

Abstract
A number of single-nucleotide polymorphisms (SNPs) have been identified in the genome of Mycobacterium bovis BCG Pasteur compared with the sequenced strain M. bovis 2122/97. The functional consequences of many of these mutations remain to be described; however, mutations in genes encoding regulators may be particularly relevant to global phenotypic changes such as loss of virulence, since alteration of a regulator's function will affect the expression of a wide range of genes. One such SNP falls in bcg3145, encoding a member of the AfsR/DnrI/SARP class of global transcriptional regulators, that replaces a highly conserved glutamic acid residue at position 159 (E159G) with glycine in a tetratricopeptide repeat (TPR) located in the bacterial transcriptional activation (BTA) domain of BCG3145. TPR domains are associated with protein–protein interactions, and a conserved core (helices T1–T7) of the BTA domain seems to be required for proper function of SARP-family proteins. Structural modelling predicted that the E159G mutation perturbs the third α-helix of the BTA domain and could therefore have functional consequences. The E159G SNP was found to be present in all BCG strains, but absent from virulent M. bovis and Mycobacterium tuberculosis strains. By overexpressing BCG3145 and Rv3124 in BCG and H37Rv and monitoring transcriptome changes using microarrays, we determined that BCG3145/Rv3124 acts as a positive transcriptional regulator of the molybdopterin biosynthesis moa1 locus, and we suggest that rv3124 be renamed moaR1. The SNP in bcg3145 was found to have a subtle effect on the activity of MoaR1, suggesting that this mutation is not a key event in the attenuation of BCG.
INTRODUCTION
The Bacille Calmette–Guérin (BCG) vaccine is a live attenuated strain of Mycobacterium bovis derived by in vitro passage of an M. bovis strain from 1908 to 1921 (Calmette, 1927). This in vitro cultivation led to the accumulation of mutations in the BCG genome that attenuated the bacillus. BCG is the only vaccine available against tuberculosis, and with more than 3 billion individuals having been immunized with BCG it is the most widely used vaccine in the world (Brosch et al., 2007). Despite the widespread use of BCG, a precise catalogue of the genetic lesions that led to attenuation has not been defined. Knowledge of these mutations would shed light not only on the attenuation of BCG, but also on virulence factors of the tubercle bacilli.
Work towards defining the attenuating genetic lesions in BCG began with the identification of the RD1–RD3 loci using subtractive genomic hybridization techniques (Mahairas et al., 1996). The RD1 locus was shown to be deleted from all BCG strains but present in all virulent strains of M. bovis and Mycobacterium tuberculosis studied. Subsequent work has shown that this deletion played a major role in the attenuation of BCG (Lewis et al., 2003; Pym et al., 2002), although restoration of RD1 by knock-in did not restore virulence to wild-type levels in immunocompetent mice (Pym et al., 2002), indicating that other attenuating mutations exist. Comparative genome analyses using genomic libraries and DNA arrays further expanded the list of deleted regions and chromosomal rearrangements between BCG and virulent M. bovis (Behr et al., 1999; Gordon et al., 1999; Mostowy et al., 2003). However, it was with the completion of the genome sequence of M. bovis BCG Pasteur, and comparison with the genomes of M. tuberculosis (Cole et al., 1998) and M. bovis (Garcia Pelayo et al., 2009), that we obtained single-nucleotide resolution of mutations present in BCG Pasteur. From genome analysis, 736 single-nucleotide polymorphisms (SNPs) were identified between BCG Pasteur and the virulent M. bovis strain AF2122/97 (Brosch et al., 2007). However, many of these SNPs may be specific to the BCG Pasteur or M. bovis AF2122/97 strains sequenced. To gain a ‘minimal’ list of SNPs that differentiate attenuated BCG from virulent M. bovis strains, Garcia Pelayo et al. (2009) screened all SNPs present in BCG against a panel of 21 M. bovis strains and 13 BCG strains. This allowed the identification of 186 SNPs between virulent M. bovis strains and all BCG strains, with 115 of these SNPs being non-synonymous (nsSNP) and hence having potential functional consequences.
Previous characterization of nsSNPs in BCG has provided key insights into gene function. The first study to link a SNP to a phenotypic change in BCG strains examined the mma3 gene, where a nsSNP abolished methoxymycolate production in ‘late’ BCG strains (derived post-1927, compared with ‘early’ strains that were derived pre-1927) (Behr et al., 2000; Belley et al., 2004). A nsSNP in the BCG gene encoding the CRP global gene regulator has been shown to affect global gene regulation, but play no role in the attenuation of BCG (Hunt et al., 2008). Furthermore a nsSNP in the pykA gene of BCG has been shown to permit growth of BCG on glycerol as a sole carbon source, a mutation that was selected during growth of the M. bovis progenitor on glycerinated potato slices (Keating et al., 2005). Hence, determining the functional consequences of nsSNPs found in BCG can reveal much about biological function of the gene product as well as any role for the gene in virulence.
One of the 186 nsSNPs between BCG and virulent M. bovis was found in the bcg3145 gene (an orthologue of rv3124 in M. tuberculosis). BCG3145 is a member of the AfsR/DnrI/SARP (Streptomyces antibiotic regulatory protein) class of transcriptional regulators. This class also contains EmbR, the regulator of three arabinosyltransferases that are the targets of the front-line tuberculosis drug ethambutol (Belanger et al., 1996). The structure of EmbR has been elucidated, revealing DNA-binding, bacterial transcriptional activation (BTA), and forkhead-associated domains (Alderwick et al., 2006). The E159G mutation in BCG3145 mutates to glycine a conserved glutamic acid residue located in the BTA domain (region T3). However, whether the E159G mutation affects the ability of BCG3145 to regulate transcription, or the identity of the genes regulated by BCG3145, was unknown. Mutations in regulatory genes may be particularly relevant to global phenotypic changes such as loss of virulence, since alteration of a regulator's function will affect the expression of a wide range of genes. In order to define the function of BCG3145/Rv3124, we describe here structural predictions, transcriptome analysis, site-directed mutagenesis, gel-shift assays and promoter reporter assays that show BCG3145/Rv3124 to be a positive transcriptional regulator of the moa1 locus.
METHODS
Bacterial strains and growth conditions.
Mycobacteria and Escherichia coli strains used in this study are listed in Table 1. Strains were grown in 7H9 Middlebrook broth (Becton Dickinson) containing albumin-glucose-catalase (ADC, Becton Dickinson) (7H9-ADC), 0.2 % glycerol and 0.05 % Tween 80. After electroporation of pSM96 and derivatives, mycobacterial transformants were selected on 7H11 medium containing 10 % (v/v) oleic acid-ADC (OADC, Becton Dickinson), with antibiotics added as appropriate: kanamycin (Sigma) at 25 μg ml−1, and hygromycin (Invitrogen) at 150 μg ml−1. E. coli strains were used as a general-purpose cloning and expression host and grown at 37 °C in Luria–Bertani (LB) medium with shaking, and on LB agar with ampicillin (Sigma; 100 μg ml−1), hygromycin (150 μg ml−1), or kanamycin (25 μg ml−1) as appropriate.
Table 1.
Bacterial strains and plasmids used in this study
| Bacterial strain/plasmid | Relevant genotype or characteristics | Reference or source |
|---|---|---|
| M. bovis BCG | Pasteur strain | VLA Weybridge, UK |
| M. bovis AF2122/97 | Virulent isolate from a cow in Cornwall, UK | VLA Weybridge, UK |
| M. tuberculosis H37Rv | Virulent strain isolated in 1905 | VLA Weybridge, UK |
| M. smegmatis mc2155 | Mutant with high plasmid transformation efficiency | Professor W. R. Jacobs, AECOM, NY, USA |
| M. bovis BCG Δbcg3145 | Knockout strain of M. bovis BCG Pasteur with bcg3145 gene replaced by hygromycin cassette | This study |
| E. coli DH5α | General cloning strain | Invitrogen |
| E. coli BL21(DE3) | Strain used for protein expression | Invitrogen |
| E. coli HB101 | General cloning strain | Invitrogen |
| E. coli XL1-Blue | General cloning strain | Invitrogen |
| pUC19 | General cloning vector | VLA Weybridge |
| pUC3124 | pUC19 containing rv3124 | This study |
| pSM96 | Mycobacterial expression vector with hsp60 promoter | VLA Weybridge, UK (Wooff et al., 2002) |
| pB1 | rv3124 in pSM96 | This study |
| pB3 | bcg3145 in pSM96 | This study |
| MBPpET | MBP–His fusion vector used in protein purification | Vircell |
| pYUB854 | Cosmid vector used for mutant construction; contains the hygromycin cassette and a multiple cloning site | Bardarov et al. (2002) |
| p3145KO | Cosmid used in the bcg3145 knockout containing the gene flanking regions into pYUB854 | This study |
| pSM128 | Mycobacteriophage L5-based vector carrying a lacZ reporter | Dussurget et al. (1999) |
| pB31 | 190 bp upstream of rv3109 in pSM128 | This study |
| pB32 | 116 bp upstream of rv3109 in pSM128 | This study |
| pB33 | 45 bp upstream of rv3109 in pSM128 | This study |
| pB10 | G90A mutation in the DNA-binding domain | This study |
| pB11 | Y91A mutation in the DNA-binding domain | This study |
| pB15 | E159I mutation in the BTA domain | This study |
| pB17 | E159W mutation in the BTA domain | This study |
| pB23 | E159D mutation in the BTA domain | This study |
| pB25 | E159Q mutation in the BTA domain | This study |
Confirmation of SNP in bcg3145.
A 500 bp fragment flanking the E159G nsSNP was amplified from a range of BCG, M. bovis and M. tuberculosis strains using Forward (ACCGTCCACACGTACATTTG) and Reverse (AGCGTGGACTTGAGTCTATG) primers (MWG). DNA from heat-killed mycobacterial cells was used as a template for the amplifications, with DNA polymerase HotStar Taq (Qiagen). The PCR cycle conditions were 95 °C for 15 min, followed by 30 cycles of 95 °C for 1 min, hybridization for 1 min at 58 °C, extension for 1 min at 72 °C and a final incubation at 72 °C for 5 min. The PCR product was purified and the point mutation was confirmed by cycle sequencing (Sequencing Service, University of Dundee, UK).
Modelling of the Rv3124 structure.
The GenTHREADER program at the PSIPRED server (Bryson et al., 2005; Jones, 1999) was used to identify and align proteins structurally related to Rv3124. Only one protein with high resemblance (approximately 50 % sequence identity) was retrieved from the search. This was EmbR, a transcriptional regulator from M. tuberculosis (PDB id: 2FF4) (Alderwick et al., 2006). Sequence alignment with EmbR was used to obtain a model of Rv3124 structure from the Swiss-model server (Kiefer et al., 2009). Protein structures were analysed with the Swiss-PDP Viewer (http://spdbv.vital-it.ch/).
Complementation of M. bovis BCG with bcg3145 and rv3124.
Using pSM96 as a vector (an E. coli–mycobacteria shuttle vector containing a hsp60 promoter upstream of a multiple cloning site, constructed by Dr S. Michell, VLA Weybridge), constructs were generated with rv3124 (pB1) or bcg3145 (pB3, Table 1). For pB1, rv3124 was amplified from M. tuberculosis H37Rv genomic DNA and for pB3, M. bovis BCG genomic DNA. PCRs used primers Rv3124BamF (ATGGGATCCGTGCAATTCAACGTCTTAGGACCACTGGAAC, BamHI site underlined) and Rv3124PstR (CATCTGCAGTTATGCCAAACCGATGGGAAGGAG, PstI site underlined). The amplification conditions were 94 °C for 15 s, then 30 cycles of 15 s at 94 °C, hybridization and extension at 68 °C for 3 min, then a final extension for 3 min at 68 °C. pSM96 and PCR products were digested with BamHI and PstI, ligated following standard protocols, and transformed into E. coli. Sequencing was used to confirm the correct sequence and orientation of the cloned fragments. Plasmid constructs were purified from E. coli and electroporated into M. bovis BCG Pasteur using standard methods, with transformants selected on 7H11 plates containing kanamycin and hygromycin. Isolated colonies were selected from the plates, checked for the presence of the plasmid by PCR, and then grown in 7H9 containing the appropriate antibiotic for transcriptome analysis.
Transcriptome analysis.
Strains were grown in 7H9-ADC medium with 0.05 % Tween 80 to an OD600 of ≈0.6, corresponding to mid-exponential growth. Each strain was cultured three times (three biological replicates), with each culture split in two and RNA extracted (two technical replicates; hence six microarrays in total per strain). Total RNA from each strain was extracted, purified, reverse-transcribed, and labelled with Cy5-dCTP (Amersham Pharmacia) as previously described (Golby et al., 2007). Cy3-labelled DNA (M. bovis AF2122/97 and M. tuberculosis H37Rv) was used as a control. Probes were hybridized to whole-genome M. bovis/M. tuberculosis composite microarrays and scanned with an Affymetrix 428 scanner. The microarrays were developed by the Bacterial Microarray Group (St Georges, University of London) in collaboration with the Veterinary Laboratories Agency (Weybridge). The array design is available in μG@Sbase (accession no. A-BUGS-31; http://bugs.sgul.ac.uk/A-BUGS-31) and also ArrayExpress (accession no. A-BUGS-31). Fully annotated microarray data have been deposited in μG@Sbase (accession no. E-BUGS-93; http://bugs.sgul.ac.uk/E-BUGS-93) and also ArrayExpress (accession no. E-BUGS-93). Image processing and data normalization steps were performed as previously described (Golby et al., 2008).
To select differentially expressed genes between strains, a t-test was applied to each gene, with the Benjamini and Hochberg False Discovery Rate applied to adjust P-values and correct for multiple testing. Those differentially expressed genes (adjusted P-value <0.05) showing an expression difference greater than threefold between strains were selected for further investigation. Results for these genes were confirmed by quantitative reverse transcription PCR (qRT-PCR) analysis, as previously described (Golby et al., 2007). Briefly, RT-PCR was carried out using the QuantiTecT SYBR Green PCR kit (Qiagen). Each reaction was carried out in a 25 μl volume containing 12.5 μl QuantiTect Master Mix, 0.4 μM of each primer and 5 μl diluted cDNA. All reactions were run in duplicate using a RotorGene 3000 instrument (Qiagen). The instrument was programmed to cycle at 50 °C for 30 min; 95 °C for 15 min; and then 50 cycles of 15 s at 95 °C, 30 s at 55 °C and 30 s at 60 °C. For each reaction the melting curve was analysed and the PCR product was run on an agarose gel in order to confirm the specificity of the RT-PCR. Expression levels were normalized using the sigA gene as an internal reference. Primers used for qRT-PCR are listed in Supplementary Table S1, available with the online version of this paper.
Construction of an M. bovis BCGΔbcg3145 knockout.
Generation of the M. bovis BCGΔbcg3145 followed a previously described phage protocol (Bardarov et al., 2002), with bcg3145 disrupted by a hygromycin cassette using a shuttle phasmid. Briefly, an approximately 1 kb region flanking bcg3145 was amplified by PCR. For the downstream region, the primers RIGHT PBCGPPEF (ATGAAGCTTCTACGCCCCTCAATGCAAGCG; HindIII site underlined) and PBCGPPER (ATACTAGTCAGCTGGCGGCCATTCAGGCTC; SpeI site underlined) were used; after HindІІІ and SpeI digestion the fragment was cloned into the cosmid pYUB854, to generate p3145right. The upstream region of bcg3145 was amplified using the primers PBCG3144F (ATTCTAGAGGCGGTCAACGAGGCGATCTGC; XbaI site underlined) and PBCG31444R (ATTCTAGAAGCACCTGCCGTCGTTGCGTTC; XbaI site underlined); after XbaI digestion this fragment was cloned into p3145right to generate the cosmid p3145KO. The correct orientation of the fragments was determined by restriction enzyme digestion and sequencing. The in vitro packaging reaction of the cosmid was done by PacI digestion and using the in vitro packaging kit Giga XL (Stratagene). After transduction of E. coli HB101 cells, transductants were selected on plates containing hygromycin and positive colonies were checked by PacI digestion.
M. smegmatis mc2155 was transformed with purified phasmid DNA by electroporation followed by selection on agar plates for 4 days at 30 °C. The phages generated were titrated, and a ratio of 10 phage per bacterium was used for the transduction of M. bovis BCG at the non-permissive temperature of 37 °C. Hygromycin-resistant transformants were screened for loss of bcg3145 by PCR and Southern blot analysis. Southern blotting (see Supplementary Fig. S1) was performed using a non-radioactive 435 bp probe and the DIG High Prime DNA Labelling and Detection Starter kit II (Roche Applied Science).
Site-directed mutagenesis.
Site-directed mutagenesis (SDM) on rv3124 was carried out using the Stratagene Quikchange II XL site-directed mutagenesis kit. The primer pairs used are listed in Supplementary Table S2. As a template for the reaction, rv3124 was cloned into the BamHI and PstI sites of pUC19 after amplification using primers Rv3124BamF (ATGGGATCCGTGCAATTCAACGTCTTAGGACCACTGGAAC) and Rv3124PstR (CATCTGCAGTTATGCCAAACCGATGG GAAGGAG), generating pUC3124. The SDM reactions were carried out in a total volume of 50 μl containing 5 μl reaction buffer, 10 ng pUC3124, 125 ng of each oligonucleotide, 1 μl dNTP mix and 2.5 units Pfu Ultra HF DNA polymerase. The thermocycling programme used was 95 °C for 30 s, followed by 16 cycles of 95 °C for 30 s, 55 °C for 1 min, and 68 °C for 5 min. After cooling the tubes on ice for 2 min, template was degraded using 10 units DpnI restriction enzyme at 37 °C for 1 h. One microlitre of each reaction was used to transform competent E. coli XL1-Blue cells and transformants were selected on LB-ampicillin plates. Mutant colonies were isolated and sequences verified (DNA Sequencing & Services, University of Dundee, UK).
Purification of recombinant Rv3124.
To purify Rv3124, MBPpET, an in-house double fusion tag vector (Vircell), was used using an MBP-Rv3124-His6 orientation with the maltose-binding protein (MBP) solubility enhancer and a 6-His tag flanking Rv3124. Fusion of rv3124 to MBPpET was accomplished using PCR to generate DNA fragments from M. tuberculosis gDNA containing BamHI and XhoI sites. The resulting DNA fragments were ligated to the BamHI and XhoI sites of the MBPpET vector. Correct nucleotide sequence was confirmed by DNA sequencing performed at the ‘Lopez Neyra’ Institute of Parasitology and Biomedicine in Granada, Spain. For expression of the MBP-Rv3124-His fusion protein, the construct was transformed into E. coli BL21 (Invitrogen), grown at 37 °C in LB containing 50 μg kanamycin ml−1 to an OD600 of 0.6, and protein expression was induced with the addition of IPTG and arabinose to a final concentration of 1 mM and 0.2 % respectively, with cultures then grown for an additional 3 h. Cells were harvested by centrifugation at 1789 g, and pellets that were not used immediately were frozen at −80 °C. For the scale-up purification procedure, frozen pellets from 1 l of BL21 cells overproducing Rv3124 were thawed and resuspended in lysis buffer [200 mM Tris/HCl (pH 7.5), 200 mM sodium chloride, 1 mM EDTA and 10 mM β-mercaptoethanol]. Cells were lysed using a French press cell disrupter (Thermo). The crude extract was centrifuged at 50 000 g for 30 min at 4 °C, and the Rv3124 protein present in the supernatant was purified using Amylose Resin matrix according to the manufacturer's instructions (New England Biolabs). Final protein concentration was determined using a Bradford Protein Assay kit (Bio-Rad) with BSA (Pierce) as a standard.
Electrophoretic mobility shift assay (EMSA).
EMSAs were performed using a 222 bp DNA fragment (Rv3109pro) containing 190 bp upstream of the moaA1 gene and the first 32 bp of the moaA1 gene. The Rv3109pro fragment was generated by PCR using the primers F1 (AACGAAAGCTCGCACATGAGTGGTC) and R3 (ATGGAGCTACCATATCAGGCAG), and end-labelled with [γ-32P]dATP (PerkinElmer) using polynucleotide kinase. Binding reactions (10 μl) contained 1 μl BSA, 1 μl poly (dI–dC), 1 μl 10× binding buffer (40 % glycerol, 50 mM DTT, 500 mM NaCl, 100 mM Tris/HCl, pH 7.5, 10 mM MgCl2, 5 mM EDTA), purified Rv3124 protein of varying concentrations and a constant concentration of labelled substrate DNA. When indicated, unlabelled Rv3109 probe was added as competitor DNA in a 10-fold and 100-fold molar excess. After incubation at 37 °C for 30 min, protein-bound and free DNA were separated by electrophoresis at ambient temperature on a 4.5 % native polyacrylamide gel running at 7 mA for 3 h. The gels were dried for 2 h and exposed to X-ray film overnight or longer at −80 °C.
Analysis of promoter activity.
rv3109 (moaA1) promoter PCR fragments were cloned upstream of lacZ in the L5-based integrating vector pSM128 (Dussurget et al., 1999), and then sequenced to verify the sequence and correct orientation of the promoter–lacZ fusions. To generate constructs, three forward primers were used, namely Fg1 (TAGTACTAACGAAAGCTCGCACATGAGTGGTC; ScaI site underlined), Fg2 (TAGTACTCCACGATGCGCCGATGCATTTCGG) and Fg3 (TAGTACTTAACCGACTGCGTCCAAAGT), and one reverse primer, Rev (TAGTACTATGGAGCTACCATATCAG). PCR products and pSM128 were ScaI digested and ligated to generate pB31, pB32 and pB33, respectively. Plasmids were electroporated into M. smegmatis mc2155 previously transformed with pB1, and kanamycin- and streptomycin-resistant transformants were isolated. Three independent transformants of each construct were selected for measurements of promoter activity. Cell extracts were prepared using a previously described method (Sala et al., 2008). β-Galactosidase assays were performed on M. smegmatis mc2155 cells grown at 37 °C to OD600 1. Cells were collected by centrifugation, resuspended in 500 μl TEDP (0.1 M Tris/HCl, 1 mM EDTA, 1 mM DTT and 1 mM PMSF), and disrupted by sonication using two pulses of 20 s at 40 % amplitude (Bandelin Sonoplus GM 70). β-Galactosidase activity was assayed as described by Miller (1972). M. smegmatis transformed with promoter-probe vector alone (pSM128) was used as a negative control. The enzyme activities were expressed as nmol ONPG converted to o-nitrophenol min−1 (mg protein)−1.
RESULTS
The bcg3145 SNP was an early event in the derivation of M. bovis BCG
The E159G SNP in bcg3145 is present in the genome-sequenced strain M. bovis BCG Pasteur 1173P2 relative to the virulent sequenced strains M. tuberculosis H37Rv and M. bovis AF2122/97 (the nucleotide sequences of the mb3147 and rv3124 orthologues are 100 % identical). In order to confirm the presence of this mutation in other BCG strains by conventional sequencing, the gene was amplified and sequenced from ‘early’ and ‘late’ BCG strains (Behr et al., 1999). The bcg3145 sequence from all M. bovis BCG strains tested (Pasteur, Denmark, Frappier, Russia, Tokyo, Sweden and Tice) differed from the M. bovis or M. tuberculosis orthologues in an adenine to guanine nsSNP that produces a glutamic acid to glycine change in amino acid residue 159 of the mature protein (E159G) relative to the orthologue in virulent strains (see Supplementary Fig. S2). Using blast we also identified the non-mutated residue in other genome-sequenced M. tuberculosis strains (CDC1551, KZN 1435, C, Haarlem, F11 and H37Ra). Therefore the E159G mutation was an early event during the derivation of BCG.
Modelling the structure of Rv3124 and predicted effects of the E159G mutation
To assess the functional consequences of the E159G mutation on Rv3124/BCG3145, the structure of Rv3124 was modelled using M. tuberculosis EmbR as the template. The sequences of these two proteins share 54 % identity and 73 % similarity. The resulting model (Fig. 1a) shows that Rv3124 is a two-domain protein composed of an amino-terminal DNA-binding domain (DBD) and a carboxy-terminal bacterial transcriptional activation (BTA) domain, the same modular arrangement as present in other members of the SARP family of transcription factors, such as Streptomyces peuceticus DnrI (Sheldon et al., 2002). Rv3124 lacks domains present in some other SARP proteins, such as forkhead-associated domains (Alderwick et al., 2006). Residue E159 is located in the third α-helix of the BTA domain of Rv3124 (Fig. 1b). The replacement of glutamic acid at position 159 by glycine perturbs the helix, and therefore would be predicted to have functional effects on the activity of BCG3145.
Fig. 1.

(a) Rv3124 structural model based on the EmbR molecular structure. The DNA-binding domain is in green, and the BTA domain in purple. Residue E159, located in the third α-helix of the BTA domain, is shown in red. (b) Close-up of the third α-helix of the BTA domain from a different angle. The side chains of residues that form the helix are in blue, except for E159, which is shown in red. Intra-chain H bonds are represented by green dashed lines. Replacement of Glu159 by Gly perturbs the helix.
Transcriptome analysis
In order to define the function of BCG3145/Rv3124, the M. tuberculosis regulator was overexpressed in M. bovis BCG Pasteur under the control of the hsp60 promoter, and the effect on the transcriptome analysed using microarrays and qRT-PCR. Overexpression of bcg3145 or rv3124 in BCG did not result in any change in the in vitro growth profile of the recombinant BCG strains in 7H9 liquid medium compared with wild-type BCG (data not shown). The results obtained from microarray analyses showed that 10 genes were significantly overexpressed and 11 genes were downregulated (minimum of threefold differential expression) when rv3124 was overexpressed in M. bovis BCG (Table 2) compared with a vector-only control. A cluster of six differentially expressed genes were localized in the moa1 locus of the M. bovis BCG genome, including the moa1 locus: moaA1 (rv3109), moaB1 (rv3110), moaC1 (rv3111) and moaD1 (rv3112). To validate the microarray data for those genes with upregulated expression, qRT-PCR analysis was performed. Fig. 2 compares the fold changes in expression in M. bovis BCG measured by microarray and qRT-PCR for the moa1 locus. The two methods displayed broad agreement in expression levels for each gene, confirming the experimental procedures and statistical approaches used in this study. Overexpression of rv3124 in M. tuberculosis H37Rv also led to upregulation of the moa1 locus as determined by microarray analysis (data not shown).
Table 2.
Microarray identification of differentially expressed genes in BCG overexpressing Rv3124 compared with BCG/pSM96 vector control
| Rv no. | Gene | Product | Fold change* |
|---|---|---|---|
| rv0991 | Hypothetical protein | 3.0 | |
| rv2428 | ahpC | Alkyl hydroperoxide reductase C | 5.4 |
| rv2636 | Hypothetical protein | 3.4 | |
| rv2931 | ppsA | Phenolpthiocerol synthesis | 3.0 |
| rv3109 | moaA1 | Molybdopterin biosynthesis | 36.8 |
| rv3110 | moaB1 | Molybdopterin biosynthesis | 21.5 |
| rv3111 | moaC1 | Molybdopterin biosynthesis | 16.4 |
| rv3112 | moaD1 | Molybdopterin biosynthesis | 9.3 |
| rv3113 | Conserved phosphatase | 15.2 | |
| rv3114 | Hypothetical protein | 5.1 | |
| rv0823c | Transcriptional regulator | 0.26 | |
| rv0824c | desA1 | Fatty acid desaturase | 0.33 |
| rv1094 | desA2 | Fatty acid desaturase | 0.31 |
| rv1798 | Conserved hypothetical protein | 0.32 | |
| rv2836c | dinF | DNA-damage-inducible protein | 0.33 |
| rv2987 | leuD | 3-Isopropylmalate dehydratase | 0.33 |
| rv2988 | leuC | 3-Isopropylmalate dehydratase | 0.16 |
| rv2989 | Transcriptional regulator | 0.18 | |
| rv3212 | Alanine-valine-rich protein | 0.32 | |
| rv3229c | desA3 | Fatty acid desaturase | 0.32 |
| rv3919 | gid | Glucose-inhibited division protein | 0.28 |
*Values <1 are downregulated in BCG overexpresssing Rv3124 compared with BCG/pSM96.
Fig. 2.
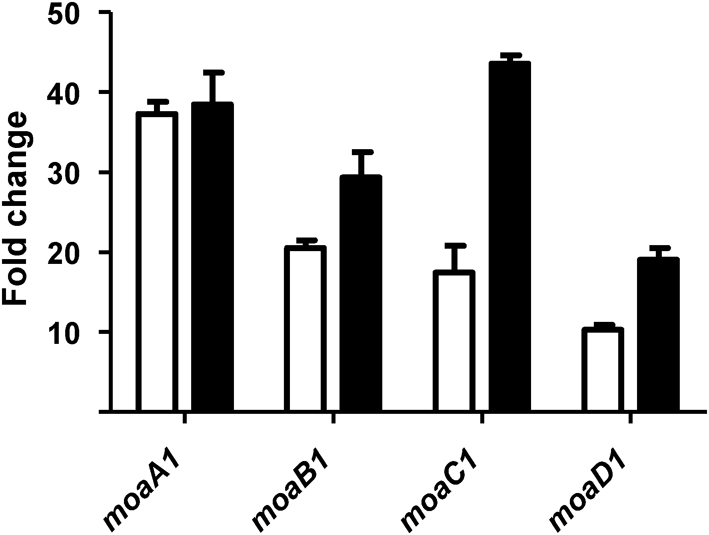
Confirmation of microarray results by qRT-PCR. The moa1 locus showed a higher level of expression in M. bovis BCG overexpressing Rv3124 than M. bovis BCG wild-type as measured by microarray (white bars) and by qRT-PCR (black bars). Fold changes are the mean ratios±sd of gene expression from two independent experiments.
M. bovis BCG Δbcg3145 mutant construction
To assess the effects of inactivation of BCG3145 on M. bovis BCG, a knockout mutant was generated by inserting a hygromycin cassette into the bcg3145 gene. Allelic exchange was confirmed in M. bovis BCG by PCR (data not shown) and Southern blotting (Supplementary Fig. S1). To initially characterize the M. bovis BCG Δbcg3145 mutant strain, the growth profiles of the knockout and the wild-type were measured under standard in vitro culture conditions and they showed similar doubling times (data not shown). As our transcriptome analysis had shown that the moa1 locus was regulated by Rv3124, we measured the expression of these genes in the wild-type and mutant by qRT-PCR, and no significant difference was found. This was probably to be expected, as BCG3145/Rv3124 both show low-level expression in mid-exponential-phase cultures of M. tuberculosis or M. bovis BCG (Brosch et al., 2007; Golby et al., 2007).
Effect of the BCG3145 mutation on transcription of the moa1 locus
To elucidate the effects of the E159G SNP on the function of the transcriptional regulator BCG3145, M. bovis BCG was complemented with a plasmid overexpressing bcg3145 or wild-type rv3124 under the hsp60 promoter. Comparison of global expression across these two strains by microarray analysis revealed a set of differentially expressed genes (Table 3). As the expression level of Rv3124 was not equal across the BCG/pB1 and BCG/pB3 recombinants, it was necessary to normalize fold changes to Rv3124 expression levels. The final normalized fold change was therefore calculated by dividing the BCG/pB1 versus BCG/pB3 fold changes by 2.84, the increased level of expression of Rv3124 seen in the BCG/pB1 compared with BCG/pB3. Overexpression of Rv3124 produced an 8.5- to 2.77-fold higher level of induction of the genes in the moa1 locus compared with BCG3145 (Table 3). The microarray results were confirmed by qRT-PCR (see Supplementary Table S3). Hence it appears that the mutation in BCG3145 reduces the ability of the regulator to induce expression of the moa1 locus.
Table 3.
Genes differently expressed in BCG overexpressing Rv3124 (BCG/pB1) or BCG3145 (BCG/pB3)
| Rv | Gene | BCG/pB1* | BCG/pB3 | Fold change without normalization to Rv3124 expression level | Normalized fold change† | ||||
|---|---|---|---|---|---|---|---|---|---|
| Expression level (log2) | Minimum level (log2) | Maximum level (log2) | Expression level (log2) | Minumum level (log2) | Maximum level (log2) | ||||
| rv2933 | ppsC | −0.45 | −0.51 | −0.35 | −2.17 | −2.41 | −2.06 | 3.39 | 1.15 |
| rv2932 | ppsB | −0.69 | −0.81 | −0.62 | −0.35 | −0.44 | −0.32 | 3.08 | 2.35 |
| rv3058 | 1.69 | 1.57 | 1.89 | 0.07 | 0.05 | 0.08 | 3.28 | 1.08 | |
| rv3109 | moaA1 | 1.87 | 1.56 | 2.02 | −2.70 | −2.86 | −2.64 | 23.91 | 8.41 |
| rv3110 | moaB1 | 2.29 | 2.16 | 2.43 | −2.31 | −2.48 | −2.08 | 24.36 | 8.57 |
| rv3111 | moaC1 | 0.97 | 0.95 | 1.12 | −2.88 | −2.94 | −2.45 | 14.51 | 5.11 |
| rv3112 | moaD1 | 1.62 | 1.43 | 1.97 | −1.35 | −1.71 | −1.06 | 7.89 | 2.77 |
| rv3113 | 0.57 | 0.35 | 0.74 | −2.89 | −3.26 | −2.83 | 11.02 | 3.88 | |
| rv3114 | 0.24 | 0.13 | 0.32 | −1.67 | −1.81 | −1.18 | 3.78 | 1.33 | |
*Expression levels for each gene are the median of three independent microarray experiments. Minimum and maximum expression levels for the three replicates are shown.
†Rv3124 was expressed 2.84-fold higher in BCG/pB1 compared to BCG/pB3.
EMSA and moaA1 promoter characterization
Specific in vitro binding of Rv3124 to the rv3108–moaA1 region was assessed by EMSAs. We evaluated binding of Rv3124 to an intergenic region of 202 bp, upstream of moaA1/rv3109, which was selected as a putative binding sequence (Fig. 3a). Purified Rv3124 bound to radiolabelled Rv3109pro promoter probe DNA and retarded its mobility (Fig. 3b). The extent of binding increased with Rv3124 protein concentration, with a mobility shift detected using 0.15 μg protein. Binding to the labelled promoter could be specifically competed with a 10 and 100-fold excess of the corresponding unlabelled DNA. Bioinformatic screens for regulatory binding motifs upstream of rv3124 and moaA1 did not reveal any significant matches.
Fig. 3.

Binding of Rv3124 to the rv3108–moa1A intergenic region. (a) The moaA1 locus showing locus organization and positions of oligonucleotides used in EMSA and lacZ fusions. Primers F1 and R3 were used to generate the Rv3109pro product used in EMSA assays; primers Fg1, Fg2, Fg3 and Rev were used to generate products for lacZ-transcriptional fusions. (b) EMSA was performed in the absence of Rv3124 (lane 1) or in the presence of 0.15, 0.3, 0.6 or 1.2 μg purified Rv3124 (lanes 2–5). Competition was performed with Rv3124 (2.4 μg) with no competitor (lane 6) or with unlabelled specific competitor rv3109 probe (10-fold and 100-fold molar excess, lane 7 and lane 8, respectively). (c) β-Galactosidase activity of promoter-probe constructs in wild-type M. smegmatis mc2155 (denoted by ‘−’) and M. smegmatis overexpressing Rv3124 (denoted by ‘+’). Activity was monitored by plating strains on medium containing X-Gal and by quantitative β-galactosidase assay. Data are shown as Miller units, and are the mean±sd of three replicates. (d) Binding assay with full-length and truncated versions of the Rv3109pro probe (190 bp, 116 bp and 45 bp) in the absence of Rv3124 (lane 1) or in the presence of 0.6 μg and 1.2 μg Rv3124 (lanes 2 and 3 respectively). Decreased Rv3124 binding to the shorter probes correlates with reduced β-galactosidase activity in the corresponding lacZ transcriptional fusions.
To further define the sequence elements recognized by Rv3124, we conducted EMSAs with 5′ deleted fragments of the rv3109 promoter, and generated promoter-reporter vector constructs. For the latter, fragments of 190 bp, 116 bp and 45 bp upstream of moaA1 were cloned into the promoterless integrative vector pSM128 (see Methods), creating transcriptional fusions with the β-galactosidase gene (Dussurget et al., 1999); these constructs were then transformed into a M. smegmatis strain overexpressing Rv3124, and reporter activity measured (Fig. 3c). β-Galactosidase activity of all constructs was significantly higher than that for the control pSM128; however, β-galactosidase background was seen in recombinants with the 190 bp fragment–lacZ fusion in the absence of Rv3124 overexpression, possibly due to read-through or to the presence of a functionally similar transcriptional regulator in M. smegmatis. Nevertheless, the presence of Rv3124 increased the promoter activity threefold. The levels of expression of the 116 bp construct were approximately 50 % lower than those showed for the 190 bp fragment. The smallest fragment (45 bp) generated only background activity levels.
EMSA was performed with Rv3124 and the same moaA1 promoter fragments as used in the β-galactosidase assays (Fig. 3d). Fragments were generated by PCR amplification using the same primern pairs as used for the promoter-probe vector construction and gel-purified. Bound protein was detected using the larger fragment Rv3109pro-190 bp. The Rv3109pro-116 bp probe was weakly bound by protein, and no binding was detectable with the Rv3109pro-45 bp probe. Thus Rv3124 binds directly to the moaA1 promoter.
Rv3124 domain characterization
The E159G mutation in BCG3145 occurs in the BTA domain of BCG3145. The transcriptome results from strains overexpressing BCG3145 or Rv3124 suggested that the E159G SNP had a subtle effect on gene regulation, so we sought to determine how other mutations at the E159 locus would affect the regulator's activity. To identify other functionally important residues, BCG3145/Rv3124 was compared with other SARP family member proteins (Supplementary Fig. S2). Previously it had been shown that alanine substitution of the conserved residues G90 and Y91 in the DBD of the related SARP-family regulator DnrI abrogated protein–DNA binding (Sheldon et al., 2002), so we sought to determine whether similar mutations in Rv3124 would block its activity.
Residues G90, Y91 and E159 in Rv3124 were subjected to site-directed mutagenesis. The range of mutations generated was G90A, Y91A, E159I, E159W, E159D, E159Q. The mutant proteins were overexpressed in the M. bovis BCGΔbcg3145 knockout strain to ensure no interference from a chromosomal copy of the gene, and alterations in the transcription levels of the moa1 locus were determined by qRT-PCR, normalizing the moa1 levels in the different strains to the mutated Rv3124 expression levels. Fig. 4(a) shows the downregulation of the moa1 locus in BCG Pasteur strains overexpressing mutated versions of Rv3124 compared with BCG overexpressing the non-mutated Rv3124.
Fig. 4.

(a) Fold change in expression of the moa1 locus in BCG strains overexpressing mutated versions of Rv3124. The expression of the moa1 locus in BCG strains complemented with the mutated Rv3124 was compared with M. bovis BCG overexpressing the non-mutated Rv3124. Data from two independent experiments for each strain were first normalized to Rv3124 and then normalized to sigA RNA levels. (b) Residues G90 and Y91 (depicted in blue) are located in one of the two β-sheets that form the DBD of Rv3124. Residue G90 seems not to be critical, explaining why its mutation had no effect on the regulator's activity. However, residue Y91 interacts with a conserved residue (H63) located in a neighbouring helix, showing why mutation of this residue in MoaR1 caused a decrease in the induction of the moa1 locus.
The mutations introduced in the DBD had divergent effects on protein function. While the G90A substitution had no effect on the transcription of the moa1 locus, the Y91A mutation significantly reduced locus transcription. This would appear to conflict with the previously reported results obtained with the DnrI regulator (Sheldon et al., 2002). To determine the structural basis for these results, the G90A and Y91A mutations were mapped to the Rv3124 structural model (Fig. 4b). The DBD domain is highly conserved among gene regulators of the OmpR/PhoB family (Alderwick et al., 2006), and is formed by a three-helix bundle flanked by two β-sheets. Residues G90 and Y91 are located in one of the two sheets. The role of G90 seems not to be critical since the bend in the short coil intersection between the two strands that form the sheet is maintained by two contiguous conserved prolines at positions 88 and 89. Therefore the mutation G90A does not have a significant effect on the activity of the protein. On the other hand, residue Y91 seems to have a more important function in the overall stability of the DBD by its interaction with another conserved residue (H63) located in a neighbouring helix. Hence the change of tyrosine to alanine in Y91A causes a perturbation of the local structure that compromises binding to DNA, thus explaining the decrease in the induction of the moa1 locus by the Y90A variant of Rv3124.
To assess the importance of residue 159 in the BTA domain, several mutations were introduced. First, we were interested in evaluating the effect of a complete change in residue properties; hence, glutamic acid was replaced with isoleucine or tryptophan. These mutations completely disrupted protein function, as shown by the dramatic decrease of RNA levels for the moa1 locus (Fig. 4a). To assess the functional effect of more subtle changes, Glu159 was substituted with aspartic acid or glutamine. qRT-PCR analysis revealed that the aspartic acid mutation did not significantly alter protein function, with the E159D Rv3124 protein being able to complement the knockout strain to wild-type levels. However, complementation with the E159Q mutated version of Rv3124 was not able to induce the moa1 locus to the same degree as the wild-type protein. Again using the Rv3124 structural model to map the mutations, it was evident that E159G, E159I and E159W caused a perturbation of the helix that affects the function of the BTA domain. Mutations E159D and E159Q have a lesser effect on the structure and hence maintain the functionality of the domain.
DISCUSSION
Comparison of the genome of M. bovis BCG Pasteur with those of M. bovis and M. tuberculosis has revealed a range of deletions and SNPs between the vaccine strain and virulent tubercle bacilli that may play a role in the virulence attenuation of BCG. However, while clear functional links have been made for some mutations in BCG, such as the RD1 deletion or SNPs in the mmaA3 and pykA genes, linking genetic differences to phenotypic changes is far from trivial.
A comprehensive survey of SNP differences across BCG vaccine strains and M. bovis strains from the UK and France revealed 186 SNPs that differentiated virulent M. bovis from BCG, with 115 of these SNPs being non-synonymous. An amplification of this mutational difference is achieved when nsSNPs occur in regulatory genes, as perforce each one affects the expression of a wide range of genes. Amongst these nsSNPs was a mutation in bcg3145, encoding a putative transcriptional regulator of the AfsR/DnrI/SARP family. The predicted secondary structure of BCG3145 revealed that the protein consists of two conserved domains. The N-terminal region is a DNA-binding domain (residues 1–96) formed by three α-helices packed against two antiparallel β-sheets forming a winged helix–turn–helix (Martinez-Hackert & Stock, 1997a), and a C-terminal region (residues 97–270) which is a BTA domain formed with seven α-helices (Alderwick et al., 2006). It has been suggested that the region of α-loop connecting the recognition helix α3 and the positioning helix α2, which are conserved in the DNA-binding domain of the SARP family, interacts with the C-terminal domain of the α subunit of RNA polymerase (Martinez-Hackert & Stock, 1997b; Tanaka et al., 2007). The E159G mutation in BCG3145 mutates to glycine a conserved glutamic acid residue located in a tetratricopeptide repeat (TRP) in the BTA domain (region T3). TRP domains are associated with protein–protein interactions (D'Andrea & Regan, 2003), while a conserved core (helices T1–T7) of the BTA domain seems to be required for proper function of SARP family proteins (D'Andrea & Regan, 2003; Sheldon et al., 2002).
In the present study we found that BCG3145/Rv3124 is a positive transcriptional regulator of the moa1 locus, promoting the expression of the moaA1B1C1D1 genes required for molybdopterin biosynthesis. Microarray results were confirmed independently by qRT-PCR, while direct binding of Rv3124 to the upstream region of moaA1, the first gene of the moa1 locus, was confirmed by EMSAs. The moaA1B1C1D1 genes encode key enzymes in the synthesis of molybdopterin, a pterin-based molybdenum-binding cofactor of molybdoenzymes such as nitrate reductase. MoaA1 and MoaC1 are predicted to catalyse the conversion of GTP into cyclic pyranopterin monophosphate (cPMP), the first step in the synthesis of molybdopterin; cPMP is then converted to the metal-binding pterin dithiolate by molybdopterin synthase, a heterotetrameric complex of MoaD1 and MoaE1 (Rv3119). MoaB1 is predicted to be involved in insertion of molybdenum into molybdopterin to form the molybdenum cofactor (Schwarz et al. 2009).
In E. coli, the moa operon shows increased expression under anaerobiosis (Baker & Boxer, 1991), a situation which requires the production of several essential molybdoenzymes for anaerobic respiration (Self et al., 1999). Oxygen limitation is considered to be a key factor affecting the metabolism of M. tuberculosis in the hypoxic environment of the granuloma (Parish & Brown, 2008). In genome-wide expression analysis of M. tuberculosis isolated from lung biopsy samples, a 13-fold upregulation of rv3124 was observed in granuloma tissues of patients with active tuberculosis compared with in vitro cultures of M. tuberculosis (Rachman et al., 2006). This condition is reproduced in vitro by the Wayne model (Wayne & Hayes, 1996), where oxygen is depleted gradually and M. tuberculosis growth shows two phases, a microaerobic state, called nonreplicating persistent stage 1 (NRP-1), and an anaerobic stage, NRP-2. rv3124 has been shown to be upregulated in both NRP-1 and NRP-2 states in M. tuberculosis compared with aerobic roller cultures (Muttucumaru et al., 2004). Hence, regulation of rv3124 appears to be modulated by oxygen availability.
Sequence alignments and protein modelling suggested that the E159G mutation in BCG3145 would have structural consequences, with microarray analysis showing that the mutation had a subtle effect on the ability of BCG3145 to activate moa1 locus transcription. Overexpression of mutated variants of Rv3124 in BCG, with subsequent qRT-PCR on the constituent genes of the moa1 locus, showed that E159I and E159W substitutions had profound effects on protein function, as would be expected, with E159Q showing more subtle effects, while mutation of the Tyr91 reside in the DNA-binding domain also abrogated the regulator's activity.
Conclusion
We have identified Rv3124 as a positive regulator of molybdopterin biosynthesis, and defined the functional consequences of a mutation in the BCG3145 orthologue. As Rv3124 regulates expression of the moa1 locus, we suggest that rv3124 be renamed moaR1. The E159G mutation in BCG3145 was shown to decrease, but not abolish, the ability of the regulator to induce expression of the moa1 locus. Given our results, and the fact that Rv3124 was not identified as a virulence factor in M. tuberculosis using saturation mutagenesis screens (Sassetti & Rubin, 2003), it would appear that the mutation in BCG3145 is not a key attenuating genetic lesion in BCG.
Acknowledgments
We wish to thank Claudia Sala, Stewart T. Cole and Juan Luis Ramos for advice and discussion, Denise Waldron and Adam Whitney (Bacterial Microarray Group, St George's) for help with depositing data in μG@Sbase and ArrayExpress, and Bill Jacobs for the supply of strains and plasmids used in the construction of the bcg3145 mutant. We wish to acknowledge Colorado State University for the provision of M. tuberculosis H37Rv genomic DNA produced under NIH contract HHSN266200400091C/ADB NO1-A1-40091 ‘Tuberculosis Vaccine and Research Materials Contract’. This work was funded by Vircell S.L., Spain, the Department of Environment, Food and Rural Affairs, GB, and the European Community's Seventh Framework Programme ([FP7/2007–2013]) under grant agreement no. 201762.
Abbreviations
BTA, bacterial transcriptional activator
DBD, DNA-binding domain
EMSA, electrophoretic mobility shift assay
nsSNP, non-synonymous SNP
qRT-PCR, quantitative reverse transcription PCR
SARP, Streptomyces antibiotic regulatory protein, SNP, single-nucleotide polymorphism
Footnotes
The array design used in this study is available in μG@Sbase (accession no. A-BUGS-31; http://bugs.sgul.ac.uk/A-BUGS-31) and also ArrayExpress (accession no. A-BUGS-31). Fully annotated microarray data have been deposited in μG@Sbase (accession no. E-BUGS-93; http://bugs.sgul.ac.uk/E-BUGS-93) and also ArrayExpress (accession no. E-BUGS-93).
Three supplementary tables and two supplementary figures are available with the online version of this paper.
References
- Alderwick, L. J., Molle, V., Kremer, L., Cozzone, A. J., Dafforn, T. R., Besra, G. S. & Futterer, K. (2006). Molecular structure of EmbR, a response element of Ser/Thr kinase signaling in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 103, 2558–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, K. P. & Boxer, D. H. (1991). Regulation of the chlA locus of Escherichia coli K12: involvement of molybdenum cofactor. Mol Microbiol 5, 901–907. [DOI] [PubMed] [Google Scholar]
- Bardarov, S., Bardarov, S., Jr, Pavelka, M. S., Jr, Sambandamurthy, V., Larsen, M., Tufariello, J., Chan, J., Hatfull, G. & Jacobs, W. R., Jr (2002). Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148, 3007–3017. [DOI] [PubMed] [Google Scholar]
- Behr, M. A., Wilson, M. A., Gill, W. P., Salamon, H., Schoolnik, G. K., Rane, S. & Small, P. M. (1999). Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284, 1520–1523. [DOI] [PubMed] [Google Scholar]
- Behr, M. A., Schroeder, B. G., Brinkman, J. N., Slayden, R. A. & Barry, C. E., III (2000). A point mutation in the mma3 gene is responsible for impaired methoxymycolic acid production in Mycobacterium bovis BCG strains obtained after 1927. J Bacteriol 182, 3394–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger, A. E., Besra, G. S., Ford, M. E., Mikusova, K., Belisle, J. T., Brennan, P. J. & Inamine, J. M. (1996). The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc Natl Acad Sci U S A 93, 11919–11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belley, A., Alexander, D., Di Pietrantonio, T., Girard, M., Jones, J., Schurr, E., Liu, J., Sherman, D. R. & Behr, M. A. (2004). Impact of methoxymycolic acid production by Mycobacterium bovis BCG vaccines. Infect Immun 72, 2803–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosch, R., Gordon, S. V., Garnier, T., Eiglmeier, K., Frigui, W., Valenti, P., Dos Santos, S., Duthoy, S., Lacroix, C. & other authors (2007). Genome plasticity of BCG and impact on vaccine efficacy. Proc Natl Acad Sci U S A 104, 5596–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson, K., McGuffin, L. J., Marsden, R. L., Ward, J. J., Sodhi, J. S. & Jones, D. T. (2005). Protein structure prediction servers at University College London. Nucleic Acids Res 33, W36–W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calmette, A. (1927). La Vaccination Preventive Contre la Tuberculose. Paris: Masson et cie.
- Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S. V., Eiglmeier, K., Gas, S. & other authors (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. [DOI] [PubMed] [Google Scholar]
- D'Andrea, L. D. & Regan, L. (2003). TPR proteins: the versatile helix. Trends Biochem Sci 28, 655–662. [DOI] [PubMed] [Google Scholar]
- Dussurget, O., Timm, J., Gomez, M., Gold, B., Yu, S., Sabol, S. Z., Holmes, R. K., Jacobs, W. R., Jr & Smith, I. (1999). Transcriptional control of the iron-responsive fxbA gene by the mycobacterial regulator IdeR. J Bacteriol 181, 3402–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia Pelayo, M. C., Uplekar, S., Keniry, A., Mendoza Lopez, P., Garnier, T., Nunez Garcia, J., Boschiroli, L., Zhou, X., Parkhill, J. & other authors (2009). A comprehensive survey of single nucleotide polymorphisms (SNPs) across Mycobacterium bovis strains and M. bovis BCG vaccine strains refines the genealogy and defines a minimal set of SNPs that separate virulent M. bovis strains and M. bovis BCG strains. Infect Immun 77, 2230–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golby, P., Hatch, K. A., Bacon, J., Cooney, R., Riley, P., Allnutt, J., Hinds, J., Nunez, J., Marsh, P. D. & other authors (2007). Comparative transcriptomics reveals key gene expression differences between the human and bovine pathogens of the Mycobacterium tuberculosis complex. Microbiology 153, 3323–3336. [DOI] [PubMed] [Google Scholar]
- Golby, P., Nunez, J., Cockle, P. J., Ewer, K., Logan, K., Hogarth, P., Vordermeier, H. M., Hinds, J., Hewinson, R. G. & Gordon, S. V. (2008). Characterization of two in vivo-expressed methyltransferases of the Mycobacterium tuberculosis complex: antigenicity and genetic regulation. Microbiology 154, 1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, S. V., Brosch, R., Billault, A., Garnier, T., Eiglmeier, K. & Cole, S. T. (1999). Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol Microbiol 32, 643–655. [DOI] [PubMed] [Google Scholar]
- Hunt, D. M., Saldanha, J. W., Brennan, J. F., Benjamin, P., Strom, M., Cole, J. A., Spreadbury, C. L. & Buxton, R. S. (2008). Single nucleotide polymorphisms that cause structural changes in the cyclic AMP receptor protein transcriptional regulator of the tuberculosis vaccine strain Mycobacterium bovis BCG alter global gene expression without attenuating growth. Infect Immun 76, 2227–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D. T. (1999). Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292, 195–202. [DOI] [PubMed] [Google Scholar]
- Keating, L. A., Wheeler, P. R., Mansoor, H., Inwald, J. K., Dale, J., Hewinson, R. G. & Gordon, S. V. (2005). The pyruvate requirement of some members of the Mycobacterium tuberculosis complex is due to an inactive pyruvate kinase: implications for in vivo growth. Mol Microbiol 56, 163–174. [DOI] [PubMed] [Google Scholar]
- Kiefer, F., Arnold, K., Kunzli, M., Bordoli, L. & Schwede, T. (2009). The SWISS-MODEL Repository and associated resources. Nucleic Acids Res 37, D387–D392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, K. N., Liao, R., Guinn, K. M., Hickey, M. J., Smith, S., Behr, M. A. & Sherman, D. R. (2003). Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette–Guérin attenuation. J Infect Dis 187, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahairas, G. G., Sabo, P. J., Hickey, M. J., Singh, D. C. & Stover, C. K. (1996). Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J Bacteriol 178, 1274–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Hackert, E. & Stock, A. M. (1997a). The DNA-binding domain of OmpR: crystal structures of a winged helix transcription factor. Structure 5, 109–124. [DOI] [PubMed] [Google Scholar]
- Martinez-Hackert, E. & Stock, A. M. (1997b). Structural relationships in the OmpR family of winged-helix transcription factors. J Mol Biol 269, 301–312. [DOI] [PubMed] [Google Scholar]
- Miller, J. M. (1972). Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Mostowy, S., Tsolaki, A. G., Small, P. M. & Behr, M. A. (2003). The in vitro evolution of BCG vaccines. Vaccine 21, 4270–4274. [DOI] [PubMed] [Google Scholar]
- Muttucumaru, D. G., Roberts, G., Hinds, J., Stabler, R. A. & Parish, T. (2004). Gene expression profile of Mycobacterium tuberculosis in a non-replicating state. Tuberculosis (Edinb) 84, 239–246. [DOI] [PubMed] [Google Scholar]
- Parish, T. & Brown, A. (2008). Mycobacterium: Genomics and Molecular Biology. Norwich, Caister Academic Press.
- Pym, A. S., Brodin, P., Brosch, R., Huerre, M. & Cole, S. T. (2002). Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 46, 709–717. [DOI] [PubMed] [Google Scholar]
- Rachman, H., Strong, M., Ulrichs, T., Grode, L., Schuchhardt, J., Mollenkopf, H., Kosmiadi, G. A., Eisenberg, D. & Kaufmann, S. H. (2006). Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect Immun 74, 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala, C., Forti, F., Magnoni, F. & Ghisotti, D. (2008). The katG mRNA of Mycobacterium tuberculosis and Mycobacterium smegmatis is processed at its 5′ end and is stabilized by both a polypurine sequence and translation initiation. BMC Mol Biol 9, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti, C. M. & Rubin, E. J. (2003). Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A 100, 12989–12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, G., Mendel, R. R. & Ribbe, M. W. (2009). Molybdenum cofactors, enzymes and pathways. Nature 460, 839–847. [DOI] [PubMed] [Google Scholar]
- Self, W. T., Grunden, A. M., Hasona, A. & Shanmugam, K. T. (1999). Transcriptional regulation of molybdoenzyme synthesis in Escherichia coli in response to molybdenum: ModE–molybdate, a repressor of the modABCD (molybdate transport) operon is a secondary transcriptional activator for the hyc and nar operons. Microbiology 145, 41–55. [DOI] [PubMed] [Google Scholar]
- Sheldon, P. J., Busarow, S. B. & Hutchinson, C. R. (2002). Mapping the DNA-binding domain and target sequences of the Streptomyces peucetius daunorubicin biosynthesis regulatory protein, DnrI. Mol Microbiol 44, 449–460. [DOI] [PubMed] [Google Scholar]
- Tanaka, A., Takano, Y., Ohnishi, Y. & Horinouchi, S. (2007). AfsR recruits RNA polymerase to the afsS promoter: a model for transcriptional activation by SARPs. J Mol Biol 369, 322–333. [DOI] [PubMed] [Google Scholar]
- Wayne, L. G. & Hayes, L. G. (1996). An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun 64, 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooff, E., Michell, S. L., Gordon, S. V., Chambers, M. A., Bardarov, S., Jacobs, W. R., Jr, Hewinson, R. G. & Wheeler, P. R. (2002). Functional genomics reveals the sole sulphate transporter of the Mycobacterium tuberculosis complex and its relevance to the acquisition of sulphur in vivo. Mol Microbiol 43, 653–663. [DOI] [PubMed] [Google Scholar]