

Abstract
Excitotoxicity, resulting from sustained activation of glutamate receptors of the N-methyl-d-aspartate (NMDA) subtype, is considered to play a causative role in the etiology of ischemic stroke and several neurodegenerative diseases. The NMDA receptor is therefore a target for the development of neuroprotective agents. Here, we identify an N-benzylated triamine (denoted as NBTA) as a highly selective and potent NMDA-receptor channel blocker selected by screening a reduced dipeptidomimetic synthetic combinatorial library. NBTA blocks recombinant NMDA receptors expressed in Xenopus laevis oocytes with a mean IC50 of 80 nM; in contrast, it does not block GluR1, a glutamate receptor of the non-NMDA subtype. The blocking activity of NBTA on NMDA receptors exhibits the characteristics of an open-channel blocker: (i) no competition with agonists, (ii) voltage dependence, and (iii) use dependence. Significantly, NBTA protects rodent hippocampal neurons from NMDA receptor, but not kainate receptor-mediated excitotoxic cell death, in agreement with its selective action on the corresponding recombinant receptors. Mutagenesis data indicate that the N site, a key asparagine on the M2 transmembrane segment of the NR1 subunit, is the main determinant of the blocker action. The results highlight the potential of this compound as a neuroprotectant.
The N-methyl-D-aspartate (NMDA) receptor activities are widely recognized to be associated with the induction of various forms of synaptic plasticity in the central nervous system including long-term potentiation and long-term depression, processes that may underlie learning and memory (1). However, prolonged activation of NMDA receptors under pathological conditions such as cerebral ischemia and traumatic injury causes neuronal cell death (2, 3). It has been hypothesized that NMDA receptor-mediated excitotoxicity may contribute to the etiology or progression of numerous neurodegenerative diseases, among them Parkinson's disease and Alzheimer's disease (4, 5). Ever since the demonstration that open-channel blockers of the NMDA receptor showed therapeutic potential in animal ischemic stroke models (6–8), the NMDA receptor has been considered an attractive therapeutic target for the development of neuroprotective agents. Unfortunately, results of human clinical trials using NMDA-receptor antagonists as neuroprotectants have been disappointing; their therapeutic usefulness is limited often by the toxic and psychotropic side effects associated with their pharmacodynamic properties such as slow dissociation from the receptor (9).
The advent of combinatorial chemistry technology is accelerating the process of drug discovery (10). Recently, a set of arginine-rich hexapeptides that potently blocked the NMDA receptor was identified from a peptide synthetic combinatorial library (SCL; ref. 11). However, concerns about the bioavailability of peptide-based leads indicated that a departure from such entities might open new avenues for the identification of novel compounds. Here, we describe the key features of a previously unidentified open-channel blocker of the NMDA receptor selected from a reduced dipeptidomimetic SCL. This compound protects cultured hippocampal neurons from NMDA excitotoxicity, suggesting its therapeutic potential as a neuroprotectant.
Methods
Synthesis of the Library and Individual N-Alkylated Triamines.
The N-alkylated triamine mixtures and individual compounds were synthesized by using the “tea-bag” solid-phase methodology (12) by selective alkylation and exhaustive reduction (13, 14). The mixtures were prepared by using the divide, couple, and recombine method (15). In brief, the first amino acid was coupled by using conventional fluorenylmethoxycarbonyl (Fmoc) chemistry, and after removal of the Fmoc group, the N-terminal amino group was tritylated by reaction with a solution of trityl chloride (5 M excess over the total free N-α-amino groups) for 3 h at 22 ± 2°C (13). Then N-alkylation was performed by treatment of the resin packet with 1 M lithium t-butoxide in tetrahydrofuran under a nitrogen atmosphere and strictly anhydrous conditions. Excess base was removed by cannulation, followed by addition of the individual alkylating agent in dimethyl sulfoxide. The solution was shaken vigorously for 2 h at 22 ± 2°C. After removal of the trityl group with 2% trifluoroacetic acid in dichloromethane (2 × 10 min), the resin was washed and neutralized, and the second amino acid was coupled and alkylated selectively as described. Exhaustive reduction was carried out in 50 ml kimax tubes under nitrogen (14) by addition of boric acid (40×) and trimethylborate (40×) followed by 1 M borane in tetrahydrofuran (40×). The solutions were heated at 65°C for 72 h, decanted, and quenched with methanol. After overnight treatment with piperidine at 65°C, the mixtures or individual compounds were cleaved from the resin with anhydrous hydrogen fluoride, extracted, and lyophilized. The individual compounds were analyzed by reverse-phase HPLC and liquid chromatography/MS (Finnigan-MAT, San Jose, CA) and purified by using a Waters Milliprep 300 preparative HPLC system.
Expression of Recombinant NMDA Receptors and Glutamate Receptors in Xenopus laevis Oocytes.
Recombinant NMDA receptor subunits, NR1 (16) and NR2A (17) were used for screening the library. Procedures for site-directed mutagenesis, preparation of cRNAs, expression of recombinant NMDA receptors and the human brain glutamate receptor GluR1 (18) in Xenopus oocytes, and data analyses were performed as described (11, 19). The blocking activities of the library were assessed at 5 μg/ml (i.e., 10 μM, assuming an average Mr ≈ 500). The assay of the 14 compounds synthesized based on the library screen was performed at 1 μM. Inward currents were elicited from a holding potential of −80 mV, except where otherwise indicated. NMDA receptors were activated by 200 μM L-Glu/20 μM Gly. GluR1 was activated by 200 μM kainate. The voltage-dependence of block was determined from the current-voltage characteristics elicited by a ramp protocol [−80 to 40 mV in 3 sec; PCLAMP 6.0 (Axon Instruments, Foster City, CA)] in the absence or presence of the identified compound. Leak currents were obtained in the absence of agonist and subtracted from the agonist-evoked currents. Concentration-response curves for identified compounds were fitted to a Hill equation:
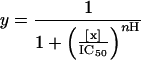 |
1 |
in which y is the ratio of current in the presence of test sample, [x] is the sample concentration, IC50 and nH represent the concentration of sample that blocks 50% of the response and the Hill coefficient, respectively. The relative location of the blocking site for N-benzylated triamine (NBTA) within the channel was inferred from the analysis of the voltage-dependent current block according to Woodhull's model (20). Data were fitted with Eq. 2:
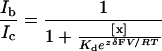 |
2 |
in which Ib and Ic are the currents recorded during the block and in control conditions, [x] is the concentration of the test sample, Kd is the dissociation constant of the compound at 0 mV, δ is the fraction of the transmembrane electric field sensed by the compound as it binds to its site, z is the valence of the test compound, and V, R, T and F have their usual meanings.
Excitotoxicity Assays in Hippocampal Neuron Cultures.
In vitro excitotoxicity assays using primary cultures of rat hippocampal neurons were performed as described (11, 21). The fraction of dead cells in cultures treated with control buffer (5 ± 2%, n = 2,000) was subtracted as background. NMDA insult alone caused significant cell death of 45 ± 6% (n = 2,000). Cell death elicited by NMDA alone was considered as 100%. Neuroprotective activity was expressed as percentage of net cell death in the absence and presence of 10 μM of known NMDA-receptor blockers or the identified compound. The procedures for the kainate-induced excitotoxicity were identical except that kainate replaced NMDA as the insult. Results were assessed by ANOVA and, if significant, group means were compared by post hoc analysis.
Results
Identification of NMDA Receptor Channel Blockers from a Reduced Dipeptidomimetic Positional Scanning (PS) SCL.
To discover novel NMDA-receptor channel blockers, an N-alkylated triamine SCL generated in a dual-defined PS format was screened for its ability to block ligand-activated currents from recombinant NMDA receptors expressed in Xenopus oocytes. This PS-SCL (Fig. 1A) consists of two separate sublibraries, each having two positions defined with a given amino acid (O1 or O3 for sublibrary 1 or 2) or a given alkyl group (O2 or O4 for sublibrary 1 or 2). The remaining two diversity positions were close to equimolar mixtures of amino acids or alkyl groups (X positions). Sublibrary 1 consists of 184 mixtures (all possible combinations of 46 amino acids and 4 alkyl groups at the O positions). Sublibrary 2 consists of 180 mixtures (all possible combinations of 45 amino acids and 4 alkyl groups at the O positions). Each mixture contains 230 individual compounds (all possible combinations of 46 amino acids and 5 alkyl groups at the X positions) for a total of 42,320 individual compounds present in the whole library.
Figure 1.

Structure of the reduced dipeptidomimetic SCL and its blocking activity on recombinant NMDA receptors expressed in amphibian oocytes. (A) Representation of the N-alkylated triamine PS-SCL in which O denotes a defined functionality derived from a single building block and X denotes a mixture of functionalities derived from a mixture of all of the building blocks used to generate the relevant diversity position. (B) Glu- and Gly-activated inward currents through NMDA receptors expressed in oocytes. (C) Block of Glu- and Gly-activated inward currents through NMDA receptors expressed in oocytes after perfusion of a mixture defined with L-isoleucine and benzyl at positions 1 and 2. Holding potential was −80 mV. Glu (200 μM)/Gly (20 μM) and mixture (10 μM) were applied for the duration indicated by the horizontal bars.
Each mixture was assayed for NMDA-receptor channel-blocking activity. Fig. 1B illustrates a typical inward current elicited on perfusion of oocytes expressing NMDA receptors with agonist: evoked currents activated rapidly to steady-state level and decayed on agonist removal. Fig. 1C shows the effect of a representative active mixture: coperfusion of the active Ile(benzyl)XX library mixture at 10 μM with the agonist drastically diminishes the evoked inward current by ≈70%. A limited number of mixtures exhibited blocking activity (Fig. 2); of note are the differences between the two sublibraries. For example, although a small number of mixtures having O4 defined with a methyl were among the most active ones (blocked responses between 70 and 80%), none of the mixtures having a methyl at O2 showed a blocked response >50%. Furthermore, all the mixtures from sublibrary 1 showing responses >60% were defined with a benzyl group at positions O2, whereas other alkyl groups defined the active mixtures from sublibrary 2. These results indicate that not only the nature of the amino acid and the alkyl group are important for activity but also their location within the molecule. Based on the screening results, the most active mixtures from sublibrary 1 and 2 were selected to carry out the deconvolution process. Thus, a series of 14 individual N-alkylated triamines were synthesized, which represented all possible combinations of the functionalities defining the selected mixtures (Fig. 3A).
Figure 2.

Profile of the blocking activity of the PS-SCL on recombinant NMDA receptors expressed in oocytes. Each graph represents a subset of mixtures from each sublibrary separated by the defined alkyl group. The bars represent the blocked response by a single mixture at 10 μM. The horizontal lines define the cutoff blocking activity. Values are given as mean ± SEM of four to six oocytes.
Figure 3.

Blocking activity of a series of 14 compounds individually synthesized based on the blocking profile of the library screening. (A) Functionalities at the four diversity positions (R1–R4) for each compound. (B) Blocking activity of the compounds assayed at 1 μM. Values are expressed as mean ± SEM of four to six oocytes.
Compound 10 was the most potent N-alkylated triamine from this series, exerting 70 ± 3% block of the NMDA-receptor current at 1 μM (Fig. 3B). The synthesis of compound 10 yielded a mixture of the monobenzylated and dibenzylated derivatives. After the separation of these two derivatives by reverse-phase HPLC, the monobenzylated triamine referred to as NBTA was found to exhibit the highest blocking activity. The structure and analytical spectra of NBTA are shown in Fig. 4. NBTA is a highly specific blocker of the NMDA receptor: 1 μM NBTA blocked ≈95% of the NMDA current but had no effect on GluR1, a glutamate α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor of the non-NMDA subtype, even at 1 mM (Fig. 5 B and C). Concentration-inhibition data fitted with a Hill equation yielded a mean IC50 of 80 ± 10 nM and a Hill coefficient close to unity (≈0.8), suggesting that NBTA binds to a single site (Fig. 5C).
Figure 4.

Structure and liquid chromatography/MS of NBTA. The total-ion counting (A), UV absorbance (B), and mass spectra and structure of NBTA (C) are shown.
Figure 5.

NBTA is a specific blocker of the NMDA receptor. Blocking activity of NBTA (1 μM) on NMDA receptors (A) or GluR1 channels (B). (C) Concentration-inhibition curves for NMDA receptors and GluR1 channels by NBTA (▪), memantine (▴), MK-801 (▾), or phencyclidine (●). (D) Blocking activity of NBTA on wild-type NMDA receptors [NR1/NR2A (■)] − IC50 = 80 ± 10 nM (n = 6), and nH = 0.8 and indicated mutants: N616R/NR2A (⧫) − IC50 = 22,000 ± 1,320 nM (n = 5), nH = 0.8; N616Q/NR2A (○) − IC50 = 240 ± 20 nM (n = 4), nH = 0.8; NR1/W606L (▾) − IC50 = 380 ± 32 nM (n = 5), nH = 0.8; NR1/N614G (▵) − IC50 = 95 ± 11 nM (n = 5), nH = 0.9; NR1/N615G (□) − IC50 = 72 ± 8 nM (n = 5), nH = 0.9; NR1/S616G (▴) − IC50 = 98 ± 9 nM (n = 5), nH = 0.8; and N616Q/N614G (●) − IC50 = 244 ± 23 nM (n = 5), nH = 0.8. Currents were elicited by 200 μM Glu/20 μM Gly at a holding potential of −80 mV.
NBTA Is a Highly Selective and Potent NMDA Receptor Antagonist.
The remarkable selectivity of NBTA for NMDA receptors and not for glutamate receptors of the non-NMDA subtype is underscored by a large body of data summarized in Fig. 5C. Phencyclidine, dizolcipine (MK-801), and memantine (1-amino-3,5-dimethyladamantane) are open-channel blockers of the NMDA receptor acting with high affinity (22–25). By contrast, non-NMDA receptors are weakly sensitive to these drugs. The potency profile of these NMDA-receptor channel blockers for recombinant NMDA receptors and AMPA receptors expressed in oocytes (Fig. 5C) shows that these three blockers have ≈1,000–10,000 higher affinity for NMDA receptors than for AMPA receptors, marginally blocking AMPA receptors in the mM concentration range. In marked contrast, NBTA sharply discriminates between these two types of receptors, displaying high blocking potency for NMDA receptors and no activity on AMPA receptors, even in the mM concentration range. We are not aware of any other open-channel blocker that would compare favorably with NBTA in terms of potency and exquisite selectivity for NMDA receptors.
NBTA Acts as an Open-Channel Blocker of the NMDA Receptor.
NMDA receptor-mediated inward currents may be attenuated by a number of mechanisms depending on the site of action of the ligand at the receptor protein. The compound may act as a competitive antagonist at the glycine or glutamate binding sites or as an allosteric modulator at the proton and polyamines sites (26). The concentration-response curves of Glu (with 20 μM Gly) obtained in the absence or presence of 0.2 μM NBTA were similar with mean 50% EC50 values of 2.08 ± 0.2 μM (n = 4) and 1.91 ± 0.3 μM (n = 4), respectively (Fig. 6A). The EC50 of Gly (with 200 μM Glu) determined in the absence and presence of 0.2 μM NBTA were 2.22 ± 0.21 μM (n = 4) and 1.95 ± 0.18 μM (n = 4), respectively (Fig. 6B). NBTA does not appear, therefore, to be a competitive antagonist of the NMDA receptor.
Figure 6.

NBTA is a reversible open-channel blocker of the NMDA receptor. (A) Concentration-response curve of Glu (with 20 μM Gly) evoked currents from NMDA receptors in the absence (■) or presence (▴) of 0.2 μM NBTA (n = 4 oocytes). (B) Concentration-response curve for Gly (with 200 μM Glu) in the absence (■) or presence (▴) of 0.2 μM NBTA (n = 4 oocytes). (C) Voltage dependence of NMDA-receptor block by NBTA. Current-voltage characteristics of responses evoked by 200 μM Glu/20 μM Gly in the absence or presence of 1 μM NBTA. The voltage-ramp protocols were performed when agonist-evoked inward currents reached a steady state. (D) Use dependence of NMDA-receptor block by NBTA. Sequentially perfused oocytes by using agonist/coagonist pulses (200 μM Glu/20 μM Gly) without and with NBTA (0.1 μM) followed by pulses of agonist/coagonist alone. Pulse duration is indicated by the horizontal bar. Evoked currents were measured at −80 mV.
We sought to investigate whether NBTA is an open-channel blocker of the NMDA receptor. A key feature of open-channel block is voltage dependence, namely at a fixed blocker concentration the fraction of current block increases with more negative membrane holding potentials. A voltage-dependent reduction of NMDA current was obtained in the presence of NBTA, which was significantly more pronounced at negative than at positive membrane potentials (Fig. 6C). A δ of 0.56 calculated from the fit of the data according to Woodhull's model suggests that the compound traverses ≈56% of the transmembrane electric field to reach its binding site in the pore. The observed current reduction presumably reflects a physical occlusion of the pore by NBTA.
A conspicuous feature of open-channel block is use dependence. Thus, at a fixed blocker concentration, the fraction of current block increases with repetitive activation of the receptors. In the presence of 0.1 μM NBTA, the extent of block increased progressively with successive pulses of Glu/Gly (Fig. 6D). Full recovery of the agonist-evoked responses was retrieved after the washout of the compound (Fig. 6D). These results demonstrate that NBTA acts as an open-channel blocker of recombinant NMDA receptors.
Site of Action of NBTA on Recombinant NMDA Receptors.
A critical asparagine, the N site of the NR1 subunit, is a structural determinant of ion permeation and channel block (26, 27). The N site is considered to be located at the narrowest constriction of the pore and to specify the action of a wide array of open-channel blockers including memantine, phencyclidine, MK-801 (23, 25), and benzylpolyamine derivatives (28). We therefore examined this site as a candidate for NBTA binding. Substitution of the N site to arginine (N616R) significantly reduces the blocking activity of NBTA by 275 fold (Fig. 5D). In contrast, replacement of N616 for glutamine, which contains a similar functional group as that of asparagine, only slightly reduces it to a mean IC50 of 0.24 μM (Fig. 5D). The results indicate that NBTA binds to the N site of NR1. A tryptophan in the M2 segment of the NR2C subunit was shown by others to specify the block of the NMDA receptor by extracellular Mg2+ (28, 29) and proposed to contribute to the narrow constriction of the channel (28, 29). A tryptophan-to-leucine mutation (W606L) at the corresponding position of the NR2A subunit reduces the blocking activity of NBTA by ≈4.7 fold (Fig. 5D), suggesting that tryptophan 606 also contributes to the binding pocket for NBTA. Three consecutive residues from the M2 segment of NR2A (N614, N615, and S616) were shown to contribute to the narrow constriction of the channel (27). Surprisingly, point mutations at any one of these three residues (N614G, N615G, and S616G) do not alter the IC50 of NBTA significantly (Fig. 5D), indicating that NBTA does not bind at these sites. Furthermore, coexpression of N616Q (NR1 mutant) with N614G (NR2A mutant) yielded an IC50 similar to that of N616Q alone. The result is in line with the notion that NBTA does not bind to asparagine 614 of the NR2A subunit (Fig. 5D).
Together, the picture that emerges is that the N site of NR1, which is exposed to the channel lumen accessed from the extracellular entryway (30–33), is the major determinant of NBTA block, and that tryptophan 606 of NR2A contributes to form the binding pocket. The fact that mutations of both consecutive asparagines at the N site and the N+1 site of NR2A do not alter the sensitivity to NBTA block establishes a major difference between block by extracellular Mg2+, which is controlled by the NR2A N− and N+1 sites, and block by NBTA, which is specified by the N site of NR1. It is not clear why mutations of residues at position 614 and 615 of NR2A, which were shown to contribute to the pore constriction, do not affect the blocking activity of NBTA. One possibility is that the side chains of these two NR2A asparagines do not lie on the same level as the NR1 N site (27).
NBTA Protects Hippocampal Neurons from NMDA Receptor-Mediated Excitotoxic Death.
To determine whether NBTA has neuroprotective activity on NMDA receptor-induced neuronal death, an in vitro model of excitotoxicity was used. Cultured hippocampal neurons exposed to 200 μM NMDA and 20 μM Gly for 20 min underwent extensive cell death as indicated by trypan blue staining (Fig. 7B: ref. 11). At 10 μM, NBTA protects neurons from NMDA receptor-mediated death (83 ± 2%; Fig. 7D) to an extent comparable to that exerted by the conventional NMDA-receptor channel-blockers memantine (93 ± 1%) and MK-801 (89 ± 1%; Fig. 7 C and E).
Figure 7.

NBTA protects cultured hippocampal neurons against NMDA receptor-mediated excitotoxic death. Trypan-blue exclusion assays of control neurons (A) and neurons exposed to 200 μM NMDA in the absence (B) or presence of 10 μM memantine (C) or 10 μM NBTA (D). (Scale bars = 3 μm.) (E) Neuronal death induced by NMDA in the absence and presence of 10 μM MK-801, 10 μM memantine, or 10 μM NBTA. (F) Neuronal death induced by kainate in the absence or presence of 20 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 10 μM MK-801, 10 μM memantine, or 10 μM NBTA. Data are mean ± SEM, n = 2,000. ***, P < 0.01 for the difference when compared with the controls (NMDA- or kainate-treated groups).
To evaluate the specificity of the neuroprotective action of NBTA, its activity against kainate-induced neuronal cell death was assessed. CNQX, an AMPA/kainate receptor antagonist, protects neurons from excitotoxic death induced by kainate (Fig. 7F). The NMDA-receptor channel blockers, memantine (48 ± 6%) and MK-801 (33 ± 5%), protect neurons from kainate-induced excitotoxicity (34) albeit not as effectively as CNQX (75 ± 4%; Fig. 7F). In sharp contrast, NBTA (4 ± 2%) has no significant effect. These results indicate that the neuroprotective effect of NBTA is specific for NMDA receptors, in agreement with its blocking activity on recombinant NMDA receptors expressed in Xenopus oocytes (Fig. 5C).
Discussion
A reduced peptidomimetic SCL was screened to discover the neuroprotectant NBTA, an N-benzylated triamine derivative that protects hippocampal neurons by blocking NMDA receptor-mediated excitotoxic death. NBTA acts as an open channel blocker of moderate affinity and high specificity for the NMDA-receptor subtype of glutamate receptors. The block is strongly voltage and use dependent, exhibiting relatively fast offset kinetics.
Then which features confer NBTA with its remarkable selectivity for NMDA receptors? The dimensions of NBTA and the identification of its binding sites shed light on the structure of the NMDA receptor channel. The diameter at the narrow constriction of the NMDA-receptor channel, the N site of NR1, is 5.5 Å (30–32). Molecular modeling of NBTA shows that the phenolic hydroxyl is distant from the benzyl and butyl groups by ≈8.8 and 14 Å, respectively. For comparison, the widest dimension of memantine is 5.5 Å, a good match to the dimensions of the pore constriction. Presumably, NBTA occludes the channel in a similar orientation as memantine with its positive-charged NH2 group pointing toward the N site of NR1. Memantine may enter deeper into the channel or even go through it and behave as a permeant cation. In contrast, NBTA is larger than memantine (10 × 14 × 8 Å), which would restrict or exclude its passage through a narrow pore of only 5.5 Å. Thus, to accommodate NBTA in the pore, the outer vestibule of the NMDA-receptor channel preceding the selectivity filter must be at least 9 Å wide. Given its size, it is likely that NBTA interacts with specific residues that contribute to the outer vestibule of the channel complex such as the region preceding the transmembrane segment M1 and possibly the N-terminal part of M4 (35) but probably not the C terminus of M3 segment, which is highly conserved among the glutamate-receptors subfamily.
These considerations imply that the higher potency of NBTA may originate from polar and van der Waals interactions with vestibule residues arising from aromatic rings being oriented as the positive amine enters into the narrow constriction of the channel. Such a location for the binding site would render NBTA less prone to being trapped within the channel and therefore more readily dissociable from the open but blocked channel, accounting for the fast reversibility of its effect after NBTA removal. Compounds with fast blocking kinetics and strong voltage dependence offer a better therapeutic profile (36). The strong voltage-dependent block of NBTA may confer it the potential to discriminate between the transient physiological and the sustained pathological activation of the NMDA receptor. Recovery from the blocked state may be prompt and complete; given the uncompetitive nature of the block, channel block would be of impact only during sustained receptor activation, as might occur during a brain insult, and not during normal synaptic activity (37). Combination of these features may endow NBTA with favorable properties to be considered as a lead compound for the development of neuroprotectants.
Today, with the exception of memantine, which has been shown in human clinical trials (ref. 38; Barry Reisberg, personal communication) to delay the progression of dementia in Alzheimer's patients and to be of therapeutic value in the management of Parkinson's disease, multiple trials of NMDA antagonists have been abandoned primarily because of their lack of specificity or efficacy. The striking selectivity of NBTA offers an advantage over other NMDA-receptor channel blockers.
Knowledge of the penetration of NBTA across the blood–brain barrier (BBB) is important to assess its therapeutic potential in the clinical arena. NBTA satisfies the minimum dual criteria considered critical for a molecule to cross the BBB, namely lipid solubility and molecular mass ≤400–600 Da (39). To evaluate this issue in detail, the method of Feher et al. (40) to predict the BBB partitioning (logBB) of NBTA was used. The method considers three predictors: the octanol-water partition coefficient, the number of solvated hydrogen bond acceptors in an aqueous environment, and the polar surface area. For NBTA, a logBB value of 0.22–0.23 was obtained. The results argue that NBTA would pass the blood–brain barrier. Thus, NBTA is a nonpeptide compound with a unique chemical structure that appears to be a realistic candidate to be used as template for drug development targeting the NMDA-receptor channel.
Acknowledgments
We thank V. Hamashin for the synthesis of the individual compounds, P. Whiting for the NR2A subunit cDNA, S. Nakanishi for the NR1 cDNA clone, S. F. Traynelis for the NR1 mutant (N616Q) cDNA, R. Dingledine for the NR1 mutant (N616R) cDNA, and J. M. Merino, X.-Y. Wang, and Ying Wang for technical advice. We are indebted to Dr. M. Feher (Nanodesign) for his expert assistance with predictions of blood–brain barrier penetration. This research was supported by grants from the U.S. Army Research Office (DAAG55-98-1-0106 to M.M.).
Abbreviations
- NMDA
N-methyl-D-aspartate
- PS-SCL
positional scanning synthetic combinatorial library
- NBTA
N-benzylated triamine
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Albright T D, Jessell T M, Kandel E R, Posner M I. Cell. 2000;18, Suppl. 100:1–55. [PubMed] [Google Scholar]
- 2.Choi D W, Rothman S M. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 3.Lee J M, Zipfel G J, Choi D W. Nature (London) 1999;399, Suppl. 6738:7–14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 4.Chase T N, Oh J D. Ann Neurol. 2000;47, Suppl. 1:122–129. [PubMed] [Google Scholar]
- 5.Heintz N, Zoghbi H Y. Annu Rev Physiol. 2000;62:779–802. doi: 10.1146/annurev.physiol.62.1.779. [DOI] [PubMed] [Google Scholar]
- 6.Gill R, Foster A C, Woodruff G N. J Neurosci. 1987;7:3343–3349. doi: 10.1523/JNEUROSCI.07-10-03343.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon R P, Swan J H, Griffiths T, Meldrum B S. Science. 1984;226:850–852. doi: 10.1126/science.6093256. [DOI] [PubMed] [Google Scholar]
- 8.Olney J W, Ikonomidou C, Mosinger J L, Frierdich G. J Neurosci. 1989;9:1701–1704. doi: 10.1523/JNEUROSCI.09-05-01701.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muir K W, Lees K R. Stroke (Dallas) 1995;26:503–513. doi: 10.1161/01.str.26.3.503. [DOI] [PubMed] [Google Scholar]
- 10.Houghten R A, Pinilla C, Appel J R, Blondelle S E, Dooley C T, Eichler J, Nefzi A, Ostresh J M. J Med Chem. 1999;42:3743–3778. doi: 10.1021/jm990174v. [DOI] [PubMed] [Google Scholar]
- 11.Ferrer-Montiel A V, Merino J M, Blondelle S E, Perez-Paya E, Houghten R A, Montal M. Nat Biotechnol. 1998;16:286–291. doi: 10.1038/nbt0398-286. [DOI] [PubMed] [Google Scholar]
- 12.Houghten R A. Proc Natl Acad Sci USA. 1985;82:5131–5135. doi: 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dorner B, Husar G M, Ostresh J M, Houghten R A. Bioorg Med Chem. 1996;4:709–715. doi: 10.1016/0968-0896(96)00067-3. [DOI] [PubMed] [Google Scholar]
- 14.Nefzi A, Ostresh J M, Houghten R A. Tetrahedron. 1999;55:335–344. [Google Scholar]
- 15.Houghten R A, Pinilla C, Blondelle S E, Appel J R, Dooley C T, Cuervo J H. Nature (London) 1991;354:84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 16.Planells-Cases R, Sun W, Ferrer-Montiel A V, Montal M. Proc Natl Acad Sci USA. 1993;90:5057–5061. doi: 10.1073/pnas.90.11.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Bourdelles B, Wafford K A, Kemp J A, Marshall G, Bain C, Wilcox A S, Sikela J M, Whiting P J. J Neurochem. 1994;62:2091–2098. doi: 10.1046/j.1471-4159.1994.62062091.x. [DOI] [PubMed] [Google Scholar]
- 18.Sun W, Ferrer-Montiel A V, Schinder A F, McPherson J P, Evans G A, Montal M. Proc Natl Acad Sci USA. 1992;89:1443–1447. doi: 10.1073/pnas.89.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrer-Montiel A V, Montal M. In: Methods in Molecular Biology: NMDA Receptor Protocols. Li M, editor. Vol. 128. Clifton, NJ: Humana; 1999. pp. 167–178. [DOI] [PubMed] [Google Scholar]
- 20.Woodhull A M. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schinder A F, Olson E C, Spitzer N C, Montal M. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huettner J E, Bean B P. Proc Natl Acad Sci USA. 1988;85:1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrer-Montiel A V, Merino J M, Planells-Cases R, Sun W, Montal M. Neuropharmacology. 1998;37:139–147. doi: 10.1016/s0028-3908(98)00007-0. [DOI] [PubMed] [Google Scholar]
- 24.Iversen L L, Kemp J A. In: The NMDA Receptor. Collingridge G L, editor. Oxford: Oxford Univ. Press; 1995. pp. 468–486. [Google Scholar]
- 25.Chen H-S, Lipton S A. J Physiol (London) 1997;499.1:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dingledine R, Borges K, Bowie D, Traynelis S F. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 27.Wollmuth L P, Kuner T, Sakmann B. J Physiol (London) 1998;506:13–32. doi: 10.1111/j.1469-7793.1998.013bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Igarashi K, Shirahata A, Pahk A J, Kashiwagi K, Williams K. J Pharmacol Exp Ther. 1997;283:533–540. [PubMed] [Google Scholar]
- 29.Williams K, Pahk A J, Kashiwagi K, Masuko T, Nguyen N D, Igarashi K. Mol Pharmacol. 1998;53:933–941. [PubMed] [Google Scholar]
- 30.Villarroel A, Burnashev N, Sakmann B. Biophys J. 1995;68:866–875. doi: 10.1016/S0006-3495(95)80263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wollmuth L P, Kuner T, Seeburg P H, Sakmann B. J Physiol (London) 1996;491:779–797. doi: 10.1113/jphysiol.1996.sp021257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuner T, Wollmuth L P, Karlin A, Seeburg P H, Sakmann B. Neuron. 1996;17:343–352. doi: 10.1016/s0896-6273(00)80165-8. [DOI] [PubMed] [Google Scholar]
- 33.Opella S J, Marassi F M, Gesell J J, Valente A P, Kim Y, Oblatt-Montal M, Montal M. Nat Struct Biol. 1999;6:374–379. doi: 10.1038/7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pringle A K, Iannotti F, Wilde G J, Chad J E, Seeley P J, Sundstrom L E. Brain Res. 1997;25:36–46. doi: 10.1016/s0006-8993(97)00089-9. [DOI] [PubMed] [Google Scholar]
- 35.Beck C, Wollmuth L P, Seeburg P H, Sakmann B, Kuner T. Neuron. 1999;22:559–570. doi: 10.1016/s0896-6273(00)80710-2. [DOI] [PubMed] [Google Scholar]
- 36.Parsons C G, Danysz W, Bartmann A, Spielmanns P, Frankiewicz T, Hesselink M, Eilbacher B, Quack G. Neuropharmacology. 1999;38:85–108. doi: 10.1016/s0028-3908(98)00161-0. [DOI] [PubMed] [Google Scholar]
- 37.Frankiewicz T, Potier B, Bashir Z I, Collingridge G L, Parsons C G. Br J Pharmacol. 1996;117:689–697. doi: 10.1111/j.1476-5381.1996.tb15245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winblad B, Poritis N. Int J Geriatr Psychiatry. 1999;14:135–146. doi: 10.1002/(sici)1099-1166(199902)14:2<135::aid-gps906>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 39.Pardridge W N. J Neurovirol. 1999;5:556–569. doi: 10.3109/13550289909021285. [DOI] [PubMed] [Google Scholar]
- 40.Feher M, Sourial E, Schmidt J M. Int J Pharm. 2000;201:239–247. doi: 10.1016/s0378-5173(00)00422-1. [DOI] [PubMed] [Google Scholar]