

Abstract
Podocyte injury has been suggested to play a pivotal role in the pathogenesis of diabetic glomerulopathy. To glean insights intomolecular mechanisms underlying diabetic podocyte injury we generated temporal global gene transcript profiles of podocytes exposed to high glucose for a time interval of 1 or 2 weeks using microarrays. A number of genes were altered at both 1 and 2 weeks of glucose exposure compared to controls grown under normal glucose. These included extracellular matrix modulators, cell cycle regulators, extracellular transduction signals and membrane transport proteins. Novel genes that were altered at both one and two weeks of high glucose exposure included Neutrophil gelatinase-associated lipocalin (LCN2 or NGAL, decreased by 3.2 fold at 1 week and by 7.2 fold at 2 weeks), Endothelial lipase (EL, increased by 3.6 fold at 1 week and 3.9 fold at 2 week), and UDP-glucuronosyltransferase 8 (UGT8, increased by 3.9 fold at 1 week and 5.0 fold at 2 weeks). To further validate these results we used real-time PCR from independent podocyte cultures, immunohistochemistry in renal biopsies and immunoblotting on urine specimens from diabetic patients. A more detailed time course revealed changes in LCN2 and EL mRNA levels as early as 6 hours and in UGT8 mRNA levels at 12 hours post-high glucose exposure. EL immunohistochemistry on human tissues showed markedly increased expression in glomeruli and immunoblotting readily detected EL in a subset of urine samples from diabetic nephropathy patients. In addition to previously implicated roles of these genes in ischemic or oxidative stress, our results further support their importance in hyperglycemic podocyte stress and possibly diabetic glomerulopathy pathogenesis and diagnosis in humans.
Keywords: diabetic glomerulopathy, podocytes, expression profiles, nephropathy
Diabetic nephropathy, a clinical syndrome of persistent microalbuminuria and a common complication of diabetes, is currently the leading cause of end-stage renal disease (ESRD) in the United States. Albuminuria indicates excessive albumin filtration from the glomerulus into the tubules which overwhelms their metabolic capacity promoting local inflammation and tubulointerstitial scarring known as diabetic nephropathy. A central initial event in the albuminuria-ESRD sequence is podocyte injury (1–3). The podocyte consists of a cell body, primary and secondary foot processes and the slit diaphragm (filtration barrier). It is proposed that hyperglycemia causes podocyte oxidative stress - defined as damage caused by reactive oxygen species- which leads to foot process effacement followed by podocyte apoptosis (4,5). For example, in the Akita model of type 1 diabetes, or the leptin receptor–deficient db/db mouse model of type 2 DKD, podocytes lose nephrin expression (the major component of the slit diaphragm), foot processes become effaced and eventually detach from the GBM undergoing death by apoptosis (4,5). Podocyte apoptosis is in part mediated by transforming growth factor-β (TGFβ ) signaling and possibly epithelial-mesenchymal transformation (EMT) (6–8). For example, Li et al show that TGF-β under conditions of high glucose suppresses expression of key slit diaphragm proteins, induces extracellular matrix protein expression (e.g., fibronectin and collagen I), and leads to secretion of matrix metalloproteinase-9 (MMP-9) (8). However, activation of other metabolic pathways, for example, the polyol pathway (9), protein kinase C (10), the hexoamine pathway (11–14), also play a role in diabetic nephropathy. Recently, VEGF-mediated signaling has also been implicated in this process (15,16) and hyperglycemia mimicking hypoxic injury to endothelial cells is proposed (17,18). These studies demonstrate that the molecular pathogenesis of diabetic podocyte injury is likely multifactorial involving a number of interrelated signaling pathways that have yet to be well understood. Understanding the molecular milieu of diabetic podocyte injury, an early event in diabetic nephropathy remains a primary target in identifying novel avenues for early intervention and prevention of severe late complications of this increasingly prevalent disease.
We have previously found that BMP7 confers podocyte resistance to hyperglycemic injury by restoring major podocyte proteins such as synaptopodin and podocin (19). Identifying new molecular changes and integrating them to known pathways are important inobtaining deeper insights into the mechanism of diabetic nephropathy, early diagnosis and possible new therapies. One approach towards this end is using high throughput methods that detect multiple molecular changes simultaneously. For example, microarray technology enables one tomeasure gene expression in whole genomes to identify new genes and pathways associated with a disease. Microarray studies on whole diabetic kidney and/or on mesangial cells, have found altered gene expression in the early phases of diabetic injury in mice (20–24). While these studies provide initial insights into early global changes in the diabetic kidney, little is known about the molecular and temporal events occurring specifically in podocytes. Recently, early global changes due to high glucose were reported in podocytes (25). Information regarding the molecular changes in podocytes due to prolonged high glucose exposure will help derive important insights into how these may lead to diabetic nephropathy, a disease that develops over a long time. This will also enhance the possibility of discovering biomarkers that can help discern podocyte dysfunction over broad range of this disease.
The aim of this study was to examine the effect of prolonged high glucose exposure on mRNA expression profiles in mouse podocytes. Using expression microarrays we discovered genes that are strongly and consistently associated with hyperglycemic podocyte stress in a time-course analysis. The in vitro podocyte injury alterations were also detected in renal biopsies and urine samples from patients with diabetic nephropathy suggesting that these may be relevant to the pathogenesis of diabetic glomerular disease in humans.
Methods
Cell Culture
Experiments were performed using a thermosensitive SV-40-transfected immortalized mouse podocyte cell line (gift from Peter Mundel, Mount Sinai School of Medicine, New York). Podocytes were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were grown at 33° C and treated with 10 U/mL of mouse recombinant -interferon (Sigma, St. Louis, Mo, USA) as previously described (19). At confluence podocytes were maintained on a bed of type I collagen at 37° C for 14 days without γ-interferon to allow differentiation (26). These conditionally immortalized cells were then either exposed to media containing normal glucose (NG) as a control (5.5 mmol/L D-glucose) or High Glucose (HG) (25 mmol/L D-glucose) for 6, 12, 18, 24, 72 hours and one or two weeks. Experiments were performed in duplicate.
To account for the osmotic effect of high glucose, podocytes were starved for 24 hours with media containing mannitol. In this model, 1 week exposure to HG was considered an arbitrary representation of sub-acute HG stress and an arbitrary representation of sustained stress was taken as HG exposure for 2 weeks. Exposure of podocytes to HG from 6–72 hours was performed to study temporal alterations and to independently validate the microarray results.
RNA extraction
Total RNA was extracted with TRIzol reagent according to manufacturer’s protocol (Invitrogen Inc.). An additional cleanup step was used by employing an RNeasy Mini Kit (QIAGEN Inc., Valencia, CA, USA). RNA concentration and purity was assessed by spectroscopy (nanodrop) with the A260/A280 ratio in the range 1.77–2.08. RNA samples were used immediately or stored at −80° C for subsequent microarray analysis or for Real-time PCR.
Microarray hybridization and data analysis
Total RNAs (1–5 μg) extracted from podocytes cultured in HG for 1 or 2 weeks and in NG for 2 weeks (in duplicate) were reverse-transcribed into cDNA and biotin-labeled cRNA targets were generated. The cRNA was fragmented and hybridized to Affymetrix GeneChip Mouse Genome 430 2.0 microarrays (Affymetrix, Santa Clara, CA, USA) at the Laboratory of Translational Pathology Microarray Core Facility of Washington University as previously described (27). Quantitative analysis of hybridization patterns and intensities was performed by Affymetrix software, and the resulting data were analyzed by Affymetrix Microarray Suite software (Version 5.0). The entire raw data are available at http://bioinformatics.wustl.edu. The data were scaled to 1500 units of signal intensity for comparisons across samples and imported into dCHIP for analysis. The data were filtered to exclude genes that were not expressed or did not vary using the coefficient of variation (standard deviation/mean) and percentage of presence calls in the arrays. First, the genes were filtered such that the coefficient of variation across samples was greater than 0.5 and less than 1000 (28,29). Second, only the genes that had a presence call percentage of ≥20% across the arrays were included for analysis. After filtering, a list of 1,790 genes was used to identify differentially expressed genes in the dataset. Using the replicate NG arrays as the baseline (B), the replicate 1W or 2W array experiments (E) were each compared for changes in gene expression using a 3-fold cutoff (B/E or E/B>3), absolute signal difference >100 (B-E or E-B>100) and a presence call percentage of ≥20% in each of the baseline and experimental datasets. The raw signal intensity data for the resulting differentially expressed genes were reviewed and only those genes that showed close agreement between the duplicates were pursued.
Real-time PCR
Total RNA extracted from podocytes cultured (duplicates) in NG or HG in various time intervals (6hrs to 2 weeks) was converted into cDNA and amplified by real-time polymerase chain reaction (PCR) in “one-step” reaction (Qiagen, OneStep RT-PCR, Germantown, MD, USA). The SYBR Green was used as fluorogenic probe system. PCR kinetics and data quantification was performed with 4000 Multiplex Quantitative PCR System Software (Stratagene, La Jolla, CA). Quantification of the target gene was performed according to the standard curve method (30). mRNA levels were normalized to β-actin. Experiments were performed in triplicate. We used the following primers: Ugt8 5’CCCACTGCCAGAAGATCTGC-3’; 3’-TGGAATAGCAAGGGCTGCTAA-5’; EL 5’-GAGCGAGCCGTACACCTCTT-3’, 3’-TGGATACGCTGGCAACTTTG-5’; LCN 5’-GATGCGCAGAGACCCAATG-3’, 3’-AGGAACGTTTCACCCGCTTT-5’.
Human tissue studies
Paraffin-embedded tissue blocks or fresh cryopreserved tissue from renal biopsies from patients (n=8) with diabetic glomerulosclerosis were randomly retrieved from the files of the Department of Pathology and Immunology at Washington University in St. Louis or George M. O’Brien Center for Kidney Disease Research Kidney Translational Research Core at Washington University. All specimens used for research were collected under protocols approved by the Institutional Review Board of Washington University School of Medicine. Light microscopy, routine immunofluorescence and electron microscopy were retrospectively reviewed to ascertain diagnosis. Seven controls consisted of histologically normal non-diabetic kidneys.
Immunohistochemistry
Immunohistochemistry on formalin-fixed paraffin-embedded sections (4μm) was performed using antigen retrieval for 15 minutes (10mM sodium citrate, 0.05% Tween20, pH 6.0) and peroxidase method. Hematoxylin-Eosin was used for counterstaining. For cryopreserved tissue, immunofluorescence was performed on 10 μm sections that were post-fixed in PBS containing 4% paraformaldehyde, washed and blocked with Image-iTTM FX signal enhancer (Invitrogen Inc.) for 30 minutes and then incubated with the primary antibody. The primary and secondary antibodies used were anti-endothelial lipase (1:50, Cayman chemical company Cat# 100030) and biotinylated anti-rabbit (1:200, JacksonImmunoresearch Inc.), respectively. WT1 antibody (1:10, Santa Cruz Biotechnology Inc.) was used to label podocytes in biopsies. The signals were visualized using streptavidin-HRP (1:400, JacksonImmunoresearch Inc.) for paraffin embedded tissue, and with streptavidin-alexa594 (molecular probes) for EL immunofluorescence, and strepatavidin-alexa488 for WT1 immunofluorescence. Slides were incubated with bis-Benzamide (Sigma) for 5 minutes to visualize nuclei. Nikon 80i upright microscope (Nikon) equipped with CoolSnapES camera (photometrics) was used to capture the images and Nikon Elements (Nikon)and Adobe Photoshop (Adobe) softwares were used for image processing.
Immunoblotting
SDS-PAGE (10%) was performed on urine specimens (15μl each) of 4 patients with diabetic nephropathy and 3 individuals with no known kidney disease (controls) using standard procedures, The proteins were transferred to PVDF membrane using Bio-Rad semi-dry apparatus. After washing twice with TBS buffer, the PVDF membrane was blocked using 2% non-fat milk and 2% BSA in TBST buffer for 1 hour at room temperature, and then incubated with rabbit anti-endothelial lipase (1:100) and mouse anti-albumin (1:2000, Invitrogen Cat#03-0700) antibodies for 16 hours at 4° C. After washing with TBST, the membranes were incubated for 1 hour at 25° C with IRDye800CW-conjugated goat anti-rabbit IgG (1:2000, red) and IRDye680-conjugated goat anti-mouse IgG (1:2000, green) secondary antibodies (LI-COR Biosciences). After washing, the respective antigens were visualized on an Odyssey Infrared Imaging System (LI-COR Biosciences) with both 700- and 800-nm channels. For confirming specificity of EL antibody different amounts of purified human Albumin (Sigma) ranging from 0.1μg to 50μg were blotted on two separate nitro-cellulose membranes and immunoblotting and detection was performed as described above for albumin and EL.
Statistical Analysis
In real-time PCR experiments a two-way analysis of variance (ANOVA) was used to compare control with experimental groups. Results are shown as the mean ± SD. P< 0.05 was considered statistically significant.
Results
To identify podocyte-specific genes potentially regulated by HG exposure we treated podocytes in culture for one or two weeks with HG and compared their expression profiles with controls grown in NG for two weeks using Affymetrix Mouse Genome 430 2.0 microarrays. A filtered set of 1,790 genes was examined for changes in gene expression. Compared to NG there were 19 transcripts that were significantly downregulated at 1 week of HG exposure (Tables 1 and 2, see methods). These include: brain expressed gene 1 (Bex1), Thyroid hormone receptor interactor 11 (Trip11) and lipocalin 2 (Lcn2). There were 19 transcripts that were up–regulated at 1 week including interleukin 1 receptor-like 1 (Il1rl1), endothelial lipase (Lipg or EL) and UDP galactosyltransferase 8A (Ugt8). Twenty five genes were downregulated in week 2, including Lcn2, matrix metalloproteinase 2 (Mmp2) and Cyclin 1 (Ccni). Thirty two genes were upregulated at 2 weeks in HG treated podocytes including Ugt8 and endothelial lipase (for detailed list and fold changes see Table 1).
Table 1.
Genes downregulated and upregulated in podocytes treated with high glucose for one week compared to podocytes cultured in normal glucose conditions. A cut off of 3 fold changes was used to filter the data (see methods).
| Probe set | Downregulated gene | Accession | fold change |
|---|---|---|---|
| 1448595_a_at | Bex1: brain expressed gene 1 | NM_009052 | −6.61 |
| 1458729_at | Mm.182696.1 | AW552255 | −5.32 |
| 1443153_at | Trip11: Thyroid hormone receptor interactor 11 | BB306866 | −4.21 |
| 1458269_at | Pcdh9: protocadherin 9 | AW048370 | −3.97 |
| 1427747_a_at | Lcn2: lipocalin 2 | X14607 | −3.82 |
| 1425339_at | Plcb4: phospholipase C, beta 4 | BB224034 | −3.6 |
| 1442704_at | Mm.214935.1 | BM250739 | −3.58 |
| 1450154_at | Folh1: folate hydrolase | NM_016770 | −3.54 |
| 1445426_at | Mm.42287.1 | BB457090 | −3.36 |
| 1444229_at | Nr2f2: nuclear receptor subfamily 2, group F, member 2 | BB053811 | −3.34 |
| 1440488_at | Mm.209825.1 | BB416028 | −3.28 |
| 1425338_at | Plcb4: phospholipase C, beta 4 | BB224034 | −3.27 |
| 1444250_at | Mm.133185.1 | AI451553 | −3.23 |
| 1456659_at | LOC552902: hypothetical LOC552902 | BM116906 | −3.2 |
| 1424375_s_at | Gimap4: GTPase, IMAP family member 4 | BC005577 | −3.19 |
| 1439224_at | Mm.133637.1 | BB373816 | −3.17 |
| 1443145_at | Apbb1ip: amyloid beta (A4) precursor protein-binding, family B, member 1 interacting protein | BB153348 | −3.12 |
| 1454589_at | 9430006E15Rik: RIKEN cDNA 9430006E15 gene | AK020405 | −3.08 |
| 1459750_s_at | Gpr123: G protein-coupled receptor 123 | AU015577 | −3.01 |
| Probe set | Upregulated gene | Accession | fold change |
| 1455930_at | Mm.28870.2 | BI651113 | 8.2 |
| 1422317_a_at | Il1rl1: interleukin 1 receptor-like 1 | NM_010743 | 5.59 |
| 1425843_at | Mrpl33: mitochondrial ribosomal protein L33 | BC027018 | 4.88 |
| 1430786_at | 1110002E22Rik: RIKEN cDNA 1110002E22 gene | BE991102 | 4.85 |
| 1449751_at | Slc6a6: Solute carrier family 6 (neurotransmitter transporter, taurine), member 6 | AA589629 | 4.37 |
| 1435330_at | Pyhin1: pyrin and HIN domain family, member 1 | BM241008 | 4.28 |
| 1437937_at | Ccbp2: chemokine binding protein 2 | AV220666 | 4.13 |
| 1442844_at | A830052D11Rik: RIKEN cDNA A830052D11 gene | BB271008 | 3.86 |
| 1431315_at | Hyls1: hydrolethalus syndrome 1 | BM570636 | 3.85 |
| 1447870_x_at | 1110002E22Rik: RIKEN cDNA 1110002E22 gene | BB099116 | 3.81 |
| 1418676_at | Isl2: insulin related protein 2 (islet 2) | NM_027397 | 3.79 |
| 1444199_at | Mm.45087.1 | AW046689 | 3.69 |
| 1425145_at | Il1rl1: interleukin 1 receptor-like 1 | D13695 | 3.58 |
| 1422691_at | Sptlc1: serine palmitoyltransferase, long chain base subunit 1 | AF003823 | 3.36 |
| 1421262_at | Lipg: lipase, endothelial | BC020991 | 3.22 |
| 1419063_at | Ugt8a: UDP galactosyltransferase 8A | NM_011674 | 3.14 |
| 1449356_at | Asb5: ankyrin repeat and SOCs box-containing 5 | NM_029569 | 3.06 |
| 1449473_s_at | Cd40: CD40 antigen | NM_011611 | 3.04 |
| 1439043_at | Tra2a: Transformer 2 alpha homolog (Drosophila) | BE982794 | 3.01 |
Table 2.
Genes downregulated and upregulated in podocytes treated with high glucose for two week compared to podocytes cultured in normal glucose conditions. A cut off of 3 fold changes was used to filter the data (see methods).
| probe set | Downregulated gene | Accession | fold change |
|---|---|---|---|
| 1427747_a_at | Lcn2: lipocalin 2 | X14607 | −7.03 |
| 1459713_s_at | Ano1: anoctamin 1, calcium activated chloride channel | AU040576 | −5.46 |
| 1434188_at | Slc16a12: solute carrier family 16 (monocarboxylic acid transporters), member 12 | AV220703 | −5.01 |
| 1419728_at | Cxcl5: chemokine (C-X-C motif) ligand 5 | NM_009141 | −4.49 |
| 1456078_x_at | Tubb2c /// Tubb2c-ps2: tubulin, beta 2C /// tubulin, beta 2c, pseudogene 2 | BB012080 | −4.32 |
| 1452014_a_at | Igf1: insulin-like growth factor 1 | AF440694 | −4.18 |
| 1423611_at | Alpl: alkaline phosphatase, liver/bone/kidney | AW319615 | −4 |
| 1435603_at | Sned1: sushi, nidogen and EGF-like domains 1 | BB487754 | −3.91 |
| 1458536_at | Ccni: Cyclin I | BB097972 | −3.69 |
| 1439364_a_at | Mmp2: matrix metallopeptidase 2 | BF147716 | −3.67 |
| 1448595_a_at | Bex1: brain expressed gene 1 | NM_009052 | −3.6 |
| 1454296_at | 4631402F24Rik: RIKEN cDNA 4631402F24 gene | AA739023 | −3.6 |
| 1450014_at | Cldn1: claudin 1 | NM_016674 | −3.5 |
| 1453550_a_at | Far1: fatty acyl CoA reductase 1 | AK011187 | −3.47 |
| 1449909_at | 2010005H15Rik: RIKEN cDNA 2010005H15 gene | NM_029733 | −3.44 |
| 1429951_at | Ssbp2: single-stranded DNA binding protein 2 | AK005150 | −3.36 |
| 1430097_at | 8430436C05Rik: RIKEN cDNA 8430436C05 gene | AU016566 | −3.35 |
| 1437405_a_at | Igfbp4: insulin-like growth factor binding protein 4 | BB787243 | −3.25 |
| 1459649_at | Mm.150125.1 | AI662750 | −3.23 |
| 1416441_at | Pgcp: plasma glutamate carboxypeptidase | BB468025 | −3.16 |
| 1440107_at | Mm.131403.1 | BB077622 | −3.15 |
| 1421239_at | Il6st: interleukin 6 signal transducer | AA717838 | −3.1 |
| 1429696_at | Gpr123: G protein-coupled receptor 123 | BE946247 | −3.1 |
| 1417625_s_at | Cxcr7: chemokine (C-X-C motif) receptor 7 | BC015254 | −3.09 |
| 1442254_at | Mm.207501.1 | BB366659 | −3.04 |
| probe set | Upregulated gene | Accession | fold change |
| 1455930_at | Mm.28870.2 | BI651113 | 13.95 |
| 1419063_at | Ugt8a: UDP galactosyltransferase 8A | NM_011674 | 5.26 |
| 1444199_at | Mm.45087.1 | AW046689 | 4.89 |
| 1431315_at | Hyls1: hydrolethalus syndrome 1 | BM570636 | 4.59 |
| 1449751_at | Slc6a6: Solute carrier family 6 (neurotransmitter transporter, taurine), member 6 | AA589629 | 4.57 |
| 1422944_a_at | Diap3: diaphanous homolog 3 (Drosophila) | NM_019670 | 4.46 |
| 1430786_at | 1110002E22Rik: RIKEN cDNA 1110002E22 gene | BE991102 | 4.39 |
| 1426278_at | Ifi27l2a: interferon, alpha-inducible protein 27 like 2A | AY090098 | 4.37 |
| 1427184_at | Tcrb-J: T-cell receptor beta, joining region | BF318536 | 3.97 |
| 1421262_at | Lipg: lipase, endothelial | BC020991 | 3.95 |
| 1422155_at | Hist2h3c2: histone cluster 2, H3c2 | BC015270 | 3.79 |
| 1455730_at | Dlgap5: discs, large (Drosophila) homolog-associated protein 5 | BM250919 | 3.67 |
| 1441757_at | 1190002F15Rik: RIKEN cDNA 1190002F15 gene | AI120476 | 3.65 |
| 1430419_at | 2310031A07Rik: RIKEN cDNA 2310031A07 gene | AK009549 | 3.64 |
| 1421350_a_at | Grip1: glutamate receptor interacting protein 1 | NM_130891 | 3.58 |
| 1439040_at | Cenpe: centromere protein E | BG068387 | 3.57 |
| 1447870_x_at | 1110002E22Rik: RIKEN cDNA 1110002E22 gene | BB099116 | 3.57 |
| 1440862_at | Mm.153468.1 | BB629079 | 3.44 |
| 1434847_at | Cnnm4: cyclin M4 | BB432741 | 3.31 |
| 1421754_at | AY036118: cDNA sequence AY036118 | NM_133243 | 3.3 |
| 1452458_s_at | Ppil5: peptidylprolyl isomerase (cyclophilin) like 5 | BC022648 | 3.28 |
| 1417587_at | Timeless: timeless homolog (Drosophila) | BM230269 | 3.26 |
| 1449171_at | Ttk: Ttk protein kinase | NM_009445 | 3.26 |
| 1439510_at | Sgol1: shugoshin-like 1 (S. pombe) | BB410537 | 3.25 |
| 1417019_a_at | Cdc6: cell division cycle 6 homolog (S. cerevisiae) | NM_011799 | 3.23 |
| 1417938_at | Rad51ap1: RAD51 associated protein 1 | BC003738 | 3.2 |
| 1452912_at | Dscc1: defective in sister chromatid cohesion 1 homolog (S. cerevisiae) | AK011162 | 3.17 |
| 1427004_at | Fbxo2: F-box protein 2 | BB311718 | 3.09 |
| 1440146_at | Vps13a: vacuolar protein sorting 13A (yeast) | BB829606 | 3.09 |
| 1420707_a_at | Traip: TRAF-interacting protein | AK012948 | 3.06 |
| 1421881_a_at | Elavl2: ELAV (embryonic lethal, abnormal vision, Drosophila)-like 2 (Hu antigen B) | BB105998 | 3.06 |
| 1418480_at | Ppbp: pro-platelet basic protein | NM_023785 | 3 |
We next focused on identifying genes that were represented by probesets showing consistent changes in response to HG at both 1 week (arbitrary representation of sub-acute HG stress) and 2 weeks (arbitrary representation of sustained HG stress) in our model. We were interested in this for two reasons. First, these could represent potential biomarkers that remain altered in diabetic glomerular disease. Second, these could provide insights into possible mechanisms underlying HG-mediated podocyte dysfunction. Therefore, we generated a gene list representing intersection of differentially expressed genes at both 1 and 2 weeks of HG compared to NG exposure (Table 3). Of the ten genes that were differentially expressed in both the 1 week and 2 week time points compared to the normal glucose control, only three genes showed consistent changes in probesets across replicates when the raw signal intensity data were examined, and that had defined annotation information. These include neutrophil gelatinase-associated lipocalin (Lcn2 also known as Ngal1) whose expression was decreased in response to high glucose, and endothelial lipase (EL) and UDP-glucuronosyltransferase 8 (Ugt8) whose expression levels increased in response to high glucose (Figure 1).
Table 3.
Genes with altered expression both in 1W and 2W array datasets, compared to controls. Columns on the right represent the raw signal intensity data for the probesets for the indicated samples. The genes highlighted in bold were prioritized for validation based on consistency of replicate data across the arrays and available annotation.
| probe set | gene | Accession | fold change | NG | NG | 1W | 1W | 2W | 2W |
|---|---|---|---|---|---|---|---|---|---|
| 1419063_at | Ugt8a: UDP galactosyltransferase 8A | NM_011674 | 5.26 | 353.8 | 163.9 | 881.9 | 715.9 | 1327 | 1251.9 |
| 1421262_at | Lipg: lipase, endothelial | BC020991 | 3.95 | 584.9 | 748.7 | 2338.1 | 1872.4 | 2285.2 | 2882.8 |
| 1427747_a_at | Lcn2: lipocalin 2 | X14607 | −7.03 | 5572.4 | 5442.6 | 1335.5 | 1546.3 | 958.8 | 563.5 |
| 1430786_at | 1110002E22Rik: RIKEN cDNA 1110002E22 gene |
BE991102 | 4.39 | 531.2 | 467.5 | 2795.8 | 2011.6 | 1458.4 | 2842.1 |
| 1431315_at | Hyls1: hydrolethalus syndrome 1 |
BM570636 | 4.59 | 212.3 | 85.9 | 691.4 | 386.7 | 642.2 | 581.3 |
| 1444199_at | Mm.45087.1 | AW046689 | 4.89 | 634.9 | 251 | 641.3 | 2593.8 | 555.9 | 3695.5 |
| 1447870_x_at | 1110002E22Rik: RIKEN cDNA 1110002E22 gene |
BB099116 | 3.57 | 1318.4 | 1917 | 6276.8 | 5591.4 | 3453 | 8163 |
| 1448595_a_at | Bex1: brain expressed gene 1 | NM_009052 | −3.6 | 8614.8 | 18678.4 | 3023.9 | 1035 | 6905.6 | 569.2 |
| 1449751_at | Slc6a6: Solute carrier family 6 (neurotransmitter transporter, taurine), member 6 | AA589629 | 4.57 | 679.1 | 271.1 | 548 | 3369.7 | 820.2 | 3314.8 |
| 1455930_at | Mm.28870.2 | BI651113 | 13.95 | 3317.7 | 2655.5 | 4788.1 | 49494.4 | 5569.2 | 89776 |
Figure 1.

Common genes (a) downregulated and (b) upregulated in podocytes cultured in high glucose for 1 and 2 weeks compared to normal glucose controls. Among the differentially expressed genes, only three were consistently altered at both time points. Refer to Table 1 for a detailed list of genes at each time point.
To further understand the temporal dynamics of these alterations, and independently validate microarray results, we isolated RNA from podocytes exposed to HG at different time intervals (6h, 12h, 18h, 24h, 72h, 1 weeks and 2weeks) and performed RT-PCR for Lcn2, EL and Ugt8 (Figure 2). Consistent with our microarray data we found that all three genes were modulated by HG. Importantly, Lcn2 levels started to decrease as early as 6 h after HG exposure and were significantly down regulated by HG at 18 hours. By 72h Lcn2 was barely detectable and remained low for the remaining time points of this experiment and up to 1 and 2 weeks (Figure 2a). EL was upregulated as early as 6 hours by HG and increased by 3 and 10 fold at 1 week and 2 weeks, respectively, compared to control (Figure 2b). Ugt8 was upregulated by HG as early as 12 hours and remained elevated at 2 weeks of HG exposure (Figure 2c). These results validate the microarray findings and demonstrate that perturbation in expression of these genes after high glucose exposure was a relatively early event (less than 18 h after HG exposure) and the expression remained downregulated (for Lcn2) or upregulated (for EL and Ugt8) at longer times of HG exposure.
Figure 2.

Effect of high glucose on lipocalin (a), endothelial lipase (b) and UDP-glucuronosyl transferase- 8 (c) mRNA expression in podocytes. Cells were incubated in normal glucose RPMI 16 with/without 25 mM D-glucose for 6, 12, 18, 24, 48, 72 h, 1 week and 2 weeks. Cells were collected and assayed for target genes mRNA levels by real time-PCR . The relative mRNA expression of the target genes were normalized to β-actin control. Each point represents the mean SD of three independent experiments performed in triplicate. * P 0.05; ** P 0.01; *** P 0.001; NS, not significant.
We next determined if the HG responsive genes found from experiments in vitro were also altered in patients with diabetic nephropathy. To obtain a clearer idea at a cell specific level we resorted to immunohistochemistry in kidneys from diabetic or non-diabetic patients (8 diabetic and 7 non-diabetic controls). Among the three differentially expressed genes, we focused on EL2 as Ugt8 antibodies were not available and Lcn2 was barely detectable in podocytes of non-diabetic patients (low abundance expression) thus rendering it unsuitable for detecting any further downregulation in situ in diabetic glomeruli. Consistent with our cell-culture results, we observed markedly higher EL2 expression in podocytes of almost all diabetic nephropathy patients (7 out of 8) compared to those from non-diabetics (normal) (Figure 3). Immunostaining with WT1 and EL antibodies using immunoperoxidase or immunofluorescence methods confirm high expression of EL in podocytes of diabetic glomeruli. Parietal cells of diabetic glomeruli also exhibit high EL immunopositivity compared to controls and non diabetic kidneys, but the significance of this is unclear.
Figure 3.
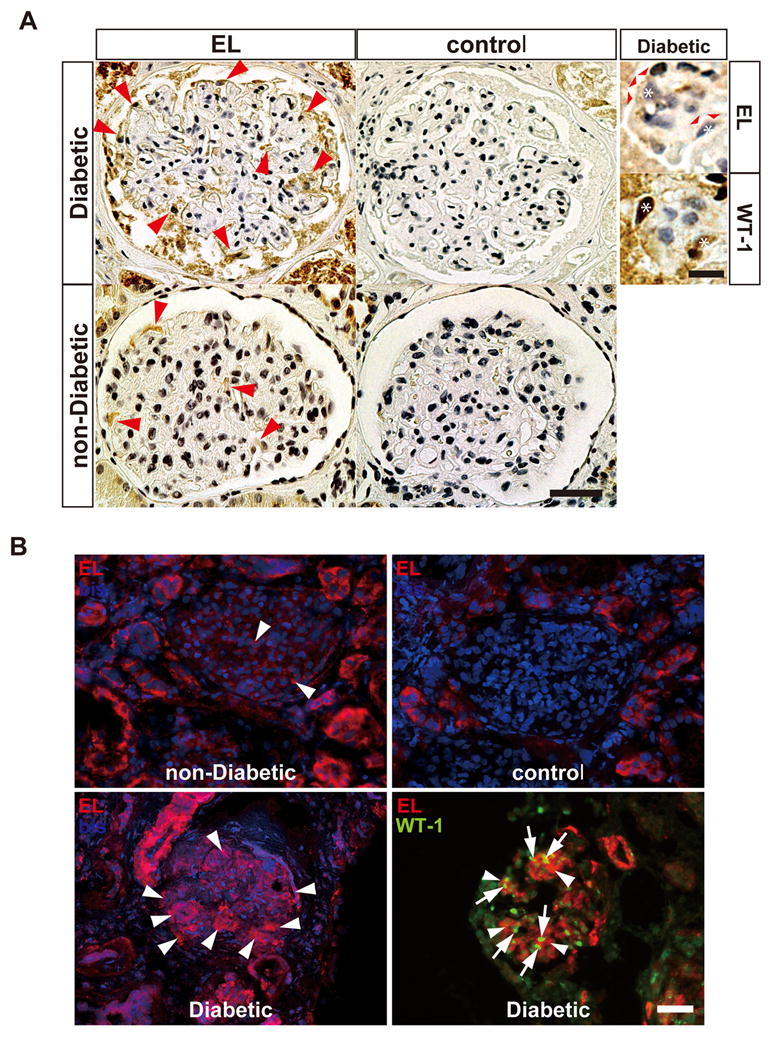
High endothethelial lipase expression in kidney sections from diabetic patients compared to non-diabetic kidneys. Endothelial Lipase (EL) immunostaining was performed on formalin-fixed paraffin embedded (A) or cryopreserved biopsies (B) from patients with diabetic nephropathy (8) or non-diabetics (7) (see methods). The primary antibody was omitted in the control panel. All diabetic patients except one had increased EL immunopositivity in podocytes. Representative images from 3 diabetic and 2 non-diabetic patients are shown. (A) Immunoperoxidase staining (brown) with anti-EL antibodies show increased EL expression (red arrows) in podocytes and parietal cells of diabetic compared to non-diabetic glomeruli. (scale bar = 50 μm). The high power images on the right (scale bar = 10 μm) are adjacent sections from a diabetic kidney stained with anti-EL or anti-WT-1 (as a podocyte marker) that show colocalization of EL (red arrowheads) and WT-1 (dark brown nuclear staining) in same cells (asterisks) supporting that EL and WT-1 are expressed in the podocytes. (B) EL immunofluorescence (red) on non-diabetic and diabetic kidney tissues also shows increased EL expression in diabetic glomeruli (lower panel), compared to non-diabetic glomeruli (upper panel). Control shows no glomerular staining. WT1 (green nuclear, arrowheads) immunostaining confirms that EL-expressing cells (arrows, cytoplasm) are podocytes. (scale bar = 50 μm). The tubulointerstitial staining is non-specific as it is present in controls in both A and B.
We further examined if these findings can have potential utility in DN diagnosis and performed EL immunoblotting in urine samples from diabetic nephropathy and non-diabetic control patients EL was readily detected in urine of a subset of DN patients (2/4), while none of the control patients (3/3) show EL expression (Fig. 4).
Figure 4.

Endothelial Lipase is readily detected in urine of diabetic nephropathy patients. (a) Immunoblot analysis for Endothelial Lipase (57KDa) (EL) and Albumin (67KDa) in urine samples of diabetic (lanes A, H, I and K) and non-diabetic controls (lanes E, F, and J). High levels of EL (green) are detected in urine samples of two diabetic patients (H and I). Controls show no EL excretion. Albumin (red) was detected in all samples. The lower panel shows distinct migration of EL and Albumin. (b) EL antibody does not cross react with albumin. Dot blot pattern of EL and Albumin immunostaining to different amounts of purified human albumin protein shows no immunostaining with EL antibody further confirming that EL detection is specific.
Discussion
We have employed microarray analysis to delineate potential molecular pathways underlying podocyte injury in diabetic glomerulopathy using an in vitro model and confirmed a subset of the observed in vitro changes in independent time-course experiments and in specimens from humans with diabetic nephropathy. Out of a total of 95 transcripts that were differentially expressed upon HG exposure of 1 or 2 weeks, we found that three remained altered at both 1 and 2 weeks (Lcn2, Ugt8 and EL) suggesting that these are promising candidates as biomarkers or potential contributors to the pathogenesis of diabetic glomerular disease. Time course RT-PCR experiments revealed that changes in mRNAs of these genes occurs shortly after exposure to high glucose and remain altered that may have implications in early and late detection and pathogenesis.
We discovered that Lcn2 expression exhibited a time-dependent decrease in podocytes beginning as early as 6 h after HG treatment. LCN2 is a member of the lipocalinsuperfamily that forms a complex with iron-binding siderophores. This complex plays a role during nephrogenesis by promoting the conversion of renal progenitorsinto tubules (31). Further, it is also highly upregulated in tubular cells after acute kidney injury (32). These results support the idea that upregulation of Lcn2 is important for survival or regeneration after injury from stress. This idea is further supported by the observation that recombinant Lcn2 reduces proximal tubular injury in an ischemia reperfusion injury model by inhibiting apoptosis (33). The decreased expression of Lcn2 observed in our study was unexpected considering its upregulation in other injury models, however, our independent validation using quantitative RTPCR at several time points supported the observed Lcn2 downregulation upon HG exposure . This observation suggests that failure of podocytes to sustain normal or high levels of Lcn2 after HG exposure may be a potential mechanism of podocyte injury and apoptosis induced by HG. LCN2 also binds to matrix metalloproteinase-9(MMP-9) and protects this extracellular matrix remodeling enzymefrom autodegradation (34). Whether reduced expression of LCN2 contributes to glomerular basement membrane (GBM) thickening and extracellular matrix accumulation (hallmark lesions of diabetic glomerulosclerosis) by increasing autodegradation of MMP-9 or other metalloproteinases remains to be seen.
Our discovery of upregulation of Endothelial lipase (EL) in vitro and in human samples have important clinical implications. EL, the most recently discovered member of the lipase gene family is an important negative modulator of high density lipoprotein (HDL). EL is also involved in inflammatory state by promoting monocyte adhesion to the vascular endothelium (35, 36). Recent data indicate that HDL inhibits apoptosis, lipid oxidation, cytokine and adhesion molecule production (37). Since in vitro and in vivo studies have linked HDL to both diabetes mellitus and inflammation, it is possible that increased EL in our study of podocyte HG exposure may be a mechanism of HDL-mediated or cytokine-mediated effects in diabetic glomerulopathy. Further, HDL is thought of as protective against formation of extrarenal vascular calcifications, a common and serious complication in patients with chronic kidney disease (38). Therefore, it is possible that elevated EL in diabetic glomerulopathy mediates low HDL-associated increased vascular calcifications. Importantly, we demonstrate for the first time that podocytes may be a significant source of EL, a protein normally secreted by vascular endothelial and smooth cells or macrophages (cell types typically not present in the glomerulus), after HG exposure. Because our studies show that EL can be readily detected in human urine samples in a subset of diabetic patients also supports its potential use in diabetic nephropathy diagnosis. Further large scale studies are warranted to determine the diagnostic utility of aberrant EL expression in diabetic and non-diabetic nephropathy at different stages of the disease.
While Lcn2 and EL can be potentially linked to HG mediated changes related to diabetic nephropathy, currently no such information is available for Ugt8. Ugt8 consists of a super family of enzymes that catalyze glucuronidation (39). In particular Ugt8 family catalyzes the transfer of galactose to ceramide, a key enzymatic step in the biosynthesis of galactocerebrosides. Galactocerebrosides are abundant sphingolipids of the myelin membrane of the central and peripheral nervous system (40), and Ugt8 is also present in the kidney during metanephric development (41). Ugt8 deficiency results in a spectrum of neurological symptoms characterized by tremors, ataxia, progressive hindlimb paralysis and vacuole formation in ventral spinal cord (42). Increased ceramide levels have also been shown to increase apoptosis in a number of systems (43). On the other hand, Ugt8 upregulation is associated with increased metastatic potential to lung by specifically enhancing the ability to metastasize to, colonize and survive within the lung (44). It should be noted that podocytes have been touted as neuronal counterparts in the kidney and the increased Ugt8 levels may be protective compensatory response to prevent podocyte degeneration by enhancing their survival, perhaps similar to its role in the nervous system by regulating lipid metabolism. Future studies would be needed to explore a potential relationship of Ugt8 to diabetic glomerulopathy or podocyte function.
In conclusion, our study identified podocyte specific genes that are regulated by acute and sustained HG exposure. While some of these have been previously associated with diabetic glomerulopathy thus validating our strategy, we found several novel ones. We confirmed a subset of these that may cause deranged lipid metabolism and impaired injury response in podocytes and contribute to diabetic nephropathy. Further, we show data for potential application of these in diagnosis of diabetic nephropathy in human specimens. Thus these studies provide new molecular insights into the role of podocytes in diabetic renal disease and support the notion that diabetes with regards to the glomerulus is another podocytopathy (45).
Acknowledgments
We thank Amanda Knoten, Angela Lluka and Adee Heiman for excellent technical assistance and Mary Hoffman, Jessica Vanwinkle and Daniel Martin for human specimen processing and acquisition. We thank Dr. George Jarad for human purified albumin and albumin antibodies. We are grateful to Mark Watson and Laboratory of Translational Pathology at Washington University School of Medicine for providing core services for microarray and laser capture microscopy studies, and to Dr. Erwin P. Bottinger for providing laboratory resources to culture podocytes for RT-PCR experiments.
This work was partly supported by Core services provided by an NIH George M. O’Brien Center for Kidney Disease Research grant (P30-DK079333) to Washington University, NIH grant DK081644 (to S. Jain) and interdepartmental research funds to H. Liapis. Part of this work was presented in an abstract form at the annual United States and Canadian Academy of Pathology (USCAP) 2006 and International Podocyte Conference (2006).
Abbreviations
- EL
endothelial lipase
- ESRD
end-stage renal disease
- HDL
high density lipoprotein
- HG
high glucose
- NG
normal glucose
Footnotes
Disclosures : None
References
- 1.Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–1634. doi: 10.2337/diabetes.54.6.1626. [DOI] [PubMed] [Google Scholar]
- 2.Reddy GR, Kotlyarevska K, Ransom RF, Menon RK. The podocyte and diabetes mellitus: is the podocyte the key to the origins of diabetic nephropathy? Curr Opin Nephrol Hypertens. 2008;17:32–36. doi: 10.1097/MNH.0b013e3282f2904d. [DOI] [PubMed] [Google Scholar]
- 3.Marshall MS. The podocyte: a major player in the development of diabetic nephropathy? Horm Metab Res. 2005;37(suppl 1):9–16. doi: 10.1055/s-2005-861397. [DOI] [PubMed] [Google Scholar]
- 4.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 5.Stitt-Cavanagh E, MacLeod L, Kennedy C. The podocyte in diabetic kidney disease. ScientificWorldJournal. 2009;9:1127–39. doi: 10.1100/tsw.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziyadeh FN. Different roles for TGF-beta and VEGF in the pathogenesis of the cardinal features of diabetic nephropathy. Diabetes Res Clin Pract. 2008;82 (Suppl 1):S38–41. doi: 10.1016/j.diabres.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 7.Schiffer M, Mundel P, Shaw AS, Bottinger EP. A novel role for the adaptor molecule CD2-associated protein in transforming growth factor-beta-induced apoptosis. J Biol Chem. 2004;279:37004–37012. doi: 10.1074/jbc.M403534200. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol. 2008;172(2):299–308. doi: 10.2353/ajpath.2008.070057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marrero B, Amy K. Banes-Berce. Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am J Physiol Renal Physiol. 2006;290:F762–F768. doi: 10.1152/ajprenal.00181.2005. [DOI] [PubMed] [Google Scholar]
- 10.Noh H, King GL. The role of protein kinase C activation in diabetic nephropathy. Kidney Int Suppl. 2007 Aug;(106):S49–53. doi: 10.1038/sj.ki.5002386. [DOI] [PubMed] [Google Scholar]
- 11.Forbes JM, Fukami K, Cooper ME. Diabetic nephropathy: where hemodynamics meets metabolism. Exp Clin Endocrinol Diabetes. 2007 Feb;115(2):69–84. doi: 10.1055/s-2007-949721. [DOI] [PubMed] [Google Scholar]
- 12.Schleicher ED, Weigert C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int Suppl. 2000 Sep;77:S13–8. doi: 10.1046/j.1523-1755.2000.07703.x. [DOI] [PubMed] [Google Scholar]
- 13.Daniels MC, McClain DA, Crook ED. Transcriptional regulation of transforming growth factor beta1 by glucose: investigation into the role of the hexosamine biosynthesis pathway. Am J Med Sci. 2000 Mar;319(3):138–42. doi: 10.1097/00000441-200003000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Goldberg HJ, Whiteside CI, Fantus IG. The hexosamine pathway regulates the plasminogen activator inhibitor-1 gene promoter and Sp1 transcriptional activation through protein kinase C 1 and. J Biol Chem. 2002;277:33833–33841. doi: 10.1074/jbc.M112331200. [DOI] [PubMed] [Google Scholar]
- 15.Müller-Deile J, Worthmann K, Saleem M, Tossidou I, Haller H, Schiffer M. The balance of autocrine VEGF-A and VEGF-C determines podocyte survival. Am J Physiol Renal Physiol. 2009;297(6):F1656–67. doi: 10.1152/ajprenal.00275.2009. [DOI] [PubMed] [Google Scholar]
- 16.Satchell SC, Harper SJ, Tooke JE, Kerjaschki D, Saleem MA, Mathieson PW. Human podocytes express angiopoietin 1, a potential regulator of glomerular vascular endothelial growth factor. J Am Soc Nephrol. 2002;13(2):544–50. doi: 10.1681/ASN.V132544. [DOI] [PubMed] [Google Scholar]
- 17.Li JM, Shah AM. ROS generation by nonphagocytic NADPH oxidase: potential relevance in diabetic nephropathy. J Am Soc Nephrol. 2003;14 (8 Suppl 3):S221–6. doi: 10.1097/01.asn.0000077406.67663.e7. [DOI] [PubMed] [Google Scholar]
- 18.Nyengaard JR, Ido Y, Kilo C, Williamson JR. Interactions between hyperglycemia and hypoxia: implications for diabetic retinopathy. Diabetes. 2004;53(11):2931–8. doi: 10.2337/diabetes.53.11.2931. [DOI] [PubMed] [Google Scholar]
- 19.De Petris L, Hruska KA, Chiechio S, Liapis H. Bone morphogenetic protein-7 delays podocyte injury due to high glucose. Nephrol Dial Transplant. 2007 Dec;22(12):3442–3450. doi: 10.1093/ndt/gfm503. [DOI] [PubMed] [Google Scholar]
- 20.Fan Q, Shike T, Shigehara T, Tanimoto M, Gohda T, Makita Y, Wang LN, Horikoshi S, Tomino Y. Gene expression profiles in diabetic KK/Ta mice. Kidney Int. 2003;64:1978–1985. doi: 10.1046/j.1523-1755.2003.00312.x. [DOI] [PubMed] [Google Scholar]
- 21.Wilson KH, Eckenrode SE, Li QZ, Ruan QG, Yang P, Shi JD, Davoodi-Semiromi A, McIndoe RA, Croker BP, She JX. Microarray analysis of gene expression in the kidneys of new- and post-onset diabetic NOD mice. Diabetes. 2003;52:2151–2159. doi: 10.2337/diabetes.52.8.2151. [DOI] [PubMed] [Google Scholar]
- 22.Susztak K, Bottinger E, Novetsky A, Liang D, Zhu Yanqing Ciccone E, Wu D, Dunn S, McCue P, Sharma K. Molecular profiling of diabetic mouse kidney reveals novel genes linked to glomerular disease. Diabetes. 2004;53:784–794. doi: 10.2337/diabetes.53.3.784. [DOI] [PubMed] [Google Scholar]
- 23.Baelde HJ, Eikmans M, Doran PP, Lappin DW, de Heer E, Bruijn JA. Gene expression profiling in glomeruli from human kidneys with diabetic nephropathy. Am J Kidney Dis. 2004;43:636–650. doi: 10.1053/j.ajkd.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 24.Morrison J, Knoll Kristen, Martin J, Liang Hessnerand Mingyu. Effect of high glucose on gene expression in mesangial cells: upregulation of the thiol pathway is an adaptational response. Physiol Genomics. 2004;17:271–282. doi: 10.1152/physiolgenomics.00031.2004. [DOI] [PubMed] [Google Scholar]
- 25.Han SH, Yang S, Jung DS, Li JJ, Kim JJ, Kwak SJ, Kim DK, Moon SJ, Lee JE, Han DS, Kang SW. Gene expression patterns in glucose-stimulated podocytes. Biochem Biophys Res Commun. 2008 Jun 6;370(3):514–8. doi: 10.1016/j.bbrc.2008.03.121. [DOI] [PubMed] [Google Scholar]
- 26.Mundel P, Reiser J, Zúñiga Mejía Borja A, et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 27.Jain S, Suarez AA, McGuire J, Liapis H. Expression profiles of Renal Dysplasia Reveal New Insights in renal Development and Disease. Pediatric nephrology. 2007;22(7):962–974. doi: 10.1007/s00467-007-0466-6. [DOI] [PubMed] [Google Scholar]
- 28.Jains A, Dubes R. Algorithms for clustering data. Prentice-Hall; Englewood Cliffs NJ: 1988. [Google Scholar]
- 29.Li, Wong, Li Cheng, Wong Wing Hung. Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. PNAS. 2001 Jan 2;98(1):31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−Δ Δ Ct method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Challen GA, Martinez G, Davis MJ, Taylor DF, Crowe M, Teasdale RD, Grimmond SM, Little MH. Identifying the molecular phenotype of renal progenitor cells. J Am Soc Nephrol. 2004;5:2344–2357. doi: 10.1097/01.ASN.0000136779.17837.8F. [DOI] [PubMed] [Google Scholar]
- 32.Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, Barasch J, Devarajan P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14:2534–2543. doi: 10.1097/01.asn.0000088027.54400.c6. [DOI] [PubMed] [Google Scholar]
- 33.Yan L, Borregaard N, Kjeldsen L, Moses MA. The high molecular weight urinary matrix metalloproteinase activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL) J Biol Chem. 2005 Oct;276 (40):37258–37265. doi: 10.1074/jbc.M106089200. [DOI] [PubMed] [Google Scholar]
- 34.Paradis ME, Badellino KO. Endothelial lipase is associated with inflammation in humans. Journal of Lipid Research. 2006 Dec;47:2808–2813. doi: 10.1194/jlr.P600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Broedl UC, Maugeais C. Effects of Nonlipolytic Ligand Function of Endothelial Lipase on High Density Lipoprotein Metabolism in Vivo. J Biol Chem. 2003 Oct;278 (42):40688–40693. doi: 10.1074/jbc.M304367200. [DOI] [PubMed] [Google Scholar]
- 36.Ruge T, Neuger L. Lipoprotein lipase in the kidney: activity varies widely among animal species. Am J Physiol Renal Physiol. 2004;287:F1131–F1139. doi: 10.1152/ajprenal.00089.2004. [DOI] [PubMed] [Google Scholar]
- 37.Rohrer L, Hersberger M, Von Eckardstein A. High density lipoproteins in the intersection of diabetes mellitus, inflammation and cardiovascular disease. Current Opinion in Lipidology. 2004;15 (3):269–278. doi: 10.1097/00041433-200406000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Torres PA. Origin of the mediacalcosis in kidney failure. J Mal Vasc. 2009 May;34(3):204–10. doi: 10.1016/j.jmv.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 39.Yoshikazu E, Koichi U, Ohnishi A, Ikushiro S, Iyanagi T. Transcriptional Enhancement of UDP-Glucuronosyltransferase Form 1A2 (UGT1A2) by Nuclear Factor I-A (NFI-A) in Rat Hepatocytes. Journal of Biochemistry. 2005;138(3):313–325. doi: 10.1093/jb/mvi128. [DOI] [PubMed] [Google Scholar]
- 40.Yurkova I, Kisel M. Free-radical fragmentation of galactocerebrosides: a MALDI-TOF mass spectrometry study. Chemistry and Physics of Lipids. 2005 Mar;134(1):41–49. doi: 10.1016/j.chemphyslip.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 41.Challen G, Gardiner B, Caruana G, Kostoulias X, Martinez G, Crowe M, Taylor DF, Bertram J, Little M, Grimmond SM. Temporal and spatial transcriptional programs in murine kidney development. Physiol Genomics. 2005;17;23(2):159–7. doi: 10.1152/physiolgenomics.00043.2005. [DOI] [PubMed] [Google Scholar]
- 42.Guttmacher AE, Collins FS. Genomic medicine: a primer. N Engl J Med. 2002;347:1512–1520. doi: 10.1056/NEJMra012240. [DOI] [PubMed] [Google Scholar]
- 43.WU BX, Clarke CJ, Hannun YA. Mammalian Neutral Sphingomyelinases: Regulation and Roles in Cell Signaling Responses. Neuromolecular Med. 2010 Jun 16; doi: 10.1007/s12017-010-8120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Landemaine T, Jackson A, Bellahcène A, Rucci N, Sin S, Abad BM, Sierra A, Boudinet A, Guinebretière JM, Ricevuto E, Noguès C, Briffod M, Bièche I, Cherel P, Garcia T, Castronovo V, Teti A, Lidereau R, Driouch K. A six-gene signature predicting breast cancer lung metastasis. Cancer Res. 2008;68(15):6092–9. doi: 10.1158/0008-5472.CAN-08-0436. [DOI] [PubMed] [Google Scholar]
- 45.Barisoni L, Schnaper HW, Kopp JB. Advances in the biology and genetics of the podocytopathies: implications for diagnosis and therapy. Arch Pathol Lab Med. 2009;133:201–216. doi: 10.1043/1543-2165-133.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]