

Abstract
The yeast-like fungus Cryptococcus neoformans favours respiration as a mechanism of energy production, and thus depends heavily on mitochondrial function. Previous studies of a C. neoformans vad1Δ mutant revealed reduced expression of the mitochondrial elongation factor TUF1 and defects in glycerol utilization, consistent with mitochondrial dysfunction. In this study, we found that in trans expression of TUF1 in the vad1Δ mutant suppressed the mitochondrial defects, including growth on respiration-dependent carbon sources and fluconazole resistance, associated with VAD1 deletion. Tetracycline, an inhibitor of mitochondrial translation, was found to confer resistance to fluconazole in the wild-type and vad1Δ mutant, whereas the fluconazole susceptibility of the TUF1-overexpressing strain was unaffected by tetracycline treatment. In the presence of fluconazole, the vad1Δ mutant exhibited increased activation of the global transcriptional regulator Sre1. TUF1 overexpression failed to alter cleavage of Sre1 in response to fluconazole in the vad1Δ mutant, suggesting that TUF1 repression in the vad1Δ mutant is distal to Sre1, or that it occurs through an independent pathway.
INTRODUCTION
Infections by the pathogenic fungus Cryptococcus neoformans are occurring with increasing frequency in regions of the world plagued by the HIV epidemic, especially in regions of Africa and South-East Asia, where C. neoformans has become the most common cause of community-acquired meningitis (Bicanic & Harrison, 2005). In addition, C. neoformans represents a significant threat to transplant recipients, as infections by C. neoformans can result in allograft loss or even death (Husain et al., 2001; Singh et al., 2005). The identification of novel therapeutic targets will aid in the treatment and prevention of cryptococcosis in these susceptible populations.
The basic mechanisms by which C. neoformans yeast cells generate energy differ significantly from those of the model yeast Saccharomyces cerevisiae. C. neoformans yeast cells rely heavily on glycolysis and aerobic respiration for energy production, with fermentation playing a minimal role (Akhter et al., 2003). Both S. cerevisiae and C. neoformans utilize the highly conserved cytochrome oxidase pathway for electron transport. Three of the major constituents of this pathway are encoded by the mitochondrial genome in C. neoformans, and are translated using mitochondrial translational machinery (Litter et al., 2005).
In addition to energy production, mitochondrial function has been linked to antifungal resistance in the pathogenic fungus Candida glabrata. Petite mutants of C. glabrata that lack functional mitochondria are resistant to azole antifungals (Brun et al., 2004). Fluconazole is an important drug in anticryptococcal therapy, as it is the only azole antifungal with adequate central nervous system penetration for treatment of cryptococcal meningoencephalitis (Perfect & Durack, 1985). In addition, it is being used as monotherapy in regions of Africa where lack of infrastructure precludes the use of amphotericin B (Bicanic et al., 2006).
In C. neoformans, fluconazole sensitivity is regulated by the sterol response element-binding protein Sre1 (Chang et al., 2007). Sre1 is required for the response to hypoxia in C. neoformans, and is activated by the accumulation of 4-methyl sterols (Hughes et al., 2007). A C. neoformans sre1Δ mutant is severely attenuated in virulence, and exhibits hypersensitivity to fluconazole (Chang et al., 2007; Chun et al., 2007). A recent insertional mutagenesis study in C. neoformans that utilized the hypoxia mimetic CoCl2 has demonstrated that Sre1, mitochondrial function and sterol metabolism are tightly linked (Ingavale et al., 2008).
We have previously described a mutant of C. neoformans that lacks the DEAD-box RNA helicase Vad1; this mutant was identified in an insertional mutagenesis screen for genes involved in production of the virulence factor laccase (Panepinto et al., 2005). The vad1Δ mutant exhibits a moderate deficiency in melanization due to reduced expression of laccase, but is severely attenuated in virulence. Forward genetic screening revealed that the vad1Δ mutant exhibited decreased expression of TUF1, a nuclear gene encoding a homologue of EF-TU, which is an elongation factor for translation within the mitochondrion (Rosenthal & Bodley, 1987). Associated with downregulation of TUF1 was impaired growth on medium containing the respiration-dependent carbon source glycerol, suggesting a defect in mitochondrial function in the vad1Δ mutant (Panepinto et al., 2005).
In this study, we set out to determine whether overexpression of TUF1 in the vad1Δ mutant was able to suppress vad1Δ mitochondrial deficiency. We determined that TUF1 overexpression could restore respiratory growth and fluconazole sensitivity, and could suppress induction of fluconazole resistance by tetracycline treatment. However, hyperactivation of Sre1 in response to fluconazole treatment, and virulence defects of the vad1Δ mutant, were unaffected by TUF1 overexpression. These data suggest that downregulation of TUF1 contributes to the mitochondrial defects of the vad1Δ mutant, and that the influence of VAD1 deletion on activation of Sre1 is either upstream of TUF1 influence or through an independent pathway. These results also demonstrate that the fluconazole sensitivity of C. neoformans is dependent on mitochondrial function, and they highlight the potential for antibiotics that target bacterial protein synthesis to antagonize the antifungal activity of fluconazole towards C. neoformans.
METHODS
Construction of the v-K, v-TUF1 and wt-TUF1 strains.
The coding sequence of the TUF1 gene was amplified from genomic DNA of C. neoformans wild-type strain H99 (J. Perfect, Duke University, Durham, NC, USA) using primers TUFOE-F (5′-GGAGGATGAATTCCATGCTCAGGAACGCCCTTCAGAG-3′) and TUFOE-R (5′-GGAGGAGAATTCTCCCTTATTCGTAGATCTCTGAAACG-3′) for cloning in-frame with the actin promoter in pORA-KUTAP to make pTUF1-KUTAP (Liu et al., 2006). Once verified by sequencing, the expression construct and the empty vector were linearized with Sce1, transformed into a previously described ura derivative of the vad1Δ mutant, and selected on asparagine salts (ASN) agar (Panepinto et al., 2005). Transformants were screened for episomal maintenance of the constructs by uncut Southern blotting. One vector control strain, v-K, and two TUF1-overexpression strains, v-TUF1-1 and v-TUF1-2, were chosen for further study, and compared with a previously described wild-type strain harbouring the pORA-KUTAP vector (Panepinto et al., 2009). Strains were maintained on ASN agar, unless otherwise indicated. As described above, the pTUF1-KUTAP vector was introduced into a ura derivative of the wild-type strain H99. Two strains were chosen for further study, wt-TUF1-1 and wt-TUF1-5.
Spot-plate assay.
Cells of each strain were grown on ASN agar for 2 days. A suspension of OD600 1.0 of each strain was made in sterile deionized water. A 5 μl volume of this suspension, and five additional 1 : 5 dilutions, were spotted on ASN medium containing the indicated carbon source. Plates were incubated at 37 °C for 4 days, and photographed.
RNA isolation, Northern blot analysis and quantitative real-time PCR (qRT-PCR).
Cells of each strain were grown to mid-exponential phase in yeast extract peptone dextrose (YPD) medium. Cells were harvested by centrifugation, and RNA was extracted using the Qiagen RNEasy mini kit, as described previously (Panepinto et al., 2005). RNA was fractionated by formaldehyde agarose electrophoresis, and transferred to a Hybond-N+ nylon membrane (GE Healthcare). TUF1 and ACT1 expression was detected using 32P-labelled probes that corresponded to the coding region of each gene. Hybridization was visualized by autoradiography using Kodak BIOMax-XR film. qRT-PCR for TUF1 expression was performed using intron-spanning primers TUF1-qRT-F (5′-CACTGCCCATGTTGAATACG-3′) and TUF1-qRT-R (5′-CATATTTTTGATGTAATCGGC-3′), which correspond to an 85 bp region of the TUF1 cDNA, and ACT1-qRT-F (5′-CTCTATGAAGTGTGATCTCG-3′) and ACT1-qRT-R (5′-CATACGGTCGGCAATACC-3′), which amplify a 100 bp region of the ACT1 reference gene cDNA. Reverse transcription was performed on DNase-treated RNA using the iScript kit (Bio-Rad Laboratories), according to the manufacturer's protocol. PCRs were set up using iQ SyberGreen SuperMix (Bio-Rad Laboratories), according to the manufacturer's protocol. Briefly, a master mix was prepared to allow each reaction to contain 12.5 μl SyberGreen SuperMix, 10.5 μl diethyl-pyrocarbonate-treated water, 0.5 μl forward primer and 0.5 μl reverse primer. A 1 μl volume of cDNA was added to each reaction. qRT-PCR was performed using either a Bio-Rad iCycler or an iQ Real-Time PCR system. TUF1 expression was calculated using the comparative Ct method, and normalized against ACT1 expression (Livak & Schmittgen, 2001).
MIC determination.
Fluconazole MICs were determined using broth microdilution and Etest (AB Biodisk). The broth microdilution assay was performed as recommended by the Clinical and Laboratory Standards Institute (CLSI), with the following modification: assays were performed in RPMI medium containing 2 % glucose, and buffered to pH 7 with 10 mM MOPS (Petrou & Shanson, 2000). The first non-turbid well was read as the MIC. Etests were carried out as described by the manufacturer, and were performed on SD agar [1× yeast nitrogen base (YNB) without amino acids, 2 % glucose].
Sterol scan analysis.
Sterol levels were measured as described by Arthington-Skaggs et al. (1999), with the following alterations. Strains were inoculated at a density of OD600 0.2 in 30 ml SD medium from an overnight starter culture. Cells were grown in a shaking incubator at 30 °C and 250 r.p.m. for 72 h. The OD600 of the cultures was measured, and approximately 80 OD units of cells [the concentration of cells (as OD units per ml) multiplied by the culture volume in ml] was subjected to heptane extraction. Extracted sterols were diluted 1 : 1 with ethanol and scanned spectrophotometrically from 230 to 300 nm on a SpectraMax M5 plate reader/spectrophotometer (Molecular Devices). Absorbance values were normalized to pellet weight.
Western blot analysis.
Cells of the wt-K, v-K and v-TUF1-1 strains were grown to mid-exponential phase in YPD broth. Following the addition of 4 μg fluconazole ml−1, cells were harvested at 0, 2 and 4 h. Total protein was extracted by mechanical disruption for 5 min, alternating vortexing and chilling every 30 s. The lysates were cleared by centrifugation at 15 000 g for 5 min, after which proteins were quantified using the Bio-Rad Protein Assay reagent. A 15 μg quantity of total protein from each time point was run on a 4–12 % SDS-PAGE gel and transferred to a nitrocellulose membrane. Full-length and cleaved Sre1 was detected using polyclonal antiserum against Sre1 (1 : 250; a generous gift from Peter Espenshade, Johns Hopkins University School of Medicine, Baltimore, MD, USA), and a peroxidase-conjugated anti-rabbit secondary antibody (1 : 1000; Pierce). Antibody reactivity was visualized using Super Signal West Pico reagent, as recommended by the manufacturer (Pierce).
Virulence studies.
wt-K, v-K and v-TUF1 strains were inoculated into the lateral tail vein of Swiss albino mice (National Institutes of Health), using 1×106 cells for each inoculation. A total of 10 mice were inoculated per group, and mouse health was monitored over 50 days. Moribund mice were sacrificed by CO2 narcosis. All animal studies were performed in accordance with the University of Illinois at Chicago Institutional Animal Care and Use Committee regulations. Melanization was assayed on ASN agar without glucose, pH 6.5, and with 50 μg ml−1 l-3,4-dihydroxyphenylalanine (l-DOPA) as a laccase substrate. The degree of melanization was observed after 24 h at 30 °C.
RESULTS AND DISCUSSION
Overexpression of TUF1 suppresses vad1Δ respiratory growth defects
Our previous studies have demonstrated that the C. neoformans vad1Δ mutant exhibits reduced expression of the mitochondrial translation elongation factor TUF1, and that it is defective in utilization of respiration-dependent carbon sources (Panepinto et al., 2005). In addition, knockout of TUF1 in wild-type C. neoformans by RNA interference completely inhibits growth on glycerol, leading us to hypothesize that the respiratory defects exhibited by the vad1Δ mutant are due to TUF1 downregulation (Panepinto et al., 2005). To further test this hypothesis, we constructed v-TUF1, which was a C. neoformans Δvad1 strain harbouring an episomal vector expressing TUF1 under the control of the ACT1 promoter in pORA-KUTAP. For the following studies, v-TUF1 was compared with a wild-type strain (wt-K) described previously (Panepinto et al., 2009) and a vad1Δ strain (v-K), harbouring the empty vector (this study). The level of TUF1 overexpression achieved by introduction of the episomal construct was measured by Northern blotting (Fig. 1a). Independent assessment of TUF1 overexpression in these strains by qRT-PCR revealed that TUF1 was overexpressed 3.5- and 4.0-fold in v-TUF1-1 and v-TUF1-2, respectively, relative to v-K, when normalized to the ACT1 reference gene. To determine whether overexpression of TUF1 could restore growth on respiration-dependent carbon sources, we compared the growth of v-TUF1-1 and v-TUF1-2 with that of wt-K and v-K on glycerol, pyruvate and lactate in a spot-plate assay. As demonstrated in Fig. 1(b), overexpression of TUF1 restored growth of the vad1Δ strain on respiration-dependent carbon sources. This is consistent with data from S. cerevisiae, in which overexpression of TUF1 is able to suppress respiratory defects of mitochondrial tRNA mutants (De Luca et al., 2006). These data suggest that the respiratory defects of the vad1Δ mutant are linked to repression of TUF1 expression.
Fig. 1.
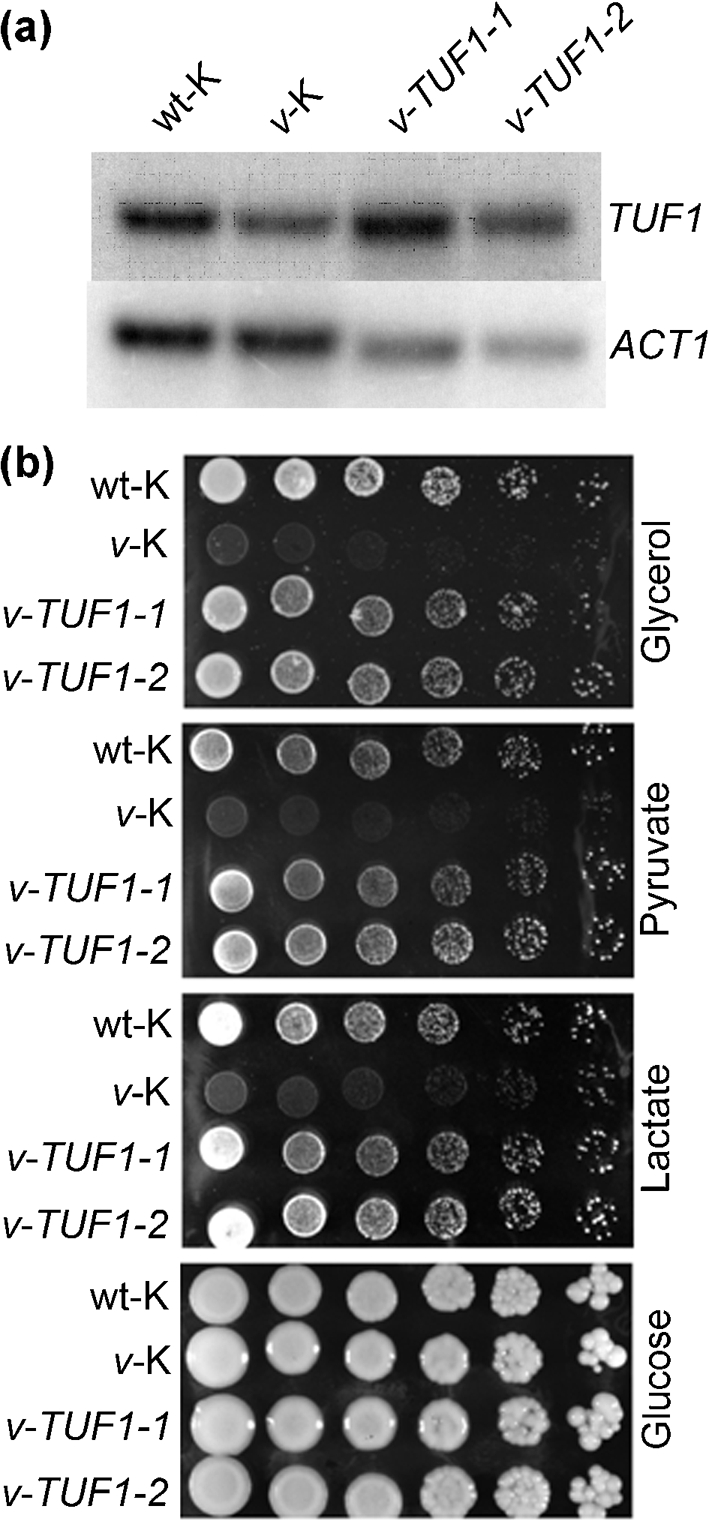
(a) Northern blot analysis of TUF1 expression in wt-K, v-K and v-TUF1 strains. RNA was harvested from cultures grown to mid-exponential phase in YPD broth at 30 °C. RNA was hybridized with TUF1 and ACT1 probes. (b) Spot-plate assay demonstrating restoration of growth on respiration-dependent carbon sources by overexpression of TUF1 in the vad1Δ mutant. Plates contained the indicated carbon sources at 2 % in ASN agar, pH 6.5. Plates were incubated at 37 °C for 4 days and then photographed.
To determine the effect of TUF1 overexpression on carbon source utilization in wild-type C. neoformans, we compared the growth of two independent isolates of wild-type C. neoformans harbouring the TUF1-overexpression construct with that of the vector control strain, as described above, by spot-plate assay. Both wt-TUF1 strains exhibited a severe growth defect on minimal medium with glucose as the carbon source (Fig. 2a). Surprisingly, the growth of both wt-TUF1 strains appeared more robust in medium with glycerol as the carbon source compared with growth in medium containing glucose, with overall growth still less than that of the wt-K strain (Fig. 2b). The level of TUF1 overexpression as measured by qRT-PCR was found to be 1.5-fold for wt-TUF1-1 and 1.6-fold for wt-TUF1-5, relative to wt-K, at 30 °C in YPD medium using ACT1 expression for normalization. These results suggest that although TUF1 overexpression is able to compensate for mitochondrial defects in a vad1Δ mutant, even modest inappropriate overexpression of TUF1 in wild-type C. neoformans has detrimental effects on growth, especially at 37 °C in the presence of glucose.
Fig. 2.
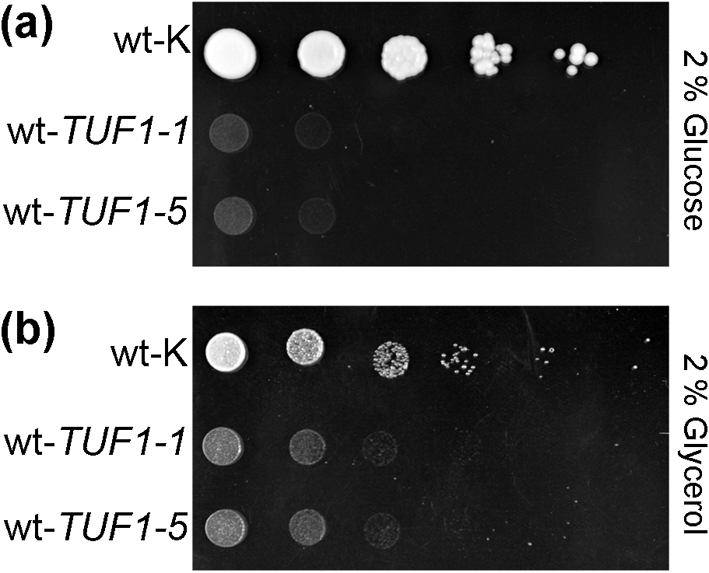
Overexpression of TUF1 in wild-type C. neoformans is growth-inhibitory. Growth of two independently derived TUF1-overexpression strains was compared with that of a wild-type strain harbouring the empty vector (wt-K) by spot-plate assay at 37 °C on minimal medium containing either glucose (a) or glycerol (b) as the carbon source.
Overexpression of TUF1 suppresses fluconazole resistance in the vad1Δ mutant
A recent study has demonstrated that inhibition of mitochondrial protein synthesis with tetracycline induces fluconazole resistance in Candida albicans and C. neoformans, with the effect on C. neoformans being much greater than on Candida albicans (Oliver et al., 2008). This led us to hypothesize that downregulation of TUF1 in the vad1Δ mutant could confer fluconazole resistance that would be suppressed by TUF1 overexpression. To test this hypothesis, the fluconazole sensitivity of wt-K, v-K, v-TUF1-1 and v-TUF1-2 was assessed by Etest. As demonstrated in Fig. 3, comparison of fluconazole MICs by the Etest method revealed an increase in MIC in v-K compared with the wt-K strain, with the v-TUF1-1 and v-TUF1-2 strains exhibiting decreased fluconazole MICs compared with the parent v-K strain. The fluconazole MIC was also measured by broth microdilution, and the MICs for the v-K and wt-K strains were found to be 8 and 2 μg ml−1, respectively, in each of three replicate assays, demonstrating further that VAD1 deletion results in fluconazole resistance. The fluconazole MICs of the v-TUF1-1 and v-TUF1-2 strains, as measured by broth microdilution, were equivalent to that of the wt-K strain, with each strain exhibiting a MIC value of 2 μg ml−1 in three replicate experiments. This is consistent with the ability of TUF1 overexpression to suppress the fluconazole resistance of the vad1Δ mutant. Disparities in the MIC results between the two methods are likely to stem from differences in growth medium. However, both assays demonstrate that the v-K strain exhibits resistance to fluconazole relative to wt-K, and that overexpression of TUF1 restores fluconazole sensitivity to the vad1Δ mutant. These data demonstrate that overexpression of TUF1 in the vad1Δ mutant is able to suppress the fluconazole-resistant phenotype. Because the effect of TUF1 overexpression in vad1Δ had the same consequences in both v-TUF1-1 and v-TUF1-2, only v-TUF1-1 was assayed in the following studies.
Fig. 3.

Etest analysis demonstrating an increased fluconazole MIC in the v-K strain compared with the wild-type control (wt-K). Fluconazole sensitivity was restored by overexpression of TUF1 (v-TUF1-1 and v-TUF1-2).
TUF1 overexpression abrogates tetracycline-induced fluconazole resistance
Tetracycline is thought to affect mitochondrial function through inhibition of mitochondrial translation, similar to the expected consequence of TUF1 downregulation (Zhang et al., 2005). If TUF1 overexpression restores fluconazole sensitivity through upregulation of mitochondrial translation, we would expect the v-TUF1-1 strain to be insensitive to tetracycline-induced fluconazole resistance. To test this hypothesis, we determined fluconazole MICs by Etest analysis on wt-K, v-K and v-TUF1-1 in the presence of increasing concentrations of tetracycline (Fig. 4). As previously reported for wild-type C. neoformans, tetracycline increased the fluconazole MIC of both wt-K and v-K (Oliver et al., 2008). However, tetracycline had little effect on the fluconazole MIC of v-TUF1-1, even at 400 μg tetracycline ml−1. This suggests that overexpression of TUF1 renders mitochondrial translation less sensitive to inhibition by tetracycline. In addition, it confirms that the effect of tetracycline on fluconazole sensitivity is a result of its activity as an inhibitor of mitochondrial translation.
Fig. 4.

Etest analysis showing fluconazole MICs in the presence of tetracycline (TET) at 50 and 400 μg ml−1 in wt-k, v-K and v-TUF1-1 strains. Etests were performed on SD agar, with and without the indicated concentration of tetracycline, incubated at 30 °C for 3 days, and then photographed.
The mechanism by which mitochondrial translation can influence fluconazole sensitivity is unknown. Studies in Candida albicans demonstrate only a modest increase in fluconazole MIC in the presence of tetracycline relative to the drastic effect seen in C. neoformans (Oliver et al., 2008). Likewise, sterol profiles of Candida albicans in the presence of tetracycline show only a modest difference from untreated controls. To determine whether sterol content was affected by tetracycline treatment, sterols were extracted from wt-K, v-K and v-TUF1-1 grown in the presence and absence of 200 μg tetracycline ml−1, and a sterol profile was obtained for each strain. As demonstrated in Fig. 5, the height of the weight-corrected sterol profiles (±sem) for each strain correlated with the fluconazole MIC, with the v-K strain exhibiting the highest-profile peaks and fluconazole resistance, and the v-TUF1 strain the lowest-profile peaks and fluconazole sensitivity. wt-K was intermediate in profile peaks and fluconazole MIC. Treatment with 200 μg tetracycline ml−1 was found to increase peak height in all three strains (Fig. 5), with the effect being most significant in the wt-K strain. It should be noted, however, that the increase in peak height in the tetracycline-treated v-TUF1-1 strain did not reach the height of the untreated wt-K profile, consistent with the lack of tetracycline-induced fluconazole resistance observed in the v-TUF1-1 strain. The magnitude of the profiles in tetracycline-treated wt-K and v-K was similar, and this might suggest that these strains were approaching a maximum in sterol synthesis. In addition, we would expect the effect of tetracycline on the v-K strain to be less, as overall mitochondrial translation is likely to be reduced owing to TUF1 repression, resulting in an increased sterol profile height in the absence of tetracycline. Taken together, these data suggest that TUF1 repression, or inhibition of mitochondrial translation with tetracycline, confers fluconazole resistance, at least in part, through an increase in sterol content. Future studies will investigate the mechanism by which sterol content is upregulated in these strains. A possible explanation for the differences in tetracycline-induced fluconazole resistance between C. neoformans and Candida albicans may stem from fundamental metabolic differences between these two pathogenic fungi. Consistent with this interpretation is that TUF1 appears to be an essential gene, since our previously described attempts to generate a deletion mutant were unsuccessful (Panepinto et al., 2005). In contrast, the S. cerevisiae TUF1 orthologue is not essential, with the deletion mutant exhibiting respiratory growth defects (Chiron et al., 2005). The pronounced effects of tetracycline on C. neoformans fluconazole susceptibility observed previously, and the essential nature of TUF1 in C. neoformans, are consistent with the minimal fermentative capacity of C. neoformans compared with the ascomycetous yeast. The reduced capacity of C. neoformans to ferment is likely to result in strong reliance on mitochondrial function, whereas Candida albicans and S. cerevisiae favour fermentative pathways to produce ATP.
Fig. 5.

(a) UV scans of heptane-extracted sterols from cultures grown in the presence and absence of tetracycline. Data shown are representative of three independent assays; vertical bars, sem. TET, tetracycline.
The vad1Δ mutant hyperactivates Sre1 in response to fluconazole
The response of C. neoformans to azole antifungal drugs includes activation of Sre1 (Chang et al., 2007; Chun et al., 2007). Sre1 responds to the accumulation of 4-methyl sterols that occurs during antifungal drug treatment and hypoxia, and deletion of SRE1 confers profound fluconazole sensitivity (Chang et al., 2007; Hughes et al., 2007). Microarray experiments have demonstrated that the hypoxic response in C. neoformans involves downregulation of TUF1, similar to that seen in the vad1Δ mutant (Chun et al., 2007). These similarities in gene expression, coupled with fluconazole resistance and increased sterol content, led us to hypothesize that the vad1Δ mutant would hyperactivate Sre1 in response to fluconazole treatment. As demonstrated in Fig. 6(a), the vad1Δ mutant exhibited increased activation of Sre1 after fluconazole treatment for 2 h, whereas wild-type Sre1 activation was barely visible after drug treatment for 2 h. However, overexpression of TUF1 in the vad1Δ mutant had no effect on the hyperactivation of Sre1 (Fig. 6b), suggesting that Sre1 regulation is upstream of TUF1 in C. neoformans, or that it functions in an independent pathway. This is also supported by the inability of tetracycline to increase resistance to fluconazole in the sre1Δ mutant (Fig. 6c). Taken together, these data support a model in which TUF1 downregulation in a vad1Δ mutant may be a direct result of Sre1 dysregulation. Future studies will test this model, and investigate a regulatory role of Vad1 in Sre1 activation.
Fig. 6.
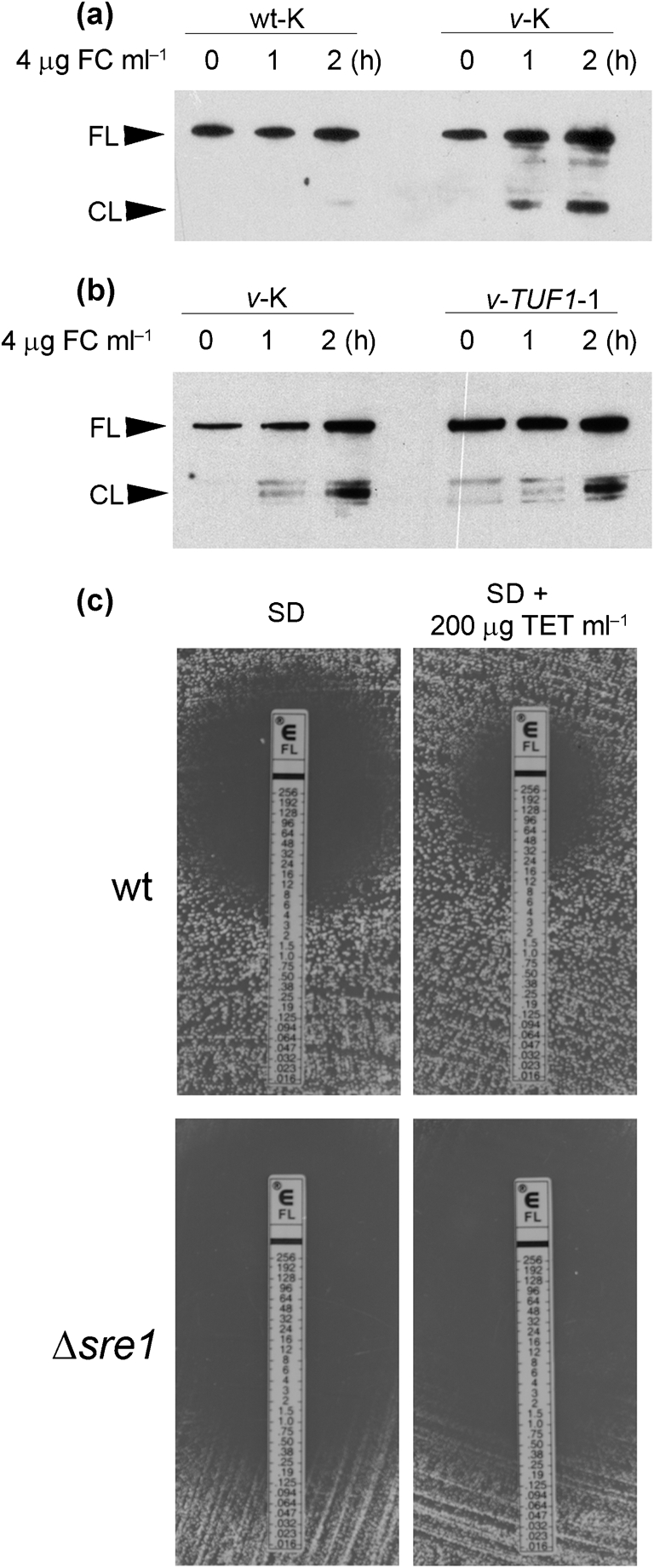
Western blot analysis of Sre1 activation in response to fluconazole treatment. (a) Time course of Sre1 activation in the presence of 4 μg ml−1 fluconazole (FC) comparing the wt-K and v-K strains. Activation of Sre1 is indicated by the appearance of the cleavage fragment (CL) in addition to the inactive, full-length Sre1 band (FL). (b) Time course of Sre1 activation in the presence of 4 μg fluconazole ml−1, comparing the v-K and TUF1-K strains. (c) Etests comparing the fluconazole sensitivity of wild-type strain H99 (wt) with that of a sre1Δ mutant in the presence and absence of 200 μg ml−1 tetracycline (TET).
Overexpression of TUF1 in the vad1Δ mutant does not restore virulence
Our previous studies have demonstrated that severe virulence attenuation accompanies deletion of VAD1 (Panepinto et al., 2005). To determine the contribution of TUF1 downregulation to vad1Δ virulence attenuation, the ability of a TUF1-expressing vad1Δ strain to cause mortality in a murine model of cryptococcosis was compared with that of the wild-type and vad1Δ mutant derivatives bearing an empty vector. Cells (1×106) of each strain were injected into the lateral tail vein of Swiss albino mice, and mortality was monitored for 7 weeks. All of the wild-type mice succumbed to infection in the first 2 weeks, whereas none of the mice infected with the vad1Δ mutant succumbed to the infection. One mouse infected with the TUF1-expressing strain succumbed to infection on day 21, with the remaining nine mice surviving to 7 weeks, at which time the experiment was terminated (data not shown). This suggests that restoration of TUF1 expression in the vad1Δ mutant is not sufficient to restore virulence, and that the virulence defects of the vad1Δ mutant are most likely due to effects on multiple genes, as Vad1 is a global regulator.
Because VAD1 was initially discovered in a screen for C. neoformans mutants defective in melanization, we assayed the effect of TUF1 overexpression on the melanin deficiency of the vad1Δ mutant. Overexpression of TUF1 did not restore wild-type melanization to the vad1Δ mutant (data not shown), consistent with the inability of TUF1 overexpression to restore virulence traits to the vad1Δ mutant.
In this study, we demonstrated that overexpression of TUF1, encoding the mitochondrial translation elongation factor, was able to suppress several aspects of the vad1Δ phenotype related to mitochondrial function. These aspects included rescuing growth on respiration-dependent carbon sources and restoring fluconazole sensitivity. This study supports a role for mitochondrial protein synthesis in the regulation of fluconazole sensitivity, demonstrating that inhibition of mitochondrial protein synthesis by tetracycline confers resistance to fluconazole in C. neoformans and increases overall sterol content. These results not only provide insight into possible mechanisms of drug resistance in C. neoformans, but also highlight potential interactions of antimicrobial agents in patients undergoing treatment for polymicrobial infections. This study raises the possibility that antibacterial agents that target protein synthesis antagonize the activity of azole antifungal drugs. Future studies using animal models of cryptococcal infection will address this question in vivo.
The study also points to the unique reliance of C. neoformans on mitochondrial respiration. Perturbation of mitochondrial function by TUF1 repression, as in the vad1Δ mutant, affects carbon metabolism, sterol metabolism and susceptibility to fluconazole. Similar effects on sterol metabolism and fluconazole sensitivity were recapitulated by treatment with tetracycline. In addition, overexpression of TUF1 in wild-type C. neoformans appears to impair glucose utilization, favouring the use of respiration-dependent glycerol under the conditions tested herein. Further studies of mitochondrial function in this important pathogen could identify targets for antifungal therapy that may confer fluconazole sensitivity by activating, rather than impairing, aspects of mitochondrial function.
Although overexpression of TUF1 was able to act as a phenotypic suppressor of vad1Δ mitochondrial defects, it was unable to restore virulence to the vad1Δ mutant. Therefore, the regulatory defects underlying the complex phenotype of the vad1Δ mutant likely arise from modulation of multiple targets, as Vad1 is a regulator of many genes and pathways. The mechanism by which TUF1 is downregulated in the vad1Δ mutant is unclear, and a possibility that TUF1 repression is secondary to defects in another regulator, such as Sre1, remains. Further studies delineating the role of Vad1 as a regulator of mRNA fate will determine the specific target transcripts regulated by Vad1, and lead to a mechanistic understanding of the vad1Δ phenotype.
Acknowledgments
This work was supported by Public Health Service Grant AI45995 to P. R. W.
Abbreviations
qRT-PCR, quantitative real-time PCR
References
- Akhter, S., McDade, H. C., Gorlach, J. M., Heinrich, G., Cox, G. M. & Perfect, J. R. (2003). Role of alternative oxidase gene in pathogenesis of Cryptococcus neoformans. Infect Immun 71, 5794–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthington-Skaggs, B. A., Jradi, H., Desai, T. & Morrison, C. J. (1999). Quantitation of ergosterol content: novel method for determination of fluconazole susceptibility of Candida albicans. J Clin Microbiol 37, 3332–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicanic, T. & Harrison, T. S. (2005). Cryptococcal meningitis. Br Med Bull 72, 99–118. [DOI] [PubMed] [Google Scholar]
- Bicanic, T., Harrison, T., Niepieklo, A., Dyakopu, N. & Meintjes, G. (2006). Symptomatic relapse of HIV-associated cryptococcal meningitis after initial fluconazole monotherapy: the role of fluconazole resistance and immune reconstitution. Clin Infect Dis 43, 1069–1073. [DOI] [PubMed] [Google Scholar]
- Brun, S., Berges, T., Poupard, P., Vauzelle-Moreau, C., Renier, G., Chabasse, D. & Bouchara, J. P. (2004). Mechanisms of azole resistance in petite mutants of Candida glabrata. Antimicrob Agents Chemother 48, 1788–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Y. C., Bien, C. M., Lee, H., Espenshade, P. J. & Kwon-Chung, K. J. (2007). Sre1p, a regulator of oxygen sensing and sterol homeostasis, is required for virulence in Cryptococcus neoformans. Mol Microbiol 64, 614–629. [DOI] [PubMed] [Google Scholar]
- Chiron, S., Suleau, A. & Bonnefoy, N. (2005). Mitochondrial translation: elongation factor Tu is essential in fission yeast and depends on an exchange factor conserved in humans but not in budding yeast. Genetics 169, 1891–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun, C. D., Liu, O. W. & Madhani, H. D. (2007). A link between virulence and homeostatic responses to hypoxia during infection by the human fungal pathogen Cryptococcus neoformans. PLoS Pathog 3, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca, C., Besagni, C., Frontali, L., Bolotin-Fukuhara, M. & Francisci, S. (2006). Mutations in yeast mt tRNAs: specific and general suppression by nuclear encoded tRNA interactors. Gene 377, 169–176. [DOI] [PubMed] [Google Scholar]
- Hughes, A. L., Lee, C. Y., Bien, C. M. & Espenshade, P. J. (2007). 4-Methyl sterols regulate fission yeast SREBP-Scap under low oxygen and cell stress. J Biol Chem 282, 24388–24396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain, S., Wagener, M. M. & Singh, N. (2001). Cryptococcus neoformans infection in organ transplant recipients: variables influencing clinical characteristics and outcome. Emerg Infect Dis 7, 375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingavale, S. S., Chang, Y. C., Lee, H., McClelland, C. M., Leong, M. L. & Kwon-Chung, K. J. (2008). Importance of mitochondria in survival of Cryptococcus neoformans under low oxygen conditions and tolerance to cobalt chloride. PLoS Pathog 4, e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litter, J., Keszthelyi, A., Hamari, Z., Pfeiffer, I. & Kucsera, J. (2005). Differences in mitochondrial genome organization of Cryptococcus neoformans strains. Antonie Van Leeuwenhoek 88, 249–255. [DOI] [PubMed] [Google Scholar]
- Liu, X., Hu, G., Panepinto, J. & Williamson, P. R. (2006). Role of a VPS41 homologue in starvation response, intracellular survival and virulence of Cryptococcus neoformans. Mol Microbiol 61, 1132–1146. [DOI] [PubMed] [Google Scholar]
- Livak, K. J. & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Oliver, B. G., Silver, P. M., Marie, C., Hoot, S. J., Leyde, S. E. & White, T. C. (2008). Tetracycline alters drug susceptibility in Candida albicans and other pathogenic fungi. Microbiology 154, 960–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panepinto, J., Liu, L., Ramos, J., Zhu, X., Valyi-Nagy, T., Eksi, S., Fu, J., Jaffe, H. A., Wickes, B. & Williamson, P. R. (2005). The DEAD-box RNA helicase Vad1 regulates multiple virulence-associated genes in Cryptococcus neoformans. J Clin Invest 115, 632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panepinto, J., Komperda, K. W., Frases, S., Park, Y. D., Djordejevic, J. T., Casadevall, A. & Williamson, P. R. (2009). Sec6-dependent sorting of fungal extracellular exosomes and laccase of Cryptococcus neoformans. Mol Microbiol 71, 1165–1176. [DOI] [PubMed] [Google Scholar]
- Perfect, J. R. & Durack, D. T. (1985). Penetration of imidazoles and triazoles into cerebrospinal fluid of rabbits. J Antimicrob Chemother 16, 81–86. [DOI] [PubMed] [Google Scholar]
- Petrou, M. A. & Shanson, D. C. (2000). Susceptibility of Cryptococcus neoformans by the NCCLS microdilution and Etest methods using five defined media. J Antimicrob Chemother 46, 815–818. [DOI] [PubMed] [Google Scholar]
- Rosenthal, L. P. & Bodley, J. W. (1987). Purification and characterization of Saccharomyces cerevisiae mitochondrial elongation factor Tu. J Biol Chem 262, 10955–10959. [PubMed] [Google Scholar]
- Singh, N., Lortholary, O., Alexander, B. D., Gupta, K. L., John, G. T., Pursell, K., Munoz, P., Klintmalm, G. B., Stosor, V. & other authors (2005). Allograft loss in renal transplant recipients with Cryptococcus neoformans associated immune reconstitution syndrome. Transplantation 80, 1131–1133. [DOI] [PubMed] [Google Scholar]
- Zhang, L., Ging, N. C., Komoda, T., Hanada, T., Suzuki, T. & Watanabe, K. (2005). Antibiotic susceptibility of mammalian mitochondrial translation. FEBS Lett 579, 6423–6427. [DOI] [PubMed] [Google Scholar]