

Abstract
Inverse docking is a relatively new technique that has been used to identify potential receptor targets of small molecules. Our docking software package MDock is well suited for such an application as it is both computationally efficient, yet simultaneously shows adequate results in binding affinity predictions and enrichment tests. As a validation study, we present the first stage results of an inverse-docking study which seeks to identify potential direct targets of PRIMA-1. PRIMA-1 is well known for its ability to restore mutant p53's tumor suppressor function, leading to apoptosis in several types of cancer cells. For this reason, we believe that potential direct targets of PRIMA-1 identified in silico should be experimentally screened for their ability to inhibit cancer cell growth. The highest-ranked human protein of our PRIMA-1 docking results is oxidosqualene cyclase (OSC), which is part of the cholesterol synthetic pathway. The results of two followup experiments which treat OSC as a possible anti-cancer target are promising. We show that both PRIMA-1 and Ro 48-8071, a known potent OSC inhibitor, significantly reduce the viability of BT-474 breast cancer cells relative to normal mammary cells. In addition, like PRIMA-1, we find that Ro 48-8071 results in increased binding of mutant p53 to DNA in BT-474 cells (which highly express p53). For the first time, Ro 48-8071 is shown as a potent agent in killing human breast cancer cells. The potential of OSC as a new target for developing anticancer therapies is worth further investigation.
Keywords: Inverse Docking, In Silico Screening, Protein-Ligand Interactions, Molecular Docking
Introduction
Inverse docking, first proposed in 2001 by Chen et al. [1] refers to computationally docking a specific small molecule of interest to a library of receptor structures. The technique may be used to identify new potential biological targets of known compounds [2–4], or to identify targets for compounds among a family of related receptors [5]. The technique has shown success in distinguishing between homology models of receptors [5]. The technique may also be used to generate a compound's predicted pharmacological profile [6], or to generate a virtual selectivity profile that characterizes the promiscuity of the inhibitors [7]. Given the multi-faceted nature of a pharmacologically active compound's biological effects, inverse docking is especially helpful, because it may generate new hypotheses for the action mechanism.
Our docking software package, MDock, can be used for inverse docking, as demonstrated in the present work (zoulab.dalton.missouri.edu/software.htm). MDock uses a novel scoring function, ITScore, which was generated using an iterative method of deriving pair interaction potentials that avoids the problem of defining a specific reference state [8]. For the first time, the full energy landscape (both native and non-native modes) is considered in the potential derivation using a physics-based global iterative function. ITScore's binding pose and affinity predictions have been extensively evaluated using diverse test sets prepared by other labs [8,9]. ITScore was also assessed using enrichment tests for virtual database screening against four target proteins [9]. In the present study, we test the ability of MDock on in silico inverse screening applications.
Specifically, we aim at searching for potential protein targets of PRIMA-1. Found from high-throughput screening, PRIMA-1 (p53 reactivation and induction of massive apoptosis, shown in Fig. 1), is a small molecule capable of activating mutant p53 protein, restoring its ability to bind to DNA as well as the tumor suppressor function associated with wild-type p53 [10,11]. This effect has been demonstrated in vitro and in vivo, and has been shown to trigger massive apoptosis in several types of human breast cancer cells [12,13]. PRIMA-1 is also known to stimulate expression of p21 and other p53-dependent promoters in mutant p53 breast cancer cell lines. p53's importance as a potential agent against cancer is well-established. Nevertheless, while specific mechanisms have been proposed for PRIMA-1's mutant p53 reactivation effect [10,14,15], none have gained wide acceptance and the question remains unsettled. For this reason, we consider PRIMA-1 well suited as the subject of an inverse docking study.
Figure 1.
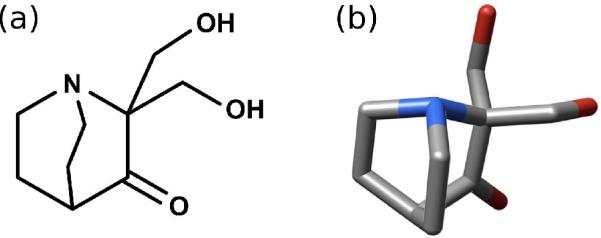
(a) Chemical structure, generated using MarvinSketch 4.1.0 (www.chemaxon.com), and (b) 3D structure of PRIMA-1. Hydrogen atoms are omitted from the 3D structure for clarity.
In this work, we used the inverse-docking approach to screen for potential molecular targets of PRIMA-1. The objective is to guide future assays of the inhibitors of these predicted targets for their efficacy in inhibiting tumor cell proliferation, as such results may lead to potential cancer treatments, as well as provide clues regarding PRIMA-1's action mechanism. We used MDock to perform this study. In support of our approach, here we present the first stage results of our assays of Ro 48-8071, a known potent inhibitor of oxidosqualene cyclase (OSC) [16,17], the highest-ranked human protein of our in silico study. We show that Ro 48-8071 is a novel potent agent in selectively reducing the viability of BT-474 cells, a mutant-p53 human breast cancer cell line. In addition, we found that Ro 48-8071 increases p53-DNA binding in BT-474 cells, an effect which is also characteristic of PRIMA-1 [11]. BT-474 cells are known to overexpress p53, even in the absence of cytotoxic stress [18].
Methods
In Silico Screening
We used our protein-ligand docking software package MDock [8,9] (zoulab.dalton.missouri.edu/software.htm) to dock PRIMA-1 into many potential drug targets. Although MDock is sufficiently computationally efficient for PDB-wide database screening, we chose to start with the well-characterized Potential Drug Target Database (PDTD), which at the time of use contained about 1100 experimentally-determined structures of 830 actual or suspected drug targets (http://www.dddc.ac.cn/pdtd) [19]. We also used the PDTD's binding site definitions, which in most cases are based on the set of amino acid residues that are within 6.5 Å of the bound ligand. OMEGA Version 2.2.1 was used to generate conformations of PRIMA-1 for flexible-ligand docking (OpenEye Scientific Software Inc., Santa Fe, NM) with the rms parameter set to 0.1 Å, maxconfs to 1000000, maxconfgen to10000000, and ewindow to 10. As PRIMA-1 has few rotatable bonds, this only resulted in 42 generated ligand conformations. Each of these conformations was docked to each protein as a rigid body.
Our docking procedure is described in detail in previous publications [8,9,20–22] and in the tutorial of MDock. Briefly, for each protein in the database, a molecular surface of the binding site was generated, along with the associated sphere points representing potential initial positions for ligand atom centers [23,24]. Ligand atoms were matched to these sphere points and orientations were sampled and ranked by our knowledge-based scoring function, ITScore [8,9]. All of MDock's default parameters were used in this work, with the exception of write_score_total, which was set to 1 so that only the highest-scoring orientation is recorded when each protein/PRIMA-1-conformation pair is docked as a rigid body. We then ranked each protein according to the lowest ITScore (corresponding to the highest predicted affinity) recorded for any of the 42 PRIMA-1 conformations that were docked to it. Because PDTD contains redundant experimental structures of the same protein [19], we clustered the resulting docked structures into groups sharing !90% sequence identity. We then ran a BLAST search [25] in order to map the PDTD proteins, which come from various species, to human gene sequences. Inhibitors of the top human or human analogue proteins were considered candidate anti-cancer agents for assay. A flowchart of our procedure is shown as Fig. 2.
Figure 2.

A flowchart illustrating the inverse docking and assay approach used in this work.
Cell Viability assay
We used the Sulforhodamine B (SRB) assay [26–29] to evaluate the effect of the OSC-inhibitor Ro 48-8071 on the viability of breast cancer cells. This cell protein dye-binding assay determines the protein content in surviving cells as an index to determine cell growth, viability, and survival [26,27]. Briefly, BT-474, T47D, and AG11132A cells were seeded into 96-well plates and incubated overnight at 37°C with 5% CO2. The culture medium was removed after 24 h and cells were washed with DMEM/F12 medium and then treated with various concentrations of Ro 48-8071 or PRIMA-1 in 5% FBS DMEM/F12 medium for 24 hours. Surviving or adherent cells were fixed in situ by withdrawing the growth medium, adding 100 ! l PBS and 100 ! l 50% trichloroacetic acid and then incubating at 4°C for one hour. Cells were washed with ice-cold water, dried at room temperature (RT), and then stained with 50 ! l 4% SRB for eight minutes at RT. Unbound dye was removed by washing five times with cold 1% acetic acid and plates were dried at RT. Bound stain was solubilized with 150 ! l of 10 ! M Tris buffer, and the absorbance of samples was read at 520nm with a SpecTRA MAX 190 microplate reader (Molecular Devices, Sunnyvale, CA). Six wells were used for each concentration and each experiment was performed twice. BT-474 and T47D breast cancer lines were obtained from ATCC (Manassas, VA), and the AG11132A normal mammary cell line was purchased from Coriell Institute for Medical Research (Camden, NJ). BT-474 and T47D cells were grown in phenol red-free DME/F12 medium (Invitrogen Corporation; Carlsbad, CA) and supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO). AG11132A cells were grown in serum free MEBM (Mammary Epithelium Basal Medium) medium (Lonza, Walkersville, MD) with supplementary 2mM L-glutamine. PRIMA-1 was purchased from Tocris Bioscience (Ellisville, MO). Ro 48-8071, sulforhodamine B, and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). The purity of Ro 48-8071 was ! 98% as determined by HPLC (Sigma-Aldrich data). The purity of PRIMA-1 was 99.8% as determined by HPLC (Tocris data sheet).
p53 Activation Assay
In preparation for the assay, BT-474 cells were grown in DMEM/F12 medium supplemented with 5% FBS overnight. Cells were washed with PBS once and treated with 50 μM PRIMA-1 or 25 μM Ro 48-8071 for 1 hour at 37°C. p53 activation was assessed using the TransAM p53 Transcription Factor Assay kit (Active Motif, Carlsbad, CA) according to the manufacturer's protocol. A summary of the procedure follows. The kit provides 96-well plates coated with an oligonucleotide that contains the p53 consensus DNA binding site. 2.5 ! g of nuclear extracts (prepared according to a nuclear extract kit provided from Active Motif) were incubated with this oligonucleotide. Bound p53 was detected by adding the anti-p53 antibody (1:1000) followed by addition of the secondary antibody (1:1000) that is conjugated to horseradish peroxidase. Absorbance was read at 450nm in a Spectra MAX 190 Microplate Reader (Molecular Device, Sunnyville, CA). MCF-7 nuclear extract treated with H2O2, provided with the TransAM kit, was used as a positive control.
Statistical Analysis
Differences among groups were tested using one-way analysis of variance (ANOVA) with repeated measures over time. Values are reported as mean ± SE. When ANOVA indicated a significant effect (F-ratio, p < 0.05), the Student-Newman-Keuls multi-range test was used to compare the means of the individual groups. The statistics were conducted using the SigmaStat software (version 3.5).
Results and Discussion
In Silico Screening
After docking PRIMA-1 to each structure of the Potential Drug Target Database (PDTD), we ranked the proteins according to their predicted affinity, based on our knowledge-based scoring function, ITScore. We searched for human proteins that are analogous to the best-scoring (i.e., lowest-scoring or tightest-binding) proteins in our docking results, using a cutoff of 30% sequence identity. Among these ten best-scoring proteins, one of them is a human protein, the X-ray crystallographic structure of human OSC (PDB entry: 1W6K) [30,31]. In Fig. 3, OSC (green) is shown docked with PRIMA-1 (magenta) along with the potent OSC-inhibitor, Ro 48-8071 (yellow) [17,32]. The binding pose indicated by docking PRIMA-1 into OSC partially overlaps the binding pose of Ro 48-8071 shown in the crystal structure. We also found that docking Ro 48-8071 to this pocket reproduces the native binding orientation shown in the crystal structure (RMSD = 0.25 Å). The score for PRIMA-1 calculated with ITScore was −45.5 and the score for Ro 48-8071 in its crystallographic position was −102.8. Approximately, this difference in score corresponds to a 6 kcal/mol difference in predicted binding affinity between the two compounds.
Figure 3.

(a) Ribbon depiction of oxidosqualene cyclase (OSC), identified as a possible target of PRIMA-1, generated using Chimera 1.4.0. PRIMA-1 (magenta) is shown in its docked position along with the partially overlapping position of the OSC inhibitor Ro 48-8071 from the crystal structure (yellow). Hydrogen atoms are omitted for clarity. (b) Chemical structure of Ro 48-8071.
To further compare the similarities and differences between the interactions involved in PRIMA-1 binding and Ro 48-8071 binding, we attempted to decompose the total energy scores into different energy components. Unfortunately this cannot be done with ITScore because the potential function in ITScore derived for each atom pair combines different energetic contributions into a single distance-dependent function. We therefore used the force field scoring function [33] provided in DOCK 6.0 (UCSF, http://dock.compbio.ucsf.edu/) [34] to analyze the natures of the interactions involved in binding of PRIMA-1 and Ro 48-8071 to OSC, by calculating the contributions of different energy terms to the total binding scores.
Specifically, the force field scoring function in UCSF DOCK 6.0 is composed of two energy terms, a van der Waals (VDW) term using Lennard-Jones 6–12 potentials and a Coulombic electrostatic energy term using a distance-dependent function for the dielectric constant of water. Table 1 lists the binding energy scores of PRIMA-1 and Ro 48-8071 and the corresponding contributions of different energy components. It can be seen from the table that Ro 48-8071 (−61.3) has a lower/better binding score than PRIMA-1 (−38.7), which is consistent with the aforementioned results calculated with ITScore. Table 1 also suggests that the VDW interactions contribute to the binding energies significantly more than the electrostatic interactions for both PRIMA-1 and Ro 48-8071, though the contribution of the VDW interaction term is more dominant for Ro 48-8071 than PRIMA-1. The strong VDW interactions for Ro 48-8071 arise from its highly hydrophobic fragments such as aromatic rings and aliphatic chains.
Table 1.
The binding energy scores and individual energy components of PRIMA-1 and Ro 48-8071 for OSC, calculated with UCSF DOCK 6.0.
| PRIMA-1 | Ro 48-8071 | |
|---|---|---|
| Binding score | −38.7 | −61.3 |
| VDW score | −24.3 | −52.2 |
| Eletrostatics score | −14.3 | −9.2 |
The details are explained in the first section of “Results and Discussion”.
Since PRIMA-1 inhibits cell growth in mutant-p53 tumor cell lines, we decided to determine whether the potent OSC-inhibitor Ro 48-8071 would have a similar anti-cancer effect.
Cell Viability assay
The SRB assay showed that Ro 48-8071 dramatically destroys BT-474 human breast cancer cells, exhibiting a dose-response relationship similar to that of PRIMA-1. IC50 was approximately 10 ! M for both compounds. The OSC-inhibitor also suppressed the growth of a second human breast cancer cell line, T47D. The data for both cell lines are shown in Fig. 4 (a) and (b). Using the same assay, we determined whether Ro 48-8071 would affect normal mammary cells. Our data showed that Ro 48-8071 exhibits significantly less inhibition of normal mammary cells from line AG11132A (Fig. 4(c)), indicating an effect that is specific to tumor cells. From our Western blot analysis, the expression of OSC was confirmed for both cancer cell lines (BT-474 and T47D), and normal mammary cells showed significantly less expression of OSC.
Figure 4.

The effect of Ro 48-8071 on breast cancer and normal mammary cell viability. BT-474 (1.0×104 / well), T47-D (0.6×104 / well), and AG11132A cells (0.7×104 / well) were seeded into a 96-well plate overnight, and cells were washed and treated with the indicated concentration of Ro 48-8071 or PRIMA-1 for 24 hours. Cell growth and viability were determined by the SRB assay described in Methods. The OSC-inhibitor Ro 48-8071 and PRIMA-1 significantly inhibit the viability of BT-474 (a) and T47-D (b) cells in a dose-dependent manner, and there is significantly less inhibition of normal mammary AG11132A cell viability shown in (c).
p53 Activation Assay
Finally, it is well known that PRIMA-1 increases the DNA-binding affinity of mutant p53, which is highly expressed in several different types of cancer cells, including BT-474 and T47D cells [11]. Since Ro 48-8071 and PRIMA-1 exhibited similar inhibition of breast cancer cells (as shown above), we examined the capacity of Ro 47-8071 to restore the DNA-binding of mutant p53 in BT-474 cells, by using a TransAM p53 Transcription Factor Assay kit (Active Motif, Carlsbad, CA). In a time-course study we found that treatment of BT-474 cells with either 25 ! M Ro 48-8071 or 50 ! M PRIMA-1 for 0.5 to 3.0 hours led to the activation of mutant p53 activity (data not shown). Fig. 5 compares the extent of mutant p53 activation in BT-474 cells following 1-hour exposure to Ro 48-8071 (25 ! M) and PRIMA-1 (50 ! M). Treatment with either Ro 48-8071 or PRIMA-1 increased the binding of mutant p53 to DNA. MCF-7 (wild-type p53) nuclear extract treated with H2O2 was provided in the TransAM kit, and used as a positive control.
Figure 5.
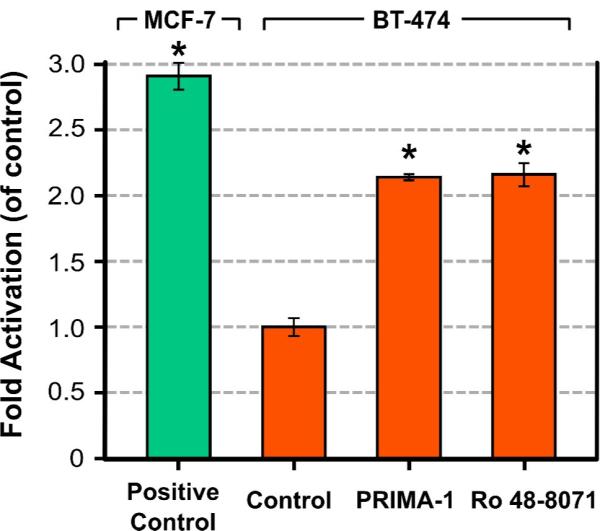
Both PRIMA-1 and Ro 48-8071 increase p53-DNA binding in BT-474 breast cancer cells. BT-474 cells were grown in DMEM/F12 medium supplemented with 5% FBS overnight. Cells were then washed with PBS once and treated with 50 μM PRIMA-1 or 25 μM Ro 48-8071 for 1 hour. Cells were harvested by scraping and nuclear extracts were prepared. 2.5 μg of nuclear extract were used for each TransAM assay and each sample was analyzed in triplicate. The fold of activation was compared to the control group (i.e., without PRIMA-1 or Ro 48-8071 treatment). MCF-7 nuclear extract treated with H2O2 provided by the TransAM kit was used as a positive control. Data are shown as the Mean ± SEM from three different determinations. Asterisk represents values differing significantly from the untreated BT-474 control (P < 0.05).
Conclusions
In this paper, we presented an application of inverse docking using our software package MDock. Our in silico screening identified OSC as one possible target of PRIMA-1. This led us to investigate whether the potent OSC-inhibitor Ro 48-8071 would selectively reduce the viability of human breast cancer cells. It does, and in addition leads to increased binding of mutant p53 to DNA. These effects of Ro 48-8071 are similar to the corresponding characteristic effects of PRIMA-1. In conjunction with our computational mechanistic study, these results lead us to suspect that these two ligands are exerting their anti-cancer effects in part due to inhibition of OSC, but it remains to be shown experimentally whether or not PRIMA-1, like Ro 48-8071, binds directly to OSC. Given the potent inhibition of breast cancer cells induced by Ro 48-8071, we consider it and other OSC inhibitors worth investigating as possible therapeutic agents against breast cancer. The present study is an onset of a series of future experimental and theoretical studies exploring OSC as a new potential target for developing anticancer therapies. Other future studies include conducting the direct binding assay of OSC for PRIMA-1 and testing the inhibitors of other proteins in the top list of our inverse docking study for their ability to inhibit cancer cell growth.
Acknowledgements
Support to XZ from OpenEye Scientific Software Inc. (Santa Fe, NM) is gratefully acknowledged. This work is supported by NIH grants R21GM088517 (XZ) and R56CA86916 (SMH), NSF CAREER Award DBI-0953839 (XZ), the COR award from College of Veterinary Medicine (SMH), and University of Missouri Research Board Grant RB-07-32 and Research Council Grant URC 09-004 (XZ). SZG is supported through the NLM Biomedical Informatics Research Training Program (T15 LM07089, PI: Caldwell). Most of the computations in this work were performed on the HPC resources at the University of Missouri Bioinformatics Consortium. Additional financial support was provided by Dell, SGI, Sun Microsystems, TimeLogic, and Intel. Fig. 3a was generated using UCSF Chimera [35].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Chen YZ, Zhi DG. Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins. 2001;43:217–226. doi: 10.1002/1097-0134(20010501)43:2<217::aid-prot1032>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- [2].Do QT, Renimel I, Andre P, Lugnier C, Muller CD, Bernard P. Reverse pharmacognosy: application of selnergy, a new tool for lead discovery. The example of epsilon-viniferin. Curr. Drug Discov. Technol. 2005;2:161–167. doi: 10.2174/1570163054866873. [DOI] [PubMed] [Google Scholar]
- [3].Muller P, Lena G, Boilard E, Bezzine S, Lambeau G, Guichard G, Rognan D. In silico-guided target identification of a scaffold-focused library: 1,3,5-triazepan-2,6-diones as novel phospholipase A2 inhibitors. J. Med. Chem. 2006;49:6768–6778. doi: 10.1021/jm0606589. [DOI] [PubMed] [Google Scholar]
- [4].Zahler S, Tietze S, Totzke F, Kubbutat M, Meijer L, Vollmar AM, Apostolakis J. Inverse In Silico Screening for Identification of Kinase Inhibitor Targets. Chem. Biol. 2007;14:1207–1214. doi: 10.1016/j.chembiol.2007.10.010. [DOI] [PubMed] [Google Scholar]
- [5].Schapira M, Abagyan R, Totrov M. Nuclear hormone receptor targeted virtual screening. J. Med. Chem. 2003;46:3045–3059. doi: 10.1021/jm0300173. [DOI] [PubMed] [Google Scholar]
- [6].Rollinger JM. Accessing target information by virtual parallel screening—the impact on natural product research. Phytochem. Lett. 2009;2:53–58. [Google Scholar]
- [7].Bissantz C, Logean A, Rognan D. High-throughput modeling of human g-protein coupled receptors: amino acid sequence alignment, three-dimensional model building, and receptor library screening. J. Chem. Inf. Comput. Sci. 2004;44:1162–1176. doi: 10.1021/ci034181a. [DOI] [PubMed] [Google Scholar]
- [8].Huang S-Y, Zou X. An iterative knowledge-based scoring function to predict protein–ligand interactions: I. derivation of interaction potentials. J. Comput. Chem. 2006;27:1865–1875. doi: 10.1002/jcc.20504. [DOI] [PubMed] [Google Scholar]
- [9].Huang S-Y, Zou X. An iterative knowledge-based scoring function to predict protein–ligand interactions: II. validation of the scoring function. J. Comput. Chem. 2006;27:1876–1882. doi: 10.1002/jcc.20505. [DOI] [PubMed] [Google Scholar]
- [10].Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002;8:282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- [11].Liang Y, Wu J, Stancel GM, Hyder SM. p53-dependent inhibition of progestin-induced VEGF expression in human breast cancer cells. J. Steroid Biochem. Mol. Bio. 2005;93:173–182. doi: 10.1016/j.jsbmb.2004.12.011. [DOI] [PubMed] [Google Scholar]
- [12].Liang Y, Besch-Williford C, Benakanakere I, Hyder SM. Re-activation of the p53 pathway inhibits in vivo and in vitro growth of hormone-dependent human breast cancer cells. Int. J. Oncol. 2007;31:777–784. [PubMed] [Google Scholar]
- [13].Liang Y, Besch-Williford C, Brekken RA, Hyder SM. Progestin-dependent progression of human breast tumor xenografts: a novel model for evaluating antitumor therapeutics. Cancer Res. 2007;67:9929–9936. doi: 10.1158/0008-5472.CAN-07-1103. [DOI] [PubMed] [Google Scholar]
- [14].Wang T, Lee K, Rehman A, Daoud SS. PRIMA-1 induces apoptosis by inhibiting JNK signaling but promoting the activation of Bax. Biochem. Biophys. Res. Commun. 2007;352:203–212. doi: 10.1016/j.bbrc.2006.11.006. [DOI] [PubMed] [Google Scholar]
- [15].Lambert JMR, Gorzov P, Veprintsev DB, Söderqvist M, Segerbäck D, Bergman J, Fersht AR, Hainaut P, Wilman KG, Bykov VJN. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376–388. doi: 10.1016/j.ccr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- [16].Morand OH, Aebi J, Guerry P, Hartman PG, Hennes U, Himber J, Ji YH, Jolidon S, Lengsfeld H. Potent inhibitors of mammalian 2,3-oxidosqualene:lanosterol cyclase are orally active cholesterol lowering agents. Atherosclerosis. 1994;109(suppl.):321. [Google Scholar]
- [17].Lenhart A, Reinert DJ, Aebi JD, Dehmlow H, Morand OH, Schulz GE. Binding structures and potencies of oxidosqualene cyclase inhibitors with the homologous squalene! hopene cyclase. J. Med. Chem. 2003;46:2083–2092. doi: 10.1021/jm0211218. [DOI] [PubMed] [Google Scholar]
- [18].Davidoff AM, Kerns BJ, Pence JC, Marks JR, Iglehart JD. p53 alteration in all stages of breast cancer. J. Surg. Oncol. 1991;48:260–267. doi: 10.1002/jso.2930480409. [DOI] [PubMed] [Google Scholar]
- [19].Gao Z, Li H, Zhang H, Liu X, Kang L, Luo X, Zhu W, Chen K, Wang X, Jiang H. PDTD: a web-accessible protein database for drug target identification. BMC Bioinform. 2008;9:104, 1–7. doi: 10.1186/1471-2105-9-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huang S-Y, Zou X. Ensemble docking of multiple protein structures: considering protein structural variations in molecular docking. Proteins. 2007;66:399–421. doi: 10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- [21].Huang S-Y, Zou X. Efficient molecular docking of NMR structures: application to HIV-1 protease. Protein Sci. 2007;16:43–51. doi: 10.1110/ps.062501507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Huang S-Y, Zou X. Inclusion of solvation and entropy in the knowledge-based scoring function for protein-ligand interactions. J. Chem. Inf. Model. 2010;50:262–273. doi: 10.1021/ci9002987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ewing TJ, Makino S, Skillman GA, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aid. Mol. Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- [24].Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- [25].Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- [26].Rubinstein LV, Shoemaker RH, Paull KD. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990;82:1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- [27].Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- [28].Liang Y, Hyder SM. Proliferation of endothelial and tumor epithelial cells by progestin-induced vascular endothelial growth factor from human breast cancer cells: paracrine and autocrine effects. Endocrinology. 2005;146:3632–3641. doi: 10.1210/en.2005-0103. [DOI] [PubMed] [Google Scholar]
- [29].Liang Y, Brekken RA, Hyder SM. Vascular endothelial growth factor induces proliferation of breast cancer cells and inhibits the anti-proliferative activity of anti-hormones. Endocr. Relat. Cancer. 2006;13:905–919. doi: 10.1677/erc.1.01221. [DOI] [PubMed] [Google Scholar]
- [30].Thoma R, Schulz-Gasch T, D'Arcy B, Benz J, Aebi J, Dehmlow H, Hennig M, Stihle M. A. Ruf: Insight into steroid scaffold formation from the structure of human oxidosqualene cyclase. Nature. 2004;432:118–220. doi: 10.1038/nature02993. [DOI] [PubMed] [Google Scholar]
- [31].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Morand OH, Aebi JD, Dehmlow H, Ji YH, Gains N, Lengsfeld H, Himber J. Ro 48-8.071, a new 2,3-oxidosqualene:lanosterol cyclase inhibitor lowering plasma cholesterol in hamsters, squirrel monkeys, and minipigs: comparison to simvastatin. J. Lipid Res. 1997;38:373–390. [PubMed] [Google Scholar]
- [33].Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy approach to macromolecule-ligand interactions. J. Comput. Chem. 1992;13:505–524. [Google Scholar]
- [34].Lang PT, Brozell SR, Mukherjee S, Pettersen ET, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]