

Abstract
The phage-shock-protein (Psp) response maintains the proton-motive force (pmf) under extracytoplasmic stress conditions that impair the inner membrane (IM) in bacterial cells. In Escherichia coli transcription of the pspABCDE and pspG genes requires activation of σ54-RNA polymerase by the enhancer-binding protein PspF. A regulatory network comprising PspF–A–C–B–ArcB controls psp expression. One key regulatory point is the negative control of PspF imposed by its binding to PspA. It has been proposed that under stress conditions, the IM-bound sensors PspB and PspC receive and transduce the signal(s) to PspA via protein–protein interactions, resulting in the release of the PspA–PspF inhibitory complex and the consequent induction of psp. In this work we demonstrate that PspB self-associates and interacts with PspC via putative IM regions. We present evidence suggesting that PspC has two topologies and that conserved residue G48 and the putative leucine zipper motif are determinants required for PspA interaction and signal transduction upon stress. We also establish that PspC directly interacts with the effector PspG, and show that PspG self-associates. These results are discussed in the context of formation and function of the Psp regulatory complex.
INTRODUCTION
The phage-shock-protein (Psp) response maintains the proton-motive force (pmf) under extracytoplasmic stress conditions (e.g. upon secretin pIV production) that impair the integrity of the inner membrane (IM) and dissipate the pmf (reviewed by Darwin, 2005; see also Jovanovic et al., 2006). This adaptation to stress has been shown to be important for growth and virulence of some enterobacterial pathogens (reviewed by Darwin, 2005, 2007; Rowley et al., 2006). Transcription of the psp genes is controlled by σ54-RNA polymerase and activated by the bacterial enhancer-binding protein PspF (reviewed by Model et al., 1997; Darwin, 2005; Wigneshweraraj et al., 2008). In Escherichia coli the PspF regulon consists of the pspABCDE operon, the adjacent pspF gene and the pspG gene (reviewed by Model et al., 1997; Darwin, 2005). In many other enterobacteria, the pspF and pspABC genes are highly conserved (Huvet et al., 2009). Under non-stress growth conditions, PspA inhibits the ATPase activity of PspF and negatively controls expression of the psp genes (Elderkin et al., 2002, 2005; Joly et al., 2009). It is proposed that under many psp-inducing stress conditions, the IM-bound sensors PspB and PspC act as positive regulators that receive and transduce the stress signal(s) to PspA via protein–protein interactions (Adams et al., 2003; Maxson & Darwin, 2006; Jovanovic et al., 2006, 2009). This ultimately results in the release of the PspA–PspF inhibitory complex, leading to psp induction and greatly elevated expression of the Psp proteins.
Recently, we provided evidence that during microaerobic growth the ArcAB system contributes to a Psp signal-transduction pathway in a PspBC-dependent manner and proposed that at least two signals could be recognized by PspBC for full pIV-dependent psp induction – caused by the mislocalization of pIV in the IM (Jovanovic et al., 2009). These data suggested that a double check-point for PspBC-specific psp induction could exist, potentially explaining the requirement for two sensor proteins. Importantly, in the absence of stress, PspBC overexpression has been shown to strongly induce psp (Weiner et al., 1991; Maxson & Darwin, 2006), implying that under these conditions PspBC may bypass upstream signalling pathways and directly release the PspA–PspF inhibitory complex. However, the mechanistic basis of this effect is unknown. In light of these findings a Psp[F–A–C–B]–ArcB regulatory complex (under non-stress conditions) was proposed (Jovanovic et al., 2009), but importantly only pairwise interactions between PspA–PspF, PspA–PspC, PspB–PspC and PspB–ArcB have been identified (Elderkin et al., 2002; Adams et al., 2003; Elderkin et al., 2005; Maxson & Darwin, 2006; Jovanovic et al., 2009). PspG is an IM effector protein not required for psp induction (Lloyd et al., 2004), but it may co-localize with PspA (Engl et al., 2009). Interestingly, it was observed that overexpression of PspG upregulates genes involved in microaerobic and anaerobic respiration (most of them ArcAB-regulated) (Jovanovic et al., 2006). The role of PspG and its potential association with the Psp regulatory complex has yet to be established.
In this work we dissect further the Psp regulatory complex in terms of protein–protein interactions and define the functional determinants in PspB and PspC that are required for pIV-dependent psp induction.
METHODS
Bacterial strains.
The bacterial strains used in this study are listed in Supplementary Table S1, available with the online version of this paper. Strains were constructed by transduction using the P1vir bacteriophage (Miller, 1992).
Media and growth conditions.
All strains were routinely grown under microaerobic conditions in Luria–Bertani (LB) broth or on LB agar plates at 37 °C (Miller, 1992). For microaerobic growth, overnight cultures of cells were diluted 100-fold (OD600 ∼0.025) and shaken at 100 r.p.m. For the bacterial two-hybrid (BACTH) assays, strains were grown in LB at 30 °C. For single-molecule fluorescence imaging of PspG–GFP, cells were grown in minimal medium as described previously (Engl et al., 2009). Induction of the pBAD ara promoter was achieved with either 0.001 % or 0.02 % (final – as indicated) l-arabinose (Ara). Antibiotics were used at the following concentrations: ampicillin, 100 μg ml−1; kanamycin, 25 or 50 μg ml−1, as indicated; chloramphenicol, 30 μg ml−1; and tetracycline, 10 μg ml−1.
DNA manipulations.
Plasmids used in this study are listed in Supplementary Table S1. All constructs were verified by DNA sequencing, and protein production was verified using Western blotting. By site-directed mutagenesis (QuikChange, Agilent Technologies) using the appropriate primers and either pAJM2 (encodes wild-type [WT] PspC) or pAJM3 (encodes WT PspBC) as a template, we constructed pAJM6–8, pAJM11–13, pGJ54, pGJ55 and pGJ57. Using either pAJM8, pAJM13 or pAJM11 as a template, we constructed pAJM5, pAJM10, pGJ60 and pGJ61. The BACTH fusion proteins were created by fusing (i) the N-terminus of the protein of interest to either pKT25 or pUT18C or (ii) the C-terminus to either pKNT25 or pUT18 (Table 1). Genes were amplified using primers that introduce either XbaI-(no stop codon)-KpnI and HindIII-(no stop codon)-KpnI or XbaI-(stop codon)-KpnI restriction sites.
Table 1.
Interactions between the Psp proteins in vivo
The BACTH system was used to detect protein–protein interactions between the Psp and ArcB proteins in vivo. Negative control, BTH101/pKT25+pUT18C vectors alone (85±9 MU); positive control, BTH101/pKT25-zip+pUT18C-zip (1405±64 MU); N-terminal (‘n’) protein fusions with T25 or T18 Cya fragments, respectively; C-terminal (‘c’) protein fusions with T25 or T18, respectively. Interaction estimates were as follows: +/−, weak interaction (283–470 MU); +, interaction (471–940 MU); ++, strong interaction (>940 MU); −, no interaction (≤282 MU); nd, not determined. See Methods for construction of fusion proteins, growth conditions and calculation of mean values.
| Protein fusion | T25–‘n’ | ‘c’–T25 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PspA | PspC | PspC1–68 | PspC40–68 | PspC40–119 | PspC69–119 | PspC69–119+PspB* | PspB | PspG | |
| T18–‘n’ | |||||||||
| PspA | + | +† | − | − | − | − | + | − | − |
| PspC | + | −†‡ | − | − | − | nd | nd | ++‡ | + |
| PspC1–68 | − | − | − | − | − | nd | nd | ++ | nd |
| PspC40–68 | nd | − | − | − | − | nd | nd | + | + |
| PspC40–119 | − | − | − | − | − | nd | nd | + | nd |
| PspCLeuZm | − | nd | nd | nd | nd | nd | nd | + | +/− |
| PspC69–119 | − | nd | nd | nd | nd | nd | nd | − | nd |
| PspC69–119+PspB* | + | nd | nd | nd | nd | nd | nd | nd | nd |
|
|
− | nd | nd | nd | nd | nd | nd | nd | nd |
| PspCG48A | − | nd | nd | nd | nd | nd | nd | ++ | − |
| PspCG74A | + | nd | nd | nd | nd | nd | nd | ++ | + |
| ArcB | +/− | +/−† | nd | nd | nd | nd | nd | +/− | − |
| ArcB* | +/− | +/−† | nd | nd | nd | nd | nd | +/− | − |
| ArcBLeuZm | +/− | +/−† | nd | nd | nd | nd | nd | +/− | − |
| ‘c’–T18 | |||||||||
| PspC40–119 | + | nd | nd | nd | nd | nd | nd | + | nd |
| − | nd | nd | nd | nd | nd | nd | + | nd | |
| PspB | − | ++† | ++ | ++ | ++ | − | nd | ++ | − |
| PspBLeuZm | − | ++† | ++ | ++ | ++ | nd | nd | ++ | nd |
| PspG | − | + | nd | nd | nd | nd | nd | − | ++ |
*PspB co-expressed from plasmid pAJM1; experimental results already presented elsewhere confirmed in this work and used as controls.
In vivo bacterial BACTH system.
The Cya-based BACTH assay was used to study in vivo protein–protein interactions (Karimova et al., 1998, 2005). Protein fusions (see Table 1 and Fig. 1) were assayed in BTH101 cells as described previously (Jovanovic et al., 2009). Interactions were quantified by measuring β-galactosidase activity in liquid cultures (see below). Chromosomal LacZ expression threefold above the negative control (vector alone) value was scored as a positive interaction signal.
Fig. 1.

Topologies of Psp proteins. (a) Schematic representation of PspA, PspB, PspC and PspG topologies according to Elderkin et al. (2005), Jones et al. (2003), Kleerebezem et al. (1996) and Engl et al. (2009), respectively. Per, periplasm; IM, inner membrane; Cyt, cytoplasm; N, N-terminus of the protein; C, C-terminus of the protein. (b) Representation of the predicted conserved domain organization of PspC: the cytoplasmic extrusion domain (Cyt, residues 1–39), the transmembrane portion (TM, residues 40–64), the periplasmic extrusion domain (Per, residues 65–119) and the conserved PspC domain (residues 1–68, light grey). The putative leucine zipper motif (LeuZ; residues 77–98) is as indicated. The numbering refers to E. coli PspC.
To test protein–protein interactions in the absence of natively produced Psp proteins that may provide or facilitate binding interactions by a bridging effect, we constructed a derivative of the BTH101 strain carrying the ΔpspF mutation – thereby preventing expression of the native Psp proteins (see Supplementary Table S2). The wild-type pspF gene was replaced by ΔpspF : : Kan, yielding MVA99 (see Supplementary Table S1). The Kan cassette from ΔpspF : : Kan was eliminated and the marker-less pspF99 variant (strain MVA100) constructed using plasmid pCP20 and the method described by Cherepanov & Wackernagel (1995) (see Supplementary Table S1). The strain was verified by PCR.
β-Galactosidase (β-Gal) assays.
Activity from a chromosomal Φ(pspA–lacZ) transcriptional fusion was assayed to gauge the level of psp expression, while activity of the native chromosomal lacZ was measured in the BACTH assays. Overnight cultures grown at 37 °C (or 30 °C for the BACTH assays) were diluted 100-fold and grown under the same conditions until mid-exponential phase. Cultures were induced (with Ara or 0.5 mM IPTG for BACTH, as indicated) for 1 h and then assayed for β-Gal activity (Miller, 1992). For all β-Gal assays, mean values from six samples taken from technical duplicates of three independently grown cultures of each strain were used to calculate activity. The data shown in the figures are the mean values with sd error bars.
Bacterial cell fractionation.
Bacterial cultures were separated at mid-exponential phase into soluble and membrane fractions by a lysozyme-EDTA-osmotic shock protocol and the inner and outer membranes were selectively extracted with Triton X-100 (Russel & Kaźmierczak, 1993). Samples were analysed by Western blotting.
Western blotting.
Bacterial cells were harvested at mid-exponential phase and resuspended in a mix of 30 μl 4 % SDS and 30 μl Laemmli buffer (Sigma). Samples were normalized according to cell growth measured as OD600, separated by SDS-PAGE and transferred onto PVDF membrane using a semidry transblot system (Bio-Rad). Western blotting was performed as described by Elderkin et al. (2002) using antibodies to PspA (1 : 10 000) (Jones et al., 2003), PspB (1 : 5000), PspC (1 : 5000), PspG (1 : 1000) (Jovanovic et al., 2006) or pIV (1 : 10 000). Proteins were detected using the ECL plus Western Blotting Detection kit (GE Healthcare). Images were captured in a FujiFilm Intelligent Dark Box by an image analyser with a charge-coupled-device camera (LAS-3000). Densitometry analysis was performed with MultiGauge 3.0 software (FujiFilm USA) and quantification (results expressed in arbitrary units) was performed using the aida software.
Confocal fluorescence microscopy to assess membrane electron potential (Δψ).
The Δψ component of pmf was measured using the JC-1 dye method (Becker et al., 2005) as described previously (Jovanovic et al., 2006; Engl et al., 2009). The green to red fluorescence emission ratio (530/590 nm) was calculated from 100 individual cells taken from technical duplicates of three independently grown cultures of each strain. The threshold to distinguish pmf differences was chosen based on the Δψ (equivalent to pmf under given experimental conditions) of the WT strain under non-stress growth conditions.
Single-molecule fluorescence imaging of PspG in vivo.
Single molecules in fluorescent PspG–GFP complexes (MG1655 ΔpspG/PspG–GFP) were imaged in vivo (Engl et al., 2009). PspG–GFP-expressing cells were mounted on a cover glass and images taken at 80 ms per frame. The fluorescent PspG–GFP complexes were analysed using the ImageJ software (http://rsb.info.nih.gov/ij/) (Abramoff et al., 2004), with GFP from MG1655/pDSW209 cell lysates as a reference.
Protein purification.
E. coli Top10 or Top10ΔpspA cells transformed with either pRD047 (encoding PspB and non-tagged PspC) or pRD047His (encoding PspB and His-tagged PspC) were grown in LB at 37 °C until they reached OD600 ∼0.5, at which point the culture was shifted to 30 °C. Overexpression was induced by addition of 0.02 % Ara and the cultures grown for a further 3 h. Cells were harvested at 8000 r.p.m. for 10 min at 4 °C; the pellet was resuspended in 20 ml buffer A (50 mM sodium phosphate pH 7, 300 mM NaCl, 5 %, v/v, glycerol) supplemented with Complete Protease Inhibitor (Roche) and lysed by sonication. The supernatant was loaded onto a 5 ml HiTrap metal-chelating column (GE Healthcare) pre-charged with NiCl2 and the protein eluted with buffer A supplemented with 1 M imidazole. Purified proteins were detected by Western blotting (see above).
Bioinformatic methods.
Multiple sequence alignment was performed using Multalin software version 5.4.1 (http://bioinfo.genotoul.fr/multalin/) (Corpet, 1988). Protein domain analysis was performed using Pfam version 21.0 (http://www.sanger.ac.uk/pfam) (Finn et al., 2006). Transmembrane (TM) helices prediction, localization and topologies were performed using tmhmm (http://www.cbs.dtu.dk/services/TMHMM/, V 2.0) (Kahsay et al., 2005), hmmtop (http://www.enzim.hu/hmmtop/, V 2.0) (Tusnády & Simon, 2001), TopPred2 (http://mobyle.pasteur.fr/cgi-bin/portal.py?form=toppred) (von Heijne, 1992) or topcons (http://topcons.net/) (Bernsel et al., 2009).
RESULTS AND DISCUSSION
PspG associates with Psp proteins known to form regulatory complexes
Previous studies suggested the presence of Psp regulatory complexes (Adams et al., 2003; Maxson & Darwin, 2006; Jovanovic et al., 2009). To determine whether PspG is able to interact with any of the proteins that constitute these regulatory complexes we performed pairwise BACTH interaction assays with PspA, PspB, PspC, ArcB and PspG (Table 1 and data not shown). The topologies of PspB, PspC and PspG (Fig. 1a) (reviewed by Darwin, 2005; see also Engl et al., 2009) were taken into account when constructing and analysing the fusion proteins (e.g. fusions to protein regions embedded in the IM could interfere with its localization). As indicated in Table 1, we found that (i) PspA interacts with itself and PspC, (ii) PspB can interact with itself, PspC and ArcB, (iii) PspC can interact with PspA, PspB and PspG, (iv) PspG can interact with itself and PspC, and (v) ArcB interacts with PspB. The interactions obtained do not appear attributable to general non-specific IM protein interactions since the IM proteins PspB and PspC show distinctive sets of positive results (see Table 1). Any bridging effects of natively produced Psp proteins however cannot be discounted, unless BACTH assays are performed in a host strain unable to express Psp (e.g. ΔpspF; see below and Supplementary Table S2).
In order to provide evidence that Psp proteins may indeed form a complex and to determine whether PspG is truly associated with PspC, we performed pull-down assays using PspC6His co-expressed with WT PspB (since overexpression of PspC acts as a psp inducer, causing a drop in pmf – see below). We specifically selected PspC given that the pairwise interaction data suggest that PspC interacts with PspA, PspB and PspG (Table 1). We reasoned that PspC would, by means of protein–protein interactions, specifically pull down Psp proteins involved in the regulatory complex during protein purification (via the His-tag on PspC). As a control, we performed the chromatography assay with non-tagged PspC to demonstrate that any Psp proteins identified in the eluted fractions are present due to a specific interaction with PspC6His. When we analysed the PspC6His elution fractions we found that PspA, PspB and PspG all co-purified with PspC6His (Fig. 2a–d) – co-purification of PspA,B,C suggests that a Psp[A–C–B] regulatory complex exists, to which PspG may associate. PspG also co-purifies with PspC6His when expressed in cells lacking PspA (Fig. 2e), further indicating that PspG interacts directly with PspC.
Fig. 2.

Co-purification of PspA, PspB and PspG with PspC6His. PspC6His co-expressed with PspB (pRD047His) was purified using metal-affinity chromatography (see Methods). As a control non-tagged PspC co-expressed with PspB (pRD047) was also purified. Peak fractions from both purifications (corresponding to fractions 2–4), were visualized using Western blotting and (a) anti-PspC, (b) anti-PspB, (c) anti-PspA or (d) anti-PspG antibodies as indicated. In (a) the positions of monomeric PspC (PspC; arrow) and an additional slower-migrating anti-PspC-reactive (double asterisk) species are as indicated. PspB (b), PspA (and non-specific band, asterisk) (c) and PspG (d) co-purified with PspC6His fractions (2–4) but not with PspC (non-tagged). (e) PspC6His co-expressed with PspB was also purified from a ΔpspA strain and the peak fractions were visualized using Western blotting and anti-PspG antibodies. The positions of molecular mass marker proteins (kDa) are indicated.
Potentially, co-expression of PspBC leads to an ‘active’ form of Psp(B)C that can specifically interact with PspA and possibly PspG. Such complexes may have significant implications in understanding the signalling cascade and activation of the Psp response as well as the effector function of PspG. Importantly, a reduced psp operon, pspACG, found in Aeromonas species (M. P. H. Stumpf, personal communication) further suggests that functional interactions occur between PspA, PspC and PspG.
Given the ability of PspG to self-associate (as suggested in the BACTH assay; Table 1) and since the oligomeric state of PspG could be important for its effector function, we analysed PspG–GFP complexes in live MG1655ΔpspG cells using single-molecule fluorescence imaging and objective-type total internal reflection fluorescence (TIRF) (see Methods, Engl et al., 2009 and e.g. Haggie & Verkman, 2008). The results illustrate that PspG–GFP can form complexes as large as pentamers/hexamers but the majority are dimers/trimers (Fig. 3).
Fig. 3.

PspG–GFP forms higher oligomers in vivo. The number of molecules in fluorescent PspG–GFP complexes was estimated using ImageJ software. The intensity of a single pixel in a fluorescent PspG–GFP (pGJ7) cluster expressed in E. coli MG1655 ΔpspG cells (MVA40) was measured and compared to GFP fluorescence in E. coli MG1655 cell lysates harbouring pDSW209 (GFP alone). (a) The mean fluorescence intensity of 50 PspG–GFP complexes within living cells was on average at least three times higher than that of GFP spots, suggesting that PspG can form at least a dimer/trimer in vivo. (b) The frequency distribution among the 50 complexes analysed illustrates that PspG–GFP self-assembles into a single major distinct oligomeric class.
Results from the BACTH assay also indicated that PspG, when expressed from the high-copy-number plasmid pUT18, interacts with ArcB (data not shown). However, when PspG was expressed from the low-copy-number plasmid pKNT25 no interaction with ArcB was detected (Table 1). As we were unable to detect an interaction between PspG and ArcB with the low-copy-number plasmid expressing PspG, we infer that the observed interaction either may be non-specific or may reflect the action of PspG as an effector when overproduced (Jovanovic et al., 2006) – the later possibility has yet to be explored.
PspC does not stably self-associate
Peak fractions from the PspBC6His purification were analysed by SDS-PAGE (Fig. 2a) and found to contain a number of PspC cross-reacting species (Supplementary Figs S1Ai, S2C, Dii, S3Bi and C) – one which migrates as a monomer and one as a putative dimer. It has been proposed that changes in pmf that occur upon extracytoplasmic stress may cause the stable self-association of PspC – reported by Adams et al. (2003) as an SDS-PAGE-stable dimeric form of PspC. Although we showed that co-expression of PspBC does not change pmf (see below), and BACTH analysis failed to detect pairwise PspC–PspC interactions in either Yersinia enterocolitica (Maxson & Darwin, 2006) or E. coli (Table 1), we cannot discount the possibility that either (i) the conformation of the fusion proteins is unfavourable for detecting PspC self-association or (ii) PspC may stably associate with an unknown protein to form the apparent ‘dimer’ species. Alternatively, PspC self-association may only occur upon pIV stress or when PspC is highly overproduced, since the apparent dimer was only observed when PspC was overproduced in a ΔpspC strain (Supplementary Fig. S3Bi and C). To investigate this further, and to address whether formation of a putative PspC dimer can be observed under physiological conditions, we analysed the expression of chromosomally encoded PspC in WT cells and cells lacking the negative regulator PspA (ΔpspA) in the presence and absence of pIV (Fig. 4). Fully consistent with its role in psp induction, pIV elevated Psp expression in WT cells (observed as an increase in monomeric PspC protein levels). Further, in cells lacking PspA, monomeric PspC expression remained high regardless of the presence of pIV (indicative of deregulated Psp expression). Notably, the intensity of the slower-migrating anti-PspC-reactive complex (the putative PspC dimer) remained unchanged under conditions where PspC was expressed at different levels (Fig. 4), suggesting that under physiological conditions PspC does not form a stable complex with either itself or PspA (since an identical anti-PspC-reactive complex, the putative PspC dimer, is observed upon overexpression of PspC in a ΔpspA strain). The lower level of PspC expression in cells encountering pIV stress versus cells lacking negative regulation (Fig. 4) is fully consistent with our findings that even under pIV stress conditions, PspA is still imposing apparent negative regulation (Engl et al., 2009; Jovanovic et al., 2009).
Fig. 4.

Oligomerization state of PspC. Western blots (using anti-PspC) illustrating PspC expression from the chromosome in either WT (MG1655) or ΔpspA (MG1655ΔpspA) cells (in the absence or presence of pIV). The positions of species that specifically cross-react with anti-PspC are highlighted as monomeric PspC (13.5 kDa; arrow) and a putative dimer (double asterisk). The positions of the marker proteins (kDa) are indicated. Below: the relative expression levels of monomeric PspC (labelled as PspC monomer) and the putative PspC dimer (double asterisk) were quantified within each strain tested, and the results expressed as a percentage of the induced corresponding protein band (+pIV; lanes 2 and 4, 100 %). ‘Proteins’ refers to the loading control. Importantly, these results demonstrate that in the absence of PspA (lanes 3 and 4), the relative expression levels of monomeric PspC are clearly highly elevated compared to WT in the presence of pIV, whereas the putative dimer expression level remains relatively unchanged, suggesting that this band corresponds to an unspecific anti-PspC cross-reacting species.
These results show that the apparent dimer species recognized by the PspC antibodies does not follow PspC expression levels, and therefore should not be considered as a stable PspC dimer. Taking these findings together with the BACTH assays, we conclude that under the physiological conditions tested PspC does not stably self-associate.
PspB and PspC act together to directly release negative regulation of psp
It is formally possible that the PspBC–PspA interactions that occur under non-stress conditions contribute to a steady-state level of a Psp[F–B]–ArcB regulatory complex. Alternatively, since the absence of PspBC does not release the negative regulation, and so cannot be required for formation of the inhibitory complex (Jovanovic et al., 2009; Gueguen et al., 2009), Psp[F–A] and Psp[C–B]–ArcB subcomplexes may communicate only in the presence of stress. Nevertheless, in both cases the stress-induced activation of psp may follow conformational changes in PspB and PspC that lead to release of negative regulation of PspA, presumably through altered protein–protein interactions. Interestingly, overexpression of PspC or PspBC, but not PspB alone, induces psp (Weiner et al., 1991; Maxson & Darwin, 2006) (also see Fig. 5a and Supplementary Fig. S1A). Since overexpression of PspC decreases pmf in ΔpspF cells (i.e. where the Psp response is absent) (Fig. 5b) it may, similarly to pIV (Jovanovic et al., 2006, 2009) or PulD (Guilvout et al., 2006), act as an inducing stress for psp (Fig. 5a). In contrast, overexpression of PspBC strongly induces psp (Fig. 5a), but does not reduce pmf (Fig. 5b). Apparently, PspB (in the context of the PspBC complex) appears to prevent PspC from changing pmf, consistent with the observation that the growth rate in exponential phase is normal in ΔpspC cells overexpressing PspBC but severely impaired (10-fold) in ΔpspC cells overexpressing PspC alone (data not shown). Since PspB counteracts the ‘toxic’ effect of PspC, the PspBC proteins may be acting as an antitoxin–toxin pair as suggested by Brown & Shaw (2003). A parallel study (Gueguen et al., 2009) showed that in Y. enterocolitica PspB stabilizes expression of WT PspC, a result differing from our E. coli data (Supplementary Fig. S1A).
Fig. 5.

psp expression in the presence of overexpressed PspBC. (a) Induction of chromosomal Φ(pspA–lacZ) in a ΔpspC strain (MVA13) by overexpression of PspB (pAJM1), PspC (pAJM2) or PspBC (pAJM3) (using 0.02 % Ara). (b) Overexpression of PspC decreases pmf while co-expression with PspB counteracts this effect. Δψ was determined in a ΔpspF strain (MG1655ΔpspF) overexpressing PspB, PspC or PspBC. (c) Overexpression of PspBC directly induces psp. PspBC (pAJM3) or PspBLeuZmC (pGJ49; PspBLeuZm does not transduce the psp-inducing signal) were co-expressed in either ΔpspBC (MVA45) or ΔpspBCΔarcB (MVA83; ΔarcB diminishes psp induction) cells (using 0.02 % Ara). PspBC were co-expressed in a ΔpspBC strain (since the arcB mutation reduces induction by pIV; using 0.02 % Ara) in the presence of pIV. Vector, pBAD18-cm. As a control, psp expression was determined in the absence of PspA (ΔpspA, MVA27; to prevent negative regulation).
Notably, overexpression of PspBC results in higher psp induction than does overexpression of PspC alone (Fig. 5a) (consistent with Weiner et al., 1991 and Maxson & Darwin, 2006) – at a level comparable to uncontrolled psp expression in ΔpspA cells (Fig. 5c). In line with this observation, co-expression of PspBC in the presence of pIV did not result in increased psp induction (Fig. 5c and Supplementary Fig. S1B). Since PspBC overexpression does not simply induce psp by decreasing pmf (i.e. the overproduced proteins do not in themselves generate a ‘stress’ signal) we suggest that overproduced PspBC resembles the ‘stressed on-state’ of the Psp response.
The effect of overproduced PspBC could directly alter the state of PspA, and hence negative regulation, thereby bypassing upstream signalling systems (e.g. the ArcAB system). To test this directly, we used a leucine zipper motif (LeuZ) mutant (LeuZm) of PspB (PspBLeuZm) that maintains the ability to interact with PspC but is no longer able to interact with ArcB and is therefore inactive in upstream signalling (Jovanovic et al., 2009). When co-expressed with PspC, this mutant form of PspB should support psp induction independent of any upstream signalling event. Significantly, overexpression of PspBLeuZmC resulted in a similar level of psp induction as overexpressed WT PspBC (Fig. 5c). In line with this, overexpression of either PspBC or PspBLeuZmC strongly induces psp in the absence of ArcB (ΔarcB strain) (Fig. 5c). These data argue that a PspBC complex is sufficient to induce the Psp response, by relieving PspA-imposed inhibition, in the absence of ArcB.
PspC residues 40–119 are required for psp induction
We next addressed which determinants were important for PspBC–PspA signalling. Recall that PspA–PspC and PspB–PspC, but not PspB–PspA, were shown to directly interact (see above), suggesting that PspC may have a central role in transducing the stress signal to PspA. We first truncated PspC based on its predicted domain structure and topology (Fig. 1) to form three PspC fragments: PspC1–68 (cytoplasmic and TM domain), PspC40–68 (TM domain) and PspC40–119 (TM-periplasmic domain). In the absence of PspC (ΔpspC), induction of the psp response upon pIV stress no longer occurs (reviewed by Darwin, 2005; see also Jovanovic et al., 2009). We initially introduced into ΔpspC cells either full-length PspC (residues 1–119) or one of the three fragments to test their activity with regard to their ability to support psp induction upon pIV stress. Each protein was expressed at a level similar to the basal level that was insufficient to induce psp in the absence of stress, and was scored for supporting pIV-dependent induction of psp (Fig. 6a). Only full-length PspC complemented the ΔpspC mutation for psp induction by pIV. Notably, similar levels of pIV expression were detected in all the samples tested (Supplementary Fig. S2A), and expression of wild-type PspC was at least 20-fold lower than that following pIV induction of chromosomal psp (Supplementary Fig. S2B). These results establish that full-length PspC, but not its fragments, supports signal transduction to PspA in pIV-dependent induction of psp.
Fig. 6.

psp expression by the PspC fragments. (a) Full-length PspC is required for signal transduction upon pIV-dependent psp-inducing stress. Induction of the chromosomal Φ(pspA–lacZ) fusion in a ΔpspC strain (MVA13) expressing a low level of PspC (1–119, pAJM2) or PspC fragments (1–68, pAJM7; 40–68, pAJM5; 40–119, pAJM8) (using 0.001 % Ara) in the absence or presence of pIV (pGJ4) (see Methods). (b) The TM-periplasmic region of PspC (PspC40–119) is sufficient for PspB-independent induction of psp. Induction of the chromosomal Φ(pspA–lacZ) fusion in a ΔpspC strain (MVA13) overexpressing PspC (1–119, pAJM2) or PspC fragments on its own (as in a) or with PspB [PspBC (pAJM3) or PspBC fragments (1–68, pAJM12; 40–68, pAJM10; 40–119, pAJM13)] (using 0.02 % Ara) (see Methods). (c) Overexpression of PspC1–68 decreases pmf while co-expression with PspB counteracts this effect in a ΔpspF strain (MG1655ΔpspF).
PspC and its fragments (tested separately or co-expressed with PspB) were strongly overexpressed in ΔpspC cells at a level that was sufficient for PspC or PspBC (see Fig. 5a) to induce psp. Under these conditions the PspC40–119 fragment induced psp expression even in the absence of co-expressed PspB (Fig. 6b). We do note that expression levels of the PspC fragments varied greatly (Supplementary Fig. S2C); however, these differences do not account for the inability of some fragments to induce psp. For example, the PspC1–68 and PspC40–119 fragments are expressed similarly in the absence of PspB (Supplementary Fig. S2Ci and Di), yet only the PspC40–119 fragment induces psp (Fig. 6b). Further, the fragments did not appear to mislocalize (Supplementary Fig. S2D). Notably, expression of the PspC40–68 and PspC40–119 fragments seemed to be stabilized in the presence of co-overexpressed PspB, potentially via interactions between the TM domains of these two proteins (see below). As an additional control, we determined Δψ in ΔpspF cells (where psp-inducing agents, such as pIV, decrease pmf in the absence of the Psp response) overexpressing the PspC fragments, including PspC40–119 (in either the absence or presence of PspB), and found that pmf was not decreased (Fig. 6c), establishing that PspC40–119 does not act as a typical psp-inducing stress agent. Importantly, overexpression of the PspC1–68 fragment in the absence of co-expressed PspB decreased pmf (Fig. 6c) and impaired cell growth in ΔpspC cells similarly to full-length PspC (data not shown), but was unable to induce psp. Therefore, even when pmf was reduced, the PspC1–68 fragment was unable to complement a ΔpspC mutation and support psp induction. These data suggest that the PspC1–68 sequence lacking the putative periplasmic domain is important for the ‘toxic’ effect of PspC (i.e. a drop in pmf, see above). The PspC putative periplasmic domain (residues 69–119, present in the PspC40–119 fragment that induces psp but not in the PspC1–68 or PspC40–68 fragments, which do not induce psp) can function independently of upstream signalling and so may be directly involved in the release of the PspA–PspF inhibitory complex and hence functions positively in psp induction.
The integrity of the putative leucine zipper motif of PspC is required for psp induction
The fragmentation approach demonstrated that the putative periplasmic region of PspC may be involved in psp induction and hence presumably might receive the inducing signal or contact PspA in order to release the negative regulation that PspA imposes on PspF.
Initially we analysed the ability of each of the PspC fragments to interact with PspB and PspA. Significantly, we observed that all the PspC fragments were capable of interacting with PspB or PspBLeuZm, demonstrating that this interaction (and also PspB self-association) does not require the PspB LeuZ (Table 1 and data not shown). These data also suggest that the determinants in PspC required for the interaction with PspB lie within the putative TM domain (residues 40–68) (Table 1 and Supplementary Fig. S2C). Importantly, and in contrast to WT PspC, two of the three PspC fragments failed to interact with PspA; only the C-terminal PspC40–119 Cya fusions interacted with PspA (Table 1 and Supplementary Table S2). This is somewhat surprising given the proposed topology of PspC, where the C-terminus is facing the periplasm (Kleerebezem et al., 1996), suggesting that in the absence of the predicted cytoplasmic domain, the PspC40–119 fragment may adopt another topology.
The sequence assigned to the PspC periplasmic domain contains a putative LeuZ (Kleerebezem et al., 1996) (Fig. 1b). When we examined the activities of LeuZ mutants (LeuZm) in the context of full-length PspC (PspCLeuZm) and the PspC40–119 fragment (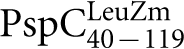 ) (see Supplementary Table S1) we found that both proteins failed to interact with PspA (Table 1 and Supplementary Table S2) but maintained an ability to interact with PspB (Table 1). Given that the LeuZ of PspC appears important for PspA interactions, we reasoned that PspC proteins that contain a disrupted LeuZ would no longer be active for psp induction (assuming that they no longer relieve the inhibition imposed by PspA that is mediated by protein–protein interactions). Since full-length PspC is strictly required for psp induction upon pIV stress (Fig. 6a), and to avoid affecting pmf (Fig. 6c), the signal transduction complementation assays were performed in ΔpspC cells co-expressing PspB and full-length PspC (Fig. 7a). PspBCLeuZm when expressed at a low level in the presence of pIV was unable to fully induce the psp response (Fig. 7a; compared to WT PspBC). The expression level of pIV under all conditions assayed was similar (Supplementary Fig. S3A), indicating that differences that arise in psp induction are unlikely to be due to variations in the level of the primary stress signal. The LeuZ of PspC appears to be important for signalling and/or the outcome of signalling to PspA.
) (see Supplementary Table S1) we found that both proteins failed to interact with PspA (Table 1 and Supplementary Table S2) but maintained an ability to interact with PspB (Table 1). Given that the LeuZ of PspC appears important for PspA interactions, we reasoned that PspC proteins that contain a disrupted LeuZ would no longer be active for psp induction (assuming that they no longer relieve the inhibition imposed by PspA that is mediated by protein–protein interactions). Since full-length PspC is strictly required for psp induction upon pIV stress (Fig. 6a), and to avoid affecting pmf (Fig. 6c), the signal transduction complementation assays were performed in ΔpspC cells co-expressing PspB and full-length PspC (Fig. 7a). PspBCLeuZm when expressed at a low level in the presence of pIV was unable to fully induce the psp response (Fig. 7a; compared to WT PspBC). The expression level of pIV under all conditions assayed was similar (Supplementary Fig. S3A), indicating that differences that arise in psp induction are unlikely to be due to variations in the level of the primary stress signal. The LeuZ of PspC appears to be important for signalling and/or the outcome of signalling to PspA.
Fig. 7.

PspC determinants involved in signal transduction and induction of psp. (a) The PspC LeuZ and residue G48 are required for pIV-induced psp expression. Induction of the chromosomal Φ(pspA–lacZ) fusion in a ΔpspC strain (MVA13) expressing low-level PspBC (pAJM3) or PspBC mutants (PspC mutants: G48A, pGJ54; G74A, pGJ55; LeuZm, pGJ57) (using 0.001 % Ara) in the absence or presence of pIV (pGJ4) (see Methods). As a control, pIV-dependent induction of psp in WT cells (MVA4) is presented. (b) High-level co-expression of PspBCG48A, PspBCLeuZm, 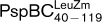 (pGJ60) and
(pGJ60) and 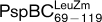 (pGJ61) mutants failed to directly induce psp expression. Induction of the chromosomal Φ(pspA–lacZ) fusion in a ΔpspC strain (MVA13) by overexpression of PspBC or PspBC mutants (using 0.02 % Ara). (c) The PspC periplasmic region may exist in two topologies: schematic illustrating the potential topologies of PspC (A, B or C) and PspC40–119 (the periplasmic region containing the LeuZ; D).
(pGJ61) mutants failed to directly induce psp expression. Induction of the chromosomal Φ(pspA–lacZ) fusion in a ΔpspC strain (MVA13) by overexpression of PspBC or PspBC mutants (using 0.02 % Ara). (c) The PspC periplasmic region may exist in two topologies: schematic illustrating the potential topologies of PspC (A, B or C) and PspC40–119 (the periplasmic region containing the LeuZ; D).
When co-expressed with PspB, the PspC LeuZm in the context of either WT PspC (PspCLeuZm) or the PspC40–119 fragment (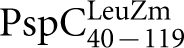 ) was either able (to some extent) or unable, respectively, to induce psp (Fig. 7b). Significantly, the expression level and localization of PspCLeuZm were similar to those of WT PspC (Supplementary Fig. S3B and C), further suggesting that a loss of productive interactions with PspA and the subsequent reduction in psp induction is not simply a consequence of protein misfolding or mislocalization.
) was either able (to some extent) or unable, respectively, to induce psp (Fig. 7b). Significantly, the expression level and localization of PspCLeuZm were similar to those of WT PspC (Supplementary Fig. S3B and C), further suggesting that a loss of productive interactions with PspA and the subsequent reduction in psp induction is not simply a consequence of protein misfolding or mislocalization.
Recent parallel studies (Gueguen et al., 2009) demonstrated that in Y. enterocolitica the periplasmic region and LeuZ of PspC were important in psp induction. Gueguen et al. (2009) proposed a model in which the periplasmic region of PspC (more specifically the LeuZ) receives the stress signal rather than directly interacting with PspA. In addition, they showed that complementation of a chromosomal pspC deletion with PspC variants harbouring cytoplasmic domain truncations led to constitutive psp expression. These outcomes resemble the activities of the PspC40–119 fragment reported here. Gueguen et al. (2009) proposed that the cytoplasmic domains of PspC and PspB are involved in the negative regulation of psp in the absence of stress, and then interact with the Psp[A–F] inhibitory complex upon stress to release PspF negative regulation. However, Gueguen et al. (2009) found that the PspC cytoplasmic domain truncations still required the putative periplasmic domain to exhibit constitutive psp expression. Our results suggest that the PspC putative periplasmic domain may function in a direct interaction with PspA for release of negative regulation rather than just in receiving the inducing signal.
Characterization of the PspC69–119 fragment
To further dissect the possibility that the putative PspC periplasmic domain (residues 69–119) directly interacts with PspA, we characterized the PspC69–119 fragment (see Supplementary Table S1), which contains the sequence predicted to reside in the periplasm. PspC69–119 expressed in the absence (Supplementary Fig. S2Ci) and presence (Supplementary Fig. S2Cii) of PspB was detected within the soluble cell fraction (Supplementary Fig. S2Dii). Overexpression of PspC69–119 does not significantly change Δψ (data not shown). Similarly to PspC40–119, overexpression of PspC69–119 in the absence of stress strongly induced psp but only when co-expressed with PspB (Fig. 7b). In addition, induction of psp, by overexpressed PspBC40–119 or PspBC69–119 is independent of ArcB (data not shown). These data clearly indicate that PspB, in concert with the periplasmic domain of PspC, directs the release of negative regulation of psp expression. The Cya–PspC69–119 fusion interacted with Cya–PspA (but not PspB–Cya) only when co-expressed with WT PspB (Table 1 and Supplementary Table S2). 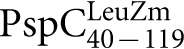 (see Supplementary Table S1 and Supplementary Fig. S2Cii), when co-overexpressed with PspB, was unable to induce psp (Fig. 7b) or interact with PspA (Table 1 and Supplementary Table S2). These results are consistent with the previously demonstrated PspA–PspB interaction observed only in the presence of PspC (Adams et al., 2003) and here we show that the PspC LeuZ is probably needed for this interaction. Taken together with results obtained with the PspC40–119 fragment (see above), direct PspC69–119 binding interactions with PspA seem to support induction of psp.
(see Supplementary Table S1 and Supplementary Fig. S2Cii), when co-overexpressed with PspB, was unable to induce psp (Fig. 7b) or interact with PspA (Table 1 and Supplementary Table S2). These results are consistent with the previously demonstrated PspA–PspB interaction observed only in the presence of PspC (Adams et al., 2003) and here we show that the PspC LeuZ is probably needed for this interaction. Taken together with results obtained with the PspC40–119 fragment (see above), direct PspC69–119 binding interactions with PspA seem to support induction of psp.
Our results show that the LeuZ located in the putative periplasmic domain of PspC is strictly required for PspA interactions and by clear inference psp induction – potentially explaining why PspC constitutive mutants still require the periplasmic domain for activation of psp expression (Gueguen et al., 2009). The results obtained with the PspC40–119 and PspC69–119 fragments in terms of psp induction and the BACTH assays, and assuming the periplasmic domain of PspC should interact with the cytoplasmic face of PspA, suggest that the periplasmic domain of PspC would favour one topology when pIV is expressed in order to directly transduce the stress signal to the Psp[A–F] regulatory complex. A phenocopy of the stress-induced ‘topology switch’ may be simply realized by mass-action effects: in the absence of stress PspC may exist in equilibrium between active and inactive conformations potentially reflecting two distinct topologies of PspC (and so controlling basal level expression of psp genes); overproduction of PspC would result in more of the activating ‘on-state’ PspC topology being present in the cell, resulting in psp induction.
PspC residue G48 is a determinant in psp induction
The possibility that the periplasmic domain of PspC may exist in two states (‘in’ and ‘out’), possibly altering its subcellular localization in response to stress, prompted us to apply the positive-inside rule (von Heijne, 1992) to the PspC sequence using different topology prediction programs (see Methods and Supplementary Fig. S4). The results were as follows: tmhmm – N-terminus inside, C-terminus outside; hmmtop – N-terminus outside, C-terminus inside; TopPred – N terminus outside, C-terminus inside; topcons – N-terminus inside, C-terminus inside with two potential TM domains and a small periplasmic loop (see Supplementary Fig. S4). These programs indicate that both topology scenarios (with respect to the periplasmic domain) are predicted for PspC, suggesting that the barrier between these two states is not high and that a topology switch potentially exists (Supplementary Fig. S4). In contrast, if we use the same programs with either PspB or PspG a single topology outcome is obtained, suggesting a relatively stable single topology state (data not shown). The localization of positive residues in extra-membrane domains is important for the positive-inside rule application and depends on Δψ, so protein topology can be dynamic (Bogdanov et al., 2002, 2008; Zhang et al., 2003, 2005). Potentially relevant for the Psp system and the PspBC sensors is that the observed reversible topological organization within some E. coli membrane proteins (e.g. LacY, PheP, GabP) is governed by a change in membrane phospholipid composition (reviewed by Bogdanov et al., 2009; Dowhan & Bogdanov, 2009). Further, kinked hydrophobic TM helices (such as the ones obtained in the PspC topology predictions; Supplementary Fig. S4), particularly those containing glycine (G) residues, are proposed to bend more easily within the membrane than those with straight TM helices (Jiang et al., 2002). Notably, these glycine residues are also thought to mediate the protein–protein interactions in polytopic membrane proteins (Javadpour, et al., 1999). PhoA fused to the PspC region predicted to reside in the periplasm showed activity (Kleerebezem et al., 1996), but it has not been discounted that this region can also at times be localized in the cytoplasm (e.g. using a fusion to β-galactosidase).
In light of the data concerning polytopic membrane proteins, we analysed the sequence of the putative TM helix (residues 40–68) of PspC and located a highly conserved glycine residue at position 48. When we mutated this residue to alanine (PspCG48A) we found that, similar to PspCLeuZm, PspCG48A failed to induce the psp response (Fig. 7a, b and Supplementary Fig. S3B, C). In agreement with these observations, the BACTH assay illustrates that PpsCG48A is no longer able to interact with PspA (or, interestingly, PspG); however, its ability to interact with PspB remains unaffected (Table 1). As a control we constructed the PspCG74A mutant, where residue G74 is predicted to lie within the periplasmic domain and as such should not affect the topology of the TM domain. As predicted, this mutant retained WT PspC activities in terms of signal transduction and psp induction (Fig. 7a, b and Supplementary Fig. S3B, C) and Psp protein interactions (Table 1). These results suggest that the highly conserved residue G48 may act as a determinant in regulating or establishing the activity of PspC in the stress signalling pathway. It is unlikely that G48 directly contacts PspA, given that this residue is predicted to lie within the IM and PspA is a peripheral IM protein. Rather, G48 might be required to establish the correct conformation and topology of PspC for an interaction with PspA.
Notably, the inability of PspCG48A to interact with PspG (in contrast to the PspCLeuZm variant) (Table 1) indicates that residue G48 may direct a specific interaction with PspG, which could have implications for the function of PspG under stress. Consistent with this view, the PspC40−68 fragment retains the ability to interact with PspG (Table 1), further suggesting that PspC IM interactions (with PspG) are critical for PspG functionality.
Concluding remarks
The analyses presented here demonstrate that PspB and PspC interact within the IM and may well serve as a scaffold within the Psp regulatory complex to which ArcB, PspA and PspG bind. We show, for the first time, that both PspB and PspG have a clear tendency to self-associate – in contrast to PspC – and identify determinants within PspB and PspC required for regulatory complex formation and psp induction. Since the experiments were conducted in the presence of co-expressed PspBC (to prevent alterations in pmf, which could ultimately lead to psp induction), and within the signalling experiments the amount of PspBC production was not elevated above basal levels, we expect that the outcomes discussed here are physiologically relevant.
In particular, we have established that residues 40–119 of PspC, which include the putative LeuZ (in the periplasmic domain) and residue G48 (in the TM domain), are important determinants required for signal transduction during pIV-dependent psp induction and consequently appear to have important effects on whether PspC interacts with PspA (and PspG for G48). We suggest that PspC may function as a membrane protein with more than one topology, possibly dynamic protein topologies (Fig. 7c). PspC topologies may respond to changes in the physical/chemical properties of the IM and Δψ. Apparently the PspC40–119 fragment (which has no predicted cytoplasmic domain), which localizes in the IM, has the ability (according to the positive-inside rule) to position the periplasmic domain within the cytoplasm, and to induce psp in the absence of PspB (Fig. 7c). Moreover, the soluble PspC69–119 fragment (carrying only the periplasmic domain) in the presence of co-expressed PspB interacts with PspA and induces psp. Since neither interaction of PspB with PspC69–119 nor interaction of PspA with PspB was detected, PspA–PspC69–119–PspB interactions may be highly co-operative, with very weak initial single component interactions. The interaction with PspA and induction of psp is dependent on the PspC periplasmic domain LeuZ. Considering that PspB–PspC interactions are both PspC LeuZ- and PspB LeuZ-independent (see Table 1) and that PspC40–119 but not 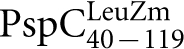 can interact with PspA in the absence of PspB (see Table 1 and Supplementary Table S2), we argue against PspC working through PspB. Additionally, we show that one ‘toxic’ effect of PspC (the drop in pmf) is counteracted by PspB. This finding indicates that PspC and PspB may be acting as a ‘toxin–antitoxin’ pair that may help to stabilize PspC in a topology or ‘active on-state’ favourable for interacting with PspA (Fig. 7c).
can interact with PspA in the absence of PspB (see Table 1 and Supplementary Table S2), we argue against PspC working through PspB. Additionally, we show that one ‘toxic’ effect of PspC (the drop in pmf) is counteracted by PspB. This finding indicates that PspC and PspB may be acting as a ‘toxin–antitoxin’ pair that may help to stabilize PspC in a topology or ‘active on-state’ favourable for interacting with PspA (Fig. 7c).
Finally, considering a potential mechanism of psp induction involving two distinct PspC topologies, we suggest that it is formally possible that another signal specifically recognized by PspC (or PspBC) may involve changes in the physical/chemical properties of the membrane, given that inhibitors of phospholipid biosynthesis (Bergler et al., 1994) or application of free fatty acids (Weiner & Model, 1994) strongly induce the psp response. In addition, transcription profiling revealed a link between PspA and use of glycerol 3-phosphate in the biosynthesis of phospholipids (Jovanovic et al., 2006). Further, PspA was shown to bind specific IM phospholipids, phosphatidylglycerol and phosphatidylserine to exhibit its effector function (Kobayashi et al., 2007). In agreement, outer-membrane secretins mislocalized within the IM or a block in protein translocation were shown to induce psp (reviewed by Model et al., 1997; Darwin, 2005). Negatively charged phospholipids were found to restore translocation of outer-membrane precursor proteins across the phosphatidyglycerol-depleted IM of E. coli cells (Kusters et al., 1991). Notably, archaea and acidophilic bacteria that have an inverted Δψ and different membrane content have a Psp system that contains only the PspA homologue and no recognizable PspBC proteins (Bidle et al., 2008; Vrancken et al., 2008), perhaps because PspBC function with a particular membrane potential and membrane lipid composition.
Acknowledgments
This work was supported by funding from the Wellcome Trust. We acknowledge D. Ladant, J. Tommassen, P. Model, M. Russel, J. Brown and J. Beckwith for providing strains, plasmids and antibodies. We would like to thank N. Joly, M. Jovanovic, E. James and T. Lenn for critical reading of the manuscript and comments.
Abbreviations
Δψ, electron potential
Ara, l-arabinose
BACTH, bacterial two-hybrid
β-Gal, β-galactosidase
IM, inner membrane
LeuZ, leucine zipper motif
LeuZm, leucine zipper mutant
pIV, protein IV
pmf, proton-motive force
Psp, phage-shock protein
Sol, soluble fraction
MU, Miller units
TM, transmembrane
Footnotes
Supplementary material is available with the online version of this paper.
References
- Abramoff, M. D., Magelhaes, P. J. & Ram, S. J. (2004). Image processing with ImageJ. Biophotonics Int 11, 36–42. [Google Scholar]
- Adams, H., Teertstra, W., Demmers, J., Boesten, R. & Tommassen, J. (2003). Interactions between phage-shock proteins in Escherichia coli. J Bacteriol 185, 1174–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, L. A., Bang, L.-S., Crouch, M.-L. & Fang, F. C. (2005). Compensatory role of PspA, a member of the phage shock protein operon, in rpoE mutant Salmonella enterica serovar Typhimurium. Mol Microbiol 56, 1004–1016. [DOI] [PubMed] [Google Scholar]
- Bergler, H., Abraham, D., Aschauer, H. & Turnowsky, F. (1994). Inhibition of lipid biosynthesis induces the expression of the pspA gene. Microbiology 140, 1937–1944. [DOI] [PubMed] [Google Scholar]
- Bernsel, A., Viklund, H., Hennerdal, A. & Elofsson, A. (2009). TOPCONS: consensus prediction of membrane protein topology. Nucl Acids Res 37, W455–W468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidle, K. A., Kirkland, P. A., Nannen, J. L. & Maupin-Furlow, J. A. (2008). Proteomic analysis of Haloferax volcanii reveals salinity-mediated regulation of the stress response protein PspA. Microbiology 154, 1436–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov, M., Heacock, P. N. & Dowhan, W. (2002). A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J 21, 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov, M., Xie, J., Heacock, P. & Dowhan, W. (2008). To flip or not to flip: lipid-protein charge interactions are a determinant of final membrane protein topology. J Cell Biol 182, 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov, M., Xie, J. & Dowhan, W. (2009). Lipid-protein interactions drive membrane protein topogenesis in accordance with the positive inside rule. J Biol Chem 284, 9637–9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J. M. & Shaw, K. J. (2003). A novel family of Escherichia coli toxin-antitoxin gene pairs. J Bacteriol 185, 6600–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov, P. P. & Wackernagel, W. (1995). Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14. [DOI] [PubMed] [Google Scholar]
- Corpet, F. (1988). Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16, 10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin, A. J. (2005). The phage-shock-protein response. Mol Microbiol 57, 621–628. [DOI] [PubMed] [Google Scholar]
- Darwin, A. J. (2007). Regulation of the phage-shock-protein stress response in Yersinia enterocolitica. In The Genus Yersinia. From Genomics to Function (Advances in Experimental Medicine and Biology, vol. 603), pp. 167–177. Edited by Perry, R. D. & Fetherston, J. D.. New York: Springer. [DOI] [PubMed]
- Dowhan, W. & Bogdanov, M. (2009). Lipid-dependent protein topogenesis. Annu Rev Biochem 78, 515–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elderkin, S., Jones, S., Schumacher, J., Studholme, D. & Buck, M. (2002). Mechanism of action of the Escherichia coli phage shock protein PspA in repression of the AAA family transcription factor PspF. J Mol Biol 320, 23–37. [DOI] [PubMed] [Google Scholar]
- Elderkin, S., Bordes, P., Jones, S., Rappas, M. & Buck, M. (2005). Molecular determinants for PspA-mediated repression of the AAA transcriptional activator PspF. J Bacteriol 187, 3238–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engl, C., Jovanovic, G., Lloyd, L. J., Murray, H., Ying, L., Errington, J. & Buck, M. (2009). In vivo localizations of membrane stress controllers PspA and PspG in Escherichia coli. Mol Microbiol 73, 382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R. D., Mistry, J., Schuster-Bockler, B., Griffiths-Jones, S., Hollich, V., Lassmann, T., Moxon, S., Marshall, M., Khanna, A. & other authors (2006). Pfam: clans, web tools and services. Nucleic Acids Res 34, D247–D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueguen, E., Savitzky, D. C. & Darwin, A. J. (2009). Analysis of the Yersinia enterocolitica PspBC proteins defines functional domains, essential amino acids and new roles within the phage-shock-protein response. Mol Microbiol 74, 619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilvout, I., Chami, M., Engel, A., Pugsley, A. P. & Bayan, N. (2006). Bacterial outer membrane secretin PulD assembles and inserts into the inner membrane in the absence of its pilotin. EMBO J 25, 5241–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggie, P. M. & Verkman, A. S. (2008). Monomeric CFTR in plasma membrane in live cells revealed by single molecule fluorescence imaging. J Biol Chem 283, 23510–23513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huvet, M., Toni, T., Tan, H., Jovanovic, G., Engl, C., Buck, M. & Stumpf, M. P. H. (2009). Model-based evolutionary analysis: the natural history of phage-shock stress response. Biochem Soc Trans 37, 762–767. [DOI] [PubMed] [Google Scholar]
- Javadpour, M. M., Eilers, M., Groesbeek, M. & Smith, S. O. (1999). Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys J 77, 1609–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., Lee, A., Chen, J., Cadene, M., Chait, B. T. & MacKinnon, R. (2002). The open pore conformation of potassium channels. Nature 417, 523–526. [DOI] [PubMed] [Google Scholar]
- Joly, N., Burrows, P. C., Engl, C., Jovanovic, G. & Buck, M. (2009). A lower-order oligomer form of phage shock protein A (PspA) stably associates with the hexameric AAA+ transcription activator protein PspF for negative regulation. J Mol Biol 394, 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S. E., Lloyd, L. J., Tan, K. K. & Buck, M. (2003). Secretion defects that activate the phage shock response of Escherichia coli. J Bacteriol 185, 6707–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic, G., Lloyd, L. J., Stumpf, M. P. H., Mayhew, A. J. & Buck, M. (2006). Induction and function of the phage shock protein extracytoplasmic stress response in Escherichia coli. J Biol Chem 281, 21147–21161. [DOI] [PubMed] [Google Scholar]
- Jovanovic, G., Engl, C. & Buck, M. (2009). Physical, functional and conditional interactions between ArcAB and phage shock proteins upon secretin-induced stress in Escherichia coli. Mol Microbiol 74, 16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsay, R. Y., Gao, G. & Liao, L. (2005). An improved hidden Markov model for transmembrane protein detection and topology prediction and its applications to complete genomes. Bioinformatics 21, 1853–1858. [DOI] [PubMed] [Google Scholar]
- Karimova, G., Pidoux, J., Ullmann, A. & Ladant, D. (1998). A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 95, 5752–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova, G., Dautin, N. & Ladant, D. (2005). Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J Bacteriol 187, 2233–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleerebezem, M., Crielaard, W. & Tommassen, J. (1996). Involvement of stress protein PspA (phage shock protein A) of Escherichia coli in maintenance of the protonmotive force under stress conditions. EMBO J 15, 162–171. [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, R., Suzuki, T. & Yoshida, M. (2007). Escherichia coli phage-shock protein A (PspA) binds to membrane phospholipids and repairs proton leakage of the damaged membranes. Mol Microbiol 66, 100–109. [DOI] [PubMed] [Google Scholar]
- Kusters, R., Dowhan, W. & de Kruijff, B. (1991). Negatively charged phospholipids restore prePhoE translocation across phosphatidylglycerol-depleted Escherichia coli inner membranes. J Biol Chem 266, 8659–8662. [PubMed] [Google Scholar]
- Lloyd, L. J., Jones, S. E., Jovanovic, G., Gyaneshwar, P., Rolfe, M. D., Thompson, A., Hinton, J. C. & Buck, M. (2004). Identification of a new member of the phage shock protein response in Escherichia coli, the phage shock protein G (PspG). J Biol Chem 279, 55707–55714. [DOI] [PubMed] [Google Scholar]
- Maxson, M. E. & Darwin, A. J. (2006). PspB and PspC of Yersinia enterocolitica are dual function proteins: regulators and effectors of the phage-shock-protein response. Mol Microbiol 59, 1610–1623. [DOI] [PubMed] [Google Scholar]
- Miller, J. H. (1992). A Short Course in Bacterial Genetics: a Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Model, P., Jovanovic, G. & Dworkin, J. (1997). The Escherichia coli phage-shock-protein (psp) operon. Mol Microbiol 24, 255–261. [DOI] [PubMed] [Google Scholar]
- Rowley, G., Spector, M., Kormanec, J. & Roberts, M. (2006). Pushing the envelope: extracytoplasmic stress responses in bacterial pathogens. Nat Rev Microbiol 4, 383–394. [DOI] [PubMed] [Google Scholar]
- Russel, M. & Kaźmierczak, B. (1993). Analysis of the structure and subcellular location of filamentous phage pIV. J Bacteriol 175, 3998–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnády, G. E. & Simon, I. (2001). The HMMTOP transmembrane topology prediction server. Bioinformatics 17, 849–850. [DOI] [PubMed] [Google Scholar]
- von Heijne, G. (1992). Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol 225, 487–494. [DOI] [PubMed] [Google Scholar]
- Vrancken, K., Van Mellaert, L. & Anné, J. (2008). Characterization of the Streptomyces lividans PspA response. J Bacteriol 190, 3475–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, L. & Model, P. (1994). Role of an Escherichia coli stress-response operon in stationary-phase survival. Proc Natl Acad Sci U S A 91, 2191–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, L., Brissette, J. L. & Model, P. (1991). Stress-induced expression of the Escherichia coli phage shock protein operon is dependent on sigma 54 and modulated by positive and negative feedback mechanisms. Genes Dev 5, 1912–1923. [DOI] [PubMed] [Google Scholar]
- Wigneshweraraj, S., Bose, D., Burrows, P. C., Joly, N., Schumacher, J., Rappas, M., Pape, T., Zhang, X., Stockley, P. & other authors (2008). Modus operandi of the bacterial RNA polymerase containing the sigma54 promoter-specificity factor. Mol Microbiol 68, 538–546. [DOI] [PubMed] [Google Scholar]
- Zhang, W., Bogdanov, M., Pi, J., Pittard, A. J. & Dowhan, W. (2003). Reversible topological organization within a polytopic membrane protein is governed by a change in membrane phospholipid composition. J Biol Chem 278, 50128–50135. [DOI] [PubMed] [Google Scholar]
- Zhang, W., Campbell, H. A., King, S. C. & Dowhan, W. (2005). Phosphatidylethanolamine is required for the proper topological organization of the gamma-aminobutyric acid permease (GabP) of Escherichia coli. J Biol Chem 280, 26032–26038. [DOI] [PubMed] [Google Scholar]