

Abstract
Synthesis of the siderophore yersiniabactin (Ybt) proceeds by a mixed nonribosomal peptide synthetase/polyketide synthase mechanism. Transcription of ybt genes encoding biosynthetic and transport functions is repressed under excess iron conditions by Fur, but is also activated by Ybt via the transcriptional regulator YbtA. While mutations in most biosynthetic genes and ybtA negate transcription activation from the regulated promoters, three biosynthetic mutations do not reduce this transcriptional activation. Here we show that two of these mutants, one lacking the putative type II thioesterase (TE) YbtT and the other with a mutation in the TE domain of HMWP1, produce reduced levels of authentic Ybt that are capable of signalling activity. Alanine substitutions in two residues of YbtT that are essential for catalytic activity in other type II TEs reduced the ability of Yersinia pestis to grow under iron-chelated conditions. The third mutant, which lacks the salicylate synthase YbtS, did not make authentic Ybt but did produce a signalling molecule. Finally, a Δpgm strain of Y. pestis, which lacks essential Ybt biosynthetic genes, also produced a signalling molecule that can activate transcription of ybt genes. The non-Ybt signal molecules from these two mutants are likely separate compounds. While these compounds are not biologically relevant to normal Ybt regulation, a comparison of the structures of Ybt and other signalling molecules will help in determining the chemical structures recognized as a Ybt signal.
INTRODUCTION
Iron is essential for nearly all pathogenic bacteria as well as for their hosts. Under normal conditions, the host binds iron to specific proteins, making free iron essentially unavailable to invading pathogens. Consequently, obtaining iron from these host proteins is one prerequisite for most pathogens to cause infection and disease (Braun, 2001, 2005; Bullen et al., 2005; Schaible & Kaufmann, 2004). Yersinia pestis, the causative agent of bubonic and pneumonic plague, encodes a large number of proven or putative iron-transport systems. Of these, the siderophore-dependent yersiniabactin (Ybt) biosynthesis and transport system has proven to be essential for the ability of the organism to cause bubonic plague.
Except for ybtD and fur, all identified genes required for the synthesis and transport of Ybt and the regulation of the entire system are encoded within a high-pathogenicity island (HPI) that is found in a number of enteric pathogens, including Yersinia enterocolitica and Yersinia pseudotuberculosis (Bobrov et al., 2002; Deng et al., 2002; Lesic & Carniel, 2004; Perry & Fetherston, 2004). Ybt synthesis occurs by a mixed nonribosomal peptide synthetase (NRPS)/polyketide synthase (PKS) mechanism (Fig. 1a). Salicylate, three cysteines, a malonyl linker group and three methyl groups are assembled into a four-ring structure made of salicylate, one thiazolidine, and two thiazoline rings with a malonyl linker separating the final thiazoline from the thiazolidine ring (Chambers et al., 1996; Crosa & Walsh, 2002; Drechsel et al., 1995; Gehring et al., 1998a; Miethke & Marahiel, 2007; Perry et al., 1999). YbtD is a putative phosphopantetheinyl (P-pant) transferase responsible for adding the phosphopantetheine tethers for the cysteine, salicylate and malonyl groups to HMWP1 and HMWP2. YbtS synthesizes salicylate from chorismate, which is then adenylated by YbtE and transferred to the HMWP2–HMWP1 assembly complex. Two cysteines are cyclized and condensed to form two thiazoline rings on HMWP2 (encoded by irp2). A malonyl linker is added and YbtU reduces the second thiazoline ring to thiazolidine before cyclization and condensation of the final thiazoline ring on HMWP1 (encoded by irp1). The YbtT putative thioesterase (TE) likely serves an editing function to remove aberrant molecules from the enzyme complex, while the TE domain of HMWP1 likely releases the completed siderophore from the enzyme complex (Fig. 1a) (Crosa & Walsh, 2002; Gehring et al., 1998a; Geoffroy et al., 2000; Kerbarh et al., 2005; Miller et al., 2002; Pelludat et al., 2003). The formation constant of this siderophore with ferric iron is 4×1036 and the crystal structure of the complex has been solved (Miller et al., 2006; Perry et al., 1999).
Fig. 1.

Models of yersiniabactin (Ybt) biosynthesis, transport and gene regulation. (a) Ybt biosynthesis. YbtS converts chorismate to salicylate and YbtE adenylates salicylate for attachment to HMWP2. YbtD transfers phosphopantetheinyl groups from coenzyme A to the indicated sites on HMWP2 and HMWP1 for attachment of salicylate, three cysteines and malonate. YbtU reduces one thiazolidine ring to a thiazoline ring, while YbtT is a TE that likely removes aberrant molecules from the Ybt synthetase enzyme complex. The TE domain of HMWP1 likely releases the completed siderophore from the enzyme complex. Asterisks in the Ybt structure indicate ferric ion coordination sites. HMWP2 and HMWP1 enzymatic domains: ArCP, aryl carrier protein; Cy, condensation/cyclization; A, adenylation; PCP, peptidyl carrier protein; KS, ketoacyl synthase; AT, acyltransferase; MT, methyltransferase; KR, β-ketoreductase; ACP, acyl carrier protein; TE, thioesterase. The model in (a) is reproduced with modifications from Perry & Fetherston (2004) with the permission of Horizon Scientific Press/Caister Academic Press, UK. (b) Model of Ybt transport and transcriptional regulation of ybtP. Synthesized Ybt is exported via an unknown mechanism that may include YbtX. Secreted Ybt can remove ferric iron from transferrin (Tf-Fe) and lactoferrin (Lf-Fe). We propose that the Ybt–Fe complex is transported through the outer membrane via the TonB-dependent receptor Psn. Ybt–Fe is transported through the inner membrane by YbtP/YbtQ. It is unclear whether this step requires a periplasmic or a membrane-spanning protein (none is shown here). In the cytoplasm, iron is released from Ybt by an unknown mechanism and is used nutritionally and for regulation of gene expression. The ybtP promoter, which controls expression of the ybtPQXS operon, is activated by YbtA, probably complexed with Ybt, and repressed by Fur in the presence of excess iron. Promoters for the irp2-irp1-ybtUTE and psn operons are similarly regulated. In contrast, YbtA negatively regulates transcription from its own promoter (Perry, 2004; Perry & Fetherston, 2004). YABS, YbtA binding site; FBS, Fur binding site; promoter region indicated by −10 and −35. Dashed arrows indicate steps or transported substrates that have not been experimentally determined.
YbtX is likely involved in, but not essential for, Ybt export from the bacterial cell (Fig. 1b). Use of iron from the Ybt–Fe complex requires the TonB-dependent outer membrane receptor Psn (termed FyuA in Y. enterocolitica) and an ABC transporter system consisting of YbtP and YbtQ: two fused-function inner membrane proteins with both permease and ATPase domains (Fig. 1b). Most ABC uptake transporters have separate polypeptides that encode permease and ATPase domains, while proteins that contain both a permease and an ATPase are usually involved in export. However, it has been demonstrated that YbtP and YbtQ are both required for uptake and are not essential for siderophore export (Fetherston et al., 1995, 1999; Perry et al., 2003b).
The four ybt operons within the HPI, (1) psn, (2) irp2-irp1-ybtU-ybtT-ybtE, (3) ybtA and (4) ybtP-ybtQ-ybtX-ybtS, are repressed by the Fur–Fe complex. In addition to Fur and iron repression, expression of the Ybt system is controlled by YbtA, a member of the AraC family of transcriptional regulators, and the Ybt siderophore. Together, YbtA and the Ybt siderophore activate transcription from the psn, irp2 and ybtP promoters while repressing transcription of the ybtA promoter (Anisimov et al., 2005; Fetherston et al., 1996, 1999; Gehring et al., 1998a; Perry et al., 2003a; Staggs et al., 1994). Repeats within the promoter regions of the ybt genes are proposed as YbtA binding sites. Mutation of the repeat in the psn promoter causes loss of transcriptional activation (Fetherston et al., 1996, 1999; Perry et al., 2003a, b). In Y. enterocolitica, Anisimov et al. (2005) have demonstrated that YbtA binds to these repeats in the irp6 (ybtP) promoter. Ybt is a potent signal molecule: concentrations 500-fold lower than that required for growth stimulation activate the ybtP promoter (Perry et al., 2003a).
A mutation in any of the seven genes encoding Ybt-biosynthetic enzymes causes a loss of siderophore production, as assessed by growth stimulation in bioassays. The ybtD, ybtE, ybtU, irp1 and irp2 mutations each cause a loss of Ybt protein expression due to lower transcription from the psn, irp2 and ybtP promoters. Addition of purified Ybt to Ybt-biosynthetic mutants restores YbtA-dependent transcriptional activation (Perry et al., 2003a). An unexpected finding was that mutations in ybtS, ybtT and the TE domain of irp1 (irp1-2086) do not affect regulation of the Ybt system (Bearden et al., 1997; Bobrov et al., 2002; Fetherston et al., 1995, 1996; Geoffroy et al., 2000; Perry et al., 2003a).
In this study we examined the means by which ybtS, ybtT and irp1-2086 mutants maintain normal regulation of the Ybt system. We found that the TE mutants ybtT and irp1-2086 produce reduced amounts of authentic Ybt. Although the ybtS mutant does not make Ybt, this mutant and a chromosomal deletion mutant lacking the entire HPI (a Δpgm strain) make Ybt-like molecules capable of transcriptionally activating the ybtP promoter. Finally, we show that substitutions in YbtT residues found to be critical for TE activity in other type II TEs cause a phenotype similar to that of a ybtT mutant.
METHODS
Bacterial strains and cultivation.
All bacterial strains and plasmids used in this study are described in Table 1. All Y. pestis strains used were avirulent due to lack of the virulence plasmid pCD1 (Perry & Fetherston, 1997; Perry et al., 1998). Cells from buffered glycerol stocks (Beesley et al., 1967) stored at –80 °C were streaked onto Congo red (CR) plates (Surgalla & Beesley, 1969) and incubated at 30 °C for 48 h to confirm retention of the pgm locus. ORFs for the Ybt system are encoded on the HPI within the pgm locus (Lesic & Carniel, 2004; Perry & Fetherston, 2004). CR+ colonies were inoculated on Tryptose Blood Agar Base (TBA; Difco Laboratories) slants and incubated at 30 °C for 24–48 h. Cells were washed off TBA slants with deferrated PMH2 medium (Gong et al., 2001), inoculated to an OD620 of 0.1, and incubated for ∼8 h at 37 °C with aeration in a New Brunswick model G76 gyratory shaker water bath (200 r.p.m.). Gong et al. (2001) has an error in the published buffer concentrations: PIPES and HEPES should be 50 mM for PMH2 and PMH, respectively. For siderophore isolation and bioassays, cultures were diluted into fresh, deferrated PMH2 to OD620 0.1 and incubated at 37 °C for ∼16 h. Both the Ybt+ strain and Ybt-biosynthetic mutants have similar growth rates and similar cell yields under these growth conditions (Fetherston et al., 2010). Thus, culture supernatants for Ybt isolation or direct use in regulation studies are from cultures incubated for the same time periods and harvested from similar cell yields. Cells were removed from spent PMH2 by centrifugation and filtration of the supernatant through 0.22 μm pore-size filters. Cell growth in liquid cultures was monitored with a Genesys 5 spectrophotometer (Spectronic Instruments). All glassware used for iron-restricted studies was soaked overnight in 5 % Micro-90 (Cole-Parmer) to remove contaminating iron and copiously rinsed in deionized water.
Table 1.
Bacterial strains and plasmids used in this study
All Y. pestis strains are Lcr− and thus avirulent due to a lack of the low-calcium-response plasmid pCD1. Strains with a plus sign possess an intact 102 kb pgm locus containing the genes for biofilm formation (hms) and the Ybt iron-transport system. All other Y. pestis strains have either a pgm deletion or a mutation within the pgm locus. Abbreviations: Apr, Cmr, Kmr, Spcr and Smr, respectively indicate resistance to ampicillin, chloramphenicol, kanamycin, spectinomycin and streptomycin.
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Y. pestis strains | ||
| KIM6+ | Pgm+ (Hms+ Ybt+) Lcr−; pMT1, pPCP1 | Fetherston et al. (1992) |
| KIM6 | Pgm− (Δpgm; Hms− Ybt−) Lcr−; pMT1, pPCP1 | Fetherston et al. (1992) |
| KIM6-2046.1 | Kmr Hms+ Ybt− (irp2 : : kan2046.1) Lcr−; pMT1, pPCP1 | Fetherston et al. (1995) |
| KIM6-2046.3 | Hms+ Ybt− (in-frame Δirp2–2046.3) Lcr−; pMT1, pPCP1 | Bearden et al. (1997) |
| KIM6-2046.6 | Kmr Hms+ Ybt− (irp2 : : kan2046.1) invA : : ybtPp : : lacZβ-gal+(ybtP : : lacZ promoter fusion) Lcr−; pMT1, pPCP1 | This study |
| KIM6-2046.7 | Cmr Hms+ Ybt− (irp2ΔS52) Δy2360 : : cam2093 Lcr−; pMT1, pPCP1 | This study |
| KIM6-2070.1 | Kmr Hms+ Ybt− (ybtS : : kan2070.1) Lcr−; pMT1, pPCP1 | Gehring et al. (1998a) |
| KIM6-2070.2 | Kmr Hms+ Ybt− (ybtS : : kan2070.1) Δy2336-2126 Lcr−; pMT1, pPCP1 | This study |
| KIM6-2072 | Hms+ Ybt− (ΔybtT2072) Lcr−; pMT1, pPCP1 | Geoffroy et al. (2000) |
| KIM6-2086 | Hms+ Ybt− (irp1-2086; HMWP1-2086, defective TE domain) Lcr−; pMT1, pPCP1 | Bobrov et al. (2002) |
| KIM6-2093 | Cmr Hms+ Δy2360 : : cam2093 Lcr−; pMT1, pPCP1 | Perry et al. (2004) |
| KIM6-2126 | Hms+ Ybt+ Δy2336–2126 Lcr−; pMT1, pPCP1 | This study |
| KIM6-2127+ | Pgm+ (Hms+ Ybt+) invA : : ybtPp : : lacZβ-gal+ (ybtP : : lacZ promoter fusion) Lcr−; pMT1, pPCP1 | This study |
| KIM10+ | Pgm+ (Hms+ Ybt+) Lcr−; pMT1 | Perry et al. (1990) |
| Plasmids | ||
| pBSlacZMCS | 7.1 kb; high-copy-number cloning vector with rrnBT1 transcriptional terminator and promoterless lacZ gene from pEU730 | Perry et al. (2004) |
| pCIRP498.8 | 17.4 kb suicide vector; irp2 : : kan2046.1; Apr Kmr | Fetherston et al. (1995) |
| pEUYbtP | 15.3 kb reporter plasmid; ybtP : : lacZ reporter fusion; Spcr | Fetherston et al. (1999) |
| pIHS1 | 7.3 kb; y2336+; 3.5 kb HindIII–SalI fragment from KIM10+ ligated into pTrueBlue-rop; Apr | This study |
| pIHS2 | 5.4 kb; Δ2336; 1.8 kb NruI–StuI fragment deleted from pIHS1; Apr | This study |
| pIHS3 | 7.1 kb; Δ2336; 1.6 kb HindIII–SalI fragment from pIHS2 ligated into pWSK29; Apr | This study |
| pKNG101 | 8.2 kb suicide vector; sacB+, R6K origin; Smr | Kaniga et al. (1991) |
| pKNGirp2ΔS52 | 9.8 kb suicide vector; irp2ΔS52; sacB+, R6K origin; Smr; ∼1600 kb BamHI–XbaI fragment from pNEBirp2S ligated into the same sites in pKNG101 | This study |
| pKNGYΔ2336 | 9.9 kb suicide vector; Δy2336, sacB+, R6K origin; Smr; 1.7 kb SalI–HindIII fragment from pIHS3 ligated into the same sites in pKNG101 | This study |
| pNEB193 | 2.7 kb; cloning vector; Apr | New England Biolabs |
| pNEBirp2S | 4.3 kb; irp2ΔS52; Apr; ∼1600 bp PCR product containing irp2ΔS52 mutation ligated into BamHI–XbaI sites of pNEB193 | This study |
| pSinvYbtP | 11.1 kb suicide vector; invA : : ybtPp : : lacZβ-gal+ (ybtP : : lacZ promoter fusion); Apr | This study |
| pSucinv | 6.7 kb suicide vector containing invA fragment from Y. pseudotuberculosis; Apr | Perry et al. (2004) |
| pTrueBlue-rop | 3.8 kb cloning vector; Apr | Genomics One |
| pYbtT-H6 | 5.5 kb YbtT-H6 IPTG-regulated expression vector; Apr | Geoffroy et al. (2000) |
| pYbtT-G92A-H6 | 5.5 kb YbtT-G92A-H6 IPTG-regulated expression vector; Apr | This study |
| pYbtT-G96A-H6 | 5.5 kb YbtT-G96A-H6 IPTG-regulated expression vector; Apr | This study |
| pYbtT-S94A-H6 | 5.5 kb YbtT-S94A-H6 IPTG-regulated expression vector; Apr | This study |
| pYbtT-H230A-H6 | 5.5 kb YbtT-H230A-H6 IPTG-regulated expression vector; Apr | This study |
| pWSK29 | 5.4 kb low-copy-number cloning vector; Apr | Wang & Kushner (1991) |
Construction of Y. pestis mutant and reporter strains.
A deletion in y2336, which encodes a protein with similarity to YbtS, was constructed, leaving the first 150 bp and last 44 bp of the gene. Total genomic DNA from KIM10+ was digested with HindIII and SalI and separated by agrose gel electrophoresis. Fragments in the 3–4 kb range were eluted from the low-temperature-melting agarose and ligated into the HindIII–SalI sites of pTrueBlue-rop. Clones with the 3.4 kb HindIII–SalI fragment carrying y2336 were detected by PCR using primers IHS-1 and IHS-2. PCR results were confirmed by restriction analysis, and one recombinant plasmid was named pIHS1. A 1777 bp deletion was made in y2336 by eliminating the NruI–StuI internal fragment, creating pIHS2. A 1.6 kb HindIII–SalI fragment from pIHS2 was cloned into pWSK29 to yield pIHS3, and the SalI–XbaI fragment from this plasmid was cloned into pKNG101, generating the suicide vector pKNGYΔ2336. The y2336 deletion was introduced into KIM6+ and KIM6-2070.1 (ybtS : : kan2070.1) by allelic exchange, as previously described (Fetherston et al., 1999). A Y. pestis mutant carrying the y2336 deletion in the chromosome (KIM6-2070.2) was verified by PCR with primers IHS-1, IHS-2 and IHS-3 (Table 2). Clones which did not show any PCR product with IHS-1 and IHS-3 (which lies inside the deleted region) were confirmed by PCR with IHS-1 and IHS-2. The resulting strains were designated KIM6-2126 (ybtS+ Δy2336–2126) and KIM6-2070.2 (ybtS : : kan2070.1 Δy2336–2126).
Table 2.
Primers used in this study
| Primer | Sequence (5′–3′) | Purpose |
|---|---|---|
| ΔirpS | CCAGGCCGGCCTGGATTCC | Check irp2ΔS52 deletion |
| G92A forward | GCTTTTACTCGCCGCGCACAGCATGGGGG | Construction YbtT-G92A-H6 |
| G92A reverse | CCCCCATGCTGTGCGCGGCGAGTAAAAGC | Construction YbtT-G92A-H6 |
| G96A forward | CGGGCACAGCATGGCGGCGCAGGTGGCG | Construction YbtT-G96A-H6 |
| G96A reverse | CGCCACCTGCGCCGCCATGCTGTGCCCG | Construction YbtT-G96A-H6 |
| S94A forward | CGCCGGGCACGCCATGGGGGCGCAGG | Construction YbtT-S94A-H6 |
| S94A reverse | CCTGCGCCCCCATGGCGTGCCCGGCG | Construction YbtT-S94A-H6 |
| H230A forward | GACGGCGATGCTTTCTATCCCATTCAACAAGC | Construction YbtT-H230A-H6 |
| H230A reverse | GCTTGTTGAATGGGATAGAAAGCATCGCCGTC | Construction YbtT-H230A-H6 |
| IHS-1 | AGAAACTGCCGAAATGGTGAGG | Clone y2336 and check y2336 deletion |
| IHS-2 | GAGAGGGAAGCAAACTGCGTATT | Clone y2336 |
| IHS-3 | TTCCCTGGAGATGTTCCCGT | Check y2336 deletion |
| S1 | CGGGATCCGCATGCACGGCGGCTTCAG | Construct irp2ΔS52 deletion |
| S2 | ATCCAGGCCGGCCTGGATCAG | Construct irp2ΔS52 deletion |
| S3 | ATAAGATTGATGAGATGGTTAC | Construct irp2ΔS52 deletion |
| S4 | GCTCTAGATTCACGTAGCGTGGCGGGTTC | Construct and check irp2ΔS52 deletion |
| YbtA-2 | GGGGTACCGACCTGGTTATCTCCCTG | Clone ybtP promoter |
| YbtP-2 | GGGGTACCGGGAGTAACTGAATTTCC | Clone ybtP promoter |
| YbtT For2 | GGCACTGCTGGCGAACGAG | Sequence ybtT mutants |
| YbtT Rev1 | GCGAGTCGTAGTGATAGC | Sequence ybtT mutants |
| YbtT Rev2 | CTTTCTGAAGTACTGGGCTG | Sequence ybtT mutants |
The 166 bp ybtP promoter region was amplified from pEUYbtP (Fetherston et al., 1999) using primers YbtP-2 and YbtA-2 (Table 2). The PCR products were digested with Asp718 and cloned into pBSlacZMCS (Perry et al., 2004) that had been digested with Asp718 and treated with alkaline phosphatase. Clones containing the lacZ gene under the control of the ybtP promoter were identified by sequence analysis. An ∼4.4 kb EagI fragment was isolated from this clone and ligated into the EagI site of pSucinv (Perry et al., 2004) to generate pSinvYbtP. The ybtP : : lacZ reporter was integrated into the Y. pestis KIM6+ inv gene by allelic exchange and confirmed by hybridization to an 814 bp EagI/EcoRI inv fragment isolated from pSucinv. The probe detected a 5242 bp BamHI fragment in wild-type strains and a 2578 bp BamHI fragment in strains containing the integrated reporter. One reporter strain was selected and designated KIM6-2127+. An irp2 : : kan2046 mutation on suicide plasmid pCIRP498.8 (Fetherston et al., 1995) was introduced into KIM6-2127+ by electroporation and allelic exchange. The resulting mutant did not synthesize Ybt and the strain was designated KIM6-2046.6.
PCR was used to delete the codon for serine 52 in the aryl carrier protein domain of HMWP2. Salicylate is attached to this residue via a phosphopantetheine cofactor (Gehring et al., 1998b; Keating et al., 2000). A 799 bp product upstream of the codon for S52 was amplified using Pfu polymerase and primer pair S1 and S2 (Table 2). Primer pair S3 and S4 (Table 2) was used to amplify an 822 bp product downstream of the S52 codon. The products were purified using Zymo columns, treated with polynucleotide kinase and ligated together overnight at 15 °C. The ligated products were used as a template for PCRs containing primers S1 and S4. The ∼1600 bp fragment resulting from the amplification of the upstream product ligated to the downstream product was isolated from a low-melting agarose gel, digested with BamHI and XbaI, and cloned into the same sites in pNEB193. A clone (pNEBirp2S) containing the correct insert with a deletion of S52 was confirmed by sequencing. The insert was transferred into the BamHI–XbaI sites of pKNG101 to generate pKNGirp2ΔS52.
The irp2ΔS52 mutation was introduced into Y. pestis KIM6-2093 (Δy2360 : : cam2093) by allelic exchange using pKNGirp2ΔS52. Sucrose-resistant presumptive second cross isolates were replica-plated after overnight growth in PMH2 at 37 °C onto plates containing PMH2 or PMH2 with 50 μM 2,2′-dipyridyl (DIP). Isolates which formed red colonies on CR agar and grew on PMH plates but not on PMH/DIP plates were screened by PCR using S4 and a primer which ends with the three deleted nucleotides (ΔirpS, Table 2). One of the strains which did not yield a PCR product was designated KIM6-2046.7. KIM6-2046.7 was grown in PMH2 for two transfers, along with KIM6+ and KIM6-2046.1 (irp2 : : kan2046.1). Filtered supernatants from these cultures were tested for the ability to stimulate the growth of KIM6-2046.1 incorporated into PMH2 plates containing 75 μM DIP. KIM6+, but not KIM6-2046.7, supernatants were able to promote the growth of KIM6-2046.1 on these plates, confirming the lack of Ybt synthesis by KIM6-2046.7.
Construction of alanine substitutions in YbtT.
Four separate amino acid changes (G92A, G96A, S94A and H230A, using a predicted start of VTQS) were made in YbtT using pYbtT-H6 (Geoffroy et al., 2000; Table 1) with appropriate primers (Table 2) and a QuikChange II Site-Directed Mutagenesis kit (Stratagene) following the manufacturer's directions. DNA sequencing (Davis Sequencing) confirmed the alanine substitutions as well as the fidelity of the entire ybtT ORF. Selected recombinant plasmids were designated pYbtT-G92A-H6, pYbtT-G96A-H6, pYbtT-S94A-H6 and pYbtT-H230A-H6 (Table 1). Western blot analysis was used to demonstrate expression levels of the YbtT substitution mutants.
Western blot analysis.
For Western blot analysis, equal amounts of Y. pestis whole cells were suspended in sample buffer and boiled for 5 min. Samples were separated by SDS-PAGE and immunoblotted to PVDF membranes. The blots were processed using a procedure modified from Towbin et al. (1979). Briefly, the membranes were blocked with 5 % non-fat dry milk in 10 mM Tris/HCl (pH 7.6), 137 mM NaCl and 0.1 % Tween 20 (TBST), and then incubated with an appropriate antibody diluted in TBST with or without 0.1 % gelatin. The blots were washed with TBST and incubated with horseradish peroxidase-conjugated protein A (Amersham Pharmacia Biotech). Immunoreactive proteins were detected with the ECL enhanced chemiluminescence Western blotting detection reagent (Amersham Pharmacia Biotech) and visualized on Kodak BioMax light film. Polyclonal rabbit sera against purified YbtE and the PCP1-Cy2-PCP2 fragment of HMWP2 (Gehring et al., 1998b; Suo et al., 1999) were generated by Animal Pharm Services. A YbtT peptide (CHAFSAMTALQKQPSTER) and corresponding polyclonal rabbit antisera against it were produced by Open Biosystems.
Bioassays for detection of Ybt synthesis.
Three bioassays were used to detect the Ybt siderophore. (1) Culture supernatants were obtained from Y. pestis cells grown for six to nine generations in deferrated PMH2 at 37 °C. Cells were pelleted by centrifugation and the supernatants filtered through a 0.2 μm pore-size filter. Cells of Y. pestis KIM6-2046.1 (irp2 : : kan2046.1), which are unable to synthesize the Ybt siderophore, were used to overlay PMH2-DIP (75 μM DIP) plates solidified with 1 % agarose as previously described, and cell-free culture supernatants (25 μl) were added to wells in the plate. Growth responses to culture supernatants were monitored for 48–72 h (Bobrov et al., 2002; Fetherston et al., 1995).
(2) Cells of Y. pestis KIM6-2072 (ΔybtT2072) carrying YbtT expression plasmids with and without alanine substitutions were assessed for their ability to promote the growth of KIM6-2046.1 at 37 °C by streaking the mutant strains adjacent to KIM6-2046.1 on solidified Brain Heart Infusion (BHI) containing 175–250 μM DIP (BHI-DIP). Prior to streaking, these Y. pestis strains were adapted to iron-deficient growth conditions as described above. KIM6-2046.1, which cannot produce the Ybt siderophore, is unable to grow on BHI-DIP at 37 °C but can be cross-fed by Ybt-producing strains.
(3) Cells of Y. pestis KIM6-2072 (ΔybtT2072) carrying YbtT expression plasmids with and without alanine substitutions were also assessed for their ability to grow under iron-chelated conditions using PMH2-DIP gradient plates (0–100 μM DIP) at 37 °C. PMH2-DIP gradient plates were poured as previously described (Bearden & Perry, 1999) using square 100 mm plates with six grids. Cells were acclimated to iron-deficient conditions as described above and spotted in each of the six grids across the DIP gradient, and growth at 37 °C was monitored daily for 48 h.
Analysis of transcription of the ybtP : : lacZ reporter.
Cells of Y. pestis KIM6-2046.6 (irp2 : : kan2046.1 inv : : ybtPp : : lacZ) were grown overnight in deferrated PMH2 at 37 °C, diluted to OD620 0.1 in fresh deferrated PMH2 and incubated at 37 °C to OD620 ∼0.3. At this point, cell-free spent supernatants from iron-deficient cultures of KIM6+ or selected ybt biosynthetic mutants were added to a final volume of 10 or 50 % (v/v). Incubation at 37 °C was continued for 30 min before harvesting cells for β-galactosidase assays. β-Galactosidase activities in each culture were measured with a Genesys 5 spectrophotometer following cleavage of OPNG. Activities were expressed in Miller units (Miller, 1992). Each assay used two to three replicate samples, and the results are means from two or more independent cultures.
Ybt purification, standardization and determination.
Ybt was extracted and purified from cell-free supernatants from iron-deficient cultures using a four-step modification of a procedure previously described (Miller et al., 2006). Supernatants and extracts were protected from light as much as possible while being handled. Approximately 1.5 l of the supernatant was extracted with three 500 ml washes of ethyl acetate. Ethyl acetate extracts were combined and the solvent was removed by rotary evaporation at 40 °C. The resulting solid material was dissolved in a total of 15 ml 100 % ethanol, diluted to 50 ml with water (distilled and de-ionized to 18 Ω) and then filtered through a 0.22 μm pore-size filter. The filter was washed with 1–2 ml ethanol.
The combined filtrate was purified with Sep-Pak C-18 mini preparative cartridges (Waters). Two cartridges for each set of samples were prepared by wetting with 5 ml methanol followed by a 5 ml water wash. The filtrate was loaded onto the cartridges in 25 ml aliquots per cartridge, washed with 5 ml water, and then eluted with two 5 ml aliquots of methanol. Loading, washing and elution of the C-18 cartridges were accomplished by gravity feed using a syringe body as a reservoir. The resulting methanol solution containing Ybt plus various impurities was reduced to 1 ml total volume with a CentriVap vacuum concentrator (Labconco).
Ybt was then isolated using a Waters 600 HPLC equipped with a Waters 996 photodiode array UV/Vis detector and a μRPC C2/C18 ST 4.6/100 column (Amersham Biosciences). Initial injection conditions were 10 mM ammonium formate, pH 8.0, for all concentrations of acetonitrile, immediately followed by 10 min with 10 % acetonitrile, 160 min of a linear gradient from 10 to 25 % acetonitrile, and ending with 5 min at 100 % acetonitrile. The flow rate was 0.5 ml min−1. Spectrophotometric data were collected from 200 to 500 nm. The baseline was monitored at 210 nm. Fractions were collected at 1 min intervals in autoclaved 1.5 ml microcentrifuge tubes and stored frozen at –20 °C for later use. The column was re-equilibrated between runs with 10 mM ammonium formate, pH 8.0.
The collected Fe–Ybt-containing fractions were combined and loaded onto a C-18 reverse-phase cartridge prepared as described above. The Fe–Ybt was eluted and the methanol removed by evaporation to dryness in a CentriVap vacuum concentrator. The resulting Ybt was diluted to 1.0 mg ml−1 in methanol. Serial dilutions of 0.05, 0.025 and 0.0125 mg ml−1 were prepared, and 50 μl of each dilution plus a blank sample was injected into the HPLC system described above and monitored at 226 nm. After elution, each peak area was integrated using Empower Pro software and a calibration curve generated by plotting peak area versus concentration with Microsoft Excel. Samples from each Y. pestis strain examined were extracted from 1.5 l iron-deficient culture supernatant, purified and concentrated to 1 ml as described above. A 100 μl volume of concentrate from various Ybt biosynthetic mutants was prepared as described above, and the resulting peaks were integrated and compared with the authentic Ybt calibration curve.
RESULTS
Transcriptional activation of the ybtP : : lacZ reporter
While most ybt biosynthetic mutants exhibit reduced transcription of a plasmid-encoded ybtP : : lacZ reporter, we previously reported that three biosynthetic mutations [ΔybtT, ybtS : : kan and irp1-2086 irp1-TE)] still result in the higher levels of ybtP transcription normally associated with Ybt siderophore synthesis. However, these three mutants are phenotypically similar to other biosynthetic mutants in that they fail to grow at 37 °C under iron-chelated conditions and culture supernatants do not stimulate growth of a siderophore-negative indicator strain (KIM6-2046.1) of Y. pestis in our Ybt bioassay. We previously proposed that the two TE mutants might make lower levels of authentic Ybt and that the ybtS : : kan mutant may produce an aberrant Ybt-like molecule with another aromatic group replacing salicylate (Bobrov et al., 2002; Geoffroy et al., 2000; Perry et al., 2003a). Since transcriptional activation of the ybtP promoter is ∼500-fold more sensitive than growth stimulation in our Ybt bioassay (Fetherston et al., 1996; Geoffroy et al., 2000; Perry et al., 2003a), we decided to use transcriptional activity of the ybtP promoter to test culture supernatants for the presence of authentic Ybt or a Ybt-like molecule capable of acting as a signal molecule but not as a growth factor for iron-starved Y. pestis cells.
We constructed KIM6-2046.6, which has an irp2 : : kan mutation so that it cannot synthesize Ybt and a chromosomally integrated ybtP : : lacZ reporter. This strain was exposed for 30 min to culture supernatants at a final concentration of 10 % (v/v). The β-galactosidase activity of cells exposed to the ΔybtT culture supernatant was equivalent to those exposed to KIM6+, which produces normal levels of Ybt (Fig. 2). However, culture supernatants from the ybtS and irp2-TE mutants caused a less than twofold activation of the transcriptional reporter compared with the KIM6 (Δpgm) negative control (Fig. 2). When we increased the concentration of our spent supernatants to 50 % (v/v), the ybtP : : lacZ reporter activity was significantly increased by both the ybtS : : kan and irp2-TE mutant culture supernatants (Fig. 2). These results indicate that all three mutants still synthesize and secrete Ybt or a Ybt-like molecule that acts in signal transduction, but that the amount of this compound(s) (or its efficacy) in the culture supernatants varies among the different mutants.
Fig. 2.
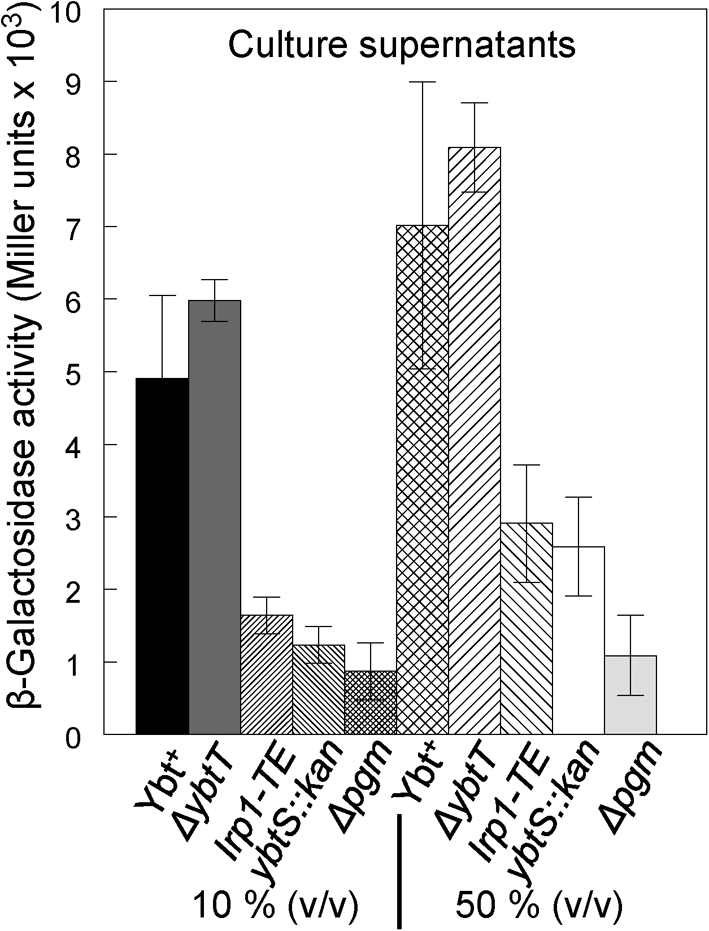
β-Galactosidase activities from a ybtP : : lacZ reporter in Y. pestis strains. Activities of a chromosomally integrated ybtP : : lacZ reporter in KIM6-2046.6 after 30 min of exposure to spent culture supernatants (10 and 50 %, v/v) from the indicated strains. Reporter strain KIM6-2046.6 has an irp2 : : kan2046.1 mutation and does not produce Ybt. Strains: Ybt+, KIM6+; ΔybtT, KIM6-2072; irp1-TE, KIM6-2086; ybtS : : kan, KIM6-2070.1; Δpgm, KIM6.
Purification of Ybt
A modified Ybt purification scheme (Methods; Miller et al., 2006) was used to determine whether ybtS, ybtT and irp1-TE mutants produce low levels of authentic Ybt. The elution profile of Ybt from KIM6+ is shown in Fig. 3(a). Elution with a 0–100 % acetonitrile gradient yielded multiple peaks (Fig. 3a). An iron-rich extract was prepared by mixing 100 μl 0.10 M FeCl3 with an equal volume of the KIM6+ extract and 800 μl methanol. The chromatogram of this sample eliminated all but the double peak at 37.5 and 40 ml, which doubled in area (Fig. 3b). The identity of the Ybt molecules was confirmed by directly extracting the UV/visible spectrum from the data provided by the Waters 996 detector using Waters Empower Pro software and by positive ion electrospray ionization (ESI)-MS (University of Kentucky Mass Spectrometry Facility). These analyses indicated that ferric–Ybt and deferrated Ybt eluted at ∼37.5–40 ml and 65 ml, respectively (arrows in Fig. 3a), with the expected UV/visible spectra (Fig. 4).
Fig. 3.

HPLC analysis of KIM6+ supernatant without (a) and with (b) added iron. Addition of iron approximately doubles the size of the peak at 37.5–40 ml (b) and eliminates the peak at 65 ml (a). Arrows in (a) show the apo (65 ml) and ferrated (37.5–40 ml) forms of Ybt.
Fig. 4.

UV/visible spectra from the peaks at approximately 65 ml (a) and 37.5–40 ml (b) in Fig. 3(a). Based on the data presented by Chambers et al. (1996), the peak at 65 ml is the apo form of Ybt while the peak at 37.5–40 ml is ferrated Ybt. The identifying characteristics of the apo form are the presence of three distinct maxima at 210, 251 and 309 nm (a), while the ferrated form has four distinct maxima at 226, 256, 306 and 388 nm (b).
KIM6+ (Ybt+) yielded 12.9 μg Ybt l−1, while KIM6-2072 (ΔybtT2072) gave a reduced yield of 3.0 μg l−1. In contrast, the irp1-TE mutant (lacking the TE activity proposed to release the completed Ybt molecule) had a greatly reduced yield of 0.37 μg l−1. Both KIM6 (Δpgm) and KIM6-2070.1 (ybtS : : kan2070) had an undetectable level of Ybt with a detection limit of 0.01 μg l−1. The amount of Ybt secreted by the two TE mutants corresponds to the level of transcriptional activity observed with crude supernatants (see above). The residual transcriptional activity of the ybtS : : kan mutant (see 50 % supernatant in Fig. 2) could be due to the production of an aberrant Ybt-like molecule with signalling activity.
YbtT and Ybt synthesis
The YbtT predicted type II TE is thought to be involved in editing and removal of incorrect precursor molecules from HMWP1 and HMWP2. YbtT would therefore contribute to the efficiency of Ybt synthesis by relieving biosynthetic bottlenecks through removal of incorrect precursor molecules from the Ybt synthetase complex. The decrease in Ybt production in a ybtT mutant indicates that this enzyme is required for optimal Ybt synthesis (see above). To more directly link reduced Ybt synthesis in the ybtT mutant with the predicted TE activity of YbtT, we mutated several residues associated with type II TE activity in other enzymes. While predicted type II TE domains span several hundred residues, alignments identified 12 invariant and 10 highly conserved (>80 %) residues (Kotowska et al., 2009; Linne et al., 2004). These include a nucleophile elbow motif (G-x-Nu-x-G; GxSxG residues 92–96 in YbtT) and a histidine residue at position 230 in YbtT. The actual importance of these residues to YbtT function has never been assessed. Consequently, we constructed separate alanine substitution mutations in each of these four residues in our YbtT-expressing plasmid pYbtT-H6, which complements the in-frame ybtT mutant strain KIM6-2072 (Geoffroy et al., 2000).
Cross-feeding studies indicated that the YbtT-H230A and YbtT-S94A variant proteins were little better than the negative control in providing Ybt siderophore for growth of a Y. pestis Ybt biosynthetic (irp2) mutant. The two individual glycine substitutions (YbtT-G92A and YbtT-G96A) provided more growth stimulation, but not as much as the YbtT-H6 positive control (data not shown). To better quantify the ability of each substitution mutant to produce Ybt, we examined the growth of KIM6+ and the YbtT− mutant bearing various YbtT-H6 plasmids across a 0–100 μM DIP gradient plate (Fig. 5). Expression of YbtT-H6 allowed growth further into the DIP gradient than the Ybt+ strain KIM6+, possibly due to increased expression from the pPROEX-1 vector. The histidine and serine substitution mutants, which had YbtT protein levels similar to those of the mutant expressing YbtT-H6 from pPROEX-1 (data not shown), showed growth similar to that of the in-frame ybtT mutant, which was significantly reduced compared with the YbtT-H6-expressing strain (Fig. 5). In contrast, KIM6+ and the two strains expressing the glycine-substituted YbtT variants had similar levels of YbtT protein expression (data not shown) and grew into similar DIP concentrations in the gradient (Fig. 5). These results correspond to the cross-feeding analysis and suggest that G92 and G96 are not essential for YbtT activity while H230 and S94 have a significant effect on Ybt synthesis.
Fig. 5.
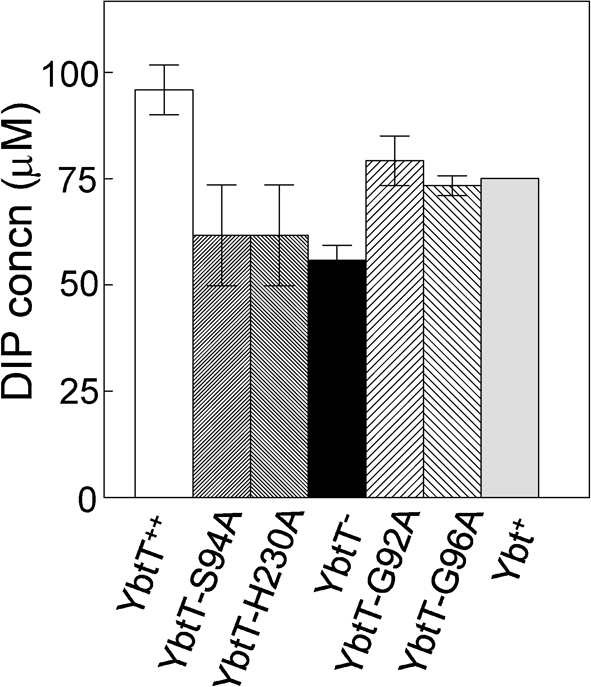
Growth of Y. pestis strains at 37 °C across PMH2-DIP gradient plates (0–100 μM DIP). The results represent the mean from two independent experiments of incremental growth across the gradient by 48 h of incubation. Error bars, sd. Strains: YbtT++, KIM6-2072(pYbtT-H6) positive control; YbtT-S94A, KIM6-2072(pYbtT-S94A-H6); YbtT-H230A, KIM6-2072(pYbtT-H230A-H6); YbtT−, KIM6-2072 (ΔybtT2072) negative control; YbtT-G92A, KIM6-2072(pYbtT-G92A-H6); YbtT-G96A, KIM6-2072(pYbtT-G96A-H6); Ybt+, KIM6+ positive control.
Aberrant Ybt-like molecules
YbtS is responsible for synthesizing salicylate, the molecule that presumably initiates Ybt synthesis. A strain with a mutation in ybtS does not produce any detectable authentic Ybt, yet ybt reporters in this mutant are fully activated (Fig. 6; Geoffroy et al., 2000; Perry et al., 2003a), a process which responds to Ybt. It is possible that another enzyme synthesizes a phenolate compound that ineffectively substitutes for salicylate. Encoded within the Y. pestis KIM pgm locus, Y2336 shows a high degree of similarity to YbtS: both proteins have anthranilate synthase domains (Deng et al., 2002). However, a ybtS : : kan Δy2336 double mutant (KIM6-2070.2) still fully activated transcription from a ybtP : : lacZ reporter (Fig. 6). Consequently, if an alternative aromatic component is incorporated by the ybtS : : kan mutant it is not dependent upon the y2336 gene product for this activity.
Fig. 6.
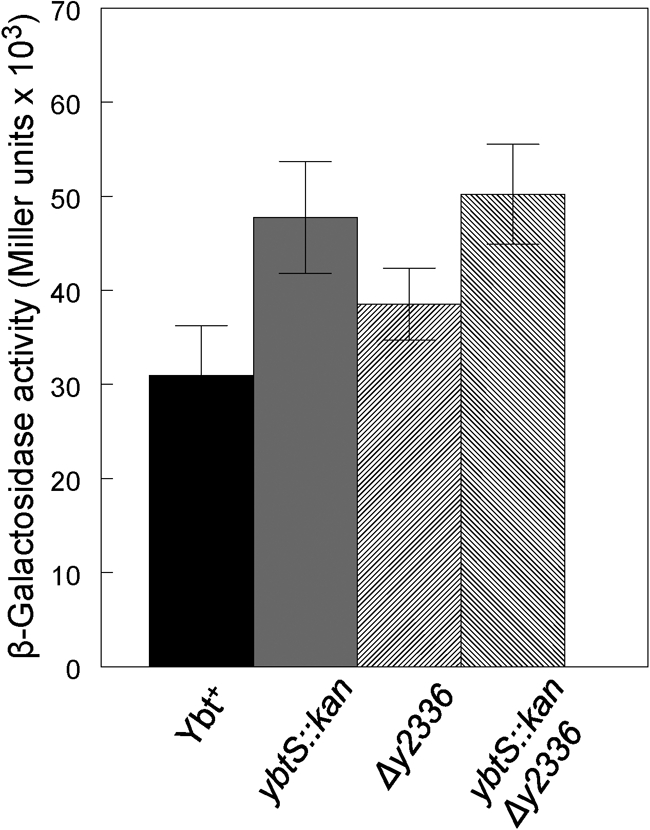
β-Galactosidase activities from a ybtP : : lacZ reporter in Y. pestis strains. Activities of a plasmid-encoded ybtP : : lacZ reporter (pEUYbtP) in the control (Ybt+) and indicated mutant strains. Endogenous synthesis of Ybt or a Ybt-like molecule is responsible for transcriptional activation. Strains: Ybt+, KIM6+; ybtS : : kan, KIM6-2070.1; Δy2336, KIM6-2126; ybtS : : kan Δy2336, KIM6-2070.2.
To test whether a Ybt-like molecule possessing only the thiazolidine and thiazoline rings without a substitute salicylate component might be capable of transcriptional activation, we deleted serine residue 52 of HMWP2 (strain KIM6-2046.7), which would eliminate tethering of salicylate or a salicylate-like component for incorporation into authentic Ybt or a Ybt-like molecule. In contrast to the ybtS : : kan Δy2336 double mutant, KIM6-2046.7 showed reduced transcription from a ybtP reporter (data not shown) and little or no YbtE or HMWP2 proteins by Western blot analysis, in the absence of added Ybt (Fig. 7). Synthesis of these two proteins in KIM6-2046.7 and the in-frame irp2 mutant, but not in the polar irp2 : : kan mutant, was restored following a 2 h exposure to purified Ybt (Fig. 7). The higher levels of HMWP2 and YbtE in Ybt+ KIM6+ and the ybtS : : kan mutant are likely due to the continuous presence of endogenous Ybt (or Ybt-like) molecules. These results suggest that the irp2ΔS52 mutant does not make a compound capable of stimulating the transcription of ybt promoters.
Fig. 7.

Western blot analysis of HMWP2 and YbtE expression in Y. pestis strains in the presence and absence of exogenous Ybt. Equal concentrations of whole-cell lysates were separated by SDS-PAGE; immunoblots were reacted with antiserum against HMWP2 or YbtE, as indicated. Strains: Ybt+, KIM6+; Δirp2 (in-frame), KIM6-2046.3; irp2ΔS52, KIM6-2046.7; ybtS : : kan, KIM6-2070.1; irp2 : : kan (polar), KIM6-2046.1.
Finally, we tested whether the signalling molecule present in the supernatant from the ybtS : : kan mutant was extracted during our purification procedure. Ethyl acetate extracts from equal volumes of culture supernatants of the ybtS : kan, ybtT and irp1-TE mutants as well as KIM6+ (Ybt+) and KIM6 (Δpgm, Ybt−) were concentrated 1000-fold. Serial 10-fold dilutions of the concentrated extracts were added to the reporter strain KIM6-2046.6. Addition of 0.01 μl of the extract from the Ybt-producing strain KIM6+ caused maximal activation of the ybtP : : lacZ reporter. Extracts from the ybtT and irp1-TE mutants required 10- and 100-fold higher volumes for maximal activation, respectively. These results approximately correspond to the amount of authentic Ybt produced by these mutants (see above). A 100-fold higher volume of the ΔybtS extract also was required for maximal activation of the reporter (data not shown).
Surprisingly, the concentrated extract from the Ybt− strain KIM6 also stimulated transcription from the reporter but required a 1000-fold higher concentration than the extract from the Ybt-producing strain to do so. Extracted and concentrated uninoculated PMH2 provided no activation of the ybtP : : lacZ reporter (data not shown). These results indicate that KIM6, which lacks all the Ybt biosynthetic genes except for ybtD, produces a non-Ybt molecule capable of signalling activity.
DISCUSSION
In the presence of Ybt, YbtA activates transcription of transport and biosynthetic genes while repressing transcription from its own promoter. The expression of several siderophore-dependent iron-transport systems is controlled by the interaction of the cognate siderophore with its outer membrane receptor, which transmits a TonB-dependent signal that activates a regulator for that system (Braun, 2001; Crosa, 1997; Venturi et al., 1995). However, in Y. pestis, three separate Δpsn mutations have no effect on YbtA-regulated gene expression. In addition, signal transduction is not affected by a Y. pestis tonB mutation (Bearden et al., 1997; Perry et al., 2003a, b). Thus, it is unlikely that binding of Ybt to the outer membrane receptor transmits a signal that then activates YbtA.
Since YbtA is a member of the AraC family of transcriptional regulators, we have proposed that YbtA binds the siderophore and that this complex directly affects the transcription of the regulated genes of the Ybt system (Fig. 1b). During the Ybt purification process, much of the siderophore is saturated with iron and more of the apo form would be converted to ferri-Ybt after addition of the purified siderophore to PMH2, which contains ∼0.3 μM iron. Consequently, we have proposed that it is the iron-chelated form of Ybt that serves as the signalling molecule. This model corresponds to the pyochelin- and PchR-dependent regulation demonstrated for that system in Pseudomonas aeruginosa (Michel et al., 2005). The Bordetella alcaligin system also requires an AraC-type regulator, AlcR, and the alcaligin siderophore for transcriptional activation of the system. In contrast to the other two systems, it appears that the iron-free form of alcaligin is the signalling molecule (Brickman & Armstrong, 2002, 2009; Brickman et al., 2001).
In an apparent contradiction to our model, ybtS, ybtT and irp1-TE mutations cause a loss of Ybt synthesis without affecting YbtA regulation. We had proposed that these three mutants produce either low levels of Ybt that are sufficient for activation or aberrant Ybt-like molecules that are not detectable by the bioassay but can bind to YbtA and activate transcription (Bearden et al., 1997; Fetherston et al., 1996; Geoffroy et al., 2000; Perry et al., 2001, 2003a, b). Our results in this study indicate that the ybtT and irp1-TE mutants do produce sufficient Ybt to activate transcription.
YbtT is a putative type II TE that likely provides an editing function to remove aberrant molecules from the enzyme complex. In this study we have demonstrated that a ybtT mutant synthesizes authentic Ybt but at levels ∼25 % of that of the parent strain. Mutations in other type II TEs cause similar reductions in the products of NRPSs and PKSs (Butler et al., 1999; Leduc et al., 2007; Schneider & Marahiel, 1998). Our results suggest that Ybt synthesis is relatively error-prone; this conclusion is supported by an apparent increase in Ybt levels due to increased expression of YbtT (Fig. 5). Miller et al. (2002) synthesized Ybt in vitro using appropriate precursors and four enzymes (YbtE, HMWP1, HMWP2 and YbtU). Including YbtT in the in vitro reaction did not increase the biosynthetic rate. However, the in vitro reaction only contained the appropriate substrates for Ybt synthesis. Perhaps in vivo, improper substrates are added to the HMWP1 and HMWP2 scaffold, in which case YbtT would be required for their removal.
A TE catalytic triad of Ser-Asp-His has been identified in type II TEs such as Bacillus subtilis SrfA and Streptomyces coelicolor ScoT. Alignments indicate that YbtT also possesses a putative catalytic triad: S94-D172-H230 (Kotowska et al., 2009; Linne et al., 2004). Alanine substitutions at S94 and H230 caused an iron-chelated growth defect similar to that of the ybtT mutant (Fig. 5). In addition to S94, the nucleophile elbow of YbtT (GxSxG) contains two invariant glycines. However, substitution of an alanine for either of these two residues did not significantly affect the ability of Y. pestis to grow under iron-chelating conditions. While these residues are likely not critical to the TE activity of YbtT, they did have an effect on YbtT protein levels. Although expressed from a moderate-copy-number plasmid, Western blot analysis indicated the two substituted proteins had levels similar to that of YbtT expressed from the single chromosomal gene (data not shown).
The irp1-TE mutant also synthesizes and secretes authentic Ybt but at levels only ∼3 % of that of the parent strain. In this mutant most of the completed Ybt molecule likely remains tethered to HWMP1. Either sufficient quantities are released to stimulate transcription or Ybt tethered to the enzyme complex can serve as a signalling molecule.
In contrast, the ybtS mutant does not make detectable levels of authentic Ybt but still produces a signalling molecule. The activity of a Ybt-activated reporter in the ybtS mutant is equivalent to that of the parental strain, although ethyl acetate extracts of culture supernatants from this mutant require 100-fold higher volumes for maximal activation of transcription from the ybtP promoter. This suggests that the aberrant Ybt-like molecule is not secreted effectively and/or not efficiently extracted into ethyl acetate. The inability of an irp2ΔS52 mutant to activate transcription of Ybt-regulated genes indicates that a Ybt-like molecule simply lacking salicylate is not made as the signalling molecule. This result is in agreement with in vitro studies that demonstrate that reactions containing purified HMWP2 with a S52A substitution do not yield a product (Keating et al., 2000). In the ybtS mutant, a different phenolate compound is most likely substituted for salicylate. We have demonstrated that the ybtS-like gene, y2336, does not encode a product that is responsible for the synthesis of this compound.
Surprisingly, a Δpgm mutant (which lacks all ybt genes except for ybtD) also produced a compound capable of activating transcription. The following reasons suggest that this compound is different from the Ybt-like signalling molecule produced by the ybtS mutant. First, the ybtS mutant produces sufficient levels of the compound to fully activate transcription of the ybtP : : lacZ reporter carried in that strain. Only background levels of activity are detected in a Δpgm mutant. However, Δpgm strains lack the transcriptional activator YbtA in addition to Ybt. Another biosynthetic mutant, such as irp2 : : kan, is a fairer comparison with the ybtS mutant. Neither of these mutants makes Ybt, but they still encode YbtA; yet the activity of a ybtP : : lacZ reporter in the ybtS mutant is about 20-fold higher than in an irp2 : : kan mutant. Indeed, if the Δpgm compound were the same as that present in the ybtS mutant then all biosynthetic mutants would be expected to show the same level of endogenous transcriptional activation, yet they do not. Second, in concentrated ethyl acetate extracts from the two mutants, the ybtS mutant compound is ∼10-fold more effective or abundant than the Δpgm compound. Note that the ybtS mutant probably produces both Ybt-like compounds. Regardless, isolation of these compounds will be required to definitively demonstrate that there are two separate non-Ybt compounds capable of Ybt signalling. While these compounds are not biologically relevant to normal Ybt regulation, a comparison of the structures of Ybt and other signalling molecules will help in determining the chemical structures recognized as a Ybt signal.
Acknowledgments
This study was supported by Public Health Service grant AI033481 from the National Institutes of Health. We thank the Chris Walsh research group (Harvard Medical School) for generously providing purified YbtE protein and the purified PCP1-Cy2-PCP2 fragment of HMWP2.
Abbreviations
CR, Congo red
DIP, 2,2′-dipyridyl
HPI, high-pathogenicity island
NRPS, nonribosomal peptide synthetase
PKS, polyketide synthase
TE, thioesterase
References
- Anisimov, R., Brem, D., Heesemann, J. & Rakin, A. (2005). Molecular mechanism of YbtA-mediated transcriptional regulation of divergent overlapping promoters ybtA and irp6 of Yersinia enterocolitica. FEMS Microbiol Lett 250, 27–32. [DOI] [PubMed] [Google Scholar]
- Bearden, S. W. & Perry, R. D. (1999). The Yfe system of Yersinia pestis transports iron and manganese and is required for full virulence of plague. Mol Microbiol 32, 403–414. [DOI] [PubMed] [Google Scholar]
- Bearden, S. W., Fetherston, J. D. & Perry, R. D. (1997). Genetic organization of the yersiniabactin biosynthetic region and construction of avirulent mutants in Yersinia pestis. Infect Immun 65, 1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beesley, E. D., Brubaker, R. R., Janssen, W. A. & Surgalla, M. J. (1967). Pesticins. III. Expression of coagulase and mechanism of fibrinolysis. J Bacteriol 94, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrov, A. G., Geoffroy, V. A. & Perry, R. D. (2002). Yersiniabactin production requires the thioesterase domain of HMWP2 and YbtD, a putative phosphopantetheinylate transferase. Infect Immun 70, 4204–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, V. (2001). Iron uptake mechanisms and their regulation in pathogenic bacteria. Int J Med Microbiol 291, 67–79. [DOI] [PubMed] [Google Scholar]
- Braun, V. (2005). Bacterial iron transport related to virulence. Contrib Microbiol 12, 210–233. [DOI] [PubMed] [Google Scholar]
- Brickman, T. J. & Armstrong, S. K. (2002). Bordetella interspecies allelic variation in AlcR inducer requirements: identification of a critical determinant of AlcR inducer responsiveness and construction of an alcR(Con) mutant allele. J Bacteriol 184, 1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickman, T. J. & Armstrong, S. K. (2009). Temporal signaling and differential expression of Bordetella iron transport systems: the role of ferrimones and positive regulators. Biometals 22, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickman, T. J., Kang, H. Y. & Armstrong, S. K. (2001). Transcriptional activation of Bordetella alcaligin siderophore genes requires the AlcR regulator with alcaligin as inducer. J Bacteriol 183, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullen, J. J., Rogers, H. J., Spalding, P. B. & Ward, C. G. (2005). Iron and infection: the heart of the matter. FEMS Immunol Med Microbiol 43, 325–330. [DOI] [PubMed] [Google Scholar]
- Butler, A. R., Bate, N. & Cundliffe, E. (1999). Impact of thioesterase activity on tylosin biosynthesis in Streptomyces fradiae. Chem Biol 6, 287–292. [DOI] [PubMed] [Google Scholar]
- Chambers, C. E., McIntyre, D. D., Mouck, M. & Sokol, P. A. (1996). Physical and structural characterization of yersiniophore, a siderophore produced by clinical isolates of Yersinia enterocolitica. Biometals 9, 157–167. [DOI] [PubMed] [Google Scholar]
- Crosa, J. H. (1997). Signal transduction and transcriptional and posttranscriptional control of iron-regulated genes in bacteria. Microbiol Mol Biol Rev 61, 319–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosa, J. H. & Walsh, C. T. (2002). Genetics and assembly line enzymology of siderophore biosynthesis in bacteria. Microbiol Mol Biol Rev 66, 223–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, W., Burland, V., Plunkett, G., III, Boutin, A., Mayhew, G. F., Liss, P., Perna, N. T., Rose, D. J., Mau, B. & other authors (2002). Genome sequence of Yersinia pestis KIM. J Bacteriol 184, 4601–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel, H., Stephan, H., Lotz, R., Haag, H., Zähner, H., Hantke, K. & Jung, G. (1995). Structure elucidation of yersiniabactin, a siderophore from highly virulent Yersinia strains. Liebigs Ann 1995, 1727–1733. [Google Scholar]
- Fetherston, J. D., Schuetze, P. & Perry, R. D. (1992). Loss of the pigmentation phenotype in Yersinia pestis is due to the spontaneous deletion of 102 kb of chromosomal DNA which is flanked by a repetitive element. Mol Microbiol 6, 2693–2704. [DOI] [PubMed] [Google Scholar]
- Fetherston, J. D., Lillard, J. W., Jr & Perry, R. D. (1995). Analysis of the pesticin receptor from Yersinia pestis: role in iron-deficient growth and possible regulation by its siderophore. J Bacteriol 177, 1824–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetherston, J. D., Bearden, S. W. & Perry, R. D. (1996). YbtA, an AraC-type regulator of the Yersinia pestis pesticin/yersiniabactin receptor. Mol Microbiol 22, 315–325. [DOI] [PubMed] [Google Scholar]
- Fetherston, J. D., Bertolino, V. J. & Perry, R. D. (1999). YbtP and YbtQ: two ABC transporters required for iron uptake in Yersinia pestis. Mol Microbiol 32, 289–299. [DOI] [PubMed] [Google Scholar]
- Fetherston, J. D., Kirillina, O., Bobrov, A. G., Paulley, J. T. & Perry, R. D. (2010). The yersiniabactin transport system is critical for the pathogenesis of bubonic and pneumonic plague. Infect Immun 78, 2045–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring, A. M., DeMoll, E., Fetherston, J. D., Mori, I., Mayhew, G. F., Blattner, F. R., Walsh, C. T. & Perry, R. D. (1998a). Iron acquisition in plague: modular logic in enzymatic biogenesis of yersiniabactin by Yersinia pestis. Chem Biol 5, 573–586. [DOI] [PubMed] [Google Scholar]
- Gehring, A. M., Mori, I., Perry, R. D. & Walsh, C. T. (1998b). The nonribosomal peptide synthetase HMWP2 forms a thiazoline ring during biogenesis of yersiniabactin, an iron-chelating virulence factor of Yersinia pestis. Biochemistry 37, 11637–11650. [DOI] [PubMed] [Google Scholar]
- Geoffroy, V. A., Fetherston, J. D. & Perry, R. D. (2000). Yersinia pestis YbtU and YbtT are involved in synthesis of the siderophore yersiniabactin but have different effects on regulation. Infect Immun 68, 4452–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, S., Bearden, S. W., Geoffroy, V. A., Fetherston, J. D. & Perry, R. D. (2001). Characterization of the Yersinia pestis Yfu ABC iron transport system. Infect Immun 69, 2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniga, K., Delor, I. & Cornelis, G. R. (1991). A wide-host-range suicide vector for improving reverse genetics in Gram-negative bacteria: inactivation of the blaA gene of Yersinia enterocolitica. Gene 109, 137–141. [DOI] [PubMed] [Google Scholar]
- Keating, T. A., Miller, D. A. & Walsh, C. T. (2000). Expression, purification, and characterization of HMWP2, a 229 kDa, six domain protein subunit of yersiniabactin synthetase. Biochemistry 39, 4729–4739. [DOI] [PubMed] [Google Scholar]
- Kerbarh, O., Ciulli, A., Howard, N. I. & Abell, C. (2005). Salicylate biosynthesis: overexpression, purification, and characterization of Irp9, a bifunctional salicylate synthase from Yersinia enterocolitica. J Bacteriol 187, 5061–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotowska, M., Pawlik, K., Smulczyk-Krawczyszyn, A., Bartosz-Bechowski, H. & Kuczek, K. (2009). Type II thioesterase ScoT, associated with Streptomyces coelicolor A3(2) modular polyketide synthase Cpk, hydrolyzes acyl residues and has a preference for propionate. Appl Environ Microbiol 75, 887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc, D., Battesti, A. & Bouveret, E. (2007). The hotdog thioesterase EntH (YbdB) plays a role in vivo in optimal enterobactin biosynthesis by interacting with the ArCP domain of EntB. J Bacteriol 189, 7112–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesic, B. & Carniel, E. (2004). The high pathogenicity island: a broad-host-range pathogenicity island. In Yersinia Molecular and Cellular Biology, pp. 285–306. Edited by E. Carniel & B. J. Hinnebusch. Wymondham, UK: Horizon Bioscience.
- Linne, U., Schwarzer, D., Schroeder, G. N. & Marahiel, M. A. (2004). Mutational analysis of a type II thioesterase associated with nonribosomal peptide synthesis. Eur J Biochem 271, 1536–1545. [DOI] [PubMed] [Google Scholar]
- Michel, L., González, N., Jagdeep, S., Nguyen-Ngoc, T. & Reimmann, C. (2005). PchR-box recognition by the AraC-type regulator PchR of Pseudomonas aeruginosa requires the siderophore pyochelin as an effector. Mol Microbiol 58, 495–509. [DOI] [PubMed] [Google Scholar]
- Miethke, M. & Marahiel, M. A. (2007). Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev 71, 413–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J. H. (1992). A Short Course in Bacterial Genetics. A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Miller, D. A., Luo, L., Hillson, N., Keating, T. A. & Walsh, C. T. (2002). Yersiniabactin synthetase: a four-protein assembly line producing the nonribosomal peptide/polyketide hybrid siderophore of Yersinia pestis. Chem Biol 9, 333–344. [DOI] [PubMed] [Google Scholar]
- Miller, M. C., Parkin, S., Fetherston, J. D., Perry, R. D. & DeMoll, E. (2006). Crystal structure of ferric-yersiniabactin, a virulence factor of Yersinia pestis. J Inorg Biochem 100, 1495–1500. [DOI] [PubMed] [Google Scholar]
- Pelludat, C., Brem, D. & Heesemann, J. (2003). Irp9, encoded by the high-pathogenicity island of Yersinia enterocolitica, is able to convert chorismate into salicylate, the precursor of the siderophore yersiniabactin. J Bacteriol 185, 5648–5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, R. D. (2004). Yersinia. In Iron Transport in Bacteria, pp. 219–240. Edited by J. H. Crosa, A. R. Mey & S. M. Payne. Washington, DC: American Society for Microbiology.
- Perry, R. D. & Fetherston, J. D. (1997). Yersinia pestis – etiologic agent of plague. Clin Microbiol Rev 10, 35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, R. D. & Fetherston, J. D. (2004). Iron and heme uptake systems. In Yersinia Molecular and Cellular Biology, pp. 257–283. Edited by E. Carniel & B. J. Hinnebusch. Wymondham, UK: Horizon Bioscience.
- Perry, R. D., Pendrak, M. L. & Schuetze, P. (1990). Identification and cloning of a hemin storage locus involved in the pigmentation phenotype of Yersinia pestis. J Bacteriol 172, 5929–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, R. D., Straley, S. C., Fetherston, J. D., Rose, D. J., Gregor, J. & Blattner, F. R. (1998). DNA sequencing and analysis of the low-Ca2+-response plasmid pCD1 of Yersinia pestis KIM5. Infect Immun 66, 4611–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, R. D., Balbo, P. B., Jones, H. A., Fetherston, J. D. & DeMoll, E. (1999). Yersiniabactin from Yersinia pestis: biochemical characterization of the siderophore and its role in iron transport and regulation. Microbiology 145, 1181–1190. [DOI] [PubMed] [Google Scholar]
- Perry, R. D., Bearden, S. W. & Fetherston, J. D. (2001). Iron and heme acquisition and storage systems of Yersinia pestis. Recent Res Dev Microbiol 5, 13–27. [Google Scholar]
- Perry, R. D., Abney, J., Mier, I., Jr, Lee, Y., Bearden, S. W. & Fetherston, J. D. (2003a). Regulation of the Yersinia pestis Yfe and Ybt iron transport systems. Adv Exp Med Biol 529, 275–283. [DOI] [PubMed] [Google Scholar]
- Perry, R. D., Shah, J., Bearden, S. W., Thompson, J. M. & Fetherston, J. D. (2003b). Yersinia pestis TonB: role in iron, heme and hemoprotein utilization. Infect Immun 71, 4159–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, R. D., Bobrov, A. G., Kirillina, O., Jones, H. A., Pedersen, L. L., Abney, J. & Fetherston, J. D. (2004). Temperature regulation of the hemin storage (Hms+) phenotype of Yersinia pestis is posttranscriptional. J Bacteriol 186, 1638–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaible, U. E. & Kaufmann, S. H. E. (2004). Iron and microbial infection. Nat Rev Microbiol 2, 946–953. [DOI] [PubMed] [Google Scholar]
- Schneider, A. & Marahiel, M. A. (1998). Genetic evidence for a role of thioesterase domains, integrated in or associated with peptide synthetases, in non-ribosomal peptide biosynthesis in Bacillus subtilis. Arch Microbiol 169, 404–410. [DOI] [PubMed] [Google Scholar]
- Staggs, T. M., Fetherston, J. D. & Perry, R. D. (1994). Pleiotropic effects of a Yersinia pestis fur mutation. J Bacteriol 176, 7614–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo, Z., Walsh, C. T. & Miller, D. A. (1999). Tandem heterocyclization activity of the multidomain 230 kDa HMWP2 subunit of Yersinia pestis yersiniabactin synthetase: interaction of the 1–1382 and 1383–2035 fragments. Biochemistry 38, 14023–14035. [DOI] [PubMed] [Google Scholar]
- Surgalla, M. J. & Beesley, E. D. (1969). Congo red-agar plating medium for detecting pigmentation in Pasteurella pestis. Appl Microbiol 18, 834–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin, H., Staehelin, T. & Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi, V., Weisbeek, P. & Koster, M. (1995). Gene regulation of siderophore-mediated iron acquisition in Pseudomonas: not only the Fur repressor. Mol Microbiol 17, 603–610. [DOI] [PubMed] [Google Scholar]
- Wang, R. F. & Kushner, S. R. (1991). Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100, 195–199. [PubMed] [Google Scholar]