

Abstract
A series of N-substituted and N′-substituted aminothiazole-derived morphinans (5) were synthesized for expanding the structure-activity relationships of aminothiazolo-morphinans. Although their affinities were somewhat lower than their prototype aminothiazolo-N-cyclopropylmorphinan (3), 3-aminothiazole derivatives of cyclorphan (1) containing a primary amino group displayed high affinity and selectivity at the κ and μ opioid receptors. [35S]GTPγS binding assays showed that the aminothiazolomorphinans were κ agonists with mixed agonist and antagonist activity at the μ opioid receptor. These novel N′-monosubstituted aminothiazole-derived morphinans may be valuable for the development of drug abuse medications.
Introduction
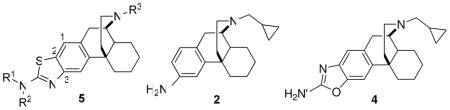
The opioid system modulates several key physiological and behavioral processes, such as: pain perception, the stress response, the immune response, and neuroendocrine function.1 With the discovery of the three different opioid receptors (κOR, μOR, and δOR), different functions and effects of the three receptor subtypes have been elucidated. Notably, it was found that the κ opioid receptor plays a role in the development of drug addiction, specifically by altering the dopamine reward pathway. Thus, the κ receptor has been implicated as a primary target for the development of pharmacotherapies for the treatment of cocaine dependence.2,3 Recent behavioral studies suggested that κ/μ opioids may be useful for the treatment of cocaine abuse and dependence.4 We reported that both acute and chronic treatment with mixed κ/μ opioids cyclorphan (1)5 and butorphan,5, 6 reduced cocaine self-administration dose-dependently and produced fewer side-effects than κ-selective agonists.7 However, the opioid derivatives are not metabolically stable: the free phenolic hydroxyl group in cyclorphan (1) and butorphan is also a potential site for metabolism, conjugation, and excretion, resulting in low oral bioavailability and short duration of action.1,8, 9 In an attempt to further extend the duration of action and to manipulate relative affinity and efficacy at κOR, modification of the phenolic hydroxyl group of cyclorphan has been performed, by incorporating 3-amino (2) 10, 3-aminothiazole (3, ATPM) 10, 2-aminooxazole (4) 11 isosteres (Figure 1).
Figure 1.

Structures of opioid ligands butorphan and 1 – 5
Among this series, one compound, 3 (Figure 1), has been identified to possess high affinity at κOR (Ki = 0.049 nM), and mixed κ agonist and μ-agonist/antagonist.10c (Table 1). Previous studies have shown that 3 inhibited morphine-induced antinociceptive tolerance, with less potential to develop tolerance and reduce heroin self-administration with lower sedative effect.12 However, recent in vivo studies of 3 in mice in the 55°C tail-flick test showed that this compound does not appear to have a longer duration of action than the phenolic compound 1.13 Aiming to extend duration of action and to improve oral bioavailability, a structure-activity relationship (SAR) study has been conducted to investigate the effect of modifications of N-substituent (R3) and N′-3-amino-substituted (R1, R2) of the morphinan 5 (Figure 1).
Table 1.
Binding Affinities of Novel Compounds to Human κ, μ and δ Opioid Receptorsa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| compound | R1 | R2 | R3 | Ki (nM) ± SEM | Selectivity | |||
| [3H]U69,593 (κ) | [3H]DAMGO (μ) | [3H]Naltrindole (δ) | μ/κ | σ/κ | ||||
| 1 b (Cyclorphan) | -- | -- | -- | 0.034 ± 0.002 | 0.062 ± 0.003 | 1.9 ± 0.1 | 2 | 56 |
| 2 b | -- | -- | -- | 0.18 ± 0.003 | 1.3 ± 0.029 | 150 ± 2.0 | ||
| 3 b (ATPM) | H | H | 0.049 ± 0.005 | 1.5 ± 0.2 | 29 ± 2 | 30 | 590 | |
| 4 b | -- | -- | -- | 52 ±1.0 | 150 ± 5.1 | 18% inh at 10 μM | 3 | >192 |
| 29 b | H | H | CH3 | 6.4 ± 0.5 | 1.1 ± 0.1 | 190 ± 10 | 0.17 | 30 |
| 10 b (ATBM) | H | H | 0.79 ± 0.02 | 7.1 ± 0.5 | 230 ± 21 | 9 | 290 | |
| 11 | H | H | 0.30 ± 0.011 | 2.7 ± 0.39 | 54 ± 1.5 | 9 | 180 | |
| 12 | H | H | 0.83 ± 0.10 | 2.4 ± 0.22 | 56 ± 7.2 | 3 | 67 | |
| 13a | H | CH3 | 0.066 ± 0.0037 | 3.0 ± 0.062 | 25 ± 1.5 | 45 | 380 | |
| 19a | H | CH2CH3 | 0.151 ± 0.0051 | 4.7 ± 0.23 | 43 ± 6.5 | 31 | 280 | |
| 15 | H | CH2CF3 | 0.84 ± 0.012 | 21 ± 0.94 | 180 ± 13 | 25 | 210 | |
| 16a | H | CH2CH2CH3 | 1.6 ± 0.096 | 17 ± 1.1 | 79 ± 3.6 | 11 | 49 | |
| 18a | H | COCH3 | 13 ± 1.1 | 57 ± 3.9 | 750 ± 64 | 4 | 58 | |
| 26 | H | C6H5 | 21 ± 0.20 | 180 ± 15 | 140 ± 11 | 9 | 7 | |
| 22 | H | C6H5CH2 | 2.1 ± 0.20 | 10 ± 1.3 | 41 ± 2.8 | 5 | 20 | |
| 21 | H | 3-CH3O-C6H5CH2 | 4.8 ± 0.51 | 9.1 ± 1.2 | 34 ± 2.8 | 2 | 7 | |
| 23 | H | 3-HO-C6H5CH2 | 3.5 ± 0.085 | 9.9 ± 0.53 | 29 ± 3.4 | 3 | 8 | |
| 24 | H | C(S)NHEt | 18 ± 1.6 | 130 ± 12 | 150 ± 13 | 7 | 8 | |
| 27 | H | 95 ± 9.7 | 610 ± 56 | > 10 μM | 6 | >110 | ||
| 28 | H | 110 ± 16 | 2700 ± 320 | > 10 μM | 25 | >91 | ||
| 17a | CH3 | CH3 | 0.45 ± 0.065 | 10 ± 1.0 | 65 ± 1.0 | 22 | 140 | |
| 14 | CH3 | COCH3 | 6.9 ± 1.0 | 110 ± 5.3 | 1100 ± 66 | 16 | 160 | |
| 20 | CH2CH3 | COCH3 | 13 ± 1.6 | 86 ± 5.1 | 740 ± 51 | 4 | 57 | |
| 13b | H | CH3 | 2.4 ± 0.10 | 1.8 ± 0.029 | 47 ± 3.9 | 1 | 20 | |
| 19b | H | CH2CH3 | 3.7 ± 0.095 | 1.1 ± 0.15 | 91 ± 4.4 | 0.3 | 25 | |
| 13c | H | CH3 | 0.71 ± 0.077 | 7.4 ± 0.93 | 50 ± 6.3 | 10 | 70 | |
| 18c | H | COCH3 | 1.2 ± 0.11 | 15 ± 1.4 | 220 ± 16 | 13 | 180 | |
| 19c | H | CH2CH3 | 0.94 ± 0.13 | 4.0 ± 0.33 | 76 ± 6.8 | 4 | 81 | |
| 16c | H | CH2CH2CH3 | 5.8 ± 0.71 | 24 ± 2.9 | 120 ± 17 | 4 | 21 | |
| 17c | CH3 | CH3 | 11 ± 1.4 | 76 ± 4.1 | 420 ± 22 | 7 | 38 | |
Membranes from Chinese hamster ovary cells, expressing either the human κ, μ, or δ opioid receptors, were incubated with 12 different concentrations of the compounds in the presence of receptor-specific radioligands at 25 °C, in a final volume of 1 mL of 50 mM Tris-HCl, pH 7.5. Nonspecific binding was determined using 10 μM naloxone.
Herein we report the synthesis and pharmacological evaluation of a series of N-substituted (R3) and N′-3-amino-substituted (R1, R2) analogs of morphinan. The highly potent (−)-3-hydroxy-N-(E)-iodoallylmorphinan14 suggested the replacement of the N-cyclopropylmethyl group in cyclorphan with a fluoropropyl group to make compounds 7c and its analogs 9c, 11, 13c, and 19c, and introduced a trifluoroethyl substituent to the amino group of the aminothiazole component in 3 to make compound 15 (Scheme 1 and Scheme 2).
Scheme 1.

areagents and conditions: (i) cyclopropylmethyl bromide, cyclobutylmethyl bromide, fluoropropyl bromide, or (−) – (s) tetrahydrofurfuryl (R)-camphor-10-sulfonate, K2CO3, DMF; (ii) PhNTf2, Et3N, CH2Cl2; (iii) Ph2C=NH, BINAP, Pd(OAc)2, Cs2CO3, THF; (iv) NaOAc, NH2OH.HCl, MeOH; (v) KSCN, Br2, AcOH.
Scheme 2.

areagents and conditions: (i) HCOOH, Ac2O, THF; (ii) LiAlH4, THF; (iii) Ac2O, pyridine; (iv) CF3CO(O)OCCF3, Et3N, toluene; (v) propionaldehyde, CH3CN; (vi) NaBH4, MeOH; (vii) (CHO)n, NaBH4, TFA, THF.
Chemistry
The synthesis of all target compounds was initiated from commercially available levorphanol tartrate, which, after conversion to its free base, could be demethylated to norlevorphanol (6). Next, 6 was alkylated with either cyclopropylmethyl bromide, cyclobutylmethyl bromide, fluoropropyl bromide, or (−)-(s) tetrahydrofurfuryl (R)-camphor-10-sulfonate to yield 7a–d, respectively. Subsequent triflation of morphinans 7a–d afforded triflates 8a–d, which were subjected to palladium-catalyzed amination to afford amines 9a–d in moderate yields. The aminothiazoles 3, 10–12 were then synthesized in 55–61% yield according to literature procedure (Scheme 1).6, 10
For the synthesis of N′-methyl substituted aminothiazolomorphinans 13a–c, aminothiazolomorphinans 3, 10 and 11 were formylated with freshly prepared formyl acetate (prepared by heating a mixture of HCOOH and Ac2O), followed by reduction, yielding the novel N′-methyl-3-aminothiazolomorphinans 13a–c in 34–45% yields (Scheme 2).15 Treatment of 13a and 13c with paraformaldehyde and NaBH4 yielded dimethyl substituted aminothiazolomorphinans 17a and 17c in 83–89% yields.16 N′-Trifluoroethyl derivative 15 was prepared in 45% yield by treating 3 with trifluoroacetic anhydride in the presence of Et3N, followed by reduction.17 N′-ethyl substituted aminothiazolomorphinans 19a–c were prepared analogously 17 in which compound 3, 10, and 11 were first acylated and then reduced. Treatment of 13a and 19a with acetic anhydride produced N′-disubstituted derivatives 14 and 20. Compounds 3 and 11 were also condensed with propionaldehyde, followed by reduction of the resulting imines to N′-propyl substituted aminothiazolomorphinans 16a and 16c in 47–55% yields18 (Scheme 2).
Using literature procedures,19 3 was reductively aminated to afford 21 and 22 in 65–68% yields. Methoxybenzylated derivative 21 was demethylated with BBr3 to give 23 in 74% yield.20 Furthermore, 3 was treated with EtSCN to yield thiourea 24 in 45% yield (Scheme 3).21
Scheme 3.

areagents and conditions: (i) benzaldehyde, p-toluenesulfonic acid, toluene; (ii) NaBH4, MeOH; (iii) m-anisaldehyde, p-toluenesulfonic acid, toluene; (iv) BBr3, CH2Cl2; (v) EtSCN, Et3N, Toluene.
For preparation of aryl substituted derivatives 26 and 27, 3 was converted to 3-bromothiazolo-N-cyclopropylmethylmorphinan 25 through the Sandmeyer reaction.22 Compound 25 was treated with aniline and 2-aminopyridine, respectively, to yield 26 and 27 in 70–72% yields. Treatment of 25 with piperazine produced 28 in 63% yield (Scheme 4). 23
Scheme 4.

areagents and conditions: (i) CuBr2/t-Butyl nitrite, CH3CN; (ii) NaH, THF; (iii) piperazine, NaH, THF.
Results and Discussion
Target compounds were screened for their affinity and selectivity for μ, κ, and δ opioid receptors with Chinese hamster ovary (CHO) cell membranes stably expressing the human opioid receptors. The data were summarized in Table 1. For comparison purposes, opioid binding affinity data for cyclorphan 1, 2, 3, 4 and N-methyl-3-aminothiazolomorphinan (29) were also included.
Previous reports from our laboratories indicated that changing N-substituted group (R3) in the aminothiazolomorphinan drastically altered potency and efficacy. Compared to the N-methyl derivative 29, the N-cyclopropyl compound 3 displayed a much higher (130-fold) affinity at κ the receptor. From the data shown in Table 1, the N-fluoropropyl derivative 11 had high affinity at κ (0.30 nM) and good selectivity for κ over μ (9-fold) and δ (180-fold) receptor. N-Tetrahydrofurylmethylmorphinan 12 also showed high affinity at κ (0.83 nM) and moderate affinity at μ (2.4 nM). Introducing a small alkyl group at N′, 13a had similar affinity with 3 at the κ receptor, with Ki value of 0.066 nM. Compound 13a also displayed high selectivity for κ over μ (45-fold) and δ (380-fold) receptors. When the size of alkyl group at N′ increased, we observed a smooth decrease in affinity at κ and μ receptors in 19a, 15, 16a. However, they still displayed high affinity at κ (Ki = 0.15–1.6 nM). N′-Acetyl aminothiazolomorphinan 18a showed low affinity at κ (13 nM) and μ (57 nM), perhaps due to the lowered basicity of nitrogen in this analogue. When the N′-substituent on amine was either benzyl (22), 3-OH-benzyl (23), or 3-MeO-benzyl (21), binding affinities were low [Ki = 2.1–4.8 nM (κ) and Ki = 9.1–10 nM (μ)]. Analogues 26 and 27, which contained N′-aryl and (hetero)aryl groups, were prepared. Compared to the alkyl substituted aminothiazole analogues (13a, 15, 19a), an unexpected decrease of affinity at κ and μ receptors was observed in 26 and 27. The N′-piperazine substituted aminothiazolomorphinan displayed very low affinity at κ (110 nM) and at μ (2700 nM). To explore the possibility that incorporation of an additional polar group into the N′-substitution would further enhance affinity, we prepared a thiourea analogue as a probe. However, the N′-ethylthiourea analogue displayed low affinity at κ (18 nM) and at μ (130 nM).
It was found that N′-disubstituted aminothiazolo-N-cyclopropylmorphinans generally had lower affinity when compared to N′-monosubstituted- aminothiazolo-N-cyclopropylmorphinans. N′-Dimethyl substituted derivative (17a) was the most potent compound in the series of N′-disubstituted compounds synthesized, with an affinity of 0.45 nM at the κ receptor and 10 nM at the μ receptor. Ki values for N′-methyl derivative (13b) and N′-ethyl derivative (19b) were 2.4 and 3.7 nM for κ, respectively. N-fluoropropyl N′-methyl (13c) and N′-ethyl (19c) morphinan analogues were very potent, with Ki values being <1 nM for binding to the κOR. N-fluoropropyl N′-propyl (16c) and N′-dimethyl (17c) morphinan analogues showed low affinity at the κ and μ receptors. From a SAR perspective, the binding affinities of substituted aminothiazolomorphinan analogues at all three receptors were generally lower than the binding affinities of the aminothiazole precursors (3, 10, 11). However, most of the N′-monosubstituted analogues showed high affinities at κ (Ki = 0.06–0.94 nM).
To characterize the relative efficacy of these ligands, 1, 3, and 10, were selected for the [35S]GTPγS assay. The stimulation and inhibition of [35S]GTPγS binding mediated by κ and μ opioid receptors are shown in Table 2 and Table 3, respectively.
Table 2.
Pharmacological properties of compounds in stimulating [35S]GTPγS binding mediated by the κ opioid receptor a
| compound | Emax ± SEM (% maximal stimulation) | EC50 ± SEM nM) |
|---|---|---|
| (−)-U50,488 | 110 ± 2.0 | 46 ± 16 |
| 1b (Cyclorphan) | 90 ±10 | 0.2 ± 0.0 |
| 3b (ATPM) | 80 ± 6 | 2.4 ± 0.6 |
| 10b (ATBM) | 80 ± 1 | 29 ± 4 |
| 11 | 100 ± 4.8 | 32 ± 5.5 |
| 12 | 190 ± 21 | 14 ± 1.5 |
| 13a | 82 ± 10 | 14 ± 3.1 |
| 13c | 170 ± 6.3 | 120 ± 6.7 |
| 15 | 120 ± 0.33 | 120 ± 16 |
| 17a | 110 ± 14 | 46 ± 8.9 |
| 19a | 190 ± 6.4 | 22 ± 5.1 |
| 19c | 150 ± 3.9 | 89 ± 8.0 |
Membranes from CHO cells that stably expressed the human κ opioid receptor were incubated with varying concentrations of the compounds in the presence of 0.08 nM [35S]GTPγS. Data are the mean values ± SEM from three experiment, performed in triplicate. None of the compounds inhibited U50,488-stimulated [35S]GTPγS binding, indicating that the compounds were agonists devoid of any antagonist properties at the κ opioid receptor.
Table 3.
Agonist and antagonist properties of compounds in stimulating [35S]GTPγS binding mediated by the μ opioid receptora
| compound | pharmacological properties | Emax (% maximal stimulation) | EC50 (nM) | Imax (% maximal inhibition) | IC50 (nM) |
|---|---|---|---|---|---|
| DAMGO | Agonist | 120 ± 12 | 110 ± 9.0 | NIa | NI |
| 1b (Cyclorphan) | Partial agonist | 40 ± 2.9 | 0.8 ± 0.1 | 50 ± 1.2 | 1.7 ± 0.40 |
| 3b (ATPM) | Agonist | 45 ± 4 | 73 ± 5 | NI | NI |
| 10b (ATBM) | Partial agonist | 26 ± 1 | > 1 μM | 29 ± 7.4 | 85 ± 5.8 |
| 11 | Partial agonist | 65 ± 1.4 | 130 ± 11 | 40 ± 1.4% inhibition at 10 μM | NA |
| 12 | Agonist | 120 ± 15 | 71 ± 4.2 | NI | NI |
| 13a | Agonist | 40 ± 0.87 | 47 ± 9.0 | NI | NI |
| 13c | Agonist | 100 ± 5.3 | 420 ± 17 | NI | NI |
| 17a | Partial agonist | 57 ± 4.7 | 140 ± 6.5 | 40 ± 0.73% inhibition at 10 μM | NA |
| 19a | Agonist | 83 ± 2.5 | 29 ± 4.6 | NI | NI |
| 19c | Agonist | 97 ± 2.9 | 240 ± 12 | NI | NI |
Membranes from CHO cells that stably expressed only the μ opioid receptor were incubated with varying concentrations of the compounds in the presence of 0.08 nM [35S]GTPγS. Data are the mean values ± SEM from three experiment, performed in triplicate. NI = no inhibition; NA = not applicable; no value could be determined because a maximal inhibition of binding was not obtained.
These ligands produced maximal stimulation of [35S]GTPγS binding (Emax) at κ comparable to that of ligand 3. Ligands 13c, 15, 19a, and 19c produced a higher Emax than that of selective agonist U50,488. None of these compounds inhibited U50,488-stimulated [35S]GTPγS at κ, demonstrating that all of these ligands were full κ agonists.
From the data shown in Table 3, ligands 11, 13a, and 17a displayed partial agonist activity at μ receptor. Ligands 12, 13c, 19a and 19c showed full agonist activity at the μ receptor; they did not inhibit DAMGO-stimulated [35S]GTPγS binding.
Conclusion
We have extended the structure-activity relationships of aminothiazolomorphinans by introducing different groups to N- and N′-positions. A series of aminothiazolomorphinans and their N′-mono- and di-substituted derivatives were synthesized, and their pharmacological properties at opioid receptors were evaluated. It was found that substituents at the aminothiazole nitrogen tended to reduce the affinity of the compounds, with the exception of the methyl group (13a), which retained high affinity at the κ receptor (0.066 nM) as well as good selectivity for κ over μ (45-fold) and δ (380-fold) receptor. N′-disubstituted aminothiazolo-N-cyclopropylmorphinan analogues 17a, 14, 20, and 17c had lower affinity at all three opioid receptors. However, N′-dimethyl aminothiazolo-N-cyclopropylmorphinan 17a was also a potent and selective compound, with an affinity of 0.45 nM at the κ receptor and 10 nM at the μ receptor. The same pattern was observed with the replacement of the cyclopropylmethyl group in 1 with the fluoropropyl group. 13a, 19a, and 17a may prove to be useful for the potential development as medications for cocaine or opioid abuse. The [35S]GTPγS binding assay revealed that all new compounds were full agonists at the κ receptors, ligands 11, 13a, and 17a were partial agonists at the μ receptors, and ligands 12, 13c, 19a, and 19c were full agonists at the μ receptors. Preliminary evaluation of 3 in non-human primates reduced self-administration and attenuated food intake, probably due to its kappa agonist properties.24
Experimental Section
General Synthetic Methods
1H (and 13C NMR) spectra were recorded at 300 MHz (75 MHz) on a Varian Mercury 300 spectrometer. Chemical shifts are given as δ value (ppm) downfield from tetramethylsilane as an internal reference. Melting points were determined on a Thomas-Hoover capillary tube apparatus and are reported uncorrected. Elemental analyses, performed by Atlantic Microlabs, Atlanta, GA, were within (0.4% of theoretical values. Analytical thin-layer chromatography (TLC) was carried out on 0.2 micrometer Kieselgel 60F-254 silica gel plastic sheets (EM Science, Newark, NJ). Flash chromatography was used for the routine purification of reaction products. Eluent systems are described for the individual compounds.
General Procedure6 for the Preparation of 3-Hydroxy-N-alkyl-morphinans 7a–d
The mixture of norlevorphanol (5 mmol), K2CO3 or NaHCO3 (7.5 mmol), and either bromomethyl cyclopropane, bromomethyl cyclobutane, 1-bromo-3-fluoropropane, or (S)-tetrahydrofurfuryl (1R)-camphor-10-sulfonate (7.5 mmol) in 20 mL anhydrous DMF were stirred at 90–95 °C for overnight. After the reaction was judged complete by TLC, the reaction mixture was cooled, poured into water, extracted with CHCl3. The organic phase washed by brine, dried over anhydrous Na2SO4, and concentrated in vacuo to give crude product, purified by flash silica gel column (DCM: MeOH = 20:1 – 5:1) to give the corresponding morphinans 7a–d. The analytical data for 7a–b, 7d was in agreement with literature values. 6
3-Hydroxy-N-fluoropropylmorphinan (7c)
White crystals (73%); M.p. 148–150 °C. 1H NMR (300 MHz, CDCl3) δ 7.04-6.88 (m, 1H), 6.71 (s, 1H), 6.66-6.55 (m, 1H), 4.64-4.53 (m, 1H), 4.49-4.37 (m, 1H), 2.97-2.83 (m, 2H), 2.71-2.48 (m, 5H), 2.33-2.24 (m, 1H), 2.15-2.01 (m, 1H), 1.97-1.58 (m, 5H), 1.54-1.07 (m, 7H). 19F NMR (282 MHz, CDCl3) δ 29.21 (m). 13C NMR (75 MHz, CDCl3) δ 154.35, 141.81, 128.72, 113.03, 111.91, 82.71 (d, J = 163.5 Hz), 56.44, 50.92 (d, J = 5.2 Hz), 45.61, 44.69, 41.63, 37.66, 36.51, 28.50 (d, J = 21.5 Hz), 26.82, 26.48, 24.20, 22.20, 22.11.
General Procedure6,10 for the Preparation of Triflates 8a–d
3-Hydroxy-N-alkylmorphinan 7a–d (3.5 mmol), was dissolved in anhydrous DCM (20 mL) and Et3N (3.5 mL). The mixture was cooled to 0 °C, and then PhNTf2 (1.94 g, 5.4 mmol) was added. The mixture was allowed to warm to rt overnight. The solution was diluted with DCM (40 mL), washed with 1N HCl followed by brine, and then dried with anhydrous Na2SO4. The solvent was removed in vacuo to afford the crude product, which was purified by flash silica gel column to give corresponding triflates. The analytical data for 8a–b was in agreement with literature values. 6
N-(Fluoropropyl)-morphinan-3-yl Trifluoromethanesulfonate (8c)
Yellow oil (99%). 1H NMR (300 MHz, CDCl3) δ 7.17 (d, J = 8.6, 1H), 7.12 (d, J = 2.5, 1H), 7.02 (dd, J = 2.6, 8.4 Hz, 1H), 4.69-4.37 (m, 2H), 2.99 (d, J = 18.6 Hz, 1H), 2.93-2.85 (m, 1H), 2.74- 2.50 (m, 4H), 2.29 (d, J = 14.1 Hz, 1H), 2.07-1.51 (m, 7H), 1.47-1.14 (m, 5H), 1.11-0.98 (m, 1H). 19F NMR (282 MHz, CDCl3) δ 29.21 (m), -73.22. 13C NMR (75 MHz, CDCl3) δ 148.38, 143.51, 138.36, 129.33, 118.76 (d, J = 318.7 Hz), 118.23, 118.15, 82.58 (d, J = 162.8 Hz), 56.16, 50.77 (d, J = 5.2 Hz), 45.01, 44.65, 41.75, 38.10, 36.44, 28.80 (d, J = 19.5 Hz), 26.66, 26.37, 24.81, 21.85.
N-((S)-tetrahydrofurfuryl)-morphinan-3-yl Trifluoromethanesulfonate (8d)
Yellow oil (95%). 1H NMR (300 MHz, CDCl3) δ 7.18 (d, J = 5.8 Hz, 1H), 7.12 (d, J = 1.8 Hz, 1H), 7.04 (m, 1H), 4.31-4.10 (m, 2H), 3.92-3.64 (m, 3H), 3.24-2.72 (m, 5H), 2.52-0.89 (m, 15H).
General Procedure6,10 for the Preparation of Aminomorphinans 9a–d
The triflate 8a–d (900 mg, 1.92 mmol) in 20 mL THF was added Pd(OAc)2 (21 mg, 0.096 mmol), BINAP (95.4 mg, 0.151 mmol), CsCO3 (936 mg, 2.88 mmol), and benzophenone imine (150 mg, 420 uL, 2.49 mmol) under N2. The mixture was heated to reflux with stirring overnight. When starting material was consumed, the solvent was removed. The residue was diluted with EtOAc, washed with brine, dried and concentrated. The crude product was purified by flash silica gel column to yield the imine intermediate as yellow oil. To a solution of the imine intermediate in MeOH (50 mL) at rt was added NaOAc (654 mg, 8.4 mmol) and hydroxylamine hydrochloride (85 mg, 3.9 mmol). The mixture was stirred at room temperature for 36h. The solvent was removed and the residue was directly purified by flash silica gel column to yield corresponding amine. The analytical data for 9a–b was in agreement with literature values. 6,10
3-Amino-N-fluoropropyl-morphinan (9c)
Yellow oil (68%); 1H NMR (300 MHz, CDCl3) δ 6.88 (d, J = 8.0, 1H), 6.61 (s, 1H), 6.55-6.44 (m, 1H), 4.69-4.32 (m, 2H), 3.52 (s, 2H), 2.87 (d, J = 18.0 Hz, 2H), 2.73-2.44 (m,, 4H), 2.30 (d, J = 9.2 Hz, 1H), 2.16-2.03 (m, 1H), 1.96-1.60 (m, 5H), 1.50 (s, 1H), 1.44-1.24 (m, 5H), 1.22-1.09 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 144.44, 141.32, 128.38, 127.88, 113.15, 111.86, 82.81 (d, J = 165.0 Hz), 56.51, 50.83 (d, J = 5.2 Hz), 45.54, 45.32, 42.03, 37.58, 36.58, 28.85 (d, J = 19.5 Hz), 26.88, 26.61, 24.23, 22.28.
3-Amino-N-(S)-tetrahydrofurfuryl-morphinan (9d)
Yellow oil (41%); 1H NMR (300 MHz, CDCl3) δ 6.88 (d, J = 5.8 Hz, 1H), 6.59 (d, J = 1.8 Hz, 1H), 6.50-6.47 (m, 1H), 4.02-3.96 (m, 1H), 3.88-3.71 (m, 2H), 3.50 (br, 2H), 3.94-2.86 (m, 2H), 2.68-2.48 (m, 4H), 2.28 (m, 1H), 2.16-1.10 (m, 15H). 13C NMR (75 MHz, CDCl3) δ 144.31, 141.30, 128.22, 127.97, 112.99, 111.71, 77.47, 67.91, 60.06, 57.04, 45.93, 44.82, 41.77, 37.27, 36.42, 30.15, 26.72, 26.50, 25.30, 24.48, 22.19.
General Procedure6,10 for the Preparation of Aminothiazolomorphinans 3, 10-12
The amine (1.1 mmol) and KSCN (426 mg, 4.4 mmol) were dissolved in 10 mL glacial acetic acid. A solution of Br2 (180 mg, 1.1 mmol) in 2 mL of glacial acetic acid was added dropwise. The mixture was stirred for 48h, then basified with 10% NaOH and extracted with CHCl3. The organic layer was washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by flash silica gel column to yield corresponding aminothiazole. The analytical data for 3, 10 was in agreement with literature values. 10c
Aminothiazolo[5,4-b]-N-fluoropropylmorphinan (11)
Slightly yellow solid (52%), M.p. 114–117 °C; 1H NMR (300 MHz, CDCl3) δ 7.46 (s, 1H), 7.32 (s, 1H), 5.33 (s, 2H), 4.68-4.37 (m, 2H), 3.04 (d, J = 18.1 Hz, 1H), 2.92-2.82 (m, 1H), 2.80-2.38 (m, 5H), 2.13-1.99 (m, 1H), 1.96-1.59 (m, 5H), 1.55-1.29 (m, 6H), 1.22-1.03 (m, 1H). 19F NMR (282 MHz, CDCl3) δ 9.72 (m). 13C NMR (75 MHz, CDCl3) δ 164.97, 151.21, 139.04, 132.40, 129.09, 119.43, 115.92, 82.77 (d, J = 163.5 Hz), 56.52, 58.85 (d, J = 5.2 Hz), 45.39, 45.20, 42.40, 37.84, 36.81, 28.85 (d, J = 18.8 Hz), 26.90, 26.60, 25.13, 22.11. Anal. Calcd. for C20H26FN3S · 3HCl · 0.5H2O: C, 50.27; H, 6.33; N, 8.79. Found: C, 50.49; H, 6.58: N, 8.63.
Aminothiazolo[5,4-b]-N-(S)-tetrahydrofurylmethylmorphinan (12)
White solid (46%), M.P. 127–130 °C; 1H NMR (300 MHz, CDCl3) δ 7.46 (s, 1H), 7.32 (s, 1H), 5.23 (s, 2H), 3.88 (m, 3H), 3.02 (m, 2H), 2.60 (m, 5H), 1.93 (m, 7H), 1.44 (m, 7H), 1.11 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 164.83, 151.13, 139.23, 132.76, 129.03, 119.41, 115.95, 77.65, 68.08, 60.22, 57.01, 45.98, 44.94, 42.30, 37.66, 36.77, 30.28, 26.87, 26.61, 25.42 (2C), 22.14. Anal. Calcd. for C22H29N3OS · 2HCl · 1.4H2O: C, 54.86; H, 7.07; N, 8.72; Found: C, 54.85; H, 7.15: N, 8.44.
General Procedure for Synthesis of N′-Methyl-aminothiazolomorphinans 13a–c
At room temperature and under nitrogen atmosphere, freshly made HCOOAc (0.7 ml, 5.0 mmol, this reagent was prepared by heating a mixture of 1.8 mL HCOOH and 3.8 mL HOAc at 50 °C for two hours) was slowly added to a solution of 3-aminothiazolomorphinan (0.88 mmol). The mixture was stirred at room temperature for 24h. The resulting mixture was then concentrated to dryness and directly separated by flash silica gel column to give the intermediate formate. The intermediate (0.65 mmol) was dissolved in 5 mL anhydrous THF followed by addition of LiAlH4 (50 mg, 1.3 mmol, added in one portion at 0 °C). Then resulting suspension was stirred at room temperature for 16h. After reaction was judged to be complete by TLC, 1 mL of water was added slowly to quench the reaction and followed by addition of 1 mL aqueous 2 N NaOH. The resulting olution was diluted with 50 mL of CH2Cl2 and washed with water and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by flash silica gel column to give corresponding morphinans.
N′-Methylaminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (13a)
Slightly yellow foam (39%), M.P. (HCl salt) 212–215 °C; 1H NMR (300 MHz, CDCl3) δ 7.50 (s, 1H), 7.30 (s, 1H), 5.27 (br, 1H), 3.19-3.07 (m, 4H), 3.05-2.94 (m, 1H), 2.81-2.64 (m, 2H), 2.58-2.28 (m, 3H), 2.11-1.97 (m, 1H), 1.96-1.74 (m, 2H), 1.69-1.59 (m, 1H), 1.55-1.32 (m, 6H), 1.25 (s, 1H), 0.96-0.80 (m, 1H), 0.58-0.45 (m, 2H), 0.19-0.07 (m, 2H).13C NMR (75 MHz, CDCl3) δ 167.26, 151.62, 139.00, 131.58, 127.90, 119.31, 115.58, 59.94, 55.86, 45.73, 45.08, 42.22, 37.90, 36.82, 31.57, 26.94, 26.65, 24.65, 22.19, 9.36, 4.09, 3.64; Anal. Calcd. for C22H29N3S · 2HCl · 1.3H2O: C, 56.96; H, 7.30; N, 9.06. Found: C, 56.99; H, 7.14; N, 8.84.
N′-Methyl-aminothiazolo[5,4-b]-N-cyclobutylmethylmorphinan (13b)
White solid (45%), M.p. (HCl salt) >217 °C (dec). 1H NMR (300 MHz, CDCl3) δ 7.49 (s, 1H), 7.32 (s, 1H), 5.33 (s, 1H), 3.14-3.01 (m, 4H), 2.89-2.37 (m, 6H), 2.15-1.58 (m, 11H), 1.53-1.27 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 167.31, 151.57, 138.98, 131.65, 127.86, 119.33, 115.54, 61.48, 55.98, 45.86, 45.01, 42.20, 37.74, 36.79, 34.85, 31.57, 27.91, 26.94, 26.61, 24.80, 22.17, 18.83; Anal. Calcd for C23H31N3S · 2HCl · 1.1H2O: C, 58.24; H, 7.48; N, 8.86. Found: C, 58.38; H, 7.47; N, 8.56.
N′-Methyl-aminothiazolo[5,4-b]-N-fluoropropylmorphinan (13c)
Slightly yellow foam (55%), M.p. (HCl salt) 205–207 °C (dec); 1H NMR (300 MHz, CDCl3) δ 7.50 (s, 1H), 7.31 (s, 1H), 5.22 (br, 1H), 4.68-4.37 (m, 2H), 3.11 (s, 3H), 3.03 (d, J = 18.1 Hz, 1H), 2.92-2.83 (m, 1H), 2.80-2.40 (m, 5H), 2.14-2.01 (m, 1H), 1.97-1.59 (m, 5H), 1.54-1.10 (m, 7H). 13C NMR (75 MHz, CDCl3) δ 167.29, 151.66, 138.93, 131.57, 127.97, 119.35, 115.62, 82.78 (d, J = 162.8 Hz), 56.59, 50.87 (d, J = 5.2 Hz), 45.44, 45.25, 42.41, 37.88, 36.83, 31.58, 28.86 (d, J = 19.5 Hz), 26.94, 26.64, 25.11, 22.20; Anal. Calcd for C21H28FN3S·2HCl·1.5H2O: C, 53.27; H, 7.03; N, 8.87. Found: C, 53.38; H, 7.20; N, 8.75.
Synthesis of N′-Acetyl-N′-methyl-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (14)
A solution of N′-Methyl-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (40mg, 0.11 mmol), acetic anhydride (0.5 mL) and pyridine (2 mL) was stirred at room temperature for 5h. After reaction was over, the volatile components were removed in vacuo. The residue was purified by flash silica gel column (Hexane:EtOAc: Et3N = 10:10:0.5) to afford a white solid (40 mg, 90%). M.p. 67–70 °C; 1H NMR (300 MHz, CDCl3) δ 7.76 (s, 1H), 7.51 (s, 1H), 3.79 (s, 3H), 3.15-3.06 (m, 2H), 2.82-2.68 (m, 2H), 2.54-2.48 (m, 2H), 2.45 (s, 3H), 2.36-2.29 (m, 2H), 2.04-1.78 (m, 3H), 1.64 (m, 1H), 1.55-1.11 (m, 6H), 0.88 (m, 1H), 0.52-0.49 (m, 2H), 0.16-0.07 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 170.55, 159.06, 147.21, 139.53, 134.33, 130.78, 119.28, 117.78, 59.97, 55.81, 45.67, 45.16, 42.57, 38.08, 36.87, 35.85, 26.95, 26.64, 24.87, 23.54, 22.19, 9.40, 4.04, 3.59; Anal. Calcd for C25H33N3OS·0.1H2O: C, 70.58; H, 7.87; N, 9.88. Found: C, 70.38; H, 7.91; N, 9.72.
Synthesis of N′-Trifluoroethyl-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (15)
Et3N (0.6 mL) was added to the solution of aminothiazolo[5,4-b]-Ncyclopropylmethylmorphinan (71 mg, 0.2 mmol) in 1.5 ml toluene. Trifluoroacetic anhydride (0.6 mL) was added to the mixture, and then stirred at 100 °C for overnight. The volatile components were removed in vacuo. The residue was purified by flash silica gel column (EtOAc: MeOH: Et3N = 60:1:1) to afford the intermediate. LiAlH4 (10 mg, 0.25 mmol) was added to a solution of intermediate (50 mg, 0.11 mmol) in 2 mL THF. The mixture was stirred at room temperature overnight. Next, 0.2 mL water was added to quench the reaction, followed by the addition of 0.2 mL of 2 N aqueous NaOH. The resulting mixture was stirred for 30 min and then filtered. The resulting solid was washed with CH2Cl2 (2×2 mL). The filtrate was concentrated in vacuo. The residue obtained was purified by flash silica gel column (Hex: EtOAc: Et3N = 10:10:1) to afford a white foam (15mg, 31%), M.p. (HCl salt) >198 °C (dec); 1H NMR (300 MHz, CDCl3) δ 7.53 (s, 1H), 7.32 (s, 1H), 4.19 (m, 2H), 3.12 (m, 1H), 3.01 (d, J = 18.2 Hz, 1H), 2.72 (m, 2H), 2.42 (m, 3H), 1.91 (m, 3H), 1.44 (m, 8H), 0.89 (m, 1H), 0.51 (dd, J = 1.4, 8.0 Hz, 2H), 0.11 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 164.93, 150.72, 139.46, 132.84, 128.06, 119.43, 116.38, 59.99, 55.82, 45.98, 45.69, 45.08, 42.25, 37.99, 36.81, 26.95, 26.64, 24.76, 22.19, 9.39, 4.09, 3.63. Anal. Calcd for C23H28F3N3S · 2HCl · 1.9H2O: C, 50.90; H, 6.28; N, 7.74. Found: C, 51.10; H, 6.21; N, 7.24.
General Procedure for Synthesis of N′-Propyl-aminothiazolomorphinans 16a and 16c
The mixture of aminothiazolomorphinan (0.3 mmol), proponialdehyde (43 μL, 0.6 mmol) in 2 mL MeOH was stirred at 60 °C for overnight. The reaction was judged to be complete by TLC. Next, NaBH4 (45.6 mg, 1.2 mmol) was added, and the resulting mixture was stirred at 60 °C for 8h. After this period, the solvents were removed in vacuo. The residue was then directly purified by flash silica gel column to give corresponding morphinans.
N′-Propyl-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (16a)
White foam (55%), M.p. (HCl salt) 212–214 °C (dec). 1H NMR (300 MHz, CDCl3) δ 7.46 (s, 1H), 7.29 (s, 1H), 5.19 (s, 1H), 3.39 (s, 2H), 3.18-3.06 (m, 1H), 3.05-2.91 (m, 1H), 2.78-2.65 (m, 2H), 2.55-2.40 (m, 2H), 2.38-2.28 (m, 1H), 2.08-1.96 (m, 1H), 1.93-1.59 (m, 5H), 1.53-1.30 (m, 6H), 1.00 (t, J = 7.4 Hz, 3H), 0.86 (d, J = 6.7 Hz, 2H), 0.56-0.45 (m, 2H), 0.18-0.05 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 166.61, 151.60, 138.96, 131.51, 127.78, 119.28, 115.49, 59.94, 55.86, 47.11, 45.74, 45.10, 42.22, 37.90, 36.83, 26.95, 26.66, 24.66, 22.86, 22.19, 11.36, 9.37, 4.09, 3.64. Anal. Calcd for C24H33N3S · 2HCl · 2.1H2O: C, 56.93; H, 7.80; N, 8.30. Found: C, 57.19; H, 7.69; N, 7.84.
N′-Propyl-aminothiazolo[5,4-b]-N-fluoropropylmorphinan (16c)
White foam (47%), M.p. (HCl salt) 215-217 °C (dec). 1H NMR (300 MHz, CDCl3) δ 7.46 (s, 1H), 7.31 (s, 1H), 5.49 (s, 1H), 4.71-4.36 (m, 2H), 3.39 (t, J = 6.6 Hz, 2H), 3.07-2.41 (m, 6H), 2.07 (t, J = 11.6 Hz, 1H), 1.97-1.58 (m, 7H), 1.54-1.23 (m, 8H), 1.06-0.95 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 166.91, 151.59, 138.76, 131.22, 127.78, 119.33, 115.37, 82.73 (d, J = 163.5 Hz), 56.55, 50.82 (d, J = 5.2 Hz), 47.21, 45.44, 45.04, 42.24, 37.81, 36.77, 28.69 (d, J = 19.5 Hz), 26.90, 26.60, 25.08, 22.85, 22.16, 11.35. Anal. Calcd for C23H32FN3S · 2HCl · 0.9H2O: C, 56.29; H, 7.35; N, 8.56. Found: C, 56.24; H, 7.48; N, 8.19.
General Procedure for Synthesis of N′, N′-Dimethyl-aminothiazolomorphinans 17a and 17c
To a stirred mixture of N′-methyl-aminothiazolomorphinan (0.15 mmol), paraformaldehyde (44 mg, 1.5 mmol), and NaBH4 (28.8 mg, 0.76 mmol) in THF (3 mL) at rt under nitrogen atmosphere was added dropwise trifluoroacetic acid (1.5 mL). The resulting mixture was stirred at rt for 24h, then poured into a mixture of 25% aqueous NaOH (5 mL) and ice to make strongly alkaline solution, which was then diluted with saturated NaCl solution (5 mL), and extracted with CH2Cl2. The combined extracts were dried by anhydrous Na2SO4, filtered, and concentrated in vacuo to afford a yellow solid. The solid was treated with 10% HCl. The aqueous layer was washed with CH2Cl2, and then added 10% NaOH to make the free base. The resulting aqueous layer was extracted with CH2Cl2. The combined extracts were washed with brine, and then dried by anhydrous Na2SO4, filtered, and concentrated in vacuo to give corresponding morphinans.
N′, N′-Dimethyl-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (17a)
Pale yellow solid (89%), M.p. (HCl salt) 193–195 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 7.51 (s, 1H), 7.31 (s, 1H), 3.19 (s, 6H), 3.15-3.10 (m, 1H), 3.04-2.93 (m, 1H), 2.82-2.64 (m, 2H), 2.57-2.26 (m, 3H), 2.11-1.74 (m, 3H), 1.70-1.58 (m, 1H), 1.54-1.30 (m, 6H), 1.26-1.10 (m, 1H), 1.00-0.79 (m, 1H), 0.61-0.42 (m, 2H), 0.19-0.06 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 168.25, 152.29, 138.99, 128.52, 119.14, 115.36, 59.99, 55.95, 45.80, 45.15, 42.26, 40.21, 37.94, 36.82, 26.97, 26.67, 24.65, 22.27, 22.21, 9.38, 4.09, 3.65; Anal. Calcd for C22H29N3S · 3HCl · H2O: C, 54.27; H, 7.13; N, 8.26. Found: C, 54.47; H, 7.18: N, 8.16.
N′, N′-Dimethyl-aminothiazolo[5,4-b]-N-fluoropropylmethylmorphinan (17c)
Pale yellow solid (83%), M.p. (HCl salt) 198–200 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 7.51 (s, 1H), 7.33 (s, 1H), 4.72-4.33 (m, 2H), 3.19 (s, 6H), 3.10-2.42 (m, 6H), 2.19-2.05 (m, 1H), 2.01-1.71 (m, 4H), 1.69-1.31 (m, 8H), 0.97-0.74 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 168.31, 152.40, 138.65, 130.46, 128.68, 119.20, 115.36, 82.66 (d, J = 163.5 Hz), 56.71, 50.89 (d, J = 5.2 Hz), 45.55, 44.90, 42.12, 40.20, 37.79, 36.70, 29.68, 28.56 (d, J = 19.5 Hz), 26.88, 26.56, 25.04, 22.21; Analysis Calcd for C22H30FN3S·2HCl · 1.3H2O: C, 54.61; H, 7.21; N, 8.68. Found: C, 54.82; H, 7.30; N, 8.35.
General Procedure for Synthesis of N′-Acetyl-aminothiazolomorphinans 18a and 18c
The mixture of aminothiazolo-morphinan (0.41 mmol), pyridine (2.1 mL), and acetic anhydride (1.1 mL) was stirred at room temperature for 24h. The volatile components were removed in vacuo. The residue was purified by flash silica gel column to afford the corresponding morphinans.
N′-Acetyl-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (18a)
Slightly yellow solid (78%), M.p. 148–151 °C; 1H NMR (300 MHz, CDCl3) δ 7.67 (s, 1H), 7.54 (s, 1H), 3.20-3.03 (m, 2H), 2.86-2.67 (m, 1H), 2.60-2.33 (m, 2H), 2.29 (s, 3H), 2.01-1.82 (m, 2H), 1.77-1.59 (m, 4H), 1.56-1.27 (m, 6H), 1.17-1.09 (m, 1H), 0.93-0.81 (m, 1H), 0.57-0.46 (m, 2H), 0.18-0.08 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 168.90, 159.47, 147.08, 140.31, 139.91, 129.48, 120.16, 117.18, 60.28, 56.03, 45.87, 45.40, 42.89, 38.40, 37.22, 27.19, 26.86, 25.17, 23.68, 22.38, 9.47, 4.32, 3.82; Analysis Calcd for C23H29N3OS · 0.1H2O: C, 69.52; H, 7.41; N, 10.57. Found: C, 69.62; H, 7.58; N, 10.22.
N′-Acetyl-aminothiazolo[5,4-b]-N-fluoropropylmorphinan (18c)
Slightly yellow solid (60%), M.p. (HCl salt) 215–217 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 11.77 (s, 1H), 7.68 (s, 1H), 7.58 (s, 1H), 4.69-4.38 (m, 2H), 3.14 (d, J = 18.1 Hz, 1H), 2.98-2.39 (m, 5H), 2.28 (s, 3H), 2.10-1.98 (m, 1H), 1.96-1.08 (m, 13H). 19F NMR (282 MHz, CDCl3) δ 9.59 (m). 13C NMR (75 MHz, CDCl3) δ 168.62, 159.10, 146.86, 139.90, 134.48, 129.27, 119.97, 116.98, 82.69 (d, J = 162.8 Hz), 56.44, 50.85 (d, J = 5.2 Hz), 45.30, 45.13, 42.66, 38.05, 36.90, 28.83 (d, J = 19.5), 26.88, 26.56, 25.31, 23.48, 22.10; Anal. Calcd for C22H28FN3OS · 2HCl · 0.7H2O: C, 54.25; H, 6.50; N, 8.63. Found: C, 54.34; H, 6.41: N, 8.47.
General Procedure for Synthesis of N′-Ethyl-aminothiazolomorphinans 19a -c
At room temperature, a solution of N′-acetyl-aminothiazolomorphinan (N′-acetyl-2′-aminothiazolo-N-cyclobutylmorphinan 18b was prepared using same procedure with 12a) (0.31 mmol) in 1 mL of dry THF was added to a suspension of LiAlH4 (24 mg, 0.62 mmol) in 2 mL of dry THF. After 24h of stirring, 0.2 mL of water was added to quench the reaction followed by the addition of 0.2 mL of 2 N aqueous NaOH. The resulting mixture was then stirred for 30 min and filtered, and the resulting solid was washed with CH2Cl2. The filtrate was concentrated in vacuo. The resulting residue was then purified by flash silica gel column to afford the corresponding morphinans.
N′-Ethyl-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (19a)
White solid (86%), M.p. 95–98 °C; 1H NMR (300 MHz, CDCl3) δ 7.47 (s, 1H), 7.30 (s, 1H), 5.81 (s, 1H), 3.54-3.43 (m, 2H), 3.21-2.90 (m, 2H), 2.83-2.64 (m, 2H), 2.57-2.30 (m, 3H), 2.07- 1.75 (m, 2H), 1.54-1.05 (m, 11H), 1.00-0.78 (m, 1H), 0.60-0.39 (m, 2H), 0.13 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 166.61, 151.57, 138.73, 131.14, 127.73, 119.24, 115.29, 59.78, 55.81, 45.66, 44.85, 42.03, 40.06, 37.78, 36.74, 26.85, 26.56, 24.61, 22.10, 14.89, 9.16, 4.06, 3.63; Anal. Calcd for C23H31N3OS · 2HCl · 1.3H2O: C, 57.80; H, 7.51; N, 8.79. Found: C, 57.88; H, 7.50; N, 8.55.
N′-Ethyl-aminothiazolo[5,4-b]-N-cyclobutylmethylmorphinan (19b)
White solid (84%), M.p. (HCl salt) >220 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 7.47 (s, 1H), 7.31 (s, 1H), 5.23 (s, 1H), 3.57-3.42 (m, 2H), 3.06 (d, J = 17.9, 1H), 2.82 (s, 1H), 2.75-2.36 (m, 6H), 2.14-1.57 (m, 11H), 1.43-1.26 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 166.40, 151.53, 139.00, 131.71, 127.73, 119.29, 115.49, 61.55, 55.97, 45.85, 45.15, 42.30, 40.09, 37.77, 36.83, 34.97, 27.91, 26.96, 26.65, 24.81, 22.18, 18.85, 14.96; Anal. Calcd for C24H33N3S · 2HCl · 1. 4H2O: C, 58.38; H, 7.72; N, 8.51. Found: C, 58.70; H, 7.60; N, 8.13.
N′-Ethyl-aminothiazolo[5,4-b]-N-fluoropropylmorphinan (19c)
White solid (93%), M.p. 113–115 °C; 1H NMR (300 MHz, CDCl3) δ 7.48 (s, 1H), 7.31 (s, 1H), 5.24 (br, 1H), 4.68-4.38 (m, 2H), 3.57-3.40 (m, 2H), 3.03 (d, J = 18.1 Hz, 1H),2.92-2.85 (m, 1H), 2.80- 2.40 (m, 5H), 2.15-2.00 (m, 1H), 1.97-1.59 (m, 5H), 1.54-1.10 (m, 11H); 19F NMR (282 MHz, CDCl3) δ 9.24 (m); 13C NMR (75 MHz, CDCl3) δ 166.42, 151.64, 138.85, 131.48, 127.86, 119.32, 115.53, 82.78 (d, J = 162.8 Hz), 56.58, 50.88 (d, J = 5.2 Hz), 45.45, 45.21,42.37, 40.08, 37.85, 36.81, 28.82 (d, J = 19.5 Hz), 26.91, 26.62, 25.07, 22.17, 14.96; Anal. Calcd for C22H30FN3S · 2HCl · 1.8H2O: C, 53.61; H, 7.28; N, 8.52. Found: C, 53.63; H, 7.25; N, 8.46.
Synthesis of N′-Acetyl-N′-ethyl-amino-thiazolo[5,4-b]-N-cyclopropylmethylmorphinan (20)
A solution of N′-ethyl-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (55mg, 0.14 mmol), acetic anhydride (0.5 mL) and pyridine (2 mL) was stirred at room temperature for 24h. After reaction was judged complete by TLC, the volatile components were removed in vacuo. The resulting residue was purified by flash silica gel column (EtOAc: Et3N = 60:1) to afford a white solid (40 mg, 65%). M.p. 88–90 °C; 1H NMR (300 MHz, CDCl3) δ 7.75 (s, 1H), 7.51 (s, 1H), 4.31 (m, 2H), 3.14-3.06 (m, 2H), 2.81-2.67 (m, 2H), 2.54-2.48 (m, 2H), 2.45 (s, 3H), 2.04-1.78 (m, 3H), 1.64 (m, 1H), 1.54 (m, 1H), 1.48-1.36 (m, 10H), 0.88 (m, 1H), 0.54-0.49 (m, 2H), 0.13-0.09 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 170.19, 147.48, 139.44, 134.31, 130.82, 119.21,117.86, 60.02, 55.85, 45.69, 45.28, 43.66, 42.62, 38.10, 36.89, 26.96, 26.66, 24.88, 22.95, 22.19, 13.77, 9.48, 4.04, 3.56; Anal. Calcd for C24H31N3OS·0.2H2O: C, 69.76; H, 7.66; N, 10.17. Found: C, 70.00; H, 7.92; N, 9.80.
Synthesis of N′-Benzyl-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (22)
A mixture of ATPM (70 mg, 0.20 mmol), benzaldehyde (84 uL, 0.8 mmol) and a crystal of p-toluenesulfonic acid (20 mg) in 25 mL dry toluene was refluxed using a Dean-Stark apparatus for 18 h. Toluene was then removed in vacuo. The resulting residue was dissolved in 8 mL MeOH and NaBH4 (31 mg, 0.8 mmol) was added. The resulting mixture was refluxed for 6 h. MeOH was then removed in vacuo. The residue was then dissolved in 10 mL 1N HCl and washed with ethyl acetate (10 mL × 2), then basified with ammonium hydroxide until pH ~ 11 was reached. The aqueous solution was then extracted with CH2Cl2 (10 mL × 3). The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was removed in vacuo and the residue was purified by flash silica gel column to give product 15 as a white foam (43 mg, 62%). M.P. (HCl salt) >216 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 7.48 (s, 1H), 7.35 (m, 6H), 5.77 (s, 1H), 4.65 (s, 2H), 3.11 (m, 1H), 2.99 (d, J = 18.3, 1H), 2.72 (m, 2H), 2.41 (m, 3H), 1.91 (m, 3H), 1.62 (s, 1H), 1.31 (m, 7H), 0.89 (m, 1H), 0.51 (m, 2H), 0.11 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 166.37, 151.42, 139.10, 137.65, 131.85, 128.76, 127.89, 127.77, 127.63, 119.33, 115.69, 59.98, 55.86, 49.16, 45.73, 45.13, 42.26, 37.92, 36.82, 26.96, 26.66, 24.69, 22.20, 9.40, 4.09, 3.62; Anal. Calc. for C29H35N3OS ·2HCl·1.2H2O: C, 62.49; H, 7.00; N, 7.81. Found: C, 62.73; H, 6.92; N, 8.00.
Synthesis of N′-(3-Methoxybenzyl)-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (21)
A mixture of 3 (135 mg, 0.38 mmol), m-anisaldehyde (93 μL, 0.76 mmol) and a crystal of p-toluenesulfonic acid (20 mg) in 25 mL dry toluene was refluxed using a Dean Stark apparatus for 18 h. Toluene was then removed in vacuo. The residue was dissolved in 8 mL MeOH and NaBH4 (31 mg, 0.8 mmol) was added. The resulting mixture was refluxed for 6 h. and then the solvent was removed in vacuo. The residue was dissolved in 10 mL 1N HCl and washed with ethyl acetate (10 mL × 2), then basified with ammonium hydroxide until pH ~ 11 was reached. The aqueous solution was extracted with CH2Cl2 (10 mL × 3). The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated and the residue was purified by flash silica gel column to give product 21 as a white foam (122 mg, 68%), M.P. (HCl salt) 205-207 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 7.48 (s, 1H), 7.28 (m, 2H), 6.95 (m, 2H), 6.84 (dd, J = 2.2, 8.0 Hz, 1H), 5.77 (s, 1H), 4.62 (s, 2H), 3.79 (s, 3H), 3.05 (m, 2H), 2.71 (m, 2H), 2.40 (m, 3H), 1.74 (m, 11H), 0.88 (m, 1H), 0.50 (m, 2H), 0.10 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 166.35, 159.89, 151.37, 139.22, 139.13, 131.91, 129.81, 127.85, 119.81, 119.32, 115.67, 113.16, 60.02, 55.84, 55.22, 49.11, 45.74, 45.18, 42.31, 37.93, 36.83, 26.96, 26.66, 24.64, 22.20, 9.45, 4.09, 3.60; Anal. Calc. for C29H35N3OS · 2HCl·1.2H2O: C, 61.30; H, 6.99; N, 7.40. Found: C, 61.33, H, 7.08; N, 7.28.
Synthesis of N′-(3-Hydroxybenzyl)-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (23)
To the solution of compound 21 (80 mg, 0.17 mmol) in 4 mL CH2Cl2 was added BBr3 (1M in CH2Cl2, 3 mL) at 0 °C, then stirred at room temperature for 3.5 h. The reaction was quenched with MeOH, and then the solvent was removed in vacuo. The resulting dark oil was redissolved in MeOH and refluxed 15 min. MeOH was removed in vacuo and the residue was dissolved in 10 mL 1M HCl, washed with ethyl acetate twice, then basified with ammonium hydroxide, extracted with CH2Cl2, and dried over Na2SO4. The crude product was purified by flash silica gel column (EtOAc/MeOH/Et3N = 50/1/1) to give product 17 as a white solid (57 mg, 74%). M.P. 180–182 °C; 1H NMR (300 MHz, CDCl3) δ 7.42 (s, 1H), 7.29 (s, 1H), 7.17 (t, J = 7.8 Hz, 1H), 6.79 (m, 3H), 4.53 (s, 2H), 3.20 (m, 1H), 3.01 (d, J = 18.3 Hz, 1H), 2.78 (m, 2H), 2.46 (m, 3H), 2.09 (m, 1H), 1.54 (m, 11H), 0.56 (m, 2H), 0.16 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 168.15, 157.66, 151.66, 140.07, 138.77, 131.42, 130.14, 128.31, 119.94, 119.24, 115.19, 114.87, 114.81, 60.08, 56.21, 46.37, 45.00, 42.14, 38.22, 37.27, 27.43, 27.07, 25.04, 22.59, 8.95, 4.35, 4.18. Anal. Calc. for C28H33N3OS · 0.6H2O: C, 71.48; H, 7.33; N, 8.93. Found: C, 71.52; H, 7.44; N, 8.89.
Synthesis of N′-ethylthiourea-thiazolo[5,4-b]-N-cyclopropylmethylmorphinan (24)
To a solution of 3 (141 mg, 0.4 mmol) in dry toluene (6 mL) were added Et3N (33 μL, 0.22 mmol) and ethyl isothiocyanate (50 mg, 0.56 mmol). The reaction mixture was stirred in the microwave reactor (150W, 130 °C) for 150 mins. After cooled, the residue was directly purified by flash silica gel column (hexanes/EtOAc/Et3N 20/20/1) to give product 24 as a white foam (62 mg, 35%). M.P. (HCl salt) >255 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 11.10 (s, 1H), 7.62 (s, 1H), 7.41 (s, 1H), 3.94 – 3.76 (m, 2H), 3.16 (s, 1H), 3.05 (d, J = 18.5 Hz, 1H), 2.84-2.65 (m, 2H), 2.59-2.26 (m, 3H), 2.03-1.77 (m, 3H), 1.74-1.06 (m, 11H), 0.94- 0.82 (m, 1H), 0.52 (ps. d, J = 7.4 Hz, 2H), 0.21-0.06 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 177.54, 159.43, 148.79, 140.02, 134.81, 127.15, 119.60, 117.28, 59.93, 55.68, 45.60, 44.96, 42.35, 40.52, 38.03, 36.79, 26.89, 26.56, 24.89, 22.19, 14.01, 9.34, 4.09, 3.65; Anal. Calc. for C24H32N4S2 · HCl · 1.5H2O: C, 57.18; H, 7.20; N, 11.11. Found: C, 57.41; H, 7.07; N, 10.72.
Synthesis of 3-Bromo-thiazolo[5,4-b]-N-cyclopropylmethylmorphinan (25)
t-Butyl nitrite (76 uL, 0.65 mmol) was added to the solution of CuBr2 (145 mg, 0.65 mmol) in dry acetonitrile (8 ml). The reaction mixture was stirred for 10 minutes at room temperature. After 10 minutes 3 (115 mg, 0.32 mmol) was added in portions at 60 °C. The reaction mixture was left to stir at 60 °C for 30 minutes, and then an additional amount of t-butyl nitrite (76 uL, 0.65 mmol) was added. After 1.5 h of stirring at room temperature the reaction mixture was poured on water (30 ml) and extracted with CH2Cl2. The organic layer was dried over Na2SO4, filtered, concentrated. The residue was purified twice by flash silica gel column (first with hexanes/EtOAc 1/3 then with hexanes/EtOAc/Et3N 20/20/1) to give product 25 as a slightly yellow oil (60 mg, 45%). 1H NMR (300 MHz, CDCl3) δ 7.90 (s, 1H), 7.51 (s, 1H), 3.18-3.02 (m, 2H), 2.82-2.67 (m, 2H), 2.54-2.42 (m, 2H), 2.37-2.26 (m, 1H), 2.02-1.79 (m, 3H), 1.70-1.22 (m, 8H), 0.95-0.82 (m, 1H), 0.57-0.48 (m, 2H), 0.17-0.06 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 151.65, 140.60, 137.61, 136.57, 134.51, 119.49, 119.12, 60.01, 55.62, 45.51, 44.94, 42.44, 38.14, 36.80, 26.93, 26.56, 24.97, 21.98, 9.44, 4.09, 3.61.
General Procedure for Synthesis of N′-Aryl-aminothiazolomorphinans (26–27) and 3-(piperazin-1-yl)-thiazolo[5,4-b]-N-cyclopropylmethylmorphinan (28)
To a solution of either aniline, 2-aminopyridine or piperazine (1.32 mmol) in dry THF (4 ml), NaH (55 mg; 1.32 mmol, ω = 0.6) was added and stirred for 30 minutes at 50 – 60 °C. Next, 2′-bromo-thiazolo[5,4-b]-N-cyclopropylmethyl-morphinan 25 (137 mg, 0.33 mmol) was added and the reaction mixture was left to stir for 4 hours. The reaction mixture was then concentrated in vacuo. The residue was then dissolved in CH2Cl2 and washed with water and brine. The organic layer was dried over Na2SO4, filtered, concentrated. The crude product was purified by flash silica gel column to give the corresponding morphinans 26–28.
N′-Phenyl-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (26)
Slightly yellow foam (72%). M.P. (HCl salt) >212 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 7.55-7.46 (m, 3H), 7.38 (m, 3H), 7.18-7.08 (m, 1H), 3.13 (s, 1H), 3.02 (d, J = 18.8 Hz, 1H), 2.74 (m, 2H), 2.58-2.44 (m, 1H), 2.43-2.25 (m, 2H), 2.11-1.74 (m, 4H), 1.62 (s, 1H), 1.42 (m, 7H), 0.89 (m, 1H), 0.51 (ps. d, J = 7.5 Hz, 2H), 0.12 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 163.45, 150.64, 139.97, 139.33, 132.59, 129.47, 127.39, 123.95, 119.86, 119.30, 116.13, 59.98, 55.86, 45.74, 45.05, 42.28, 37.96, 36.78, 26.95, 26.64, 24.77, 22.21, 9.37, 4.11, 3.66. Anal. Calc. for C27H31N3S · HCl · 1.6H2O: C, 65.53; H, 7.17; N, 8.49. Found: C, 65.34; H, 6.87; N, 8.30.
N′-(Pyridin-2-yl)-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan (27)
Slightly yellow foam (39%); M.P. (HCl salt) >223 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 8.41 (d, J = 3.9, 1H), 7.63 (s, 1H), 7.57-7.48 (m, 2H), 6.99 (d, J = 8.3 Hz, 1H), 6.94-6.87 (m, 1H), 3.17 (s, 1H), 3.10 (d, J = 18.4 Hz, 1H), 2.89-2.68 (m, 2H), 2.59-2.50 (m, 1H), 2.42-2.32 (m, 2H), 2.11-1.23 (m, 11H), 0.96-0.85 (m, 1H), 0.55-0.49 (m, 2H), 0.16- 0.13 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 161.20, 151.72, 147.90, 146.89, 139.14, 137.73, 132.47, 129.54, 119.56, 116.78, 115.66, 111.11, 59.93, 55.89, 45.76, 45.03, 42.47, 38.02, 36.79, 26.96, 26.66, 24.83, 22.39, 9.33, 4.10, 3.66. Anal. Calcd. for C26H30N4S · 2HCl · 2H2O: C, 57.88; H, 6.73; N, 10.38. Found: C, 58.21; H, 6.95; N, 9.97.
N′-(Piperazin-1-yl)-thiazolo[5,4-b]-N-cyclopropylmethylmorphinan (28)
White foam (63%); M.P. (HCl salt) >245 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 7.50 (s, 1H), 7.33 (s, 1H), 3.63-3.55 (m, 4H), 3.23 (s, 1H), 3.02-2.95 (m, 4H), 2.85-2.73 (m, 2H), 2.70-2.37 (m, 4H), 2.17-1.79 (m, 3H), 1.70-1.30 (m, 8H), 0.98-0.95 (m, 1H), 0.62-0.47 (m, 2H), 0.24-0.11 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 168.61, 151.77, 138.81, 130.94, 128.07, 119.24, 115.68, 59.59, 55.81, 49.50, 45.66, 45.44, 44.50, 41.82, 37.78, 36.65, 26.85, 26.53, 24.74, 22.17, 8.90, 4.08, 3.75. Anal. Calcd. for C25H34N4S · 3HCl · 3.1H2O: C, 51.08; H, 7.41; N, 9.53. Found: C, 51.37; H, 7.49; N, 9.05.
Opioid binding to the human κ, δ, and μ opioid receptors
Chinese hamster ovary (CHO) cells stably transfected with the human κ opioid receptor (hKOR-CHO), δ-opioid receptor (hDOR-CHO) were obtained from Dr. Larry Toll (SRI International, Palo Alto, CA), and the μ-opioid receptor (hMOR-CHO) were obtained from Dr. George Uhl (NIDA Intramural Program, Baltimore, MD). The cells were grown in 100-mm dishes in Dulbecco’s modified Eagle’s media (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin–streptomycin (10,000 U/mL) at 37 °C in a 5% CO2 atmosphere. The affinity and selectivity of the compounds for the multiple opioid receptors were determined by incubating the membranes with radiolabeled ligands and 12 different concentrations of the compounds at 25 °C in a final volume of 1 mL of 50 mM Tris–HCl, pH 7.5. Incubation times of 60 min were used for the κ-selective peptide [3H]DAMGO and the j-selective ligand [3H]U69,593. A 3-h incubation was used with the δ-selective antagonist [3H]naltrindole.
[35S]GTPγS binding studies to measure coupling to G proteins
Membranes from CHO cells stably expressing either the human κ or μ opioid receptor were used in the experiments. Cells were scraped from tissue culture plates and then centrifuged at 1000g for 10 min at 4 °C. The cells were resuspended in phosphatebuffered saline, pH 7.4, containing 0.04% EDTA. After centrifugation at 1000g for 10 min at 4 °C, the cell pellet was resuspended in membrane buffer, which consisted of 50 mM Tris–HCl, 3 mM MgCl2, and 1 mM EGTA, pH 7.4. The membranes were homogenized with a Dounce homogenizer, followed by centrifugation at 40,000g for 20 min at 4 °C. The membrane pellet was resuspended in membrane buffer, and those transfected with the centrifugation step was repeated. The membranes were then resuspended in assay buffer, which consisted of 50 mM Tris–HCl, 3 mM MgCl2, 100 mM NaCl, and 0.2 mM EGTA, pH 7.4. The protein concentration was determined by the Bradford assay using bovine serum albumin as the standard. The membranes were frozen at −80 °C until used.
CHO cell membranes expressing either the human κ opioid receptor (15 μg of protein per tube) or μ opioid receptor (7.5 μg of protein per tube) were incubated with 12 different concentrations of the agonist in assay buffer for 60 min at 30 °C in a final volume of 0.5 mL. The reaction mixture contained 3 μM GDP and 80 pmol of [35S]GTPγS. Basal activity was determined in the presence of 3 μM GDP and in the absence of an agonist, and nonspecific binding was determined in the presence of 10 μM unlabeled GTPγS. Then, the membranes were filtered onto glass fiber filters by vacuum filtration, followed by three washes with 3 mL of ice-cold 50 mM Tris–HCl, pH 7.5. Samples were counted in 2 mL of Ecoscint A scintillation fluid. Data represent the percent of agoniststimulation [35S]GTPγS binding over the basal activity, defined as [(specific binding/basal binding) × 100] − 100. All experiments were repeated at least three times and were performed in triplicate. To determine antagonist activity of a compound at the μ opioid receptors, CHO membranes expressing the μ opioid receptor were incubated with the compound in the presence of 200 nM of the agonist DAMGO. To determine antagonist activity of a compound at the κ opioid receptors, CHO membranes expressing the κ opioid receptor were incubated with the compound in the presence of 100 nM of the κ agonist U50,488.
Acknowledgments
This work was supported by NIH grants R01-DA14251(J. L. N.), K05-DA00360 (J. M. B.) and T32 DA 007252 (A. W. S.).
Abbreviations
- κOR
kappa opioid receptor
- μOR
mu opioid receptor
- δOR
delta opioid receptor
- SAR
structure-activity relationship
- ATPM
aminothiazolo-N-cyclopropylmorphinan
- ATBM
aminothiazolo-N-cyclobutylmorphinan
Footnotes
Reported in part at the 72th College on Problems of Drug Dependence in Scottsdale Arizona, June, 2010.
References
- 1.Aldrich JV, Vigil-Cruz SC. Narcotic Analgesics. In: Abraham D, editor. Burger’sMedicinal Chemistry and Drug Discovery. 6 Chapter 7. John Wiley & Sons; New York: 2003. pp. 329–481. [Google Scholar]
- 2.(a) Neumeyer JL, Negus SS, Bidlack JM. Kappa Opioid Agonists as Targets for Pharmacotherapies in Cocaine Abuse. Pharm Acta Helv. 2000;74:337–344. doi: 10.1016/s0031-6865(99)00044-8. [DOI] [PubMed] [Google Scholar]; (b) Schenk S, Partridge B, Shippenberg T. Effects of the Kappa-Opioid Receptor Agonist, U69593 on the Development of Sensitization and on the Maintenance of Cocaine Self-Administration. Neuropsychopharmacology. 2001;24:441–450. doi: 10.1016/S0893-133X(00)00190-1. [DOI] [PubMed] [Google Scholar]; (c) Chefer V, Moron JA, Hope B, Rea W, Shippenberg TS. Kappa-Opioid Receptor Activation Prevents Alterations in Mesocortical Dopamine Neurotransmission That Occur during Abstinence from Cocaine. Neuroscience. 2000;101:619–627. doi: 10.1016/s0306-4522(00)00417-6. [DOI] [PubMed] [Google Scholar]
- 3.Archer S, Glick SD, Bidlack JM. Cyclazocine Revisited. Neurochem Res. 1996;21:1369–1373. doi: 10.1007/BF02532378. [DOI] [PubMed] [Google Scholar]
- 4.(a) Negus SS, Mello NK, Portoghese PS, Lin CE. Effects of Kappa Opioids on Cocaine Self-administration by Rhesus Monkeys. J Pharmacol Exp Ther. 1997;282:44–55. [PubMed] [Google Scholar]; (b) Mello NK, Negus SS. Effects of Kappa Opioids Agonists on Schedule-Controlled Behavior and Cocaine Self-Administration by Rhesus Monkeys. J Pharmacol Exp Ther. 1998;286:812–824. [PubMed] [Google Scholar]
- 5.(a) Gates M, Montzka TA. Some Potent Morphine Antagonists Possessing High Analgesic Activity. J Med Chem. 1964;7:127–131. doi: 10.1021/jm00332a002. [DOI] [PubMed] [Google Scholar]; (b) Gates M. U.S. 3,285,922, 1966; CAN 66:28955, AN 1967:28955 N-Cyclopropylmethyl- and -cyclobutylmethylmorphinans. ; (c) Zhang A, Xiong W, Bidlack JM, Hilbert JE, Knapp BI, Wentland MP, Neumeyer JL. 10-Ketomorphinan and 3-Substituted-3-desoxymorphinan Analogues as Mixed κ and μ Opioid Ligands: Synthesis and Biological Evaluation of Their Binding Affinity at Opioid Receptors. J Med Chem. 2004;47:165–174. doi: 10.1021/jm0304156. [DOI] [PubMed] [Google Scholar]
- 6.Neumeyer JL, Bidlack JM, Zong R, Bakthavachalam V, Gao P, Cohen DJ, Negus SS, Mello NK. Synthesis and Opioid Receptor Affinity of Morphinan and Benzomorphan Derivatives: Mixed k Agonists and μ Agonists/Antagonists as Potential Pharmacotherapeutics for Cocaine Dependence. J Med Chem. 2000;43:114–122. doi: 10.1021/jm9903343. [DOI] [PubMed] [Google Scholar]
- 7.Bowen CA, Negus SS, Zong R, Neumeyer JL, Bidlack JM, Mello NK. Effects of Mixed-Action Kappa/Mu Opioids on Cocaine Self-Administration and Cocaine Discrimination by Rhesus Monkeys. Neuropsychopharmacology. 2003;28:1125–1139. doi: 10.1038/sj.npp.1300105. [DOI] [PubMed] [Google Scholar]
- 8.(a) Fries DS. Opioid Analgesics. In: Williams DA, Lemke TL, editors. Foye’s Principles of Medicinal Chemistry. Lippincott Williams & Wilkins; Philadelphia, PA: 2002. pp. 453–479. [Google Scholar]; (b) Zimmerman DM, Leander JD. Selective opioid receptor agonists and antagonists: research tool and potential therapeutic agent. J Med Chem. 1990;33:895–902. doi: 10.1021/jm00165a002. [DOI] [PubMed] [Google Scholar]
- 9.Hori M, Iwamura T, Morita T, Imai E, Oji H, Kataoka T, Shimizu H, Ban M, Nozaki M, Niwa M, Fujimura H. Facile Synthesis of 8-Benzoylthio-2,6-methano-3-benzazocines and 3-Benzoylthiomorphinans having Small-Ring Substituents. Chem Pharm Bull. 1989;37:2222–2224. doi: 10.1248/cpb.37.2222. [DOI] [PubMed] [Google Scholar]
- 10.(a) Zhang A, Van Vliet S, Neumeyer JL. Synthesis of Aminothiazole Derived Morphinans. Tetrahedron Lett. 2003;44:6459–6462. [Google Scholar]; (b) Zhang A, Neumeyer JL. Microwave-Promoted Pd-Catalyzed Cyanation of Aryl Triflates: A Fast and Versatile Access to 3-Cyano-3-desoxy-10-ketomorphinans. Org Lett. 2003;5:201–203. doi: 10.1021/ol027256p. [DOI] [PubMed] [Google Scholar]; (c) Zhang A, Xiong W, Hilbert JE, DeVita EK, Bidlack JM, Neumeyer JL. 2-Aminothiazole-Derived Opioids. Bioisosteric Replacement of Phenols. J Med Chem. 2004;47:1886–1888. doi: 10.1021/jm049978n. [DOI] [PubMed] [Google Scholar]; (d) Neumeyer JM, Zhang A. Mixed kappa/mu opioids and uses thereof. PCT. Int Appl. 2004:56. [Google Scholar]
- 11.Peng X, Knapp BI, Bidlack JM, Neumeyer JL. In Vitro Investigation of Oxazol and Urea Analogues of Morphinan at Opioid Receptors. Bioorg Med Chem. 2007;15:4106–4112. doi: 10.1016/j.bmc.2007.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y-J, Tao Y-M, Li F-Y, Wang Y-H, Xu X-J, Chen J, Cao Y-L, Chi Z-L, Neumeyer JL, Zhang A, Liu J-G. Pharmacological Characterization of ATPM [(−)-3-Amino-thiazolo[5,4-b]-N-cyclopropylmorphinan hydrochloride], a novel mixed κ-Agonist and μ-agonist/antagonist that attenuates morphine antinociceptive tolerance and herion self-administration behavior. J Pharmacol Exp Ther. 2009;329:306–313. doi: 10.1124/jpet.108.142802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang A, Yan Z-H, Neumeyer JL, Hilbert JE, DeVita EK, Bidlack JM. Further Modification of Aminothiazolomorphinans. Abstracts of Papers, 232nd ACS National Meeting; San Francisco, CA, United States. Sept. 10–14, 2006; p. MEDI-459. [Google Scholar]
- 14.Neumeyer JL, Gu X-H, van Vliet LA, DeNunzio NJ, Rusovici DE, Cohen DJ, Negus SS, Mello NK, Bidlack JM. Mixed κAgonists and μ Agonists/Antagonists as Potential Pharmacotherapeutics for Cocaine Abuse: Synthesis and Opioid Binding Affinity of N-Substituted Derivatives of Morphinan. Bioorg Med Chem Lett. 2001;11:2735–2740. doi: 10.1016/s0960-894x(01)00543-1. [DOI] [PubMed] [Google Scholar]
- 15.Krishnamurthy S. A Highly Efficient and General N-Monomethylation of Functionalized Primary Amines via Formylation and Borane-Methyl Sulfide Reduction. Tetrahedron Letters. 1982;23(33):3315–3318. [Google Scholar]
- 16.Gribble GW, Nutaitis CF. Reactions of Sodium Borohydride in Acidic Media. XVI. N-Methylation of Amines with Paraformaldehyde/Trifluoroacetic Acid. Synthesis. 1987;8:709–711. [Google Scholar]
- 17.Akendengue B, Uriac P, Huet J. Quelques Derives de l’Amino-11 Vinburnine a Proprietes Protectrices Cerebrales. Eur J Med Chem. 1987:511–520. [Google Scholar]
- 18.Baxter EW, Reitz AB. Organic Reactions. Vol. 59 John Wiley & Sons, Inc; 2002. Reductive Aminations of Carbonyl Compounds with Borohydride and Borane Reducing Agents. [Google Scholar]
- 19.Duggan PJ, Lewis RJ, Lok YP, Lumsden NG, Tuck KL, Yang AJ. Low Molecular Weight Non-Peptide Mimics of ω-Conotoxin GVIA. Bioorg Med Chem Lett. 2009;19:2763–2675. doi: 10.1016/j.bmcl.2009.03.130. [DOI] [PubMed] [Google Scholar]
- 20.Neumeyer JL, Kula NS, Baldessarini RJ, Gao YD. (R)- and (S)-Enantiomers of 11-Hydroxy- and 10,11-Dihydroxy-N-allylnoraporphine: Synthesis and Affinity for Dopamine Receptors in Rat Brain Tissue. J Med Chem. 1991;34:24–28. doi: 10.1021/jm00105a005. [DOI] [PubMed] [Google Scholar]
- 21.Anzini M, Chelini A, Mancini A, Cappelli A, Frosini M, Ricci L, Valoti M, Magistretti J, Castelli L, Giordani A, Makovec F, Vomero S. Synthesis and Biological Evaluation of Amidine, Guanidine, and Thiourea Derivatives of 2- Amino(6-trifluoromethoxy)benzothiazole as Neuroprotective Agents Potentially Useful in Brain Diseases. J Med Chem. 2010;53:734–744. doi: 10.1021/jm901375r. [DOI] [PubMed] [Google Scholar]
- 22.Porcari AR, Devivar RV, Kucera LS, Drach JC, Townsend LB. Design, Synthesis, and Antiviral Evaluations of 1-(Substituted benzyl)-2-substituted-5,6-dichlorobenzimidazoles as Nonnucleoside Analogues of 2,5,6-Trichloro-1-(β,-dribofuranosyl) benzimidazole. J Med Chem. 1998;41:1252–1262. doi: 10.1021/jm970559i. [DOI] [PubMed] [Google Scholar]
- 23.Ćaleta I, Kralj M, Marjanović M, Bertoša B, Tomić S, Pavlović G, Pavelić K, Karminski-Zamola G. Novel Cyano- and Amidionbenzothiazole Derivatives: Synthesis, Antitumor Evaluation, and X-ray and Quantitative Structure-Activity Relationship (QSAR) Analysis. J Med Chem. 2009;52:1744–1756. doi: 10.1021/jm801566q. [DOI] [PubMed] [Google Scholar]
- 24.Bidlack JM, Knapp BI, Zhang T, Neumeyer JL. 3-Aminothiazole derivatives of cyclorphan and morphinan: Affinity, selectivity, and pharmacological properties. 72nd Annual Meeting on the College on Problems of Drug Dependence; 2010. http://www.cpdd.vcu.edu/Pages.Meetings/Meetings_PDFs/2010AbstractBook.pdf. [Google Scholar]