

Abstract
This review summarizes some of the recent developments and identifies critical challenges associated with in vitro and in silico representations of the liver and assesses the translational potential of these models in the quest of rationalizing the process of evaluating drug efficacy and toxicity. It discusses a wide range of research efforts that have produced, during recent years, quantitative descriptions and conceptual as well as computational models of hepatic processes such as biotransport and biotransformation, intra‐ and intercelular signal transduction, detoxification, etc. The abovementioned research efforts cover multiple scales of biological organization, from molecule–molecule interactions to reaction network and cellular and histological dynamics, and have resulted in a rapidly evolving knowledge base for a “systems biology of the liver.” Virtual organ/organism formulations represent integrative implementations of particular elements of this knowledge base, usually oriented toward the study of specific biological endpoints, and provide frameworks for translating the systems biology concepts into computational tools for quantitative prediction of responses to stressors and hypothesis generation for experimental design.
Keywords: model, virtual liver, toxicity
Introduction
As the major site of xenobiotic metabolism, the liver plays a central role in preventing accumulation of a wide range of compounds by converting them into a form suitable for elimination. As the process of xenobiotic metabolism requires multiple biochemical transformations, and the fact that some intermediates mediate toxic responses, the liver is potentially susceptible to injury 1 during the act of performing its function. An improved quantitative understanding of the balance between functional xenobiotic metabolism and hepatic damage would be of great utility in forming guidelines for safe exposure levels in both the pharmaceutical and the toxicological contexts. In particular, the ability to predict the toxicity profile of lead candidates 2 is critical to streamlining pharmaceutical drug development, and a better understanding of the onset of liver toxicity is an avenue to realizing the “personalized medicine” concept, wherein drugs are selected and dosed in accordance with the genetics, active biomarkers, and environment of the individual patient. 3 Furthermore, improved descriptions of the cellular‐level pharmacokinetics of xenobiotics are needed for integration into whole‐body physiologically based pharmacokinetic (PBPK) models to improve their accuracy and translation potential.
Advances in genomics and molecular and cell biology are providing a much improved view of the molecular players and pathways involved in xenobiotic metabolism, yet this information alone is limited in its translational potential. To fully exploit this wealth of information, one needs a framework for integrating various forms of data and utilizing them for predictive pharmacology 4 These include data on drugs and toxicants, including biodistribution profiles in various species or individuals, and patient data, including medical and drug history, gene amplifications and deletions, liver enzyme levels, etc. It is of high clinical priority to use these data to make predictions regarding human dosing in phase I clinical trials based on preclinical data, to refine dosing regimens based on human biodistribution data, and, ultimately, to stratify and individualize dosing. Furthermore, a better understanding of xenobiotic interactions in the liver will aid in diagnosing chronic liver disease at an earlier stage, when more treatment options are available.
PBPK models provide a framework to integrate, interpret, and make predictions regarding the overall host response to xenobiotics. 5 PBPK modeling is a special approach to pharmacokinetics analysis where the physiology and anatomy of the human body and the biochemistry of the chemical or chemicals of interest are incorporated into a conceptual model for computer simulation ( Figure 1 ). In PBPK models, the body is treated as a set of compartments with the concentrations of species related among them by formal material balance, thus establishing internal consistency. The rates of metabolism or other processes within compartments can be modeled with whatever level of detail available, providing flexibility and the means to incorporate multiple types of data. Thus, unlike classical pharmacokinetics, PBPK modeling is a powerful tool for many types of extrapolations, including species‐to‐species, route‐to‐route, and dose‐to‐dose extrapolations. In the context of drug development and toxicity predictions, PBPK models offer a promising approach toward a mechanistic understanding of undesired drug effects and present a promising avenue for rational drug design and screening with significant translational potential. 6 , 7 , 8 Therefore, in these integrated host models, the accuracy of the representation of the biology as well as the anatomy of the liver is a critical issue for their translational success.
Figure 1.
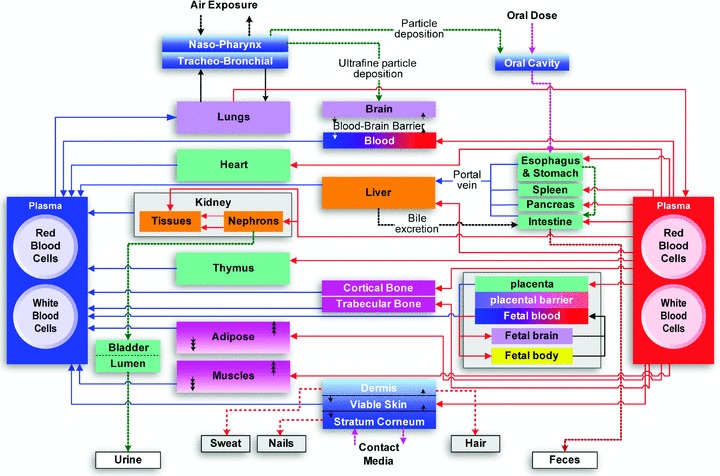
Typical structure of a generalized PBPK model 94 (figure adapted and used with kind permission of Springer Science and Business Media).
Because of the increasing sophistication of PBPK models and, in particular, the incorporation of molecular‐level information, the importance of in vitro experiments is being emphasized more heavily 6 , 7 Such systems allow direct measurement of metabolism in hepatocytes, separated from the effects of transport, distribution, and metabolism by other tissues. Thus, they provide important data for the parameterization and validation of PBPK models. This review summarizes some of the recent developments and identifies critical challenges associated with in vitro and in silico representations of the liver and assesses the translational potential of these models in the quest of rationalizing the process of evaluating drug efficacy and toxicity.
Development of “virtual” (or “in silico”) tissues and organs represents a critical step in the continuing advancement of the state of the science in physiologically based pharmacokinetic and pharmacodynamic (PBPK/PD) modeling. Particular attention in such developmental efforts, undertaken by various research groups, has been given to the liver, due to its critical role in central metabolism and detoxification and in the synthesis and metabolism of hormones and other compounds necessary for maintaining critical body functions and its ability to regenerate. As the key metabolic organ for degrading xenobiotics (entering the body through oral ingestion, inhalation, dermal absorption, or injection), the liver often shows the earliest signs of injury due to pharmaceuticals and environmental chemicals. Understanding and quantifying the mechanisms of such injury would allow rational assessments of safe dosage levels for humans and characterization of interindividual response variability and susceptibility with respect to these levels. 8 , 9 The ability to simulate hepatic processes through comprehensive mechanistic in silico representations of the liver—incorporated, as necessary, in relevant whole‐body model formulations—will substantially affect rational tissue engineering and personalized medicine and risk assessment and can lead to improved strategies for targeted intervention, for reduction of animal testing in drug development and environmental chemical toxicity assessment, and for more efficient clinical testing.
Liver Models
The granularity of any model is what controls the accuracy of its representation and, to a great extent, its predictions. When modeling physiological systems, an often invoked assumption is that of apparent homogeneity. In other words, all cells comprising a tissue are the same; they all experience the same conditions, and metabolic processes are indistinguishable across the tissue. In fairness, the main reason for this is that the complexity of the representation increases dramatically when spatial and functional heterogeneities are considered, as has been shown previously 10 Liver is known to be metabolically zonated, 11 , 12 , 13 , 14 , 15 , 16 , 17 and so is often characterized for simplicity in terms of two tissue regions (the periportal zone and the perivenous zone) that differ in enzyme activity levels and content ( Figure 2 ). Functions related to central metabolism and xenobiotic transformations are affected by the gradients of oxygen 18 and hormones 19 , 20 expressed across the liver tissue. Although static cultures of hepatocytes under periportal versus perivenous conditions can be used to identify a number of changes in gene expression and enzyme activity, the use of hepatocyte cell culture analogs to estimate pharmacodynamic parameters requires a bioreactor configuration more akin to the in vivo environment. Another assumption is that of “lumped” kinetics. The metabolic rates are represented through overall Michaelis‐Menten‐type kinetics, with the rate parameters (V max,Km) usually fitted from experimental data.
Figure 2.
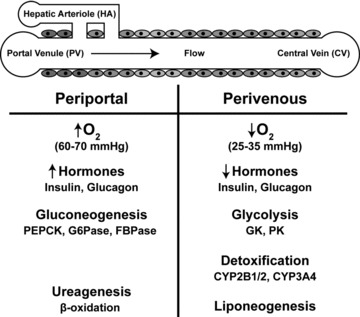
Zonation effects.
Therefore, most widely applicable models make two critical assumptions, i.e. (i) spatial homogeneity and (ii) apparent metabolic rates. However, advances in our ability to visualize tissues and successfully model liver physiology, 21 recent advances in physicochemical modeling that allow the descriptions of pathways involving “signals” rather than involving metabolites, 22 as well as recent advances in high‐throughput genomic and metabolic engineering 23 can potentially enable a more detailed representation of the liver as well as of the kinetics of the metabolic reactions involved in drug metabolism.
In vitro hepatocyte models
Without a doubt, inherent difficulties exist in translating biological information from animal studies, and significant effort has been invested in analyzing the causes of this disparity. 24 Among others, animal‐to‐animal variability has been associated with the limitations of such models. As such, researchers have also focused on in vitro studies that, while defining a far less general model, allow for a much tighter control of extraneous conditions and limit their potential implications.
Given the critical role of the liver, it comes as no surprise that numerous in vitro models have been proposed and have all played a critical role in advancing the understanding of hepatic metabolism. 25 The cell models, slices, and mainly primary hepatocyte cultures appear to be the most powerful in vitro systems, as liver‐specific functions and the responsiveness to inducers are retained for a few days or several weeks depending on culture conditions. Maintenance of phase I and phase II xenobiotic‐metabolizing enzyme activities allows various chemical investigations to be performed, including determination of kinetic parameters, metabolic profiling, interspecies comparison, inhibition and induction effects, and drug‐drug interactions. In vitro liver cell models also have various applications in toxicology: screening of cytotoxic and genotoxic compounds, evaluation of chemoprotective agents, and determination of characteristic liver lesions and associated biochemical mechanisms induced by toxic compounds. 26 Extrapolation of the results to the in vivo situation remains a matter of debate; 25 however, several studies have shown that hepatocye cultures are good models to qualitatively predict in vivo metabolic profiles. Such models significantly helped in assessing the impact of P450s in xenobiotic metabolism as well as in establishing the critical contribution of receptors such as aryl hydrocarbon receptor (AhR), pregnane X receptor (PXR) and constitutive androstane receptor (CAR) in controlling the expression of specific Cytochrome P450s (CYPs), such as CYP1A (Ahr) and CYP2 and CYP3A (PXR and CAR families). 27 In addition to transcriptional regulation, transport‐mediated uptake and efflux are also known to impact the transport of relevant regulators and hence drive regulation of gene expression. Given the importance of such transporters in the pharmacokinetics of xenobiotics, the study of such transport processes becomes of critical importance. 25 Given the close relationship between the metabolizing enzymes and the transporters regulating their trafficking, it is no surprise that both share common nuclear factors. 28
Physiological hepatocyte bioreactors
One limitation of traditional in vitro cultures for studying the biology of liver function is that they do not capture the physiology of liver and instead present an environment of homogeneity in the culture. Zonation, the variability in morphology and function of hepatocytes with position along the liver sinusoids, is a critical concept. Metabolic functions, such as oxidative energy metabolism, carbohydrate, lipid, and nitrogen metabolism, conjugation, and xenobiotic metabolism, are all known to be localized to specialized zones throughout the organ. 16 , 29 , 30 Often, complete chemical conversions require successive transformations across zones, leading to a net reaction that would not be possible in either zone individually, as in ammonia detoxification. 12 , 13 , 30
The intimate relationship between gene expression, enzymatic activity, and metabolic function also manifests itself in the impact of zonation on the expression of hepatic genes. 17 , 18 This spatial variability has been shown to play an important role in all of the functions of the liver. 31 Using in vitro bioreactors that mimic the microenvironment of these zones, allowing for subspecialization within each zone, offers a more powerful tool for the investigation of hepatic function in the context of xenobiotic metabolism. 32 Quantification of the spatial distribution of gene expression, induction of relevant CYPs, activation of transcriptional regulators as a function of oxygen availability, 33 and xenobiotic concentration will substantially improve our fundamental understanding of the cellular processes underlying xenobiotic metabolism.
Researchers have explored the possibility of developing biomimetic reactors that induce localization through appropriate oxygen transport. Most notably, Allen et al. 32 coupled an intricate experimental design with the solution of the associated reaction‐diffusion equation in order to evaluate oxygen gradients across a bioreactor. The system was used to evaluate acetaminophen toxicity to enable toxicological evaluations. One can envision the system being explored to characterize the toxicity effects of a host of xenobiotic compounds. Despite simplifications, this was a powerful demonstration of the possibilities of an in vitro system that begins to reproduce spatial and temporal inhomogeneities. Extension of this concept via miniaturization to make a “liver chip” has been receiving ever‐increasing recognition from the medical community 34 , 35 , 36 , 37 , 38
Particularly fascinating is the “cell culture analog” (CCA) introduced by Shuler and coworkers 39 , 40 , 41 , 42 in order to bridge the gap between multicompartment computer models and single‐compartment in vitro cell cultures. PBPKs offer an alternative that mimics the potential interactions between tissues and chemicals resulting from various dynamic (time‐dependent) exposure scenarios and provide a potentially rational basis to extrapolate across species, particularly to humans. However, the large number of parameters, particularly for metabolic processes, poses a significant problem because the quality and quantity of available data do not readily permit independent estimates of parameters. Consequently, most PBPK models either employ an empirical description of metabolic functions or a more mechanistic description but with a large number of adjustable parameters. A CCA system combines the advantages of in vitro and PBPK approaches and circumvents many of the limitations associated with either in vitro or PBPK systems. For example, the CCA system dosing can be done on the same milligram per kilogram basis as used in whole animals and PBPKs. Tissues will potentially experience the same dynamic (time‐dependent) exposure that would occur within whole animals. The goal in creating a CCA is to make the compartments exact analogs of those in a PBPK model so that any discrepancy between PBPK prediction and CCA response would result from incomplete or incorrect assumptions about biological mechanisms or, possibly, from poor estimates of parameters. In reality, the accuracy of the CCA will relate to the fidelity with which a tissue is captured by the cell culture. Advances in tissue engineering are resulting in improved cell cultures; however, CCA tissues should be validated for their individual accuracy in replicating tissue functions of interest before incorporation into a CCA. Nonetheless, an advantage of a CCA over a whole animal is the ability to alter the system arbitrarily (e.g., increase the size of a compartment or remove a compartment) to test mechanistic hypotheses directly.
In silico liver models
Even though in vitro liver models are a promising surrogate for the actual organ, they are still cumbersome to work with in the sense that lead hypothesis generation needs to be rapid and needs to enable the development, testing, and screening of multiple “what‐if” types of scenarios, rationalize experimental observations, generate a dynamic view of a response, and, in general, make quantitative predictions aiming at the development of testable hypotheses. As such, there has been significant interest in developing in silico representations, that is, computer models, of the liver in order to evaluate the implications of the presence of xenobiotics in the human body. Major challenges lie in modeling both the physiology and the metabolism of liver and, in particular, in integrating these two levels. The recent paper of Ohno et al. 43 presents an excellent example of how such a model can be constructed. Blood enters through branches of the portal vein and hepatic artery and then flows through small channels called sinusoids, which are lined, predominantly, with parenchymal liver cells (hepatocytes) ( Figure 3). The hepatocytes remove toxic substances from the blood, which subsequently exits the lobule through the central vein. Because the concentration of nutrients and metabolites along the sinusoid as the blood moves from the upper reach (periportal zone) area to the lower reach (perivenous zone) area are graded (zonation), it is also expected that the metabolic responses along the sinusoid would differ.
Figure 3.
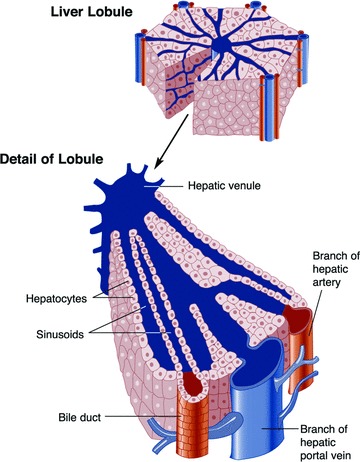
Structure of the liver's lobules (taken from http://www.niaaa.nih.gov/Resources/GraphicsGallery/Liver/lobulep295.htm and Ref. 95).
For a particular metabolic process of interest, one can construct appropriate networks of metabolic reactions using available databases of metabolic pathways 44 , 45 and identify for each reaction relevant metabolic enzymes. 46 , 47 , 48 Ohno et al. propose to simulate the zonation across the liver sinusoid by “stacking” a number of compartments, each of which experiences a stratified level of concentration of enzymes and thus of metabolic activities ( Figure 4 ). Yan et al. 49 explore the concept of agent‐based models (ABMs) to treat all elements of the hepatic components as interacting entities. Complementary to these activities, researchers consider alternative descriptions of liver physiology in order to improve the computational aspects of the simulation. 21 Successive iterations of model building and testing are employed. 50
Figure 4.
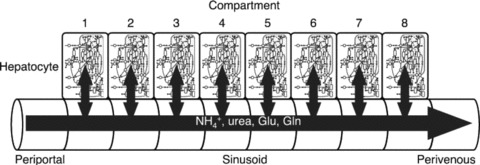
Multicompartment liver model incorporating the effects of zonation on metabolic reactions (figure adapted from Ref. 43).
The various modeling options encompass a wide range of levels of detail with regard to liver physiology. The simplest formulation involves a single well‐mixed compartment, denoted as a continuous stirred tankreactor (CSTR), in which the chemical in the liver blood is assumed to be in equilibrium with the chemical in the tissue, and the concentration of the chemical is assumed to be uniform throughout the liver. The hepatic micro circulatory network provides a high degree of mixing, 51 which, in conjunction with relatively low concentrations of environmental toxins and a slow rate of uptake and metabolism in the liver, justifies the well‐mixed assumption. Usually, kinetic parameters of such a model are estimated in vitro through liver microsomes or in vivo through biomarker data and parameter estimation techniques.
The spatial variations (zonation) within a single compartment can also be modeled as a plug flow reactor, effectively approximated as a number of CSTRs in series, in parallel, or in combinations of both, 52 whereas dispersion models can be used to represent an intermediate degree of mixing. 53 Although the well‐stirred model is successful in describing the “apparent” (phenomenological) kinetics of many xenobiotics (drugs as well as environmental chemicals), it often “lumps together” various physiological and biochemical processes, thus resulting in the “filtering” of the mechanistic information regarding chemical‐tissue interactions. This may not be critical in pharmacokinetic calculations (i.e., calculations of “what the body does to the chemical”), but it is often very important in the mechanistic interpretation of toxicodynamic processes (i.e., “what the chemical does to the body”). The more complex formulations of the liver compartments include zonal or segmental models, where different zones of the liver are denoted as subcompartments to include heterogeneity of transporters and enzymes; 54 , 55 circulatory models, which account for concentration differences within the vascular space; 56 fractal models, which represent the heterogeneity of the flow within the organ in terms of fractal concepts; 21 , 55 , 57 and, finally, ABMs, 49 which involve a large number of parameters that are, however, difficult to estimate from biomarkers or in vitro data. The advantage of ABMs compared with traditional modeling approaches, based on ordinary or partial differential equation formalisms, is that more intricate detail can be more easily incorporated; however, the dynamics of the response may be harder to rationalize. The model of Hunt et al. 49 was used to evaluate hepatic disposition and metabolism of antipyrine, atenolol, labetalol, and diltiazem as typical examples of cationic drugs.
An additional complexity deals with hepatic clearance, which also depends on binding proteins, transporters, and metabolic enzymes, which may not be homogeneously distributed through the liver. This is another reason why it is necessary to revisit the well‐mixed assumption. For example, it has been demonstrated that cytochrome P450 is induced heterogeneously in the liver and is present at higher concentration in the centrolobular and midzone regions than in the periportal region, 58 and that the metabolism of enalapril (an angiotensin‐converting enzyme [ACE] inhibitor used in the treatment of hypertension and some types of chronic heart failure) is greater in the perivenous region rather than in the periportal region. 59 Finally, from a modeling point of view, two general types of approaches have been developed to model spatial heterogeneities. The first employs multicompartment models that divide the liver into separate homogeneous zones, each with its own parameters to describe local events, such as protein expression. 54 The second approach uses distributed parameter models that describe the observed heterogeneities with spatially dependent functions, governed by partial differential equations. 60 , Table 1 presents an overview of different mathematical descriptions of the liver in simulating toxicokinetics and toxicodynamics of xenobiotics.
Table 1.
An overview of different mathematical descriptions of the liver in simulating toxicokinetics and toxicodynamics. 8
| Model type | Schematic depiction | Assumptions/applicability | References |
|---|---|---|---|
| One‐compartment models | |||
| Well‐stirred (CSTR) |

|
Well‐mixed (both macro‐ and micro‐mixing). Uniform metabolic and biochemical properties through‐out the liver. | 59, 86 |
| Plug flow reactor (PFR) |

|
Flow is uniform with no mixing, and metabolism is fast. | 86, 87 |
| Dispersion flow |

|
Highly nonuniform flow patterns; incomplete mixing. Uniform chemical and biochemical properties. | 86, 87 |
| Distribution‐based models | |||
| Residence time distribution (RTD)‐based circulatory models |
|
Nonmechanistic study of distribution of toxicants through residence time analysis. | 55, 56, 88 |
| Statistical distribution‐based model |
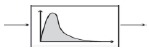
|
Representation of heterogeneity through a statistical distribution. Useful when heterogeneity is due to limited set of factors. | 85 |
| Stochastic/fractal models |

|
Heterogeneity in the liver modeled through stochastic terms or fractal descriptions. | 55, 89, 90 |
| Multicompartment models | |||
| CSTRs in series |

|
Multiple regions of the liver, with each region well mixed. Flow is uniform and from one region to the next. | 52, 59, 86 |
| Multizonal (multicompart‐mental) model |
|
Multiple regions of the liver with different uptake and metabolic properties; metabolism occurring in deep tissue. | 54, 59 |
| Back‐mixing plus fixed lag times and slow perfused sinusoids |

|
Zonal model with significant back mixing. | 52 |
| Compartmental model with cellular compartments |

|
Zonal model with significant back mixing, with variation across bulk tissue, deep tissue, and cellular space. | 52, 91 |
| Discrete, agent‐based models | |||
| Agent‐based |

|
Bottom‐up synthetic, nonmechanistic* description of multilevel processes within the liver; computationally and data‐intensive. | 5, 49, 65 |
| “Higher‐dimensione 1” models | |||
| Continuous, interconnected tubes |

|
Variation in uptake and metabolic properties across the cross‐section and along the direction of uniform flow. | 87, 92, 93 |
| Distributed zones |
|
Variation in uptake and metabolic properties across the cross‐section and along the direction of nonuniform flow. | 87 |
| Discrete, interconnected tubes |

|
Same as distributed zones but with intermittent mixing. | 87 |
| Fluid mechanics modeling of liver lobules |
|
Computational fluid dynamics‐based, detailed realistic modeling of individual liver lobules; computationally and data‐intensive. | 72 |
*Mechanistic is defined here as derived from the first principles of thermodynamics and kinetics (i.e. conservation laws and constitutive equations).
Translational Opportunities and Challenges
This review has attempted only to scratch the surface of the exciting opportunities and the outstanding difficulties that need to be overcome in order to develop appropriate in vitro and in silico liver models. The idea of engineering a construct or mathematically simulating a proxy that reliably mimics the function of an organ is quite extraordinary, and one can only imagine the implications that the success of such an endeavor may have. Although an enormous amount of information has been accumulated over the years in cellular and molecular biology, the translational potential of the accumulation of this knowledge has been hampered by the fact that this information needs to be put in the context of a higher‐level organization, either tissue or whole‐body. Therefore, the potential avenues to be explored are not related, necessarily, to the use of liver surrogates for further deciphering the biology of hepatic functions but rather to the evaluation of the possibility of integrating diverse pieces of information and either assessing hepatic response or treating the liver as a critical component of a host (whole‐body) response.
Traditional applications of physiologically based toxicokinetic/toxicodynamics models simulate the absorption, distribution, metabolism elimination, and toxicity of chemicals, with compartments representing major organs of interest, including all sites of metabolism. 61 By developing explicitly mechanistic models and with the advances in computational processing power, it will be possible to model important organs in significantly more detail. 62 In that respect, virtual organs, either in vitro or in silico, can greatly increase the quantitative insight into the response of organisms following toxic insults. Virtual organs can be used not only in assessing the impact of toxicant exposures on biological responses but also in assessing past exposures based on different exposure to biological response biomarkers. In addition, they can also be used to study toxicokinetic and toxicodynamic interactions among chemicals within major sites of metabolism. Because the liver is a complex organ and a major target for metabolism and chronic toxicity, it is an appropriate starting point for detailed modeling analysis. A virtual liver can enable much faster testing of environmental and xenobiotic species for toxicity. An in silico liver can be utilized in conjunction with measured pharmacokinetic data and hepatic disposition events in order to refine and optimize the parameters associated with drug clearance phenomena 50 ( Figure 5), which are critical for evaluating regulation and signaling of phase I and II metabolizing enzymes ( Figure 6 ).
Figure 5.

Framework illustrating interactions between in silico and in vitro liver model (figure adapted from Yan et al. 50 ).
Figure 6.
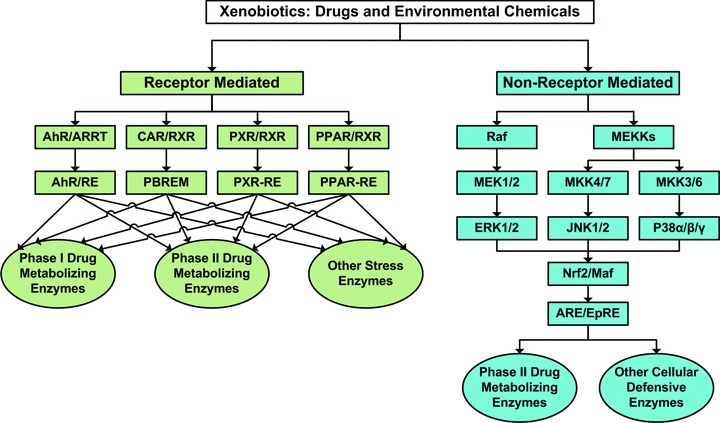
Pharmacogenomic/toxicogenomic regulation and signaling of phase I and phase II metabolizing enzymes participating in hepatic metabolic and detoxification processes for xenobiotics (figure adapted from Rushmore and Kong 81 ).
One can envision opportunities at two levels of complexity: the organ and the host. At the organ level, toxicity screening and population studies can significantly benefit from the ability to simulate, and estimate, toxic side effects of xenobiotic metabolism by characterizing the appropriate activation of metabolic pathways, which act as precursors to detrimental events, and/or by assessing the potential for the synthesis of toxic by‐products. Either in vitro or in silico models coupled with the emerging compendia of liver responses 63 can serve as a template for characterizing expected liver toxicities. In silico models with realistic descriptions of the physiology can evaluate the impact of local inhomogeneities on cellular and molecular events and describe the heterogeneity of xenobiotics metabolism ( Figure 7).
Figure 7.
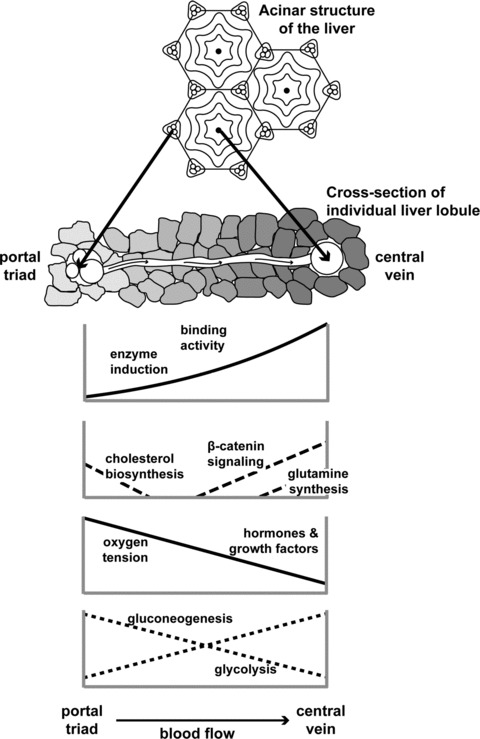
Schematic representation of various factors contributing to heterogeneity in toxicokinetics and toxicodynamics of xenobiotics in the liver 85 .
However, one can argue that possibly more exciting are the prospects of integrating in silico liver models with whole‐body physicochemical models. The idea of a human reconstruction in silico may still seem an unattainable goal. However, the first attempts have already materialized, as exemplified by the physiome.jp project whose ultimate goal is “... to provide building blocks useful to develop in silico human. The blocks will include mathematical models and experimental data representing physiological functions.” Physiome.jp is a part of the Worldwide Integrative Biomedical Research Cooperation to promote physiome and systems biology (http://wwwphysiome.jp/index.html). The initial attempts were recently discussed. 64 , 65 Whole‐body PBPK models have already been demonstrated in the context of assessing individual variability in drug effects to assist in the planning and design of clinical trials. 66 , 67 , 68
Arguably, however, a leading challenge is in quantifying the metabolic effects, manifested through changes in fluxes across key reactions and pathways. For many compounds, the primary biotransformation pathways can be reconstructed and integrated into pathway models of metabolism, allowing a direct connection between xenobiotic and central metabolism, such as the relationships between sulfur/glucose metabolism and sulfation/glucuronidation conjugations, respectively. Toxic intermediates and adducts can be incorporated also and their interrelationships within the larger metabolic network can be modeled and quantified. The current practice is to evaluate variants of Michaelis‐Menten kinetics, 43 with apparent rates fitted from experimental data. However, constraint‐based modeling approaches are emerging, which allow for stoichiometric and thermodynamic consistency across a metabolic network. 69 , 70 The geometry and physiology of the organ can be ascertained and modeled based on currently available imaging‐ and data‐processing techniques. 71 Furthermore, the flow problems associated with blood circulation along the sinusoids of the hepatic lobule, albeit not solved, are well studied, and computational fluid dynamics questions are already well posed. 72
Greater challenges and opportunities lie in the connections among signaling, regulation of gene expression, 73 , 74 , 75 , 76 , 77 protein synthesis, and metabolic fluxes. 78 , 79 A major milestone will be reached, without a doubt, when gene regulation models will be coupled with rate expression in order to assess the true impact of the emergence of local conditions and their implication in the spatial and temporal function variability throughout the liver. Although efforts in that respect are ongoing, at this point, the current state of the art is mostly driven by either sensitivity analyses or correlation‐type modeling. 80 Such capabilities would be of considerable utility in advancing in silico models to estimate the longer‐term effects of xenobiotic exposure. These include, for example, inflammatory sequelae resulting from the prolonged activation of stress signaling pathways downstream of aromatic receptor binding and generation of reactive oxygen species 81 as well as mutation rates and carcinogenesis. 82
Concluding Remarks
The idea of using the virtual liver to assess toxicity effects and screen compounds is slowly making its appearance. Entelos (http://entelos.com) was recently awarded the first patent for applying predictive technologies and “virtual humans” to find better drugs and more tailored health‐related products. This enabling technology, entitled “Cholestasis Signature,” helps researchers to screen novel compounds more rapidly and efficiently during preclinical studies for cholestasis, a specific type of drug‐induced liver damage. 83 The search for better models to predict drug‐induced human liver damage has led the US Food and Drug Administration (FDA) to adopt similar approaches toward the design of a “virtual liver” to guide biomarker and assay development as part of their critical path activities to better understand the causes of drug‐induced liver injury; similar efforts are underway at the Environmental Protection Agency (EPA) (http://www.epa.gov/ncct/virtual_liver). Finally, a major international activity is currently emerging in the European Union under the general framework of Hepatosys (http://www.systembiologie.de/doc/070416MilestonesHepatoSysII_Text.pdf). HepatoSys focuses on the dynamical processes of detoxification, endocytosis, iron regulation, and regeneration in primary hepatocytes. The final long‐term goal of HepatoSys is to translate the insights into the systems behavior of hepatocytes obtained from mechanism‐based mathematical models in clinical and pharmaceutical practice. Understanding patient‐specific drug metabolism paves the way for predictive and personalized medicine. Understanding the regulation of liver regeneration will have a major impact on the prediction of clinical outcomes of liver injury and developing strategies for the targeted regeneration of hepatocytes, thus reducing the need for animal testing in drug development. Comprehensive mathematical models will be applied to in silico drug screening, resulting in a significant reduction in the time to market and costs in drug development, especially in identifying toxic side effects, which present the main reason for dropout in late clinical phases.
Advances in basic biological understanding, biological databases, and computer simulation tools, all integrated in the form of an overall computational framework, appear to be gaining acceptance as a rational approach for evaluating hypotheses related to hepatic toxicity and, eventually, for exploring the clinical translational potential of in vitro or in silico liver models.
Acknowledgments
Support for this work has been provided by the USEPA‐funded Environmental Bioinformatics and Computational Toxicology Center (ebCTC), under STAR grant GAD R 832721‐010, and by the National Science Foundation (NSF) Metabolic Engineering Initiative (BES‐0519563). Further support was provided by the National Institute of Environmental Health Sciences (NIEHS)‐sponsored UMDNJ Center for Environmental Exposures and Disease (NIEHS P30ES005022). This work has not been reviewed by and does not represent the opinions of the funding agencies.
References
- 1. Merrick BA. Toxicoproteomics in liver injury and inflammation. Ann N YAcod Sci. 2006; 1076 707–717. [DOI] [PubMed] [Google Scholar]
- 2. Dome JL, Skinner L, Frampton GK, Spurgeon DJ, Ragas AM. Human and environmental risk assessment of pharmaceuticals: differences, similarities, lessons from toxicology. Anal Bioonol Chem. 2007; 387(4): 1259–1268. [DOI] [PubMed] [Google Scholar]
- 3. Burke W, Psaty BM. Personalized medicine in the era of genomics. JAMA. 2007; 298(14) 1682–1684. [DOI] [PubMed] [Google Scholar]
- 4. Liu ET. Expression genomics and drug development: towards predictive pharmacology. Brief Fund Cenomic Proteomic. 2005; 3(4): 303–321. [DOI] [PubMed] [Google Scholar]
- 5. Hunt CA, Ropella GE, Yan L, Hung DY, Roberts MS. Physiologically based synthetic models of hepatic disposition. J Pharmacokinet Pharmacodyn. 2006; 33(6): 737–772 [DOI] [PubMed] [Google Scholar]
- 6. Pelkonen O, Turpeinen M, Uusitalo J, Rautio A, Raunio H. Prediction of drug metabolism and interactions on the basis of in vitro investigations. Basic Ciin Pharmacol Toxicol. 2005; 96(3): 167–175. [DOI] [PubMed] [Google Scholar]
- 7. Pelkonen O, Turpeinen M. In vitro‐in vivo extrapolation of hepatic clearance: biological tools, scaling factors, model assumptions and correct concentrations. Xenobiotica. 2007; 37(10–11) 1066–1089. [DOI] [PubMed] [Google Scholar]
- 8. Jaeschke H. Toxic responses of the liver In: Klaassen CD, ed. Casarett & Doull's Toxicology—The Basic Science of Poisons. New York : McGraw‐Hill; 2008: 471–490. [Google Scholar]
- 9. Sahu SC. Hepatotoxicity from Genomics to In Vitro and In Vivo Models. Chichester , England John Wiley & Sons; 2007. [Google Scholar]
- 10. Lauffenburger DA, Kennedy CR. Localized bacterial infection in a distributed model for tissue inflammation. J Math Biol. 1983; 16(2): 141–163. [DOI] [PubMed] [Google Scholar]
- 11. Beier K, Volkl A, Metzger C, Mayer D, Bannasch P, Fahimi HD. Hepatic zonation of the induction of cytochrome P450 IVA, peroxisomal lipid beta‐oxidation enzymes and peroxisome proliferation in rats treated with dehydroepiandrosterone (DHEA). Evidence of distinct zonal and sex‐specific differences. Carcinogenesis. 1997; 18(8): 1491–1498 [DOI] [PubMed] [Google Scholar]
- 12. Haussinger D, Lamers WH, Moorman AF. Hepatocyte heterogeneity in the metabolism of amino acids and ammonia. Enzyme. 1992; 46(1–3): 72–93 [DOI] [PubMed] [Google Scholar]
- 13. Jungermann K. Metabolic zonation of liver parenchyma. Semin Liver Dis. 1988; 8(4): 329–341. [DOI] [PubMed] [Google Scholar]
- 14. Jungermann K. Metabolic zonation of liver parenchyma: significance for the regulation of glycogen metabolism, gluconeogenesis, and glycolysis. DiabetesMetab Rev. 1987; 3(1): 269–293 [DOI] [PubMed] [Google Scholar]
- 15. Kietzmann T, Jungermann K. Modulation by oxygen of zonal gene expression in liver studied in primary rat hepatocyte cultures. Cell Biol Toxicol. 1997; 13(4–5): 243–255 [DOI] [PubMed] [Google Scholar]
- 16. Guzman M, Castro J. Zonation of fatty acid metabolism in rat liver. Biochem J. 1989; 264(1) 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jungermann K. Zonation of metabolism and gene expression in liver. Histochem Cell Biol 1995; 103(2): 81–91. [DOI] [PubMed] [Google Scholar]
- 18. Jungermann K, Kietzmann T. Role of oxygen in the zonation of carbohydrate metabolism and gene expression in liver. Kidney Int. 1997; 51(2): 402–412 [DOI] [PubMed] [Google Scholar]
- 19. Oinonen T, Nikkola E, Lindros KO. Growth hormone mediates zone‐specific gene expression in liver. FEBS Lett. 1993; 327(2): 237–240. [DOI] [PubMed] [Google Scholar]
- 20. Kinlaw WB, Tron P, Witters LA. Thyroid hormone and dietary carbohydrate induce different hepatic zonation of both “spot 14” and acetyl‐coenzyme‐A carboxylase: a novel mechanism of coregulation. Endocrinology. 1993; 133(2): 645–650. [DOI] [PubMed] [Google Scholar]
- 21. Lin DW, Johnson S, Hunt CA. Modeling liver physiology: combining fractals, imaging and animation. Conf Proc IEEE Eng Med Biol Soc. 2004; 5:3120–3123. [DOI] [PubMed] [Google Scholar]
- 22. Aidridge BB, Burke JM, Lauffenburger DA, Sorger PK. Physicochemical modelling of cell signalling pathways. Nat Cell Biol. 2006; 8(11): 1195–1203. [DOI] [PubMed] [Google Scholar]
- 23. Yarmush ML, Banta S. Metabolic engineering: advances in modeling and intervention in health and disease. Annu Rev Biomed Eng. 2003; 5: 349–381 [DOI] [PubMed] [Google Scholar]
- 24. Von Herrath MG, Nepom GT. Lost in translation: barriers to implementing clinical immunotherapeutics for autoimmunity J Exp Med. 2005; 202(9): 1159–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hewitt NJ, Lechon MJG, Houston JB, Hallifax D, Brown HS, Maurel P, Kenna JG, Gustavsson L, Lohmann C, Skonberg C, Guillouzo A, Tuschl G, Li AP, LeCluyse E, Groothuis GM, Hengstler JG. Primary hepatocytes: Current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metabolism Reviews. 2007; 39(1): 159–234. [DOI] [PubMed] [Google Scholar]
- 26. Guillouzo A. Liver cell models in in vitro toxicology. Environ Health Perspect. 1998; 106(Suppl2) 511–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kullak‐Ublick GA, Becker MB. Regulation of drug and bile salt transporters in liver and intestine. Drug Metab Rev. 2003; 35(4): 305–317 [DOI] [PubMed] [Google Scholar]
- 28. Elferink MG, Olinga P, Draaisma AL, Merema MT, Faber KN, Slooff MJ, Meijer DK, Groothuis GM. LPS‐induced downregulation of MRP2 and BSEP in human liver is due to a posttranscriptional process. Am J Physiol Castrointest Liver Physiol. 2004; 287(5): G1008–1016. [DOI] [PubMed] [Google Scholar]
- 29. Jungermann K, Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev Nutr. 1996; 16: 179–203. [DOI] [PubMed] [Google Scholar]
- 30. Jungermann K, Thurman RG. Hepatocyte heterogeneity in the metabolism of carbohydrates. Enzyme. 1992; 46(1–3): 33–58. [DOI] [PubMed] [Google Scholar]
- 31. Gebhardt R. Metabolic zonation of the liver: regulation and implications for liver function. Pharmacol Ther. 1992; 53(3): 275–354. [DOI] [PubMed] [Google Scholar]
- 32. Allen JW, Khetani SR, Bhatia SN. In vitro zonation and toxicity in a hepatocyte bioreactor. Toxicol Sci. 2005; 84(1): 110–119. [DOI] [PubMed] [Google Scholar]
- 33. Lindros KO. Zonation of cytochrome P450 expression, drug metabolism and toxicity in liver. Gen Pharmacol. 1997; 28(2): 191–196. [DOI] [PubMed] [Google Scholar]
- 34. Yates C, Shepard CR, Papworth G, Dash A, Stolz DB, Tannenbaum S, Griffith L, Wells A. Novel three‐dimensional organotypic liver bioreactor to directly visualize early events in metastatic progression. Adv Cancer Res. 2007; 97: 225–246. [DOI] [PubMed] [Google Scholar]
- 35. Yates C, Beer‐Stolz D, Griffith L, Wells A. Direct visualization of prostate cancer progression utilizing an organotypic liver bioreactor. Am J Clin Pathol. 2005; 124(3): 455–456. [Google Scholar]
- 36. Griffith L. The liver chip. Technol Rev. 2003; 106(2): 64–67 [Google Scholar]
- 37. Griffith L, Sivaraman A, Domansky K, Tannenbaum SR, Capitano A, Roberts J. Microfabricated liver for metabolism and toxicity. Chem Res Toxicol. 2002; 15(12): 1657–1657 [Google Scholar]
- 38. Khetani SR, Bhatia SN. Microscale culture of human liver cells for drug development. Nat Biotechnol. 2008; 26(1): 120–126. [DOI] [PubMed] [Google Scholar]
- 39. Ghanem A, Shuler ML. Combining cell culture analogue reactor designs and PBPK models to probe mechanisms of naphthalene toxicity. Biotechnol Prog. 2000; 16(3): 334–345 [DOI] [PubMed] [Google Scholar]
- 40. Ghanem A, Shuler ML. Characterization of a perfusion reactor utilizing mammalian cells on microcarrier beads. Biotechnol Prog. 2000; 16(3): 471–479. [DOI] [PubMed] [Google Scholar]
- 41. Shuler ML, Ghanem A, Quick D, Wong MC, Miller P. A self‐regulating cell culture analog device to mimic animal and human toxicological responses. Biotechnol Bioeng. 1996; 52(1) 45–60. [DOI] [PubMed] [Google Scholar]
- 42. Sweeney LM, Shuler ML, Babish JG, Ghanem A. A cell‐culture analog of rodent physiology‐application to naphthalene toxicology. Toxicol In Vitro. 1995; 9(3): 307–316. [DOI] [PubMed] [Google Scholar]
- 43. Ohno H, Naito Y, Nakajima H, Tomita M. Construction of a biological tissue model based on a single‐cell model: a computer simulation of metabolic heterogeneity in the liver lobule. Artif Life. 2008; 14(1): 3–28. [DOI] [PubMed] [Google Scholar]
- 44. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004; 32(Database issue): D277‐D280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kanehisa M, Goto S, Hattori M, Aoki‐Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006; 34(Database issue): D354‐D357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Caspi R, Karp PD. Using the MetaCyc pathway database and the BioCyc database collection. Curr Protoc Bio informatics. 2007; 20: Chapter 1: Unitl .17. [DOI] [PubMed] [Google Scholar]
- 47. Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, Paley S, Rhee SY, Shearer AG, Tissier C, Walk TC, Zhang P, Karp PD. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2008; 36(Database issue): D623‐D631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Caspi R, Foerster H, Fulcher CA, Hopkinson R, Ingraham J, Kaipa P, Krummenacker M, Paley S, Pick J, Rhee SY, Tissier C, Zhang P, Karp PD. MetaCyc: a multiorganism database of metabolic pathways and enzymes. Nucleic Acids Res. 2006; 34(Database issue): D511‐D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yan L, Ropella GE, Park S, Roberts MS, Hunt CA. Modeling and simulation of hepatic drug disposition using a physiologically based, multi‐agent in silico liver. Pharm Res. 2008; 25(5): 1023–1036. [DOI] [PubMed] [Google Scholar]
- 50. Yan L, Sheihk‐Bahaei S, Park S, Ropella GE, Hunt CA. Predictions of hepatic disposition properties using a mechanistically realistic, physiologically based model. Drug Metab Dispos. 2008; 36(4): 759–768. [DOI] [PubMed] [Google Scholar]
- 51. Weiss M. On the degree of solute mixing in liver models of drug elimination. J Pharmacokinet Biopharm. 1997; 25(3): 363–375. [DOI] [PubMed] [Google Scholar]
- 52. Anissimov YG, Roberts MS. A compartmental model of hepatic disposition kinetics: 1. Model development and application to linear kinetics. J Pharmacokinet Pharmacodyn. 2002; 29(2): 131–156. [DOI] [PubMed] [Google Scholar]
- 53. Roberts MS, Rowland M. Hepatic elimination‐dispersion model. J Pharm Sci. 1985; 74(5): 585–587. [DOI] [PubMed] [Google Scholar]
- 54. Andersen ME, Eklund CR, Mills JJ, Barton HA, Birnbaum LS. A multicompartment geometric model of the liver in relation to regional induction of cytochrome P450s. Toxicol AppI Pharmacol. 1997; 144(1): 135–144. [DOI] [PubMed] [Google Scholar]
- 55. Pang KS, Weiss M, Macheras P. Advanced pharmacokinetic models based on organ clearance, circulatory, and fractal concepts. AAPS J. 2007; 9(2): E268‐E283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weiss M. Pharmacokinetics in organs and the intact body: model validation and reduction. Eur J Pharm Sci. 1999; 7(2): 119–127. [DOI] [PubMed] [Google Scholar]
- 57. Weiss M. A note on the interpretation of tracer dispersion in the liver. J Theor Biol. 1997; 184(1): 1–6. [DOI] [PubMed] [Google Scholar]
- 58. Andersen ME, Mills JJ, Jirtle RL, Greenlee WF. Negative selection in hepatic tumor promotion in relation to cancer risk assessment. Toxicology. 1995; 102(1–2): 223–237 [DOI] [PubMed] [Google Scholar]
- 59. Abu‐Zahra TN, Pang KS. Effect of zonal transport and metabolism on hepatic removal: enalapril hydrolysis in zonal, isolated rat hepatocytes in vitro and correlation with perfusion data. Drug Metab Dispos. 2000; 28(7): 807–813. [PubMed] [Google Scholar]
- 60. Banks HT, Musante CJ, Raye JK. Predictions for a distributed parameter model describing the hepatic processing of 2,3,7,8‐TCDD. Math Comput Model. 2001; 33(1–3): 49–64. [Google Scholar]
- 61. Watanabe KH, Chen C. The role of physiologically based toxicokinetic models in biologically based risk assessment. Folia Histochem Cytobiol. 2001; 39(Suppl 2): 50–51. [PubMed] [Google Scholar]
- 62. Mekenyan O, Dimitrov S, Dimitrova N, Dimitrova G, Pavlov T, Chankov G, Kotov S, Vasilev K, Vasilev R. Metabolic activation of chemicals: in‐silico simulation. SAR QSAR Environ Res. 2006; 17(1): 107–120. [DOI] [PubMed] [Google Scholar]
- 63. Natsoulis G, Pearson Cl, Gollub J, Eynon BP, Ferng J, Nair R, Idury R, Lee MD, Fielden MR, Brennan RJ, Roter AH, Jarnagin K. The liver pharmacological and xenobiotic gene response repertoire. Mol Syst Biol. 2008; 4: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kawazu T, Nakanishi M, Suzuki Y, Odai S, Nomura T. A platform for in silico modeling of physiological systems. Conf Proc IEEE Eng Med Biol Soc. 2007; EMBS 2007: 1394–1397. [DOI] [PubMed] [Google Scholar]
- 65. Kawazu T, Odai S, Shibuya N, Nomura T Development of a support tool for multi‐agent‐based biological modeling. Conf Proc IEEE Eng Med Biol Soc. 2006; 1: 4163–4166. [DOI] [PubMed] [Google Scholar]
- 66. Bassingthwaighte JB. Strategies for the physiome project. Ann Biomed Eng. 2000; 28(8): 1043–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bassingthwaighte JB. Back to fundamentals: anatomy‐based physiological bioengi nee ring. Ann Biomed Eng. 2000; 28(7): 701–703. [DOI] [PubMed] [Google Scholar]
- 68. Bassingthwaighte JB, Chizeck HJ. The physiome projects and multiscale modeling. IEEE Signal Proc Mag. 2008; 25(2): 121–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jamshidi N, Palsson BO. Formulating genome‐scale kinetic models in the post‐genome era. Mol Syst Biol. 2008; 4: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Smallbone K, Simeonidis E, Broomhead DS, Kell DB. Something from nothing: bridging the gap between constraint‐based and kinetic modelling. FEBS J. 2007; 274(21): 5576–5585. [DOI] [PubMed] [Google Scholar]
- 71. Florin C, Paragios N, Funka‐Lea G, Williams J. Liver segmentation using sparse 3D prior models with optimal data support. Inf Process Med Imaging. 2007; 20: 38–49. [DOI] [PubMed] [Google Scholar]
- 72. Rani HP, Sheu TW, Chang TM, Liang PC. Numerical investigation of non‐Newtonian microcirculatory blood flow in hepatic lobule. J Biomech. 2006; 39(3): 551–563. [DOI] [PubMed] [Google Scholar]
- 73. Sun YN, McKay LI, DuBois DC, Jusko WJ, Almon RR. Pharmacokinetic/pharmacodynamic models for corticosteroid receptor down‐regulation and glutamine synthetase induction in rat skeletal muscle by a receptor/gene‐mediated mechanism. J PharmacolExp Ther. 1999; 288(2): 720–728. [PubMed] [Google Scholar]
- 74. Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ. Pharmacodynamics and pharmacogenomics of methylprednisolone during 7‐day infusions in rats. J Pharmacol Exp Ther. 2002; 300(1): 245–256. [DOI] [PubMed] [Google Scholar]
- 75. Jusko WJ. Pharmacokinetics and receptor‐mediated pharmacodynamics of corticosteroids. Toxicology. 1995; 102(1–2): 189–196. [DOI] [PubMed] [Google Scholar]
- 76. Mager DE, Jusko WJ. Pharmacodynamic modeling of time‐dependent transduction systems. Clin Pharmacol Ther. 2001; 70(3): 210–216. [DOI] [PubMed] [Google Scholar]
- 77. Mager DE, Jusko WJ. Development oftranslationalpharmacokinetic‐pharmacodynamic models. Clin Pharmacol Ther. 2008; 83(6): 909–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rossell S, Van Der Weijden CC, Lindenbergh A, Van Tuijl A, Francke C, Bakker BM, Westerhoff HV. Unraveling the complexity of flux regulation: a new method demonstrated for nutrient starvation in Saccharomyces cerevisiae . Proc Natl Acad Sci USA. 2006; 103(7): 2166–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rossell S, Van Der Weijden CC, Kruckeberg AL, Bakker BM, Westerhoff HV. Hierarchical and metabolic regulation of glucose influx in starved Saccharomyces cerevisiae . FEMS Yeast Res. 2005; 5(6–7): 611–619. [DOI] [PubMed] [Google Scholar]
- 80. Li Z, Chan C. Extracting novel information from gene expression data. Trends Biotechnol. 2004; 22(8): 381–383. [DOI] [PubMed] [Google Scholar]
- 81. Rushmore TH, Kong AN. Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr Drug Metab. 2002; 3(5): 481–490. [DOI] [PubMed] [Google Scholar]
- 82. Clewell HJ, Thomas RS, Gentry PR, Crump KS, Kenyon EM, El‐Mash HA, Yager JW. Research toward the development of a biologically based dose response assessment for inorganic arsenic carcinogenicity: a progress report. Toxicol AppI Pharmacol. 2007; 222(3): 388–398. [DOI] [PubMed] [Google Scholar]
- 83. Natsoulis G, Pearson C, Tugendreich S, Furness M, Natsoulis G, Pearson C, Tugendreich S, Furness M, Natsoulis G, Pearson C, Tugendreich S, Furness MS. Cholestasis signature US patent 7396645. July 8, 2008.
- 84. US FDA . Key FDA Critical Path Activities Under Way in 2007. Silver Spring , MD : U.S. Department of Health and Human Services, Food and Drug Administration; 2008. [Google Scholar]
- 85. Isukapalli SS, Brinkerhoff CJ, Sasso AF, Georgopoulos PG. Progress in developing infrastructure for a virtual liver V: PBTK/TD models with zonal and distributed parameters for metabolism of environmental toxins. The 2nd Annual Systems Toxicology Symposium: Multiscale Modeling, from Molecules to Organisms, Piscataway , NJ , 2008.
- 86. Ridgway D, Tuszynski JA, Tarn YK. Reassessing models of hepatic extraction. J Biol Phys. 2003; 29(1): 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Anissimov YG, Bracken AJ, Roberts MS. Interconnected‐tubes model of hepatic elimination. J Theor Biol. 1997; 188(1): 89–101. [DOI] [PubMed] [Google Scholar]
- 88. Weiss M, Krejcie TC, Avram MJ. Circulatory transport and capillary‐tissue exchange as determinants of the distribution kinetics of inulin and antipyrine in dog. J Pharma Sci. 2007; 96(4): 913–926. [DOI] [PubMed] [Google Scholar]
- 89. Fuite J, Marsh R, Tuszynski J. Fractal pharmacokinetics of the drug mibefradil in the liver. Phys Rev E Stat Nonlin Soft Matter Phys. 2002; 66(2): 021904. [DOI] [PubMed] [Google Scholar]
- 90. Ditlevsen S, De Gaetano A. Stochastic versus deterministic uptake of dodecanedioic acid by isolated rat livers. Bull Math Biol. 2005; 67(3): 547–561. [DOI] [PubMed] [Google Scholar]
- 91. Tanaka C, Kawai R, Rowland M. Physiologically based pharmacokinetics of cyclosporine A: reevaluation of dose‐nonlinear kinetics in rats. J Pharmacokinet Biopharm. 1999; 27(6): 597–623. [DOI] [PubMed] [Google Scholar]
- 92. Banks HT, Musante CJ, Tran HT. A dispersion model for the hepatic uptake and elimination of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Math Comput Model. 1998; 28(1): 9–29. [Google Scholar]
- 93. Chalhoub E, Xie L, Balasubramanian V, Kim J, Belovich J. A distributed model of carbohydrate transport and metabolism in the liver during rest and high‐intensity exercise. Ann Biomed Eng. 2007; 35(3): 474–491. [DOI] [PubMed] [Google Scholar]
- 94. Georgopoulos P. A multiscale approach for assessing the interactions of environmental and biological systems in a holistic health risk assessment framework. Water Air Soil Pollut. 2008; 8(1): 3–21. [Google Scholar]
- 95. Cunningham CC, Van Horn CG. Energy availability and alcohol‐related liver pathology. Alcohol Res Health. 2003; 27(4): 291–299. [PMC free article] [PubMed] [Google Scholar]