by dephosphorylating Src-family kinases (Thomas and Brown, 1999; Penninger et al., 2001). CD45 plays additional roles in regulating selectin expression (Stibenz et al., 1996; Wroblewski and Hamann, 1997) and integrin function (Roach et al., 1997; Shenoi et al., 1999). CD45 has also been shown to negatively regulate cytokine receptor-mediated signaling via Janus associated kinases (Irie-Sasaki et al., 2001), revealing yet another role of CD45 in dampening overly exuberant immune responses.
Resting microglia constitutively express CD45 in vitro, which is further inducible at the cell surface during activation (Sedgwick et al., 1991; Carson et al., 1998). Importantly, microglia in the frontal cortex and hippocampus of normal aging individuals express CD45, and expression abundance is markedly increased in close vicinity of β-amyloid plaques in AD patient brains (Masliah et al., 1991) and in transgenic mouse models of the disease (Wilcock et al., 2001; Maier et al., 2008). Stimulation of microglial CD45 opposes CD40 ligand (CD40L)-induced activation of the Src-family kinases Lck and Lyn, which are key transducers of proinflammatory innate immune responses (Tan et al., 2000a). Cotreatment of microglia with CD40L and agonistic CD45 antibody abrogates microglial tumor necrosis factor-α (TNF-α) production via inhibiting p44/42 mitogen-activated protein kinase (MAPK) activity, a downstream signaling event resulting from Src-family kinase activation (Tan et al., 2000a,b). Thus, stimulation of the CD45 signaling pathway suppresses proinflammatory microgliosis that is etiologically implicated in neurodegenerative disorders, including AD (Akiyama et al., 2000; Tan et al., 2000b; Penninger et al., 2001).
To elucidate the role of CD45 in AD-like pathology, we took a genetic approach to cross doubly transgenic PSAPP mice [bearing mutant human amyloid precursor protein (APP) and mutant human presenilin-1 (PS1) transgenes], which develop accelerated cerebral amyloidosis (Jankowsky et al., 2001), with animals deficient in CD45 (Benatar et al., 1996). We then examined AD-like pathology in bigenic mice as detailed to follow.
Materials and Methods
Mice.
All mice were housed and maintained in the College of Medicine Animal Facility at the University of South Florida (USF), and all experiments were conducted in compliance with protocols approved by the USF Institutional Animal Care and Use Committee. Double transgenic “Swedish” APPK595N/M596L (APPswe) + PS1ΔE9 B6C3-Tg 85Dbo/J strain (PSAPP mice) and CD45-deficient mice (B6.129-Ptprctm1Holm/J) were purchased from The Jackson Laboratory. PSAPP mice were maintained as heterozygotes by crossing transgenic mice to wild-type B6C3F1/J mice as described in the original report (Jankowsky et al., 2001). We interbred first filial offspring resulting from crossing heterozygous PSAPP mice with homozygous CD45-deficient mice and analyzed four groups of mice at 4 and 8 months of age: nontransgenic/CD45 wild-type (wild-type), PSAPP/CD45 wild-type (PSAPP), nontransgenic/CD45−/− (CD45−/−), and PSAPP/CD45−/− offspring. Animals were screened for PSAPP and CD45 genotypes by PCR from genomic DNA. CD45 genotype was further confirmed by flow cytometry. Because sex differences can impact Aβ deposition (Jankowsky et al., 2001), we used only females in our analyses.
Protein extraction.
For specific extraction of extracellular versus intracellular proteins, hemibrains were harvested and placed in 500 μl of solution containing 50 mm Tris-HCl, pH 7.6, 0.01% NP-40, 150 mm NaCl, 2 mm EDTA, 0.1% SDS, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (Sigma) as described (Lesné et al., 2006). Soluble, extracellular proteins were collected from mechanically homogenized lysates after centrifugation for 5 min at 3000 rpm. Cytoplasmic proteins were extracted from cell pellets mechanically dissociated with a micropipettor in 500 μl of TNT buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, and 0.1% Triton X-100) after centrifugation for 90 min at 13,000 rpm. Insoluble material was incubated with 20 μl of 70% formic acid, mechanically dissociated with a micropipette, gently agitated for 1 h, and buffered with 380 μl of 1 m Tris-HCl, pH 8.0. Samples were centrifuged for 90 min at 13,000 rpm, and supernatants were collected for analysis.
For total protein extraction, brains were removed and hemibrains were snap-frozen on dry ice and stored at −80°C. Samples were subsequently homogenized in immunoprecipitation assay buffer containing the following: 150 mm NaCl, 50 mm Tris, pH 7.4, 0.5% deoxycholic acid, 0.1% SDS, 1% Triton X-100, 2.5 mm EDTA, and protease inhibitor cocktail. Protein concentration was measured in the supernatant by BCA Protein Assay (Pierce).
Western blotting.
Following the sample preparation as described above, an aliquot corresponding to 40 μg of total protein was electrophoretically separated using 10% Tris-SDS gels or 10–20% Tris-tricine gels (Bio-Rad) and transferred to polyvinylidene fluoride membranes (Bio-Rad). As a positive control, Aβ oligomers were prepared from synthetic human Aβ1–42 according to published methods (Walsh et al., 2000; Lesné et al., 2006). Membranes were blocked for 1 h at room temperature in Tris-buffered saline (TBS) (containing 0.1% Tween 20 with 5% nonfat dry milk) and were then incubated with primary antibodies including mouse monoclonal neuronal-specific nuclear protein (NeuN) (1:1000; Millipore Corporation), rabbit polyclonal Bcl-xL or Bax (1:1000; Cell Signaling Technology), mouse monoclonal 6E10 (1:2000; Covance), or mouse monoclonal β-actin (1:4000; Sigma). Afterward, membranes were immunoblotted with anti-mouse (1:2000; Cell Signaling Technology) or anti-rabbit (1:10,000; Pierce) IgG secondary antibodies conjugated with horseradish peroxidase. Proteins were detected with Super Signal West Femto Maximum Sensitivity Substrate (Pierce) and BIOMAX-MR Film (Thermo Fisher Scientific).
ELISA.
For TNF-α and interleukin-1β (IL-1β) ELISAs, total proteins were extracted from mouse brain homogenates as described above. We then collected supernatants and diluted them at 1:4 in lysis buffer for detection of TNF-α (R & D Systems) or IL-1β (eBioscience), in accordance with the instructions of the manufacturer. Total protein concentrations were determined for each brain sample before quantification of cytokines by ELISA to allow for sample normalization. For Aβ ELISA, separate extracts of extracellular and intracellular proteins were prepared from mouse brain homogenates as described above. Quantification of total Aβ species (including Aβ1-40, 42) was performed according to published methods (Rezai-Zadeh et al., 2005). Total soluble Aβ species in blood plasma and extracellular/intracellular Aβ in brain homogenates were detected at 1:4 and 1:20 dilutions, respectively. Detergent-insoluble total Aβ species were detected in brain by extracting pellets in 5 m guanidine HCl buffer, followed by a 1:20 dilution in lysis buffer. Aβ1-40, 42 was quantified in all samples using Aβ1-40, 42 ELISA kits (IBL-America) in accordance with the instructions of the manufacturer, except that standards included 0.25 m guanidine HCl buffer in some cases.
Tissue preparation.
Mice were killed under isoflurane anesthesia, and 0.5 ml of blood was collected from the heart. Plasma was then separated and stored at −80°C for later analysis of Aβ levels. Animals were then transcardially perfused with ice-cold PBS. Brains were rapidly isolated and the right hemisphere was snap-frozen on dry ice and stored at −80°C before protein extraction. The left hemisphere was placed in 4% paraformaldehyde (PFA) in 0.1 m PBS overnight and then transferred to a graded series of sucrose solutions (10, 20, and 30%, each at 4°C overnight) for cryoprotection. Sequential 25 or 40 μm frozen coronal sections were cut using a sliding microtome. Free-floating sections were then stored at 4°C in 24-well plates containing PBS with 100 mm of sodium azide.
Histochemistry.
Brain sections were incubated for 5 min in a 1% thioflavin S (ThioS) (Sigma) solution dissolved in distilled water containing 70% ethanol. We then rinsed tissue sections twice with distilled water and mounted them with fluorescence mounting medium containing 4′,6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Nissl staining was performed to assay neuronal morphology. Briefly, free-floating frozen sections were mounted on slides and air dried before overnight incubation with a 1:1 solution of alcohol and chloroform. Afterward, sections were rehydrated through a graded series of alcohols and distilled water and stained with 0.1% cresyl violet solution for 5–10 min. Slides were then rinsed in distilled water and dehydrated in 95% ethanol. After dehydration, slides were mounted with mounting medium and visualized in bright field. Congo red staining was performed as described previously (Shankar et al., 2008).
Immunohistochemistry.
Brain sections were stained with rat anti-mouse CD11b (1:1000; Serotec), fluorescein isothiocyanate (FITC)-conjugated hamster anti-mouse CD40 (1:100; BD Biosciences Pharmingen), rabbit anti-mouse ionized calcium binding adaptor molecule 1 (Iba1) (1:1000; Wako Pure Chemicals), hamster anti-mouse CD11c (1:50; Pierce), rat anti-mouse chemokine receptor Ccr2 (1:100; Novus Biologicals), mouse anti-human Aβ (clones 4G8 or 6E10; 1:500; Covance), or mouse anti-NeuN (1:3000; Millipore). We incubated brain sections with species-specific Alexa Fluor 488- and 594-conjugated secondary antibodies (Invitrogen) for 1 h at room temperature, followed by staining with the VECTASTAIN Elite ABC kit (Vector Laboratories) coupled with 3,3′-diaminobenzidine substrate. Sections were analyzed in independent channels with an Olympus FV1000 laser scanning confocal microscope equipped with Fluoview SV1000 imaging software.
Quantification of Aβ deposits.
We quantified Aβ deposits by immunofluorescence using six 25 μm free-floating sections spaced 200 μm apart through each anatomic region of interest (hippocampus and cerebral cortex) as described previously (Tan et al., 2002; Town et al., 2008). Brain sections were immunostained with rabbit polyclonal oligomer/conformational (OC) Aβ antibody (Kayed et al., 2007) (provided by S.R. and C.G.G.), using Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (Invitrogen). Amyloid burden was determined at 20× magnification by quantitative image analysis using an automated Zeiss Observer Z1 inverted microscope with an attached Axiocam MRm CCD camera and Axiovision software version 4.6 (Carl Zeiss). For measurement of microglial distances from Aβ deposits, brain sections were double labeled with Iba1 antibody and Congo red, and the distance of microglia from the center of the nearest Aβ plaque was blindly measured in 8-month-old PSAPP/CD45 or PSAPP/CD45−/− mice using SimplePCI software (Hamamatsu Photonics). Quantitative image analysis was performed by a single examiner (T.M.) blinded to sample identities. Data are reported as percentage of positive pixels divided by total pixels captured for each region of interest.
Murine primary cell culture.
Murine primary cultured microglia were isolated from mouse cerebral cortices and grown in complete RPMI 1640 medium according to previously described methods (Zhu et al., 2008). Briefly, cerebral cortices from newborn mice (1–2 d old) were isolated under sterile conditions and kept at 4°C before mechanical dissociation. Cells were grown in RPMI 1640 medium supplemented with 5% heat-inactivated FCS, 2 mm glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 nm 2-mercaptoethanol. Primary cultures were kept for 14 d so that only glial cells remained. Astrocytes were separated from microglial cultures using a mild trypsinization protocol as described (Saura et al., 2003). More than 98% of these glial cells stained positive for Mac-1 (Roche Diagnostics) by flow cytometry.
Microglial phagocytosis assay.
Primary mouse microglia were seeded at 1 × 105 cells per well (n = 6 for each condition) in 24-well tissue culture plates containing 0.5 ml of complete RPMI 1640 medium for fluorometric analysis of Aβ. These cells were treated for 2 h with “aged” Aβ1-42 [500 nm; preaggregated for 24 h at 37°C in complete medium as described by Chung et al. (1999) and conjugated with FITC (FITC–Aβ1-42) (Bachem Americas)]. Microglial cells were then cotreated with agonistic CD45 antibody or isotype control IgG (2.5 μg/ml) for 2 h in the presence of FITC–Aβ1-42. Cells were then rinsed three times in Aβ-free complete medium, and the medium was exchanged with fresh Aβ-free complete medium for 10 min to allow for removal of non-incorporated Aβ and to promote concentration of Aβ into phagosomes. Extracellular (in cell culture media) and cell-associated (in cell lysates) FITC–Aβ were quantified using an MSF SpectraMax spectrophotometer (Molecular Devices) with an emission wavelength of 538 nm and an excitation wavelength of 485 nm. A standard curve from 0 to 600 nm FITC–Aβ was generated for each plate. Total cellular proteins were quantified by BCA Protein Assay. The mean fluorescence values for each sample at 37°C and 4°C at the 2 h time point were determined by fluorometric analysis. Relative fold change values were calculated as follows: mean fluorescence value for each sample at 37°C/mean fluorescence value for each sample at 4°C. Considering nonspecific adherence of Aβ to the plastic surface of culture plates, an additional control without cells was performed in parallel for each experiment above. An incubation time of <4 h did not change the amount of Aβ peptide detected in the supernatant, consistent with a previous report (Mitrasinovic and Murphy, 2002). To determine whether cell death influenced Aβ uptake in the various treatment groups, we performed lactate dehydrogenase release assays but did not detect significant (p > 0.05) cell death over the 3 h experimental timeframe in any of the treatment groups (data not shown).
Confocal microscopy.
“Aged” FITC–Aβ1-42 was prepared according to methods described above. Microglial cells were cultured at 1 × 105 cells per well in 24-well tissue culture plates with glass inserts. These cells were treated for 2 h with aged FITC–Aβ1-42. Separate groups of microglial cells were incubated in parallel at 4°C (control). After treatment, cells were washed five times with ice-cold PBS to remove non-incorporated FITC–Aβ1-42 and fixed for 10 min at 4°C in 4% PFA, followed by three rinses in PBS. Finally, sections were mounted with fluorescence mounting media containing DAPI (ProLong Gold; Invitrogen) and viewed with a Leica SP5 confocal microscope (Leica Microsystems). Excitation wavelengths of 488 nm (to reveal FITC–Aβ1-42) and 405 nm (for DAPI-counterstained nuclei) were used. Images were captured and analyzed using LAS AF software version 1.6.0 (Leica Microsystems). Normarski optic (differential interference contrast) images were captured in wide field to accompany each confocal image.
Stereological analysis.
We stained 40 μm free-floating serial brain sections in plastic multiwell carriers with nylon net bottoms using NeuN antibody as described above. To prepare for unbiased stereologic estimation of neuronal counts, an initial tissue section was randomly selected at one anatomic border of the brain region to be examined. Thereafter, every sixth section throughout the anatomic region of interest was used for each counting series. NeuN-positive cells were examined with a Nikon Eclipse 600 microscope and quantified using Stereo Investigator software, version 6 (MicroBrightField). Cells were counted in the entorhinal cortex using the optical fractionator method of unbiased stereologic cell counting (West et al., 1991). Our sampling method was optimized to count at least 250 cells per animal with error coefficients of <0.07. Each counting frame (50 × 50 μm) was placed at an intersection of the lines forming a virtual grid (200 × 200 μm), which was randomly generated and placed by the software within the outlined structure.
Statistical analysis.
All data were normally distributed; therefore, in instances of single mean comparisons, Levene's test for equality of the variance followed by t test for independent samples was used to assess significance. In instances of multiple mean comparisons, ANOVA was used, followed by post hoc comparison using Bonferroni's method. α levels were set at 0.05 for all analyses. The Statistical Package for the Social Sciences release 10.0.5 (SPSS) was used for all data analysis.
Results
Cerebral amyloidosis is increased in aged PSAPP mice deficient in CD45
Brain Aβ deposition is a pathognomonic feature of AD (Selkoe, 2001), and oligomeric Aβ species are thought to be a driving force in AD-type neurodegeneration (Klyubin et al., 2005; Walsh et al., 2005; Shankar et al., 2008). The double-transgenic PSAPP mouse (Jankowsky et al., 2001) is a widely used model of cerebral amyloidosis, and we bred PSAPP mice with animals deficient in CD45 and killed offspring at either 4 or 8 months of age to evaluate changes in AD-like pathology. We reacted mouse brain sections with OC antibody directed against oligomeric/conformational Aβ species (Kayed et al., 2007; Glabe, 2008) and counterstained cell nuclei with DAPI (Fig. 1a) (supplemental Fig. 1a, available at www.jneurosci.org as supplemental material) and also stained for fibrillar Aβ with ThioS (Fig. 1b). Aβ burden was calculated for OC or ThioS stains (within cingulate cortex, hippocampus, and entorhinal cortex) by quantitative image analysis. Data are represented as mean ± SD with n = 8 females per group at 4 or 8 months of age. Quantitative image analysis revealed significantly (**p < 0.01) increased OC reactivity at 4 months of age and ThioS burden at 8 months of age by 56–60% when comparing PSAPP/CD45−/− to PSAPP/CD45 littermates. By 8 months of age, PSAPP/CD45−/− animals had only modest elevation in OC immunoreactivity (supplemental Fig. 1b, available at www.jneurosci.org as supplemental material), suggesting that CD45 deficiency accelerates cerebral amyloidosis as opposed to having a cumulative effect on Aβ pathology. As expected, altered cerebral Aβ abundance in PSAPP/CD45−/− versus PSAPP/CD45 mice was not attributable to differences in APP transgene expression or Aβ metabolism (data not shown).
Figure 1.
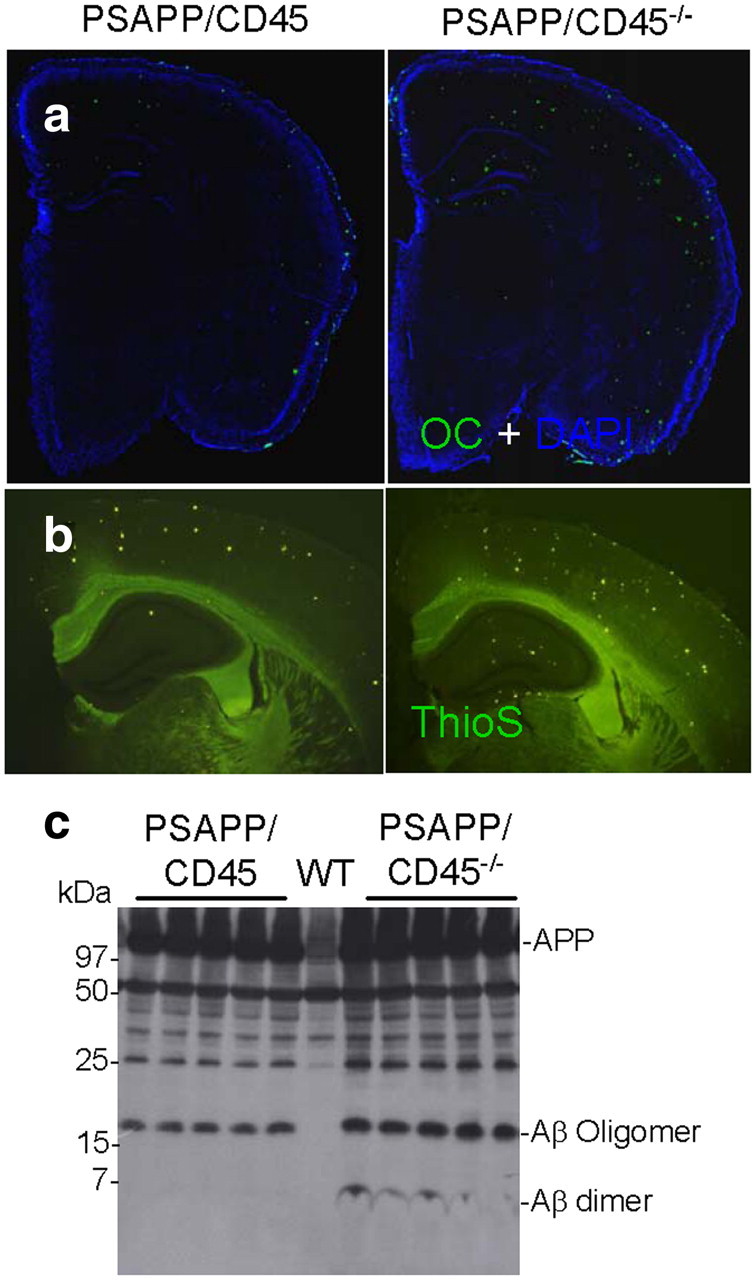
Accelerated cerebral amyloidosis in PSAPP/CD45−/− mice. a, b, Mouse brain sections from PSAPP/CD45 and PSAPP/CD45−/− mice stained with Aβ OC antibody (green signal) and DAPI (blue signal) at 4 months of age (a) or ThioS at 8 months of age (b). c, Brain homogenates were prepared from 8-month-old wild-type (WT), PSAPP/CD45, or PSAPP/CD45−/− mice. Western blot by antibody 6E10 shows increased abundance of dimeric and oligomeric Aβ species in brain homogenates from PSAPP/CD45−/− versus PSAPP/CD45 or wild-type mice.
Aβ peptides are metastable and can exist as monomeric, dimeric, and higher-molecular-weight oligomeric forms both in vitro and in vivo (Selkoe, 2001; Klyubin et al., 2005; Walsh et al., 2005; Shankar et al., 2008). It is becoming clear that Aβ dimers and oligomers are likely the neurotoxic species, because direct in vivo administration of these Aβ conformers injures neurons (Shankar et al., 2008). To determine whether CD45 deficiency impacted the metastable equilibrium of Aβ, we probed brain homogenates from PSAPP/CD45 or PSAPP/CD45−/− mice at 4 and 8 months of age by Western immunoblot. Aβ oligomers were increased in PSAPP/CD45−/− mice at 4 months of age (supplemental Fig. 1c, available at www.jneurosci.org as supplemental material). Strikingly, both dimeric and oligomeric Aβ species were elevated in PSAPP/CD45−/− versus PSAPP/CD45 mice at 8 months of age (Fig. 1c). Together, these results indicate that cerebral Aβ pathology is overrepresented in CD45-deficient PSAPP mice.
Impaired brain-to-blood Aβ clearance in aged PSAPP/CD45−/− mice
It has been proposed that cerebral Aβ is cleared across the blood–brain barrier (BBB) via a “peripheral sink,” and there is evidence of dysfunctional brain-to-blood Aβ clearance in AD patients and in transgenic mouse models of the disease (DeMattos et al., 2001; Deane et al., 2003). To determine whether CD45 deficiency impacted relative Aβ abundance in cerebral and systemic compartments, we probed brains and plasma from CD45-deficient and -sufficient PSAPP mice using a biochemical approach. We assessed total insoluble Aβ species (including Aβ1-40 and Aβ1-42) in PSAPP/CD45−/− and PSAPP/CD45 mouse brain homogenates at 4 and 8 months of age by ELISA. Analysis of 4-month-old mouse brains revealed significantly (**p < 0.01) elevated abundance of insoluble Aβ species in CD45-deficient versus -sufficient PSAPP mice (Fig. 2a), although this difference was not evident in 8-month-old brains (supplemental Fig. 2a, available at www.jneurosci.org as supplemental material). Correspondingly, cerebral detergent-soluble Aβ species were increased whereas plasma-soluble Aβ abundance was diminished by a similar magnitude at both 4 and 8 months of age in PSAPP/CD45−/− versus PSAPP/CD45 animals (Fig. 2b) (supplemental Fig. 2b, available at www.jneurosci.org as supplemental material). Together, these results suggest that PSAPP/CD45−/− mice have impaired brain-to-blood Aβ clearance.
Figure 2.
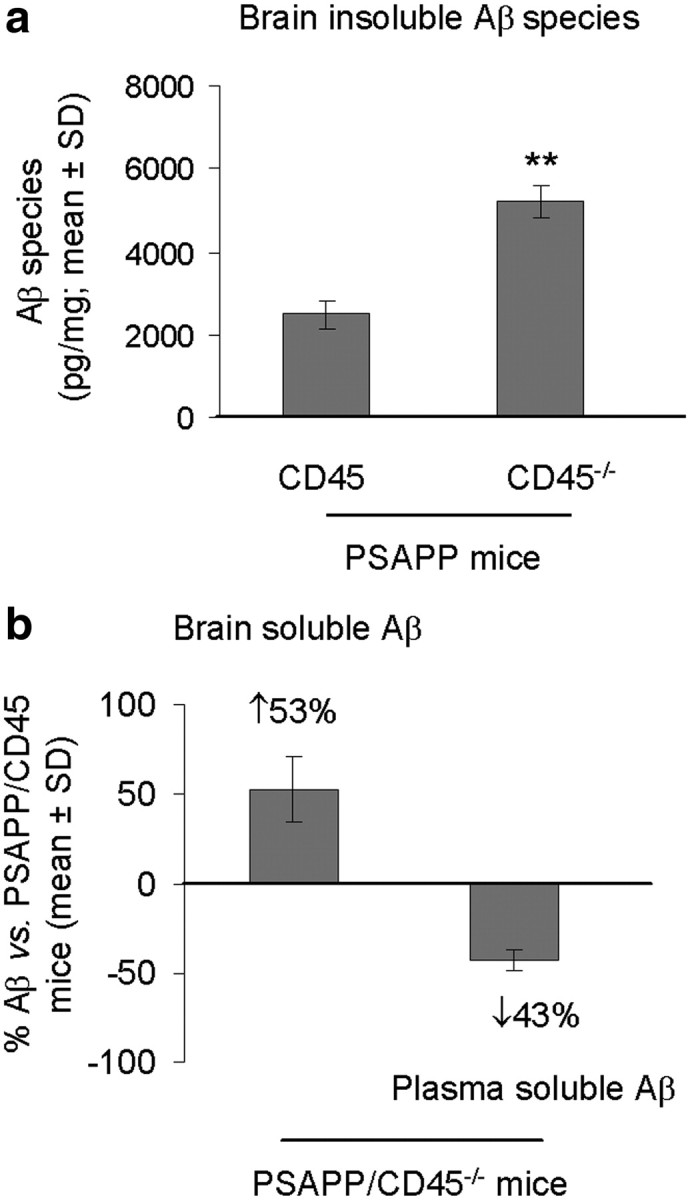
Simultaneously increased central and reduced peripheral Aβ in PSAPP/CD45−/− mice. Mouse brain homogenates were prepared from PSAPP/CD45−/− and PSAPP/CD45 mice at 4 months of age. a, Detergent-insoluble (5 m guanidine-soluble) total Aβ species (including Aβ1-40, 42; picograms per milligrams of protein) were biochemically assessed in brain homogenates by ELISA. Data are presented as mean ± SD (n = 8 females per group). b, We also analyzed steady-state cerebral and plasma total soluble Aβ species (including Aβ1-40, 42) at the time of death by ELISA. Aβ levels are represented as percentages versus PSAPP/CD45 mice with mean ± SD (n = 8 females per group). **p < 0.01.
CD45 deficiency promotes inflammatory microglia in PSAPP mice
Microglia are activated in close vicinity of β-amyloid plaques in AD patient brains and in transgenic mouse models of the disease (Benzing et al., 1999; Jimenez et al., 2008; Mandrekar-Colucci and Landreth, 2010). Although it was once thought that microglial “activation” was a single phenotype, we now know that multiple forms of functionally distinct reactive microglia exist (Town et al., 2005; Colton, 2009; Colton and Wilcock, 2010). To determine whether CD45 deficiency impacted microglial phenotype in PSAPP mice, we stained brain sections from PSAPP/CD45 and PSAPP/CD45−/− mice with antibodies directed against the activated microglial markers Iba1, CD11b, or CD40 (Tan et al., 1999; Townsend et al., 2005; Ahmed et al., 2007), in combination with Aβ antibody 4G8 and DAPI as a nuclear counterstain. Because microglia activate in response to Aβ deposits and 4-month-old PSAPP/CD45−/− mice had elevated β-amyloid plaque load versus controls (Fig. 1), we wanted to avoid this confounder and therefore focused on analyzing our 8-month-old cohort with minimal or no differences on insoluble Aβ abundance (supplemental Fig. 2a, available at www.jneurosci.org as supplemental material). As shown in Figure 3a, Iba1-positive microglia were generally found in close spatial proximity to cortical Aβ plaque centers in PSAPP/CD45 mice, whereas PSAPP/CD45−/− animals displayed a more random and diffuse pattern of parenchymal Iba1 reactivity. Furthermore, the distance between each microglial cell to the center of the nearest Congo red-positive Aβ plaque was measured in brain sections from 8-month-old PSAPP/CD45−/− versus PSAPP/CD45 mice. At the quantitative level, Iba1-positive cells were significantly (**p < 0.01) farther away from Aβ deposits when comparing PSAPP/CD45−/− with PSAPP/CD45 mice. The diffuse nature of Iba1-positive microglia in PSAPP/CD45−/− mice seems consistent with a “runaway” proinflammatory state that is poorly directed toward amyloid plaques in these mice. Similar results were observed in PSAPP/CD45−/− mice at 4 months of age, despite imbalance in cerebral amyloidosis in this cohort (data not shown).
Figure 3.
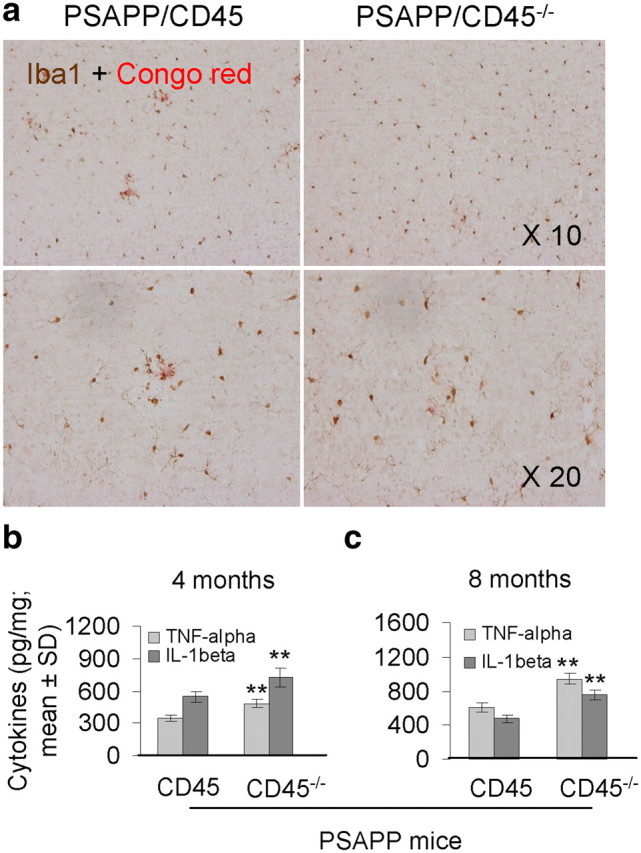
PSAPP/CD45−/− mice have an inflammatory microglial phenotype. a, Mouse brain sections from 8-month-old PSAPP/CD45 or PSAPP/CD45−/− mice were double-labeled with Iba1 antibody and Congo red. b, c, The microglial proinflammatory cytokines TNF-α and IL-1β were quantified in brain homogenates from both PSAPP/CD45 and PSAPP/CD45−/− mice at 4 (b) and 8 (c) months of age by ELISA. Data are represented as mean ± SD (n = 8 female mice per group) for each cytokine (picograms per milligrams of protein). **p < 0.01.
We have shown that immunological costimulation via the CD40 pathway enables microglial inflammatory responses after Aβ peptide stimulation and also reduces Aβ clearance responses by these cells (Tan et al., 1999, 2002; Townsend et al., 2005). Consistent with a proinflammatory but anti-Aβ phagocytic microglial phenotype, Aβ plaque-associated microglia in PSAPP/CD45−/− brains loose CD11b signal but gain expression of CD40 (supplemental Fig. 3a, available at www.jneurosci.org as supplemental material). Quantification of these results revealed a statistically significant (***p < 0.005) reduction in CD11b but significantly (**p < 0.01) increased CD40 signal in PSAPP/CD45−/− versus PSAPP/CD45 mouse brains (supplemental Fig. 3b, available at www.jneurosci.org as supplemental material). To further assess brain inflammation, we measured the microglial proinflammatory cytokines TNF-α and IL-1β in brain homogenates from PSAPP/CD45 and PSAPP/CD45−/− mice at 4 and 8 months of age. Consistent with our histological observations, data revealed significantly (**p < 0.01) increased levels of both cytokines in CD45-deficient versus -sufficient PSAPP mice at both ages (Fig. 3b,c). When taken together with the above Aβ plaque microglial localization findings, these results suggest that CD45 deficiency promotes an inflammatory microglial phenotype that is inefficient at restricting cerebral amyloidosis and promotes buildup of neurotoxic Aβ oligomers.
To better characterize whether CD45 deficiency affected microglia or blood-borne monocytes/macrophages (or both), we took an immunofluorescence approach based on morphologic and immunophenotypic criteria to critically examine brain sections for any evidence of hematogenously derived immune cells (El Khoury et al., 2007; Town et al., 2008). This methodology was chosen over irradiation/bone marrow chimeras, because the latter have become controversial. Specifically, the act of irradiating mice artificially sensitizes the CNS to large-scale infiltration and engraftment of the adoptively transferred peripheral macrophages (Ahmed et al., 2007; Mildner et al., 2007). Despite careful determination of CD3, CD4, CD45 (data not shown), Iba1, CD11c, and Ccr2 expression and inclusion of an experimental autoimmune encephalomyelitis-positive control, we were unable to detect blood-derived immune cells in any of the four mouse groups analyzed (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
CD45−/− microglia have impaired Aβ1-42 phagocytosis
Although there has been much recent debate about whether microglia are efficient Aβ phagocytes (Grathwohl et al., 2009), microglial Aβ phagocytosis has nonetheless been suggested to occur to a limited extent in the AD brain (Familian et al., 2007), and we have shown recently that peripherally derived mononuclear phagocytes can clear oligomeric Aβ species (Town et al., 2008). Our in vivo results suggested that CD45 deficiency promoted a proinflammatory yet anti-Aβ phagocytic microglial phenotype (Town et al., 2005). To determine whether CD45 agonism could produce the converse in vitro, we prepared CD45-deficient and -sufficient microglia from neonates as described previously (Zhu et al., 2008) and challenged them with agonistic CD45 antibody or isotype-matched control IgG in the presence of aged FITC–Aβ1-42. As shown in Figure 4a, ablation of CD45 significantly (**p < 0.01) diminished microglial phagocytosis of FITC–Aβ1-42, and addition of agonistic CD45 antibody significantly (*p < 0.05) elevated this effect in CD45-sufficient cells (but had no effect on CD45-deficient control cells). Microglia treated with a nonrelevant isotype-matched IgG control antibody did not differ from untreated cells (data not shown). To validate this result, we treated primary microglia as described above and then imaged them by confocal microscopy. Data revealed FITC–Aβ1-42 peptide within the cytoplasm of CD45-sufficient primary microglial cells, whereas the peptide remained on the surface of CD45-deficient cells (Fig. 4b).
Figure 4.

CD45-deficient primary microglia have impaired Aβ1-42 phagocytosis. CD45-sufficient or -deficient primary microglial cells were prepared from neonatal mice and treated with agonistic CD45 antibody (2.5 μg/ml) or isotype control IgG (data not shown) in the presence of 1 μm aged FITC–Aβ1-42 for 60 min. a, Cellular supernatants and lysates were analyzed for cell-associated (left) and extracellular (right) FITC–Aβ1-42 using a fluorometer. Data are represented as the relative fold of mean fluorescence change (mean ± SD), calculated as the mean fluorescence for each sample at 37°C divided by mean fluorescence at 4°C (n = 6 for each condition presented). b, In parallel experiments, primary microglia were treated as in a but were then fixed and imaged in independent channels using a confocal microscope equipped with Normarski optics. Merged images shown on the right reveal localization of Aβ1-42 within the cytoplasm of CD45-sufficient microglia but on the cell surface of CD45-deficient cells. *p < 0.05, **p < 0.01.
Interestingly, unlike the more ramified appearance of wild-type cells that typically indicates a “resting” state, CD45-deficient microglia had a unique morphology denoted by an ovoid cytoplasm and relatively few cytoplasmic processes compared with wild-type cells (Fig. 4b). This morphological phenotype of CD45-deficient microglia occurred in concert with strikingly increased expression of CD40 (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), a key costimulatory protein required for proinflammatory innate immune activation of antigen presenting cells. Furthermore, as shown in Figure 4b, ovoid CD45-deficient microglia were unable to take up fluorescently tagged Aβ peptide in vitro. We conclude that CD45 deficiency leads to a functional switch in microglial phenotype characterized by morphologic and immunophenotypic changes consistent with an activated, proinflammatory state that is incompatible with Aβ clearance. Although this particular microglial phenotype seems to be deleterious in the context of AD, it is important to note that not all forms of microglial activation are detrimental; this is underscored by findings from Aβ “immunotherapy” approaches, in which microglia could be stimulated to phagocytose and clear Aβ deposits decorated with Aβ-specific antibodies (Schenk et al., 1999; Bard et al., 2000).
Increased neuronal intracellular Aβ in aged PSAPP/CD45−/− mice
Aβ can exist in both secreted and intracellular pools within the brain (Watson et al., 2005). APP is normally metabolized to Aβ via an endocytosis-dependent, pH-sensitive pathway (Shoji et al., 1992; Koo and Squazzo, 1994), and intracellular Aβ has been found in degenerating neurons in the AD brain (Probst et al., 1991). If CD45-deficient microglia were unable to effectively clear cerebral Aβ, then one might expect intracellular buildup of the peptide. To evaluate this hypothesis, we investigated intracellular Aβ in 4- and 8-month-old PSAPP/CD45 and PSAPP/CD45−/− brain sections by immunostaining. Regardless of age, CD45-deficient mouse brains showed a marked increase in intraneuronal 6E10 reactivity (supplemental Fig. 5a,b, available at www.jneurosci.org as supplemental material). To confirm the Aβ identity of these signals, we performed Western immunoblot by 6E10 antibody and observed that extracellular and intracellular dimeric and oligomeric Aβ species were increased in PSAPP/CD45−/− versus PSAPP/CD45 mice at 8 months (Fig. 5a), and a similar pattern of results was also likely the case at 4 months of age (supplemental Fig. 5c, available at www.jneurosci.org as supplemental material), although we were working at the detection limit for the assay at this earlier age. Additionally, we quantified extracellular and intracellular soluble Aβ from PSAPP/CD45 and PSAPP/CD45−/− mouse brains by ELISA. Data revealed significantly (*p < 0.05; **p < 0.01) increased abundance of total soluble intracellular Aβ species in PSAPP/CD45−/− versus PSAPP/CD45 mice at both ages and in both fractions (Fig. 5b).
Figure 5.

Increased intracellular Aβ in PSAPP/CD45−/− mice. a, Extracellular and intracellular proteins were prepared from 8-month-old wild-type (WT), PSAPP/CD45, and PSAPP/CD45−/− mouse brain homogenates. Western blot analysis by antibody 6E10 shows increased abundance of Aβ dimers and oligomers in brain extracts from PSAPP/CD45−/− versus PSAPP/CD45 or wild-type mice. b, Total detergent-soluble Aβ species (including Aβ1-40, 42) in extracellular or intracellular fractions were assayed in 4- and 8-month-old PSAPP/CD45 and PSAPP/CD45−/− mouse brain extracts by ELISA. Results are represented as mean ± SD (n = 8 females per group) of total soluble Aβ species (picograms per milligrams of protein). *p < 0.05, **p < 0.01.
PSAPP/CD45−/− mice have neuronal loss
An important hallmark of AD is loss of neurons, resulting in significant atrophy of the cerebral cortex and certain subcortical regions, including the temporal lobe, parietal lobe, parts of the frontal cortex, and the cingulate gyrus (Wenk, 2003). As detailed above, intraneuronal Aβ is increased in PSAPP/CD45−/− mice, and we sought to investigate whether this form of Aβ pathology was accompanied by neuronal loss. We reacted brain sections from 8-month-old wild-type (data not shown), CD45−/−, PSAPP/CD45, or PSAPP/CD45−/− mice with Nissl or NeuN antibody and performed stereological analysis for NeuN-positive cells. Nissl staining revealed neuronal dysmorphology suggestive of neuronal degeneration (Fig. 6a). Furthermore, NeuN immunohistochemistry disclosed a more rarefied pattern of neurons in PSAPP/CD45−/− mice (Fig. 6b), and stereological analysis demonstrated significantly (*p < 0.05) decreased NeuN-positive cells in the medial entorhinal cortex (Fig. 6c) of PSAPP/CD45−/− versus PSAPP/CD45 or control CD45−/− mice at 8 months of age, but this was not yet evident in 4-month-old animals (data not shown).
Figure 6.

a, b, PSAPP/CD45−/− mice have neuronal injury and loss. Mouse brain sections from 8-month-old CD45−/−, PSAPP/CD45, and PSAPP/CD45−/− mice were stained with Nissl (dysmorphic neurons are indicated by arrows) (a) or NeuN antibody (b), and representative entorhinal cortex images are shown. c, Stereological analysis for NeuN-positive cells in the medial entorhinal cortex (MEA) (n = 6 female mice per group; mean ± SD) is graphically represented. d, e, Brain homogenates were prepared from 8-month-old control CD45−/−, PSAPP/CD45, and PSAPP/CD45−/− mice and probed by Western blot using antibodies against NeuN, Bcl-xL, or Bax (d) total and cleaved (active) caspase-3 (e). Note reduced expression of NeuN and Bcl-xL and increased abundance of Bax protein and cleaved caspase-3 in PSAPP/CD45−/− versus PSAPP/CD45 or CD45−/− mouse brains. WT, Wild type. *p < 0.05.
To validate these results, we prepared brain homogenates from each group of mice at 8 months of age. Western blot analysis revealed decreased levels of NeuN relative to actin (Fig. 6d) in PSAPP/CD45−/− versus PSAPP/CD45 or control CD45−/− mice. A similar pattern of results was noted when considering expression ratio of the neuronal anti-apoptotic regulator Bcl-xL (Parsadanian et al., 1998) to the proapoptotic protein Bax (Fig. 6d). Furthermore, another index of apoptosis, cleaved caspase-3, was overrepresented in PSAPP/CD45−/− mice compared with the other two mouse groups, whereas unprocessed caspase-3 did not differ between groups (Fig. 6e). When taken together, these results demonstrate neuronal loss in PSAPP/CD45−/− mice, likely as a result of apoptosis.
Mitochondrial dysfunction in PSAPP/CD45−/− mice
The brain is highly dependent on aerobic metabolism, and mitochondria are responsible for cellular respiration. To investigate whether neuronal loss in PSAPP/CD45−/− mice was accompanied by loss of mitochondrial function, we isolated mitochondria from cortical regions (including frontal, entorhinal, and cingulate cortices) and hippocampi of 8-month-old wild-type, CD45−/−, PSAPP/CD45, and PSAPP/CD45−/− mice. We then enumerated respiratory rates for each brain region in all mouse groups. We observed significantly (*p < 0.05; **p < 0.01) reduced basal (state II) respiration (supplemental Fig. 6a, left panels, available at www.jneurosci.org as supplemental material) and attenuated maximum respiratory rate (supplemental Fig. 6a, right panels, available at www.jneurosci.org as supplemental material) in PSAPP/CD45−/− mice versus the three other groups for all brain regions examined. Furthermore, mitochondrial membrane potential (supplemental Fig. 6b, left panels, available at www.jneurosci.org as supplemental material) and reactive oxygen species abundance (supplemental Fig. 6b, right panels, available at www.jneurosci.org as supplemental material) were significantly (*p < 0.05; **p < 0.01) reduced in PSAPP/CD45−/− compared with wild-type, CD45−/−, or PSAPP/CD45 mice for mitochondria isolated from either cortical or hippocampal brain regions. These results indicate that PSAPP/CD45−/− mice exhibit mitochondrial dysfunction, which dovetails with shift from anti-apoptotic to proapoptotic proteins and neuronal loss in these animals.
Discussion
There has been considerable recent debate surrounding the relationship between microglia and AD-like pathology. Although microglia in brains of healthy elderly individuals are uniformly distributed, these cells appear in tight temporal and spatial proximity to amyloid plaques in brains of AD patients and in transgenic mouse models of the disease (McGeer et al., 1987; Benzing et al., 1999; Akiyama et al., 2000; Heneka and O'Banion, 2007). These pathological observations have prompted the conclusion that microglia are etiological participants in AD, although this remains controversial (Grathwohl et al., 2009). In support of this notion, studies that impair microglial or mononuclear phagocyte functions by (1) treatment with nonsteroidal anti-inflammatory drugs (Lim et al., 2000, 2001), (2) interrupting CD40–CD40L interaction (Tan et al., 1999,2002), or (3) genetically ablating transforming growth factor-β (TGF-β) receptor signaling (Town et al., 2008) mitigate AD-like pathology in transgenic AD mice. Additionally, immunotherapy approaches that rely on Aβ-specific antibodies to stimulate microglial clearance of Aβ deposits resolve AD-like pathology in mouse models (Schenk et al., 1999; Bard et al., 2000). However, deficiency of the Ccr2 chemokine receptor reduces microglial recruitment to brains of AD model mice and causes accumulation of cerebral amyloid plaques (El Khoury et al., 2007), whereas genetic ablation of the Cx3cr1 fractalkine receptor impairs microglial migration to neurons “marked for death” and prevents neuronal loss in 3xTg–AD mice (Fuhrmann et al., 2010). A parsimonious conclusion that arises from these results is that multiple forms of microglial activation exist, some being deleterious and others, beneficial (Town et al., 2005; Wyss-Coray, 2006).
CD45 is the most abundant membrane-bound protein tyrosine phosphatase and functions to dampen overly exuberant immune responses (Justement, 1997). Furthermore, microglial CD45 abundance is increased in brains of AD patients and in mouse models of the disease (Masliah et al., 1991; Licastro et al., 1998). Although multiple variants of CD45 are generated by alternate mRNA splicing, the CD45RB isoform is most highly expressed by microglia (Townsend et al., 2004). Microglial CD45 functions to inhibit nitric oxide and TNF-α production induced by Aβ peptides (Tan et al., 2000b), CD40L, or bacterial endotoxin by dephosphorylating Src-family kinases and thereby inactivating p44/42 and p38 MAPKs (Zhu et al., 2008). In vitro, we have suggested that CD45RB on microglia likely acts as a molecular switch to turn phagocytosis “off” and damaging proinflammatory response “on” in the presence of exogenous Aβ (Zhu et al., 2008).
To explore the functional consequences of CD45 deficiency in vivo, we crossed transgenic mice overproducing Aβ with animals deficient in CD45. PSAPP/CD45−/− mice manifest accelerated cerebral amyloidosis, characterized by elevated abundance of β-amyloid plaques and both intracellular and extracellular pools of soluble, oligomeric, and insoluble Aβ. Although soluble intraneuronal forms of Aβ are produced under physiologic conditions, a tight balance exists between peptide production and clearance (Shoji et al., 1992; Koo and Squazzo, 1994); yet, abnormally high amounts of intracellular Aβ are present in degenerating neurons in brains of AD and Down's syndrome patients (Allsop et al., 1989; Probst et al., 1991), in monkey and rodent models of Aβ deposition (Martin et al., 1994), and in human immunodeficiency virus patients with dementia (Green et al., 2005). Intracellular Aβ is produced in the endoplasmic reticulum and Golgi complex in neuronal cells (Wertkin et al., 1993; Wild-Bode et al., 1997; Xu et al., 1997), and Aβ immunoreactivity within lysosomes of degenerating neurons has been found in both aging macaques (Martin et al., 1994) and in Aβ-infused rats (Frautschy et al., 1996). Our results from PSAPP/CD45−/− mice lend support to the idea that intraneuronal Aβ accumulation precedes neuronal loss, as recently suggested by another group (Fuhrmann et al., 2010).
But what form of Aβ is neurotoxic? Recent focus has shifted toward soluble oligomeric Aβ species as the agents of neurotoxicity. Administration of Aβ oligomers directly isolated from AD patient cerebral cortices to normal rats impaired long-term potentiation, enhanced long-term depression, and reduced dendritic spine density. Furthermore, these deleterious effects were specifically attributable to Aβ dimers (Shankar et al., 2008), and it is likely that Aβ-directed immunotherapy works by clearing oligomeric species of Aβ (Klyubin et al., 2005). However, do mononuclear phagocytes including microglia impact steady-state Aβ oligomer abundance? We previously showed that blockade of TGF-β–Smad 2/3 signaling promotes uptake and clearance of Aβ oligomers by cells of mononuclear phagocyte origin (Town et al., 2008), and the present work demonstrates that CD45 deficiency drives accumulation of cerebral Aβ dimers and oligomers in a transgenic mouse model of AD. Results presented here place microglia on the Aβ oligomer clearance pathway and suggest that ablating CD45 and thereby inhibiting this clearance machinery causes buildup of neurotoxic Aβ oligomers and neuropathology downstream of the amyloid cascade (Hardy and Allsop, 1991).
A number of studies have shown that the BBB is responsible for elimination of human Aβ from the brain into the blood (Shibata et al., 2000; Shiiki et al., 2004; Terasaki and Ohtsuki, 2005). Some have even harnessed this peripheral sink to clear cerebral amyloid by passive Aβ immunotherapy (DeMattos et al., 2001; DeMattos et al., 2002). Aβ transport across the BBB is bidirectional because, when exogenous human Aβ1-40 is systemically injected, it is transported into the brain (Martel et al., 1997; Wengenack et al., 2000; Deane et al., 2003). As such, Aβ BBB transport homeostasis is likely an important factor governing accumulation of cerebral Aβ (Ito et al., 2006). In addition to elevated cerebral amyloidosis, PSAPP/CD45−/− mice also demonstrate decreased plasma-soluble Aβ abundance, likely reflective of diminished brain-to-blood Aβ efflux. Given the exquisite microglial specificity of CD45 expression in the brain, it is unlikely that CD45 deficiency directly impacts Aβ clearance at the BBB. A more likely possibility consistent with our observations is that failure in microglial Aβ clearance in PSAPP/CD45−/− mice overloads brain-to-blood Aβ efflux machinery, leading to increased cerebral amyloid and reduced circulating Aβ.
In addition to accelerated cerebral amyloidosis, we also find increased abundance of TNF-α and IL-1β [which are neurotoxic at high levels (Meda et al., 1995; Barger and Harmon, 1997; Tan et al., 1999)], mitochondrial dysfunction, and neuronal loss in PSAPP/CD45−/− mice. Association among these three observations leads to a model wherein CD45 deficiency endorses a proinflammatory but anti-Aβ phagocytic form of microglial activation. Because of failed microglial clearance of cerebral Aβ and overexpression of neurotoxic cytokines, downstream events in this model would include dysfunctional mitochondrial respiration and ultimately neuronal loss. However, it is unclear whether mitochondrial dysfunction brought on by loss of CD45 is a cause or consequence of neurotoxicity. In this regard, oxidative phosphorylation—and in particular cytochrome c oxidase activity—is deficient in AD patient brains (Cottrell et al., 2001; Fukui et al., 2007). However, falloff in cytochrome c oxidase activity is likely related to global decline in numbers of mitochondria as a result of neurotoxicity. A number of factors might contribute to the observed reduction in oxidative phosphorylation activity in AD, including failed mitochondrial transport through axonal and dendritic processes, compromised regulatory feedback mechanisms responsible for individual complex-subunit synthesis, and impaired complex assembly (Mancuso et al., 2008).
It is well established that CD45 has multiple splice variants (chiefly, -RA, -RB, -RC, and -RO), that are variously expressed by different immune cells. CD45 isoforms may functionally differ, and this explains why gross CD45 deficiency can lead to both hypo- and hyper-responsive immunological defects. We previously demonstrated that, in the case of microglia, ∼90% of CD45 can be accounted for by the CD45RB isoform (Townsend et al., 2004). Furthermore, although agonistic antibodies directed against CD45RA or CD45RC isoforms had minimal effects on lipopolysaccharide-induced microglial activation, antibody-mediated stimulation of CD45RB resulted in almost complete shutdown of lipopolysaccharide-induced TNF-α release in cultured microglia (Townsend et al., 2004; Zhu et al., 2008). Finally, stimulation of CD45RB specifically enhanced Aβ uptake that was dependent on inhibition of the p44/42 MAPK signaling cascade (Zhu et al., 2008). Nonetheless, future studies including phenotypic analyses of each CD45 isoform in the absence of alternative variants will be needed to determine the functional implications of the various CD45 isoforms vis-à-vis microglial biology.
Our present work demonstrates that genetic loss of CD45 (1) accelerates cerebral amyloidosis, (2) causes brain accumulation of soluble oligomeric Aβ species and reduction in plasma-soluble Aβ, (3) promotes proinflammatory and anti-Aβ phagocytic microglial activation, and (4) leads to mitochondrial dysfunction and neuronal loss in PSAPP/CD45−/− mice. If this mouse model translates to the clinical syndrome, then a pharmacotherapeutic strategy aimed at promoting CD45-mediated microglial Aβ clearance should be beneficial for AD treatment.
Footnotes
This work was supported by the National Institutes of Health (NIH)/National Institute on Aging (NIA) Grants AG04418/Project-2 (J.T.) and R01AG032432 (J.T.) and NIH/National Institute of Neurological Disorders and Stroke Grant R01NS048335 (J.T.). B.G. is supported by NIH/National Institute of Mental Health Clinical Scientist Award 1 K08 MH082642-01A1. T.T. is supported by NIH/NIA “Pathway to Independence” Awards 5R00AG029726-03, 5R00AG029726-04, and 3R00AG029726-04S1, is the recipient of Alzheimer's Association Zenith Fellows Award ZEN-10-174663, and is the inaugural holder of the Ben Winters Endowed Chair in Regenerative Medicine. We thank David Morgan (University of South Florida) for helpful advice. Mosaic images were taken by the Analytic Microscopy Core Facility at H. Lee Moffitt Cancer Center.
References
- Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickson DW. Actin-binding proteins coronin-1a and IBA-1 are effective microglial markers for immunohistochemistry. J Histochem Cytochem. 2007;55:687–700. doi: 10.1369/jhc.6A7156.2007. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allsop D, Haga SI, Haga C, Ikeda SI, Mann DM, Ishii T. Early senile plaques in Down's syndrome brains show a close relationship with cell bodies of neurons. Neuropathol Appl Neurobiol. 1989;15:531–542. doi: 10.1111/j.1365-2990.1989.tb01252.x. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- Benatar T, Carsetti R, Furlonger C, Kamalia N, Mak T, Paige CJ. Immunoglobulin-mediated signal transduction in B cells from CD45-deficient mice. J Exp Med. 1996;183:329–334. doi: 10.1084/jem.183.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging. 1999;20:581–589. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Chung H, Brazil MI, Soe TT, Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells. J Biol Chem. 1999;274:32301–32308. doi: 10.1074/jbc.274.45.32301. [DOI] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9:174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- Cottrell DA, Blakely EL, Johnson MA, Ince PG, Turnbull DM. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology. 2001;57:260–264. doi: 10.1212/wnl.57.2.260. [DOI] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Familian A, Eikelenboom P, Veerhuis R. Minocycline does not affect amyloid beta phagocytosis by human microglial cells. Neurosci Lett. 2007;416:87–91. doi: 10.1016/j.neulet.2007.01.052. [DOI] [PubMed] [Google Scholar]
- Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Calderón L, Cole GM. Rodent models of Alzheimer's disease: rat A beta infusion approaches to amyloid deposits. Neurobiol Aging. 1996;17:311–321. doi: 10.1016/0197-4580(95)02073-x. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui H, Diaz F, Garcia S, Moraes CT. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2007;104:14163–14168. doi: 10.1073/pnas.0705738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS. 2005;19:407–411. doi: 10.1097/01.aids.0000161770.06158.5c. [DOI] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, Griffiths E, Krawczyk C, Richardson CD, Aitken K, Iscove N, Koretzky G, Johnson P, Liu P, Rothstein DM, Penninger JM. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature. 2001;409:349–354. doi: 10.1038/35053086. [DOI] [PubMed] [Google Scholar]
- Ito S, Ohtsuki S, Terasaki T. Functional characterization of the brain-to-blood efflux clearance of human amyloid-beta peptide (1-40) across the rat blood-brain barrier. Neurosci Res. 2006;56:246–252. doi: 10.1016/j.neures.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–11661. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justement LB. The role of CD45 in signal transduction. Adv Immunol. 1997;66:1–65. doi: 10.1016/s0065-2776(08)60595-7. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Licastro F, Mallory M, Hansen LA, Masliah E. Increased levels of alpha-1-antichymotrypsin in brains of patients with Alzheimer's disease correlate with activated astrocytes and are affected by APOE 4 genotype. J Neuroimmunol. 1998;88:105–110. doi: 10.1016/s0165-5728(98)00096-4. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28:6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso M, Orsucci D, Siciliano G, Murri L. Mitochondria, mitochondrial DNA and Alzheimer's disease. What comes first? Curr Alzheimer Res. 2008;5:457–468. doi: 10.2174/156720508785908946. [DOI] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2010;9:156–167. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W, McComb JG, Frangione B, Ghiso J, Zlokovic BV. Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer's amyloid beta. J Neurochem. 1997;69:1995–2004. doi: 10.1046/j.1471-4159.1997.69051995.x. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Pardo CA, Cork LC, Price DL. Synaptic pathology and glial responses to neuronal injury precede the formation of senile plaques and amyloid deposits in the aging cerebral cortex. Am J Pathol. 1994;145:1358–1381. [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R, Shapiro P, Sundsmo M, Saitoh T. Immunoreactivity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer's disease. Acta Neuropathol. 1991;83:12–20. doi: 10.1007/BF00294425. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Mitrasinovic OM, Murphy GM., Jr Accelerated phagocytosis of amyloid-beta by mouse and human microglia overexpressing the macrophage colony-stimulating factor receptor. J Biol Chem. 2002;277:29889–29896. doi: 10.1074/jbc.M200868200. [DOI] [PubMed] [Google Scholar]
- Parsadanian AS, Cheng Y, Keller-Peck CR, Holtzman DM, Snider WD. Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J Neurosci. 1998;18:1009–1019. doi: 10.1523/JNEUROSCI.18-03-01009.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penninger JM, Irie-Sasaki J, Sasaki T, Oliveira-dos-Santos AJ. CD45: new jobs for an old acquaintance. Nat Immunol. 2001;2:389–396. doi: 10.1038/87687. [DOI] [PubMed] [Google Scholar]
- Probst A, Langui D, Ipsen S, Robakis N, Ulrich J. Deposition of beta/A4 protein along neuronal plasma membranes in diffuse senile plaques. Acta Neuropathol. 1991;83:21–29. doi: 10.1007/BF00294426. [DOI] [PubMed] [Google Scholar]
- Rezai-Zadeh K, Shytle D, Sun N, Mori T, Hou H, Jeanniton D, Ehrhart J, Townsend K, Zeng J, Morgan D, Hardy J, Town T, Tan J. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci. 2005;25:8807–8814. doi: 10.1523/JNEUROSCI.1521-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach T, Slater S, Koval M, White L, Cahir McFarland ED, Okumura M, Thomas M, Brown E. CD45 regulates Src family member kinase activity associated with macrophage integrin-mediated adhesion. Curr Biol. 1997;7:408–417. doi: 10.1016/s0960-9822(06)00188-6. [DOI] [PubMed] [Google Scholar]
- Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44:183–189. doi: 10.1002/glia.10274. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoi H, Seavitt J, Zheleznyak A, Thomas ML, Brown EJ. Regulation of integrin-mediated T cell adhesion by the transmembrane protein tyrosine phosphatase CD45. J Immunol. 1999;162:7120–7127. [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiiki T, Ohtsuki S, Kurihara A, Naganuma H, Nishimura K, Tachikawa M, Hosoya K, Terasaki T. Brain insulin impairs amyloid-β1-40 clearance from the brain. J Neurosci. 2004;24:9632–9637. doi: 10.1523/JNEUROSCI.2236-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- Stibenz D, Bührer C, Laufer D, Obladen M. CD45 engagement induces L-selectin down-regulation. Scand J Immunol. 1996;44:37–44. doi: 10.1046/j.1365-3083.1996.d01-282.x. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M. Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science. 1999;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Mullan M. CD45 inhibits CD40L-induced microglial activation via negative regulation of the Src/p44/42 MAPK pathway. J Biol Chem. 2000a;275:37224–37231. doi: 10.1074/jbc.M002006200. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Mori T, Wu Y, Saxe M, Crawford F, Mullan M. CD45 opposes β-amyloid peptide-induced microglial activation via inhibition of p44/42 mitogen-activated protein kinase. J Neurosci. 2000b;20:7587–7594. doi: 10.1523/JNEUROSCI.20-20-07587.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat Neurosci. 2002;5:1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- Terasaki T, Ohtsuki S. Brain-to-blood transporters for endogenous substrates and xenobiotics at the blood-brain barrier: an overview of biology and methodology. NeuroRx. 2005;2:63–72. doi: 10.1602/neurorx.2.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas ML, Brown EJ. Positive and negative regulation of Src-family membrane kinases by CD45. Immunol Today. 1999;20:406–411. doi: 10.1016/s0167-5699(99)01506-6. [DOI] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend KP, Vendrame M, Ehrhart J, Faza B, Zeng J, Town T, Tan J. CD45 isoform RB as a molecular target to oppose lipopolysaccharide-induced microglial activation in mice. Neurosci Lett. 2004;362:26–30. doi: 10.1016/j.neulet.2004.01.082. [DOI] [PubMed] [Google Scholar]
- Townsend KP, Town T, Mori T, Lue LF, Shytle D, Sanberg PR, Morgan D, Fernandez F, Flavell RA, Tan J. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur J Immunol. 2005;35:901–910. doi: 10.1002/eji.200425585. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Shankar GM, Townsend M, Fadeeva JV, Betts V, Podlisny MB, Cleary JP, Ashe KH, Rowan MJ, Selkoe DJ. The role of cell-derived oligomers of Abeta in Alzheimer's disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33:1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- Watson D, Castaño E, Kokjohn TA, Kuo YM, Lyubchenko Y, Pinsky D, Connolly ES, Jr, Esh C, Luehrs DC, Stine WB, Rowse LM, Emmerling MR, Roher AE. Physicochemical characteristics of soluble oligomeric Abeta and their pathologic role in Alzheimer's disease. Neurol Res. 2005;27:869–881. doi: 10.1179/016164105X49436. [DOI] [PubMed] [Google Scholar]
- Wengenack TM, Whelan S, Curran GL, Duff KE, Poduslo JF. Quantitative histological analysis of amyloid deposition in Alzheimer's double transgenic mouse brain. Neuroscience. 2000;101:939–944. doi: 10.1016/s0306-4522(00)00388-2. [DOI] [PubMed] [Google Scholar]
- Wenk GL. Neuropathologic changes in Alzheimer's disease. J Clin Psychiatry. 2003;64(Suppl 9):7–10. [PubMed] [Google Scholar]
- Wertkin AM, Turner RS, Pleasure SJ, Golde TE, Younkin SG, Trojanowski JQ, Lee VM. Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular beta-amyloid or A4 peptides. Proc Natl Acad Sci U S A. 1993;90:9513–9517. doi: 10.1073/pnas.90.20.9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Gordon MN, Ugen KE, Gottschall PE, DiCarlo G, Dickey C, Boyett KW, Jantzen PT, Connor KE, Melachrino J, Hardy J, Morgan D. Number of Abeta inoculations in APP+PS1 transgenic mice influences antibody titers, microglial activation, and congophilic plaque levels. DNA Cell Biol. 2001;20:731–736. doi: 10.1089/10445490152717596. [DOI] [PubMed] [Google Scholar]
- Wild-Bode C, Yamazaki T, Capell A, Leimer U, Steiner H, Ihara Y, Haass C. Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42. J Biol Chem. 1997;272:16085–16088. doi: 10.1074/jbc.272.26.16085. [DOI] [PubMed] [Google Scholar]
- Wisniewski HM, Frackowiak J. Commentary to “Differences between the pathogenesis of senile plaques and congophilic angiopathy in Alzheimer disease” [J Neuropathol Exp Neurol (1997) 56:751–761] J Neuropathol Exp Neurol. 1998;57:96–98. doi: 10.1097/00005072-199801000-00012. [DOI] [PubMed] [Google Scholar]
- Wroblewski M, Hamann A. CD45-mediated signals can trigger shedding of lymphocyte L-selectin. Int Immunol. 1997;9:555–562. doi: 10.1093/intimm/9.4.555. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Xu H, Sweeney D, Wang R, Thinakaran G, Lo AC, Sisodia SS, Greengard P, Gandy S. Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc Natl Acad Sci U S A. 1997;94:3748–3752. doi: 10.1073/pnas.94.8.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Hou H, Nikolic WV, Ehrhart J, Rrapo E, Bickford P, Giunta B, Tan J. CD45RB is a novel molecular therapeutic target to inhibit Abeta peptide-induced microglial MAPK activation. PLoS One. 2008;3:e2135. doi: 10.1371/journal.pone.0002135. [DOI] [PMC free article] [PubMed] [Google Scholar]