

Abstract
Poly(N-isopropylacrylamide)(pNIPAm) microgels were synthesized by precipitation polymerization at temperatures ranging from 37 °C to 45 °C using the redox initiator system ammonium persulfate (APS)/N,N,N’,N’-tetramethylethylenediamine (TEMED), or the photoinitiator 2,2′-azobis(amidinopropane) dihydrochloride (V50). Photon correlation spectroscopy (PCS) and atomic force microscopy (AFM) studies revealed that spherical microgels with narrow size dispersities can be obtained with these methods, and that the resultant microgels have volume phase transition behaviors expected from their compositions. Additionally, the low temperature, redox initiator strategy produces microgels devoid of self-cross-linking, thereby permitting the synthesis of completely degradable microgels when using N,N’-(1,2-dihydroxyethylene)bisacrylamide (DHEA) as a cleavable cross-linker. We also demonstrate the potential utility of the approach in bioconjugate syntheses; in this case avidin immobilization is demonstrated by one-pot copolymerization at low temperature.
Keywords: degradable microgels, N-isopropylacrylamide, protein immobilization, self cross-linking
Introduction
As the field of responsive hydrogel particles (microgels) has grown, the pursuit of potential applications for these fascinating materials continues to gain momentum.1-6 Microgels share a common thread with most areas of materials chemistry, wherein the development of new synthetic approaches has often enabled new domains of application.7-10 In this paper, we demonstrate a very simple route to the synthesis of thermoresponsive poly(N-isopropylacrylamide) (pNIPAm) microgels at temperatures lower than those typically employed. Whereas the synthetic modifications that enable this approach are individually very minor, the changes in the material properties and potential for broader applicability of the resultant microgels are significant.
PNIPAm microgels are typically prepared by classical precipitation polymerization or emulsion polymerization. In a standard approach, the polymerization of NIPAm monomer and N,N’-methylenebis(acrylamide) (BIS) cross-linker is initiated by a water soluble, free-radical initiator such as ammonium persulfate (APS) or 2,2′-azobis(amidinopropane)dihydrochloride (V50) at 60 to 70 °C.11 This method offers numerous advantages, not the least of which are the production of dispersions with very low polydispersity and the ability to control parameters such as particle size, charge, and cross-link density.12-18 However, others and we have found that using these high temperatures can result in unpredictable or poorly controlled microgel network structure. The most common manifestation of this is the presence of self-cross-linking,19,20 which leads to a poorly predictable network structure and inability to synthesize ‘degradable’ microgels that are truly degradable. The fact that non-degradable microgels are produced even when cleavable cross-linkers are used in their preparation will largely limit their potential applications in drug delivery and tissue engineering. Additionally, the elevated temperatures used in standard syntheses are not appropriate for the direct encapsulation of delicate cargo such as enzymes, antibodies, or any other thermally unstable moieties. To address this issue, efforts have been undertaken to lower the synthesis temperature by using a different microgel formation mechanism. Kuckling et al. have successfully prepared nanogels by UV irradiation of NIPAm and dimethylmaleinimido acrylamide copolymer solution at 45 °C,21,22 and Kazakov et al. synthesized nanogels by UV-induced polymerization of monomers within the liposomes at room temperature.23,24 Here, we consider methods by which the synthesis temperature can be lowered while maintaining the very functional synthetic framework of precipitation polymerization, as an initial step in our larger efforts to expand the application sphere of poly(alkylacrylamide) microgels.
To tackle this issue, an understanding of microgel formation is needed. In precipitation polymerization, pNIPAm microgels are formed by homogeneous nucleation,5 with the polymerization being carried out at 60 to 70 °C to both decompose the initiator and to induce collapse of the propagating oligoradicals to give precursor particles.25 However, given that the lower critical solution temperature (LCST) of pNIPAm (~31 °C) is much lower than the synthesis temperature,22,26-28 particle nucleation should be possible at temperatures lower than 60 °C, as long as that temperature is above the LCST.
We have previously shown that for APS-based syntheses, the initiator concentration, and hence microgel formation, can be controlled using temperature,13 as the decomposition rate and hence the radical yield of APS decreases with decreasing temperature. Considering this, our approach was relatively straightforward, as we borrowed from methods used in bulk hydrogel preparation, wherein the redox initiator system of APS and N,N,N’,N’-tetramethylethylenediamine (TEMED) is used to initiate the polymerization below the decomposition temperature of APS.29 Here, we present data wherein a redox initiator system is employed instead of thermal initiation to synthesize pNIPAm microgels at either 37 °C or 45 °C. In an alternate approach, we have induced the decomposition of V50, a cationic initiator with a similar structure to that of 2,2′-azobis(2-methylpropionitrile) (AIBN), using ultraviolet (UV) radiation instead of temperature.30 Using these straightforward approaches, we demonstrate the ability to synthesize totally degradable microgels via cross-link scission, and the immobilization of the protein avidin in microgels by one-pot copolymerization.
Experimental Section
Materials
All materials were purchased from Sigma-Aldrich unless otherwise noted. The monomer NIPAm (TCI) was purified by recrystallization from n-hexane (J. T. Baker). Reagents N,N’-(1,2-dihydroxyethylene)bisacrylamide (DHEA), BIS, ammonium persulfate (APS), TEMED, V50, N-acryloxysuccinimide (NAS), and avidin (Invitrogen) were used as received. Water was produced by distillation followed by deionization to a resistance of 18 MΩcm (Barnstead E-Pure system), followed by filtration through a 0.2 μm filter to remove particulate matter.
Microgel Synthesis
Precipitation polymerizations were conducted in a 100 mL three-necked round bottom flask fitted with a reflux condenser, a thermometer and a N2 inlet/outlet under continuous magnetic stirring. For all microgel syntheses, the total monomer concentration was 140 mM, with a molar composition of 98% NIPAm and 2% BIS (Table 1). In a typical synthesis, 50 mL of a filtered, aqueous solution of NIPAm and BIS was added to the flask and heated to the desired temperature. The solution was purged with N2 gas and stirred vigorously until the temperature stabilized. For anionic microgel synthesis, the reaction was initiated by adding TEMED immediately followed by the addition of APS. For cationic microgel synthesis, the reaction was initiated by V50 under UV irradiation (Blak-Ray@B-100A, UVP). All reactions were allowed to proceed for 6 h under a N2 blanket with constant stirring. After synthesis, the solution was filtered through glass wool to remove any coagulum. Microgels were purified by centrifugation, decantation and redispersion in deionized water to remove remnant monomer, oligomer and initiator. The microgel dispersion was then lyophilized and stored for future use. Degradable microgels were synthesized by replacing the BIS cross-linker with 2 mol% DHEA. APS and TEMED were used for initiation, with the remaining synthetic conditions remaining as described above. DHEA-based microgel degradation was performed by soaking substrate-supported microgel monolayers (vide infra) in a 1 mM aqueous solution of sodium periodate (NaIO4) overnight. Addition of NaIO4 to the films leads to the cleavage of the 1,2-glycol bond in DHEA.31-33
Table 1.
Synthesis conditions used and the resultant microgel properties
| Code | Initiator Concentration (mM) |
Initiator | Synthesis Temperature (°C) |
Rh at 22 °C (nm) |
Microgel Yield |
ζ-potential (mV) |
|
|---|---|---|---|---|---|---|---|
| 22 °C | 40 °C |
||||||
| 45A1 | 1 | APS | 45 | - | 13% | - | - |
| 70A1 | 1 | APS | 70 | 336 | 79% | −15.4 | −33.0 |
| 37AT1 | 1 | APS+TEMED | 37 | 771 | 35% | −2.36 | −29.7 |
| 45AT1 | 1 | APS+TEMED | 45 | 358 | 72% | −9.16 | −28.2 |
| 45AT0.5 | 0.5 | APS+TEMED | 45 | 366 | 51% | −2.46 | −23.9 |
| 45AT2 | 2 | APS+TEMED | 45 | 358 | 60% | −7.76 | −32.8 |
| 55AT1 | 1 | APS+TEMED | 55 | 317 | 70% | −7.18 | −32.2 |
| 70AT1 | 1 | APS+TEMED | 70 | 278 | 60% | −6.29 | −36.1 |
| 45V | 1 | V50 | 45 | - | 18% | - | - |
| 37VL | 1 | V50+UV | 37 | 597 | 63% | 8.65 | 23.5 |
| 45VL | 1 | V50+UV | 45 | 436 | 78% | 11.2 | 32.2 |
To prepare avidin-laden microgels, avidin was modified with acrylate groups by standard protocols via coupling with NAS in a phosphate buffer solution (0.02 M, pH 7.4). A 10 μL aliquot of a 50 mM NAS solution in DMF was added to a phosphate buffer solution containing 5 mg avidin (NAS/avidin molar ratio of 6:1), and the reaction was left at room temperature overnight. The dispersion was purified by dialysis against deionized water. Avidin-containing microgels were synthesized by adding the above acrylate-avidin solution (2 mL) to 25 mL of monomer solution (70 mM, 98% NIPAm and 2% BIS). For this synthesis, V50 (1 mM) was used as an initiator at 45 °C. The remaining synthesis conditions were carried out in the same fashion as described above.
Characterization
Particle Sizing
Microgel sizes in deionized water were measured by photon correlation spectroscopy (PCS, Protein Solutions, Inc.) using an instrument equipped with an integrated Peltier temperature control device, which gives temperature accuracy within ±0.1 °C. Prior to taking measurements, the microgel dispersions were allowed to thermally equilibrate at each temperature for 30 min. All correlogram analyses were performed with software supplied by the manufacturer (Dynamics v.5.25.44, Protein Solutions, Inc.). The data presented below are the averaged values of 25 measurements, with a 10 s integration time for each measurement. The swelling ratio (Rs/Rd)3 of the microgels is the cube of the ratio of PCS-measured particle radius at a particular temperature (Rs) to the radius measured at 40 °C (Rd). The swelling volume ratio is used to define the swollen state of a particle in comparison to its collapsed state (40 °C), although the particles will not be completely collapsed at 40 °C as thermally “deswollen” microgels typically contain ~20% water by volume.
ζ-potential determination
The ζ-potential was measured with a Zetasizer Nano-ZS (Malvern). Before the measurements, the 1 mL microgel dispersion in pure water was thermally equilibrated between parallel electrodes in the cuvette for 10 min. The reported ζ-potentials are the average of at least three successive measurements.
Atomic Force Microscopy (AFM)
Microgels immobilized on glass coverslips were imaged using an Asylum Research MFP-3D Instrument (Santa Barbara, CA). Imaging was performed and processed using the MFP-3D software under the IgorPro (WaveMetrics Inc., Lake Oswego, OR) environment. Non-contact mode aluminum-coated silicon nitride cantilevers were purchased from NanoWorld (force constant = 42 N/m, resonance frequency = 320 kHz). All images were taken in air under ambient conditions. Glass coverslips (22 mm × 22 mm) were placed in a ceramic holder and cleaned using a sequential solvent sonication method in a Bransonic 2510 Ultrasonicator: 30 min in dilute soapy (Alconox) water, 15 min in deionized water, 15 min in acetone, 15 min in isopropyl alcohol, and 15 min in absolute ethanol. Afterward, each cleaned slide was functionalized by exposure to a 1 v/v% solution of 3-aminopropyltrimethoxysilane (APTMS) in absolute ethanol (200 proof) for ~3 h.34 The glass was then rinsed with ethanol and dried under a gentle stream of N2. Cleaned and dried glass coverslips were individually placed at the bottom of 6-well plates, which were then filled with 0.05 mg/mL microgel aqueous solution. The well plates were placed immediately across from a counter-weighted well plate in an Eppendorf 5804R centrifuge equipped with a plate-holding rotor.35 Microgels were centrifugally deposited at a relative centrifugal force (rcf) of 2250 × g for 5 min. After deposition, the films were gently rinsed with deionized water and dried under a gentle stream of N2. For the cationic microgels, the amine-functionalized coverslips were further modified by immersion for 20 min in a 0.1 mg/mL poly(sodium styrenesulfonate) solution before centrifugal deposition.
Steady-state fluorescence
Fluorescence spectra were recorded with a steady-state fluorescence spectrophotometer (Photon Technology International), equipped with a Model 814 PMT photon-counting detector. The slits were set to achieve a spectral bandwidth of 2 nm. Fluorescein fluorescence was measured at ambient temperature, using an excitation wavelength of 493 nm.
Results and Discussion
The synthetic conditions explored and some properties of the thus produced microgels are listed in Table 1. When APS is used alone as thermal initiator at 45 °C (45A1), an excessive amount of coagulum was generated resulting in a very low microgel yield. PCS measurements reveal that the 45A1 microgels have a very broad size distribution, which is also evident in the AFM height image (Figure 1a). This result stands in contrast with microgels prepared via traditional, APS-initiated precipitation polymerization at 70 °C (70A1), which have a narrow dispersity and are formed in much higher yield (Figure 1d and Table 1). Presumably the poor results at the lower temperature arise from slower decomposition of APS, a correspondingly lower radical yield, and a lower sulfate-associated charge density on the growing particles. The lower charge density results in poorer colloidal stability and hence the formation of more coagulum during synthesis. To increase the radical yield at low temperature, TEMED was added, thereby forming a redox initiator system.36 This strategy was used at 45 °C (45AT1) producing microgels with a hydrodynamic radius of 360 nm at 72% yield with significantly improved monodispersity and uniformity (Figure 1b). Lowering the temperature to 37 °C (37AT1), just above the pNIPAm lower critical solution temperature (LCST), still produced uniform microgels (Figure 1c), albeit with a larger hydrodynamic radius and lower yield (770 nm, 35% yield). Variation of the initiator concentration between 0.5 mM and 2 mM produced essentially identical microgel radii with small variations in yield (Table 1), suggesting that the redox initiation approach is well-behaved and is not overly sensitive to initiator concentration.
Figure 1.
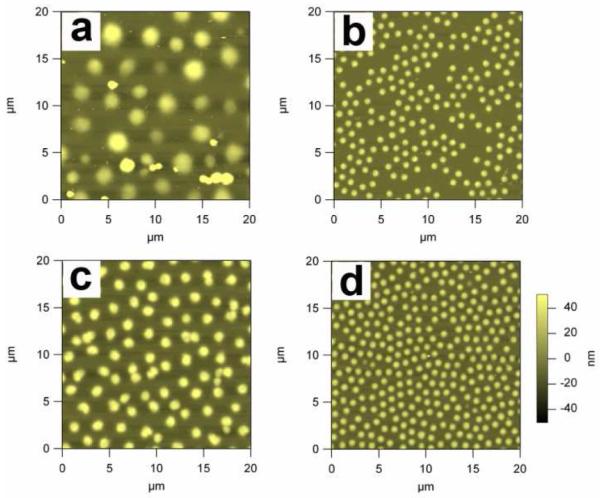
AFM height images of (a) 45A1, (b) 45AT1, (c) 37AT1 and (d) 70A1 microgels on glass substrates. The z-scale is the same for all images and is shown next to panel (d).
This approach is valid for other initiator systems such as V50, which can also be decomposed thermally, but does not produce high quality particles when used alone at 45 °C (Table 1 and Figure 2a). In this case, the decomposition rate of V50 can be increased by ultraviolet irradiation of the strong absorption of V50 at ~365 nm (Figure S1, Supporting Information). Using this approach, microgels with hydrodynamic radii of 600 nm and 440 nm were obtained in good yield at 37 °C and 45 °C, respectively (Table 1 and Figure 2b,c).
Figure 2.

AFM height images of (a) 45V, (b) 37VL and (c) 45VL microgels on glass substrates. The z-scale is the same for all images and is shown next to panel (c).
For all microgels the obtained ζ-potentials at 22 °C and 40 °C are presented in Table 1 to illustrate the presence of charged initiator fragments. As expected, APS- and V50-based syntheses produce anionic and cationic microgels, respectively, and all microgels display a higher ζ-potential value at 40 °C than that at 22 °C due to an increase in charge/surface area during microgel shrinkage. It is tempting to correlate the absolute value of the ζ-potential with the number of initiator fragments per particle, but we caution against such correlations. The measured ζ-potential is a convolution of actual microgel charge density, particle topology (e.g. ‘hairiness’), and the degree to which the charges are surface localized. Without independent determination of the exact charge distribution within the particles, attempts to quantitatively correlate ζ-potential and microgel composition are unwise.
The thermally-induced volume phase transition is a characteristic property of microgels based on pNIPAm. At temperatures below the LCST of pNIPAm, which is ~32 °C, the microgel is swollen due to strong polymer-solvent interactions. At temperatures above the LCST of pNIPAm there is an entropically-favored release of water from the interior of the microgel and the polymer-polymer interactions become stronger, thereby deswelling the microgel.5 A wide range of studies have been devoted to this process and associated phenomena.37 In one particularly illuminating study, Shibayama et al. used small-angle neutron scattering to study the volume phase transition of pNIPAm gels and found that all the critical exponents supported a classification of the gel transition in the three dimensional Ising model.38 Here, we have determined the phase transition behavior of the particles prepared by these methods to investigate whether the different synthetic approaches produce microgels with similar thermodynamic properties. Importantly, we find that the measured microgel volume phase transition temperatures (VPTT) determined by PCS for all microgels are nearly identical (Figure 3a), illustrating that the different synthetic conditions do not produce particles with dramatically different phase transition properties. The differences in swelling ratio (Figure 3b) likely reflect differences in network structure, density, and/or distribution of charged groups between the different particles. The exact origins of these differences are currently under investigation.
Figure 3.

Variation in radius (a) and swelling ratio (b) as a function of temperature for the pNIPAm microgels described in the text.
As mentioned above, typical precipitation polymerization of NIPAm at 70 °C results in stable microgels even in the absence of cross-linker due to the presence of self-cross-linking reactions.19,20 In other words, the presence of cross-linking monomers is not required to produce cross-linked network polymers; chain transfer reactions have been proposed as the likely source of chain branching and cross-linking. However, it is well known that completely degradable polyacrylamide bulk hydrogels can be prepared using a redox initiator method at 25 °C.39 To investigate if self-cross-linking could be suppressed by judicious choice of initiator system, we carried out three reactions similar to the 70A1, 70AT1 and 45AT1 microgel syntheses but without the addition of cross-linker. The cross-linker-free syntheses are denoted as 70A1-B, 70AT1-B and 45AT1-B.
The differences between these three syntheses are evident upon visual inspection (Figure S2, Supporting Information). At room temperature immediately following synthesis, the 70A1-B solution appears quite turbid, as is typical for standard microgel preparations. However, the 45AT1-B and 70AT1-B solutions are transparent and colorless. Note, however, that vials (b) and (c) become turbid above the pNIPAm LCST, indicating the presence of polymer chains in solution (data not shown). PCS measurements reveal that particles with a radius of ~450 nm are present in the 70A1-B solution whereas no particles are detectable in the other two solutions. These results suggest that self-cross-linking occurs under standard precipitation polymerization conditions (70A1-B), resulting in intact microgel particles even in the absence of traditional cross-linking monomers. However, this self-cross-linking can be prevented under both low and high temperature conditions by using a redox initiator system; pNIPAm synthesis under these conditions results in linear polymer without any clear evidence of intact, cross-linked microgels. This illustrates that the two different initiator systems can result in pNIPAm microgels with different network structures, even when the same amount of BIS is used in each synthesis. The initiation by APS alone results in pNIPAm microgels cross-linked by both BIS and self-cross-linking reactions, while the redox initiator results in pNIPAm microgels cross-linked only by BIS.
The origin of self-cross-linking has been attributed to chain transfer to polymer chains during polymerization.19 Transfer to the polymer results in the formation of a radical site on a polymer chain, and continuous polymerization of remaining monomer at this site leads to the production of a branched polymer by disproportionation termination, or leads to cross-linking by coupling termination with another macromolecular free radical.36 Thus, these chain transfer reactions can result in the formation of a self-cross-linked network structure (Figure 4a). However, these pathways appear to be less favored under redox initiation conditions. It is well known that electron-rich chain transfer agents such as triethylamine have enhanced reactivity with electron-poor (electron-acceptor) monomers, such as acrylonitrile and methyl acrylate.36 In our reaction system, TEMED (similar to triethylamine) should also act as a transfer reagent40 with NIPAm (electron-poor monomer). Thus, the propagating pNIPAm chain will likely prefer to undergo transfer with residual TEMED as opposed to undergoing a chain transfer reaction with other propagating chains (Figure 4b).
Figure 4.

Schematic depiction of the formation process of self-cross-linked network (a), step 1: chain transfer to polymers; step 2: monomer addition, coupling termination and network formation. (b) Chain transfer to TEMED, which will limit chain branching.
These results suggest that completely degradable microgels can be successfully prepared if a degradable cross-linker is used, as opposed to previous reports from our group wherein self-cross-linking resulted in non-degradable microgels.41 Here, we use the cross-linker DHEA instead of BIS to carry out the synthesis. The cross-linker DHEA contains a 1,2-glycol bond that can be cleaved by NaIO4, which should result in network destruction and particle dissolution.31,33 To test this hypothesis, pNIPAm microgels cross-linked with DHEA were synthesized using APS alone at 70 °C and APS together with TEMED at 45 °C; these reactions are denoted 70A1-C and 45AT1-C, respectively.
After depositing 70A1-C or 45AT1-C microgels on glass slides (see Experimental Section), the supported microgel monolayer was soaked in a 1 mM aqueous solution of NaIO4 overnight and then dried under a gentle N2 stream before AFM measurements. As expected, 70A1-C microgels do not undergo complete dissolution upon exposure to periodate (Figure 5b), an observation we have made previously.41 Since the internal microgel connectivity is in part due to the DHEA cross-linker, the periodate-treated microgels are flatter, presumably due to a decrease in particle rigidity. In contrast, no microgels can be found on the glass slide after the 45AT1-C microgels were exposed to periodate, suggesting that the 45AT1-C microgels can be totally degraded (Figure 5d). This result was confirmed by PCS measurements. After treating a dispersion of 70A1-C microgels with periodate, the radius of those microgels was determined to be 563 nm, in contrast to a radius of 484 nm before exposure, suggesting a decrease in network connectivity and a concomitant swelling. However, no detectable microgels were found in solution for the 45AT1-C microgels after exposure to periodate. This observation was also made previously by our group in a degradation study of DHEA cross-linked poly(N-isopropylmethacrylamide) microgels, where linear chains with molecular weights 3-4 orders of magnitude smaller than that of the original microgels were formed after the degradation.41
Figure 5.

AFM height images of substrate-supported 70A1-C (a, b) and 45AT1-C (c, d) microgels before (a, c) and after (b, d) periodate-induced degradation. The z-scale is the same for all images and is shown next to panel (d).
In addition to degradable particles, bioconjugates between synthetic colloids and biomacromolecules are an important construct for current biotechnology applications.42-44 However, direct immobilization of proteins in microgels during precipitation polymerization can be difficult due to the thermal instability of proteins. Such syntheses are potentially enabled via a lower temperature microgel synthesis strategy. As a proof-of-concept, one-pot immobilization of the protein avidin is demonstrated. Here, due to the fact that avidin is positively charged at pH 7 in water (pI~10), V50 was selected as the initiator since an anionic initiator (APS) would likely induce colloidal instability in the reaction. Avidin-containing microgels were synthesized by adding a solution of acrylate-modified-avidin solution (2 mL, 2.5 mg/mL avidin) to 25 mL of monomer solution (70 mM, 98% NIPAm and 2% BIS) using V50 (1 mM) as an initiator at 45 °C. The remaining synthesis conditions were carried out in the same fashion as described above. To confirm the presence of immobilized avidin, and to confirm that the immobilized avidin retained its ability to bind ligand, 10 μL of fluorescein-labeled biotin solution (2 mM in pH 7.4 PBS buffer) was incubated with 5 mL of purified avidin-microgel solution at room temperature overnight. Following removal of unbound biotin by repeated centrifugation, analysis of the dispersion by steady-state fluorescence revealed the presence of bound fluorescein-biotin (Figure 6), which suggests retention of avidin bioactivity. In contrast, microgels synthesized in the presence of native avidin (not acrylate-modified) did not reveal any significant degree of biotin association, suggesting that the presence of protein-localized polymerizable groups is required for efficient incorporation of protein into the microgels. No measureable binding of fluorescein-biotin was observed for microgels synthesized in the absence of avidin (Figure S3, Supporting Information). If we assume that the fluorescence intensity observed from the avidin microgels is not modulated by self-quenching of fluorescein, we can estimate the biotin concentration as ~420 nmol/g of dry microgel. Assuming a 4:1 biotin:avidin stoichiometry, and a particle molecular weight of ~1×109 g/mol,45 we estimate that each particle contains a few hundred avidin molecules. We are currently performing more detailed characterization of these architectures in order to more precisely determine protein loading and binding stoichiometry.
Figure 6.
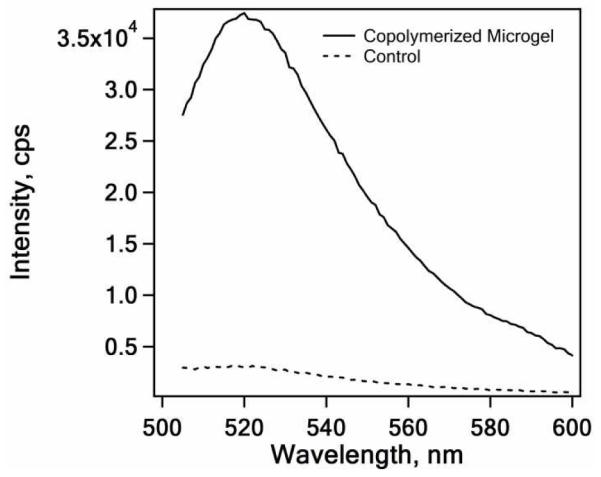
Fluorescence spectra of fluorescein-biotin treated microgels synthesized by using NAS-modified (solid line) or native (dotted line) avidin.
Conclusions
Microgels composed of pNIPAm can be synthesized near room temperature by accelerating the decomposition rate of APS through the addition of TEMED or that of V50 by ultraviolet irradiation. Importantly, we find that the redox initiator strategy results in microgels with a different network structure from that of microgels prepared by classic precipitation polymerization, as the degree of chain-transfer associated self-cross-linking is significantly reduced in the redox approach. This, in turn, permits the synthesis of completely degradable microgels when a cleavable cross-linker (DHEA) is used in microgel synthesis. Finally, a one-pot copolymerization approach to protein bioconjugate synthesis was illustrated, wherein direct copolymerization of avidin is enabled by the low temperature synthesis approach. Admittedly, avidin is an extremely robust protein that represents a relatively straightforward demonstration of direct protein incorporation. We are currently exploring more challenging bioconjugate targets for specific applications in drug delivery and bioanalysis.
Supplementary Material
Acknowledgments
This work was partially supported by the National Institutes of Health (1 R01 GM088291-01). X. Hu thanks China Scholarship Council (CSC) for fellowship support. We thank the Kröger group at GT for the use of their ζ-potential equipment.
Footnotes
Supporting Information Available: Absorption spectrum of an aqueous V50 solution, photograph of microgel dispersions, and fluorescence data for control experiments. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1).Fernandez-Barbero A, Suarez IJ, Sierra-Martin B, Fernandez-Nieves A, de las Nieves FJ, Marquez M, Rubio-Retama J, Lopez-Cabarcos E. Adv. Colloid Interface Sci. 2009;147-48:88. doi: 10.1016/j.cis.2008.12.004. [DOI] [PubMed] [Google Scholar]
- (2).Motornov M, Roiter Y, Tokarev I, Minko S. Prog. Polym. Sci. 2010;35:174. [Google Scholar]
- (3).Li YY, Dong HQ, Wang K, Shi DL, Zhang XZ, Zhuo RX. Sci. China Chem. 2010;53:447. [Google Scholar]
- (4).Hendrickson GR, Smith MH, South AB, Lyon LA. Adv. Funct. Mater. 2010;20:1697. [Google Scholar]
- (5).Nayak S, Lyon LA. Angew. Chem. Int. Ed. 2005;44:7686. doi: 10.1002/anie.200501321. [DOI] [PubMed] [Google Scholar]
- (6).Lyon LA, Meng ZY, Singh N, Sorrell CD, John AS. Chem. Soc. Rev. 2009;38:865. doi: 10.1039/b715522k. [DOI] [PubMed] [Google Scholar]
- (7).Oh JK, Bencherif SA, Matyjaszewski K. Polymer. 2009;50:4407. [Google Scholar]
- (8).Semsarilar M, Perrier S. Nat. Chem. 2010;2:811. doi: 10.1038/nchem.853. [DOI] [PubMed] [Google Scholar]
- (9).Franc G, Kakkar AK. Chem. Soc. Rev. 2010;39:1536. doi: 10.1039/b913281n. [DOI] [PubMed] [Google Scholar]
- (10).Tanaka Y, Gong JP, Osada Y. Prog. Polym. Sci. 2005;30:1. [Google Scholar]
- (11).Pelton RH, Chibante P. Colloids Surf. 1986;20:247. [Google Scholar]
- (12).Blackburn WH, Lyon LA. Colloid. Polym. Sci. 2008;286:563. doi: 10.1007/s00396-007-1805-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Meng ZY, Smith MH, Lyon LA. Colloid. Polym. Sci. 2009;287:277. [Google Scholar]
- (14).Ito S, Ogawa K, Suzuki H, Wang BL, Yoshida R, Kokufuta E. Langmuir. 1999;15:4289. [Google Scholar]
- (15).Lopez-Leon T, Ortega-Vinuesa JL, Bastos-Gonzalez D, Elaissari A. J. Phys. Chem. B. 2006;110:4629. doi: 10.1021/jp0540508. [DOI] [PubMed] [Google Scholar]
- (16).Varga I, Gilanyi T, Meszaros R, Filipcsei G, Zrinyi M. J. Phys. Chem. B. 2001;105:9071. [Google Scholar]
- (17).Nur H, Pinkrah VT, Mitchell JC, Benee LS, Snowden MJ. Adv. Colloid Interface Sci. 2010;158:15. doi: 10.1016/j.cis.2009.07.008. [DOI] [PubMed] [Google Scholar]
- (18).Senff H, Richtering W. Colloid. Polym. Sci. 2000;278:830. [Google Scholar]
- (19).Gao J, Frisken BJ. Langmuir. 2003;19:5212. [Google Scholar]
- (20).Gao J, Frisken BJ. Langmuir. 2003;19:5217. [Google Scholar]
- (21).Vo CD, Kuckling D, Adler HJP, Schohoff M. Colloid. Polym. Sci. 2002;280:400. [Google Scholar]
- (22).Kuckling D, Vo CD, Adler HJP, Volkel A, Colfen H. Macromolecules. 2006;39:1585. [Google Scholar]
- (23).Kazakov S, Kaholek M, Teraoka I, Levon K. Macromolecules. 2002;35:1911. [Google Scholar]
- (24).Kazakov S, Kaholek M, Kudasheva D, Teraoka I, Cowman MK, Levon K. Langmuir. 2003;19:8086. [Google Scholar]
- (25).Pelton R. Macromol. Symp. 2004;207:57. [Google Scholar]
- (26).Deng YL, Pelton R. Macromolecules. 1995;28:4617. [Google Scholar]
- (27).Qiu XP, Kwan CMS, Wu C. Macromolecules. 1997;30:6090. [Google Scholar]
- (28).Chan K, Pelton R, Zhang J. Langmuir. 1999;15:4018. [Google Scholar]
- (29).Hu XB, Xiong LJ, Wang T, Lin ZM, Liu XX, Tong Z. Polymer. 2009;50:1933. [Google Scholar]
- (30).Lime F, Irgum K. Macromolecules. 2007;40:1962. [Google Scholar]
- (31).Hu XB, Tong Z, Lyon LA. J. Am. Chem. Soc. 2010;132:11470. doi: 10.1021/ja105616v. [DOI] [PubMed] [Google Scholar]
- (32).Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego: 1995. [Google Scholar]
- (33).Nayak S, Gan DJ, Serpe MJ, Lyon LA. Small. 2005;1:416. doi: 10.1002/smll.200400089. [DOI] [PubMed] [Google Scholar]
- (34).Serpe MJ, Lyon LA. Chem. Mater. 2004;16:4373. [Google Scholar]
- (35).South AB, Whitmire RE, Garcia AJ, Lyon LA. ACS Appl. Mater. Interfaces. 2009;1:2747. doi: 10.1021/am9005435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Odian G. Principles of Polymerization. Fourth Edition ed. John Wiley & Sons, Inc; 2004. [Google Scholar]
- (37).Shibayama M, Tanaka T. Adv. Polym. Sci. 1993;109 [Google Scholar]
- (38).Shibayama M, Tanaka T, Han CC. J. Chem. Phys. 1992;97 [Google Scholar]
- (39).Aliyar HA, Hamilton PD, Ravi N. Biomacromolecules. 2005;6:204. doi: 10.1021/bm049574c. [DOI] [PubMed] [Google Scholar]
- (40).Cleland RE, Grace SC. Febs Letters. 1999;457:348. doi: 10.1016/s0014-5793(99)01067-4. [DOI] [PubMed] [Google Scholar]
- (41).Michael HS, Herman ES, Lyon LA. Submitted. [Google Scholar]
- (42).Pollak A, Blumenfeld H, Wax M, Baughn RL, Whitesides GM. J. Am. Chem. Soc. 1980;102:6324. [Google Scholar]
- (43).Welsch N, Wittemann A, Ballauff M. J. Phys. Chem. B. 2009;113:16039. doi: 10.1021/jp907508w. [DOI] [PubMed] [Google Scholar]
- (44).Ogawa K, Nakajima-Kambe T, Nakahara T, Kokufuta E. Biomacromolecules. 2002;3:625. doi: 10.1021/bm025512f. [DOI] [PubMed] [Google Scholar]
- (45).Sorrell CD, Lyon LA. Langmuir. 2008;24:7216. doi: 10.1021/la800092q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.