

Summary
To study the regulation of cardiac muscle contraction by the myosin essential light chain (ELC) and the physiological significance of its N-terminal extension, we generated transgenic (Tg) mice partially replacing the endogenous mouse ventricular ELC with either the human ventricular ELC wild type (Tg-WT) or its 43 amino acid N-terminal truncation mutant (Tg-Δ43) in the murine hearts. The mutant protein is similar in sequence to the short ELC variant present in skeletal muscle and the ELC protein distribution in Tg-Δ43 ventricles resembles that of fast skeletal muscle. Cardiac muscle preparations from Tg-Δ43 mice demonstrate reduced force per cross-sectional area of muscle, which is likely caused by a reduced number of force generating myosin cross-bridges and/or by decreased force per cross-bridge. As the mice grow older, the contractile force per cross-sectional area further decreases in Tg-Δ43 mice and the mutant hearts develop a phenotype of non-pathologic hypertrophy while still maintaining normal cardiac performance. The myocardium of older Tg-Δ43 mice also exhibits reduced myosin content. Our results suggest that the role of the N-terminal ELC extension is to maintain the integrity of myosin and to modulate force generation by decreasing myosin neck region compliance and promoting strong cross-bridge formation and/or by enhancing myosin attachment to actin.
Keywords: Cardiac myosin ELC, Transgenic mice, ATPase, Force in skinned papillary muscles, MRI
Introduction
Muscle myosin is a hexamer composed of two heavy chains (MHC) and two pairs of myosin light chains (MLC) 1; 2; 3; 4. The C-terminal coiled-coil MHC constitute the myosin thick filaments while the N-termini fold into two globular heads (S1) containing a nucleotide binding site, an actin binding domain and an α-helical lever arm, where one essential (ELC) and one regulatory (RLC) light chains are attached 5. The major actin binding site, as depicted by crystallography data, is in the 50 kDa domain of the myosin head 6; 7. However, extensive biochemical, electron microscopy and NMR studies have demonstrated an additional binding site for actin, located at the N-terminus of the myosin ELC 8; 9; 10; 11; 12; 13; 14; 15; 16; 17. These studies however have not yet been confirmed structurally, likely owing to the highly disordered structure of the N-terminus of the ELC 5; 18. The recent modeling study by Morano's group illustrates the N-terminal domain of ELC as a rod-like 91 Å-long extension that could function as a bridge between the ELC and the actin filament 19. Using electron cryomicroscopy in conjunction with light-scattering and fluorescence analyses, Lowey et al. demonstrated the binding of the N-terminal extension of the ELC to the SH3 domain of MHC and subsequently to actin 20.
Across the muscle types, the ELC can exist as a long variant (cardiac muscle), short (smooth muscle) or appear as a combination of both forms (skeletal muscle) 12. Murine fast skeletal muscle for instance contains a long MLC1 and a 38 amino acids shorter MLC3 (Fig. 1A). On the other hand, cardiac muscle contains only the long ELC isoform, both in the atria (ELCa) and ventricles (ELCv) (Fig. 1B) 4; 21; 22. The functional importance of the ELCv (long isoform) is highlighted by recent identification of several missense mutations in the human ventricular ELC (huELCv), shown to cause Familial Hypertrophic Cardiomyopathy (FHC) (reviewed in 23 and 4). FHC is an autosomal dominant disease characterized by left ventricular and septal hypertrophy, myofibrillar disarray and sudden cardiac death. The mechanisms by which these mutations in huELCv lead to FHC are not known.
Figure 1.

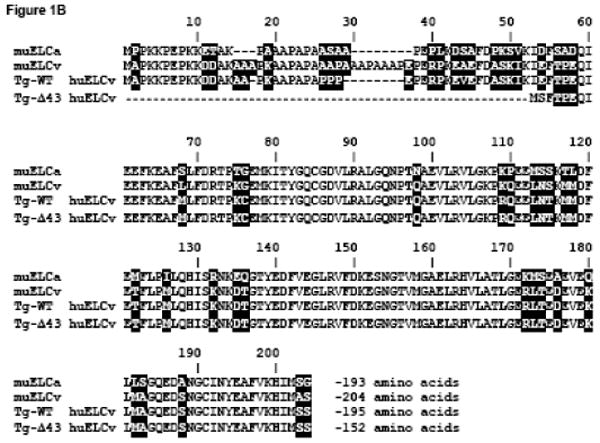
Sequence comparison of muscle myosin ELC isoforms. (A) Sequence overlap of the murine (mu) fast skeletal ELC isoforms, mu skMLC1 (NCBI # NP_067260) and mu skMLC3 (NCBI # NP_001106858). The amino acid sequence of mu skMLC3 is 38 amino acid residues shorter than that of mu skMLC1. (B) Sequence comparison of cardiac ELC isoforms: murine atrial ELC (muELCa, NCBI # NP_034988), murine ventricular ELC (muELCv, NCBI # P09542), human (hu) ventricular ELC (huELCv, NCBI # P08590) expressed in mice in this study (Tg-WT) and its 43 amino acid N-terminal truncation mutant, Tg-Δ43.
Despite extensive effort by many, the physiological significance of the ELC and its N-terminal extension remains obscure. Various mechanisms have been put forward in attempts to explain the function of the N-terminus of ELC in skeletal and cardiac muscle contraction. The general hypothesis derived from numerous studies comprehensively summarized in the recent reviews by Timson 22 and Hernandez et al. 4 suggests that the N-terminal ELC extension regulates myosin motor function by changing binding of the myosin head to actin and thereby influencing the cross-bridge cycling kinetics. Presumably this fine tuning of myosin cross-bridge kinetics by the N-terminal ELC extension occurs in an isoform and/or tissue specific manner. To elucidate the molecular mechanism underlying the ELC-specific contractile physiology and the importance of the N-terminus of the ventricular ELC for cardiac muscle function, we generated transgenic (Tg) mice expressing the human ventricular huELCv wild type (Tg-WT) or its 43 amino acid N-terminal truncation mutant (Tg-Δ43) in murine hearts. Our long term interests in understanding the ELC mediated biology of the heart in health and disease including the effects of FHC mutations found in huELCv on cardiac muscle function prompted transgenic expression of the human rather than mouse ELC isoform. Thus, Tg-WT mice generated simultaneously with Tg-Δ43 lines for this study can be utilized in other experiments investigating FHC ELC transgenic animal models.
The huELCv-Δ43 protein is similar in sequence to the short MLC3 present in skeletal muscle (Fig. 1), and the ELC protein distribution in Tg-Δ43 ventricles resembles that of fast skeletal muscle (Fig. 2). Our results demonstrate that a partial replacement of the endogenous mouse ventricular muELCv with transgenic truncated huELCv-Δ43 results in reduced contractile force measured in Tg-Δ43 skinned papillary muscle fibers compared to Tg-WT and non-transgenic (NTg) control cardiac muscle preparations. As the mice mature, the hearts of the Tg-Δ43 mice hypertrophy, but the ventricles do not show a pathologic phenotype. An MRI (magnetic resonance imaging) assessment also demonstrates normal cardiac function. Our results suggest that the N-terminal ELC extension regulates force generation in muscle through its direct contacts with actin and/or through influencing the conformation of the MHC and its interaction with actin, both mechanisms promoting strong cross-bridge formation. While N-terminal ELC signaling is prevented in the genetically manipulated mice, we conclude that the myocardium of Tg-Δ43 animals remodels to maintain normal cardiac performance.
Figure 2.
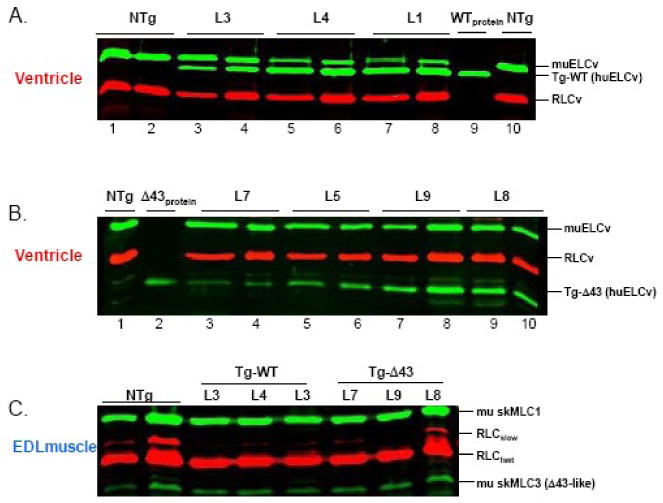
(A) Human ventricular ELC (huELCv) expression in Tg-WT mice. Lanes 1 and 2, NTg LV samples; lanes 3 and 4, expression in Tg-WT L3 determined in LV extracts (36.4±1.0, n=13) and in myofibrils (36.4±1.8, n=14); lanes 5 and 6, Tg-WT L4 - LV (36.4±1.0, n=13), MF (36.4±1.8, n=14); lanes 7 and 8, Tg-WT L1 - LV (77.5±1.5, n=8), MF (74.8±1.8, n=8); lane 9, WTprotein standard; lane 10, NTg MF. (B) Expression of human ventricular Δ43 mutant in Tg-Δ43 mouse hearts. Lane 1, NTg sample; lane 2, Δ43protein standard; lanes 3 and 4, expression in Tg-Δ43 L7 determined in LV extracts (12.2±2.3, n=10) and in MF (12.2±2.5, n=9); lanes 5 and 6, Tg-Δ43 L5 - LV (29.8±0.9, n=10), MF (37.7±0.8, n=6); lanes 7 and 8, Tg-Δ43 L9 - LV (34.6±1.9, n=14), MF (39.1±1.6, n=12); lanes 9 and 10, Tg-Δ43 L8 - LV (37.2±1.5, n=13), MF (40.1±1.4, n=8); (C) Fast skeletal MLC1 and MLC3 expression in extensor digitorum longus (EDL) muscle from Tg-Δ43 and Tg-WT mice compared to NTg mice. Abbreviations: LV, left ventricular extracts; MF, myofibrils; NTg, non-transgenic; Tg-WT, transgenic wild type; Tg-Δ43, transgenic mutant; L, mouse line; huELCv, human ventricular ELC protein = (wild-type) WTprotein; Δ43protein, mutant protein standard; muELCv, mouse ventricular ELC; RLCv mouse ventricular RLC; RLCslow, mouse slow skeletal RLC; RLCfast, mouse fast skeletal RLC; mu skMLC1, mouse skeletal long ELC; mu skMLC3, mouse skeletal short ELC (Δ43-like).
Results
Three lines of Tg-WT (L1, L3, and L4) expressing the full length huELCv (Fig. 2A) in mouse myocardium and multiple lines of Tg-Δ43 mice (L2, L4, L5, L7, L8 and L9) expressing a 43 amino acid truncation mutant (Δ43) of huELCv (Fig. 2B) were generated and subjected to functional, histological and MRI assessments. The amino acid sequences of Tg-WT and Tg-Δ43 proteins are shown in Fig. 1B along with the sequence of the endogenous mouse ventricular muELCv. The studies were performed with age and gender matched Tg-Δ43 and Tg-WT mice and non-transgenic (NTg) littermates. Female (F) and male (M) mice were used in protein expression, MRI and histopathology determinations while the majority of the functional studies on papillary muscle fibers were performed with age matched male mice. The results obtained with Tg-Δ43 lines expressing 30-40% of huELCv-Δ43 protein (L2, L4, L5, L8 and L9) were compared with Tg-WT L3 expressing ∼ 36% of huELCv (Fig. 2). Tg-WT, L1 and L4 expressing ∼78 and 71 % of huELCv, respectively were used as another set of controls.
Transgenic protein expression
Transgenic protein expression in all Tg-WT and Tg-Δ43 mouse lines was determined in left ventricular extracts and verified in myofibrils purified from transgenic hearts excluding atria. Due to the larger molecular weight of muELCv vs. huELCv (Fig. 1B), the gel mobility of these two proteins differs enabling direct estimation of transgenic WT protein replacement in mouse ventricles (Fig. 2A). Despite repeated breeding among siblings, we were not able to achieve higher than about 40% transgene expression in Tg-Δ43 mouse lines (Fig. 2B). The lowest expressing line was Tg-Δ43 L7 with 12% replacement of the endogenous muELCv with transgenic huELCv-Δ43 protein. The preparations from this line were withheld from functional experiments. Interestingly, the resulting distribution of the ELC proteins (endogenous mouse and transgenic human) in Tg-Δ43 ventricles (Fig. 2B) resembled that present in the fast extensor digitorum longus (EDL) skeletal muscle (Fig. 2C), expressing a combination of a long murine skeletal MLC1 (mu skMLC1) and a short mu skMLC3 24 (Fig. 1A). No alterations in skeletal endogenous ELC (mu skMLC1 and mu skMLC3) protein distribution due to ventricular expression of the WT and Δ43 proteins was observed in the EDL muscle of Tg-WT and Tg-Δ43 mice compared to NTg mice (Fig. 2C). The percent of mu skMLC3 (Δ43-like) content to total endogenous ELC (mu skMLC3 + mu skMLC1) in the EDL muscle was 21±2% in Tg-Δ43 mice, 17±3% in Tg-WT mice compared to 17±5% in NTg mice (Fig. 2C).
Transcript expression assessed with real time PCR
Table I presents the gene expression profile in the left ventricular tissue (LV) and in the EDL muscle from ∼ 3 month old Tg-Δ43 and Tg-WT mice compared to NTg control. The data were generated with 3-4 hearts per mouse group and the individual RNA samples were run in duplicate. The fold change (FC) expression of all tested genes in Tg-Δ43 and Tg-WT mice was compared to NTg mice (FC=1). As indicated in Table I, the pattern of gene expression was similar among the three groups of mice (p>0.05). Interestingly, Actc 1 encoding for cardiac α-actin was down-regulated in Tg-Δ43 compared to NTg mice (p=0.008); however, when compared to α-actin expression in Tg-WT (FC=1), the FC of Tg-Δ43=-1.14 and the difference was not statistically significant (p=0.072). No changes in skeletal endogenous ELC mRNA expression (Myl 1) due to ventricular expression of the WT and Δ43 proteins was observed in the EDL muscle of Tg-WT and Tg-Δ43 mice compared to NTg mice (Table I).
Table I.
Gene expression profile in the left ventricular tissue and EDL muscle from ∼3 month old Tg-Δ43 and Tg-WT compared with NTg mice.
| Gene (protein) | Tg-Δ43 | Tg-WT | NTg | ||
|---|---|---|---|---|---|
| FC | P-value | FC | P-value | FC | |
| Myl4 (ELC atrial) / LV | -2.85 | 0.228 | 3.16 | 0.394 | 1 |
| Myh6 (α-MHC) / LV | -1.83 | 0.239 | -1.22 | 0.740 | 1 |
| Myh7 (β-MHC) / LV | -2.40 | 0.239 | -2.48 | 0.094 | 1 |
| Actc1 (α-actin) / LV | -1.53 | 0.008 | -1.34 | 0.452 | 1 |
| Atp2a2 (SERCA2) / LV | -2.29 | 0.077 | -1.51 | 0.377 | 1 |
| GAPDH (GAPDH) / LV | -2.40 | 0.285 | -2.48 | 0.910 | 1 |
| Myl1 (ELC fast skeletal) / LV | 1.29 | 0.864 | 2.65 | 0.597 | 1 |
| Myl1 (ELC fast skeletal) / EDL | -1.19 | 0.485 | -1.68 | 0.154 | 1 |
Abbreviations: LV – mouse left ventricular tissue, EDL – mouse extensor digitorum longus muscle, MHC – myosin heavy chain, FC – fold change.
Actin-activated myosin ATPase activity
The kinetics of the actomyosin interaction in transgenic mutant, WT and control mice were assessed at the molecular level in actin activated myosin ATPase activity assays (Fig. 3). Myosin from at least two different preparations isolated and purified from the ventricles of ∼ 4 (Fig. 3A) and ∼ 8 (Fig. 3B) month old NTg, Tg-WT and Tg-Δ43 mice were used and each myosin preparation was obtained from 5-6 hearts per group of mice. A slight decrease in the Vmax was observed for myosin from both young and older Tg-Δ43 (0.27 ± 0.02 s-1 for ∼ 4 month old and 0.29 ± 0.01 s-1 for ∼ 8 month old) mice compared to Tg-WT (0.31 ± 0.01 s-1 for ∼ 4 month old and 0.32 ± 0.02 s-1 for ∼ 8 month old) and NTg (0.34 ± 0.02 s-1 for ∼ 4 month old and 0.35 ± 0.02 s-1 for ∼ 8 month old) mice (Fig. 3). There was a significant decrease in the Vmax of the ATPase activity of myosin from ∼4 month old Tg-Δ43 vs. NTg mice (p=0.048), whereas there were no statistically significant changes in Vmax of the actomyosin interactions between ∼4 month old Tg-Δ43 vs. Tg-WT mice (Fig. 3A, p>0.05) or between ∼8 month old mutant vs. control mice (Fig. 3B, p> 0.05). There was also no change in Vmax between younger and older Tg-Δ43 mice. Likewise, no significant differences in Km were observed between myosins from all groups of mice. The respective Km values for ∼4 and ∼8 month old animals were: Tg-Δ43, 2.10 ± 0.70 μM and 1.68 ± 0.34 μM; Tg-WT, 2.27 ± 0.43 μM and 2.56 ± 0.51 μM; NTg, 2.62 ± 0.53 μM and 2.83 ± 0.50 μM. The data are the average of n=4 individual experiments ± SE. These results suggest that the genetic ablation of the N-terminal ELC extension does not affect the Vmax and Km parameters of the ATP-driven interaction of myosin with actin.
Figure 3.
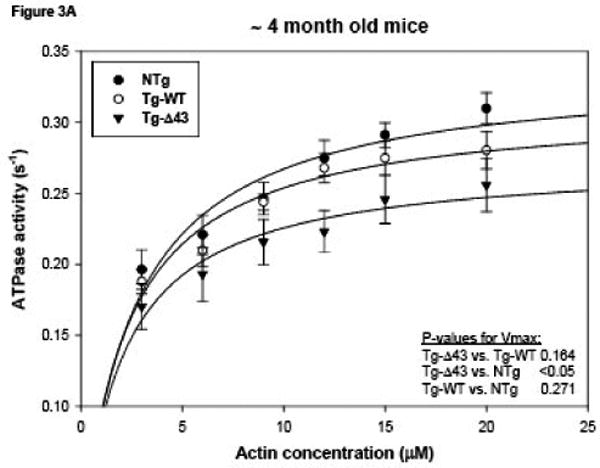
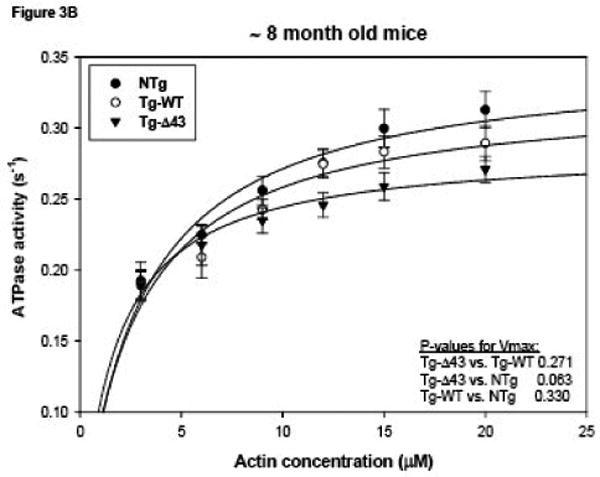
Actin activated myosin ATPase activity performed on purified myosin from ∼4 month old (A) or ∼8 month old (B) NTg, Tg-WT and Tg-Δ43 mice. The data points are the average of n=4 individual experiments ± SE. Data were analyzed using the Michaelis-Menten equation (Sigma Plot 11).
Myofibrillar ATPase activity
Since no age related differences were observed in the actin activated myosin ATPase activity, we have examined the Ca2+ regulation of the actomyosin interaction in the unloaded myofibrillar ATPase assay for only ∼4 month old NTg, Tg-WT and Tg-Δ43 mice. The assays represent the actin activated activity of myosin in the presence of the regulatory system containing tropomyosin (Tm) and troponin (Tn). Fig. 4A shows the ATPase activity of cardiac myofibrils in the presence of 0.5 mM CaCl2 or the absence of Ca2+ (1 mM EGTA), while Fig. 4B shows the myofibrillar ATPase activity as a function of increasing Ca2+ concentrations 25. No differences among myofibrils from all tested groups of mice were observed at -Ca2+ (1 mM EGTA) conditions (Fig. 4A). In the presence of Ca2+, the activity of Tg-Δ43 myofibrils was slightly lower compared to control groups, with the value for 20 μg of Tg-Δ43 myofibrils (utilized in the ATPase-pCa assay) significantly lower than Tg-WT (p=0.02, Fig. 4A). The Ca2+ sensitivity of myofibrillar ATPase (Fig. 4B) was similar among all groups of myofibrils, with Tg-Δ43 showing lower cooperativity (Hill coefficient, nH=1.49) compared to control NTg (nH=1.67) and Tg-WT (nH=1.73) myofibrils. The maximal myofibrillar activity of Tg-Δ43 mice was not different from that of Tg-WT but was significantly lower compared with NTg mice (p<0.05). On the other hand, the myofibrillar ATPase activity at low and submaxiaml calcium concentrations (6<pCa<8) was slightly elevated in Tg-Δ43 myofibrils compared with Tg-WT preparations (p<0.05) (Fig. 4B). This result suggests that the N-terminal ELC extension might be important for the regulation of the Tm-Tn dependent interaction of myosin cross-bridges with actin at low and submaximal Ca2+ concentrations and that the ablation of the N-terminus of ELC affects this interaction and impairs cross-bridge turnover.
Figure 4.
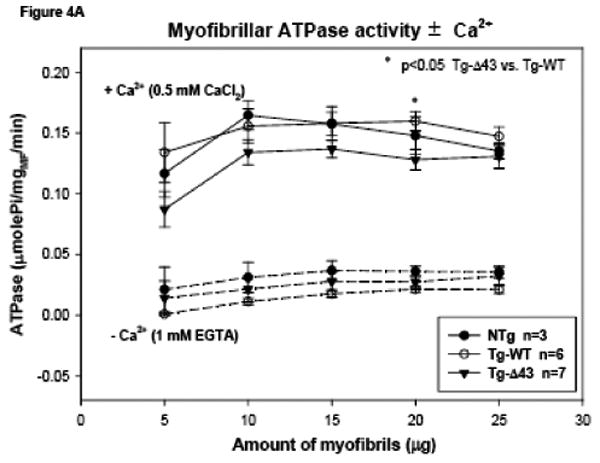
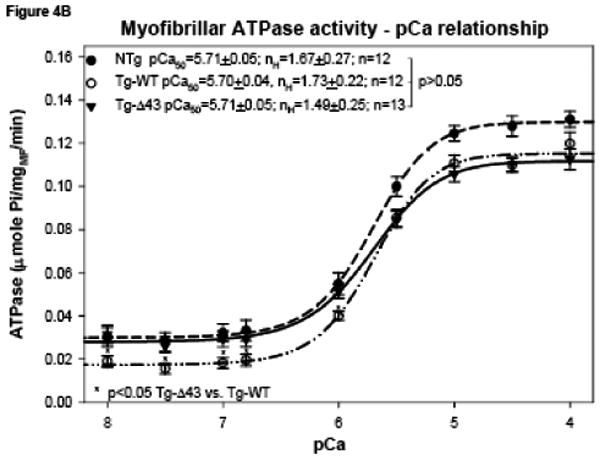
Myofibrillar ATPase activity of myosin isolated and purified from the ventricular tissue (ventricles and inter-ventricular septum) from NTg, Tg-WT and Tg-Δ43 mice. (A) The myofibrillar ATPase activity plotted as a function of increasing concentrations of myofibrils and ±Ca2+. (B) The Ca2+ sensitivity of myofibrillar ATPase activity in NTg, Tg-WT and Tg-Δ43 myofibrils.
Skinned muscle fiber studies
The functional studies in skinned papillary muscle fibers from ∼3 and ∼7 month old NTg, Tg-WT and Tg-Δ43 mice were performed as described in Materials and Methods and in our recent work 26; 27. The data are presented in Table II. Simultaneous ATPase-pCa and force-pCa measurements under isometric conditions were performed using all generated Tg-Δ43 lines expressing 30-40% protein (Fig. 2). The results were compared to Tg-WT L3 and L4 expressing ∼36% and ∼71% of huELCv, respectively and to NTg fibers. Two Tg-WT mouse lines (L3 and L4) were also compared against each other and no statistical differences (p>0.05) in ATPase and/or force measurements were found between ∼3 and ∼7 month old Tg-WT L3 vs. ∼3 and ∼7 Tg-WT L4 mice.
Table II.
Functional studies in skinned papillary muscle fibers from Tg-Δ43, Tg-WT and NTg mice.
| Skinned Muscle Fiber Measurements | ∼3 month old mice | ∼7 month old mice | ||||
|---|---|---|---|---|---|---|
| Tg-Δ43 | Tg-WT | NTg | Tg-Δ43 | Tg-WT | NTg | |
| Max Force (kN/m2) | 60.8±1.5 | 78.6±2.2 | 71.9±2.2 | 38.1±1.6 | 80.9±3.0 | 67.8±1.8 |
| pCa50 | 5.05±0.02 | 5.11±0.01 | 5.12±0.01 | 5.05±0.01 | 5.06±0.02 | 5.08±0.01 |
| nH | 2.60±0.15 | 2.74±0.26 | 3.10±0.14 | 2.63±0.09 | 2.74±0.20 | 3.08±0.13 |
| fXB (maxCa2+) | 0.699±0.007 | 0.706±0.010 | 0.701±0.007 | 0.594±0.008 | 0.690±0.006 | 0.700±0.007 |
| n | 17 | 12 | 11 | 14 | 19 | 16 |
Abbreviations: XB- cross-bridge, fXB (maxCa2+) – Fractional (f) cross-bridge attachment at maximal calcium activation, nH – Hill coefficient, n- number of individual fibers tested. One heart yielded 3-5 individual papillary muscle fibers.
Fiber force
The most pronounced effect of the Δ43 truncation monitored in this study was in the maximal force per cross-sectional area of muscle (Table II). A 23% and 15% decrease in maximal force was observed in skinned muscle fibers from ∼3 month old Tg-Δ43 mice compared to age matched Tg-WT and NTg mice, respectively (p<0.01). Interestingly, the difference in force/cross-sectional area between the mutant and control muscles was larger in older mice (Table II). Maximal force measured in fibers from ∼7 month old Tg-Δ43 mice (38.1 kN/m2) was 53% lower than that in Tg-WT mice (80.9 kN/m2), and 44% lower than that measured in NTg fibers (67.8 kN/m2) (p<0.01) (Table II). Furthermore, a slight but significant decrease in the Ca2+ sensitivity of force determined in muscle fibers from ∼3 month old Tg-Δ43 compared with age matched Tg-WT (p=0.035) and NTg fibers (p=0.014) was observed (Table II). The statistically significant difference in calcium sensitivity in ∼7 month old animals was only between Tg-Δ43 and NTg mice (p=0.047) (Table II). In addition, a Δ43 induced decrease in the Hill coefficient (nH) of the force-pCa dependence was observed in Tg-Δ43 muscle fibers compared to NTg fibers, in both ∼3 (p=0.03) and ∼7 (p=0.08) month old animals (Table II). Less cooperativity in the mutant fibers might be responsible for a shift in the force – pCa relationship to higher Ca2+ concentrations in Tg-Δ43 mice. A lower number of myosin heads available to interact with actin would also lead to lower cooperativity along the thin filament due to the greater distances between the active regulatory units.
Cross-bridge attachment at maximal Ca2+ activation
To get more insight into the nature of the force related phenotype in Tg-Δ43 mice, we have pursued the effect of the Δ43 mutation on the fraction of force generating myosin cross-bridges that can attach to actin at maximal calcium activation. At the end of the force/ATPase-pCa measurements, the fibers were perfused with the Ca2+ activating solution (pCa 3.4), in which 2 mM ATP was replaced with 10 mM MgADP to bring all cycling myosin cross-bridges into the force generating state and the level of tension determined in this solution was the maximal force that the fiber could develop 28; 29; 30. Once the maximum force was measured in the presence MgADP, the fibers were retired from any further experiments. The ratio of the maximal Ca2+ activated force divided by the maximum force the muscle fiber can develop in the presence of 10 mM MgADP represents the fraction of cross-bridges (fXB) that can attach to actin at maximal Ca2+ activation (maxCa2+) (Table II). As demonstrated, there was a significant 15% decrease in fXB at maximum calcium activation in ∼7 month old Tg-Δ43 mice compared to age matched Tg-WT and NTg control mice (p< 0.05). There was also a significant 15% decrease in fXB at maxCa2+ in ∼7 month old Tg-Δ43 mice compared to ∼3 month old Tg-Δ43 (Table II). These results indicate that as mice grow older, the fraction of total cross-bridges that can attach to actin during maximal Ca2+ activation is decreased approximately 15% in the Δ43 truncated myocardium compared to control Tg-WT and NTg mice and to ∼3 month old Tg-Δ43 mice.
To elucidate why force per cross-sectional area of muscle was so profoundly decreased in older Tg-Δ43 mice we have studied the age dependent changes in the thick-filament protein content (ELCtotal and RLCtotal) relative to the thin filament troponin I (TnI) in papillary muscle fibers from Tg-Δ43 mice. Two age groups of mice, 5 and 12 month old Tg-Δ43, Tg-WT and NTg mice were compared in five independent experiments. Fig. 5 demonstrates representative Western blots of papillary muscle extracts from transgenic mice blotted with ELC and RLC antibodies (upper panel) and subsequently with TnI antibody (lower panel). The ratios of ELCtotal /TnI and RLCtotal /TnI determined in papillary muscle (n=4) and left ventricular wall (n=1) preparations from Tg-Δ43 mice showed a 26±7% decrease in ELCtotal and a 34±8% decrease in RLCtotal as the mice developed from 5 to 12 months of age (Fig. 5A). In contrast, preparations from control Tg-WT animals showed slight age dependent increases of 6±9% in ELCtotal and 5±13% in RLCtotal (Fig. 5B). Likewise, NTg preparations showed a slight increase of 9±17% in ELCtotal but a slight decrease of 4±21% in RLCtotal content in 12 month old mice compared to 5 month old NTg animals. Notably, the ratio of the transgenic ELC proteins (WT and/or Δ43) to the total ELC (endogenous + transgenic) remained the same in both age groups indicating that the level of transgenic protein expression did not change with age. These results suggest that a greater Δ43 induced decrement in force observed in older animals compared to younger groups (Table II) might be due to a lower total protein content in older Tg-Δ43 mice.
Figure 5.
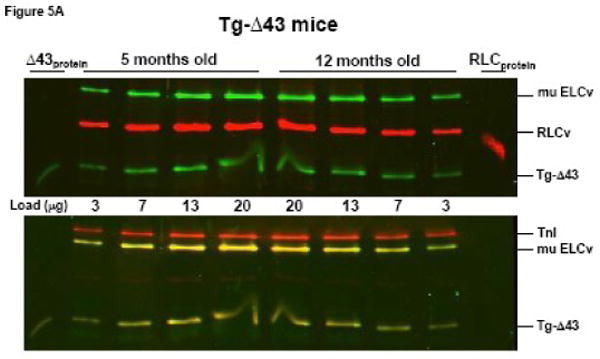
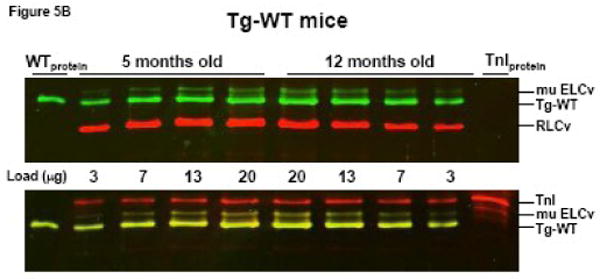
The effect of aging on the thick filament protein content in Tg-Δ43 compared to control Tg-WT mice. Representative Western blots of papillary muscle samples from Tg-Δ43 (A) and Tg-WT (B) mice. 3, 7, 13 and 20 μg of papillary muscle samples from 5 or 12 month old animals were loaded on SDS-PAGE and blotted with monoclonal antibody, clone H22.1 for ELC detection while RLC content was assessed with CT-1 antibodies (as described in Materials and Methods). Total troponin I (TnI) was detected with the 6F9 antibody (Research Diagnostics Inc.). Band intensities were plotted as a function of sample load and the ratios of ELCtotal/TnI and RLCtotal/TnI were determined in five independent experiments. Abbreviations as in Fig. 2.
Cross-bridge kinetics and the energy cost of contraction
The transition from the non-force-generating state to the force-generating state in muscle can be characterized by the cross-bridge attachment rate “f” while the rate of the cross-bridge return to the non-force-generating state is described by “g” 31; 32; 33. In our study, we have determined the cross-bridge detachment rate as g = ATPase/ concentration of cross-bridges attached at all levels of force activation, where the denominator = normalized force × concentration of myosin cross-bridges × fraction of cross-bridges attached at maximal Ca2+ activation 32. The total intracellular myosin cross-bridge (S1) concentration in muscle was assumed as 154 μM 34. As shown in Fig. 6A, the rate of cross-bridge dissociation g was slightly lower in papillary muscle fibers from ∼3 month old Tg-Δ43 compared with Tg-WT (p<0.05). No statistically significant differences in g were observed in ∼7 month old Tg-Δ43 vs. control mice (Fig. 6B). To account for a possible decrease in myosin cross-bridge concentration as it was determined in the papillary muscles from ∼12 month old Tg-Δ43 mice (Fig. 5), we have recalculated the g values for Tg-Δ43 fibers assuming a hypothetical 10% (green), 20% (blue) and 30% (red) decrease in S1 concentration and plotted as a function of fractional cross-bridge attachment (Fig. 6B, color lines). As shown in Fig. 5, the latter percent of decrease in [S1] was experimentally determined in 12 month old Tg-Δ43 mice. The color curves demonstrate that, as expected, the calculated g values would increase as the myosin cross-bridge concentrations decrease (Fig. 6B). Although the experimental “older” Tg-Δ43 mice were ∼7 and not ∼12 month old we observe a correlation between the g values illustrated by the red curve in Fig. 6B and a 15% decrease in the fraction of cross-bridges attached at maximal Ca2+ measured in the fibers from ∼7 month old Tg-Δ43 vs. control mice (0.594 vs. 0.690, Table II). This is because at maximal calcium activation the fraction of attached cross-bridges fXB = f/(f+g) 32. Thus, the myocardium of ∼7 month old Tg-Δ43 most likely contains a 30% lower S1 concentration compared to age matched control mice and to all groups of younger animals.
Figure 6.
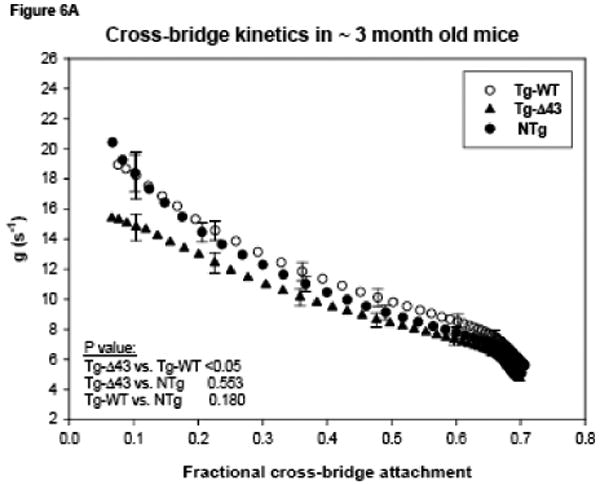
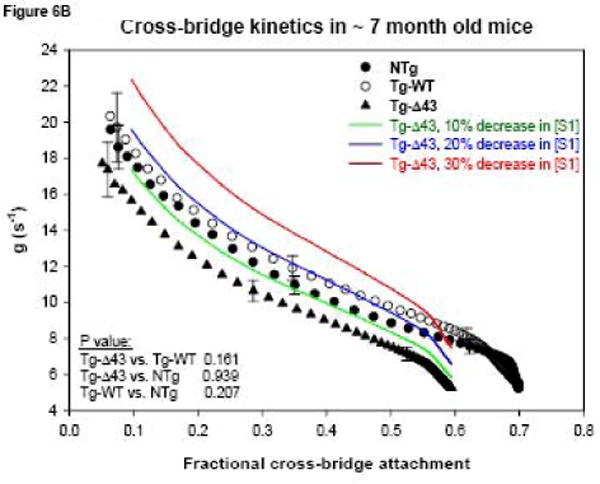
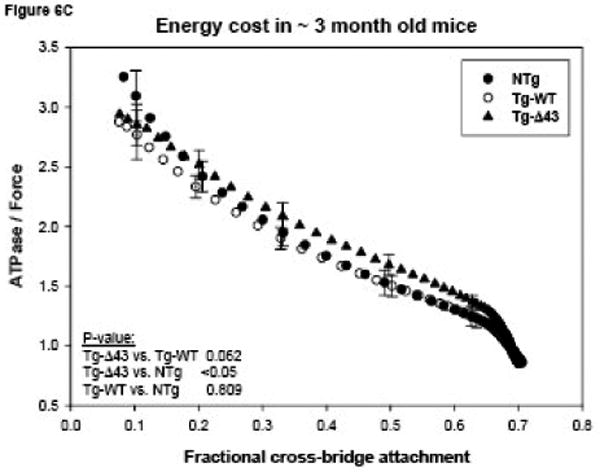
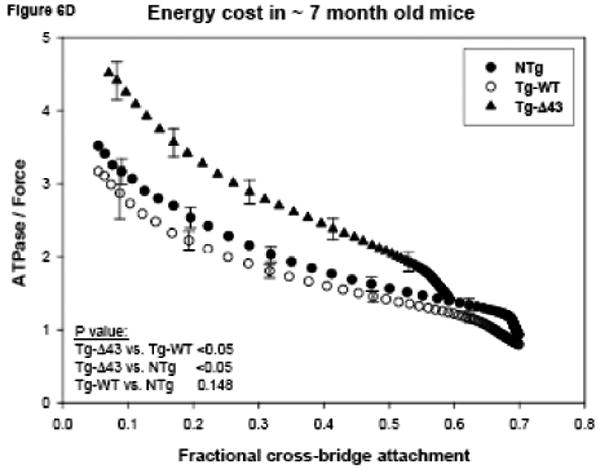
Functional measurements in freshly skinned papillary muscle fibers from ∼ 3 (A, C) and ∼ 7 (B, D) month old Tg-Δ43, Tg-WT and NTg mice. (A, B) The rate of dissociation of force generating myosin cross-bridges, g (in s-1). The color lines in Fig. 6B demonstrate the hypothetical g assuming 10% (green), 20% (blue) and 30% (red) decreased cross-bridge (S1) concentration. (C, D) The energy cost, ATPase/force (in 10-11 s-1 N-1 moles). Data are the average of n (as depicted in Table II) individual fibers ± SE.
Figs. 6C and D demonstrate the energy cost (fiber ATPase/force) of isometric contraction in Tg-Δ43 compared with control Tg-WT and NTg muscle fibers plotted as a function of the fractional cross-bridge attachment. A small but significant increase in energy cost was present in fibers from younger Tg-Δ43 mice compared with NTg fibers (p<0.05) (Fig. 6C). Since the ratio of fiber ATPase/force is proportional to g/Fav 31; 32, where Fav is the average force per cross-bridge, one can hypothesize that in order for the energy cost to increase slightly (Fig. 6C) with a small decrease or no change in g (Fig. 6A), the force per myosin cross-bridge, Fav, has to decrease. Interestingly, similar to the force phenotype in maturing Tg-Δ43 mice, the energy cost in the fibers from ∼7 month old mutant mice was also profoundly affected compared to controls (Fig. 6D). A large increase in ATPase/force (Fig. 6D) with no change or small increase in g (Fig. 6B) indicates that Fav in ∼7 month Tg-Δ43 fibers is markedly decreased. Since the ratio of fiberATPase/force plotted as a function of fractional cross-bridge attachment does not depend on [S1], the fact that fewer cross-bridges might be available in older Tg-Δ43 animals has no bearing on the strikingly high energy cost observed in contracting fibers from ∼7 month old Tg-Δ43 vs. control Tg-WT and NTg mice.
Taken together, we show that the Δ43 ELC truncation most likely results in a lower force per myosin cross-bridge in Tg-Δ43 mice of both ages. In addition, as the mutant mice grow older, fewer cross-bridges are available to interact with actin contributing to the decreased force per cross-sectional area of muscle in Tg-Δ43 mice. Reduced contractile protein mass in the hearts of older Tg-Δ43 mice is likely responsible for a greater mutant induced decrement in force observed in older mice compared to younger animals. In addition, there is inefficient energy use in the fibers from older Tg-Δ43 mice. Our findings highlight the importance of the N-terminal ELC extension not present in Tg-Δ43 mice for cardiac muscle physiology.
Magnetic Resonance Imaging
To examine whether the Δ43 phenotype observed in skinned muscle fibers could be manifested in vivo, ∼7 month old Tg-Δ43 mice and age matched Tg-WT and NTg controls were subjected to high resolution magnetic resonance imaging (MRI) (Fig. 7). Measurements of the left ventricular volumes in diastole (EDV) and systole (ESV) were done for each slice by tracing the blood pool in the left ventricles at the end-diastole and end-systole. Total ventricular volumes were calculated by summing the volumes of each slice according to Simpson's rule 35. The respective following equations were used to assess the stroke volume (SV), ejection fraction (EF) and cardiac output (CO): SV = EDV – ESV, EF = SV / EDV, CO = SV × Heart rate, where EDV and ESV were end diastolic and systolic volumes, respectively. Heart rates were monitored during imaging and were read 7-9 times for each mouse.
Figure 7.
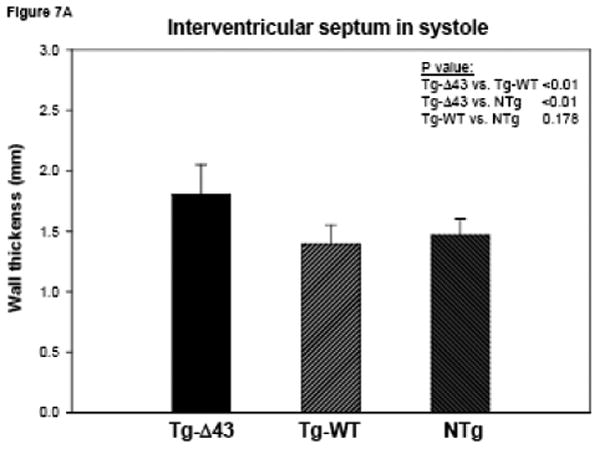
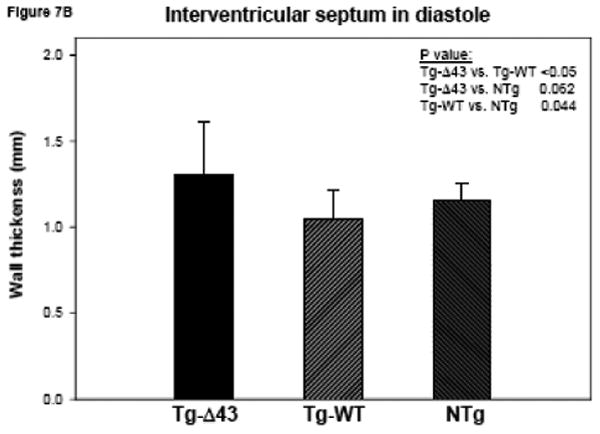
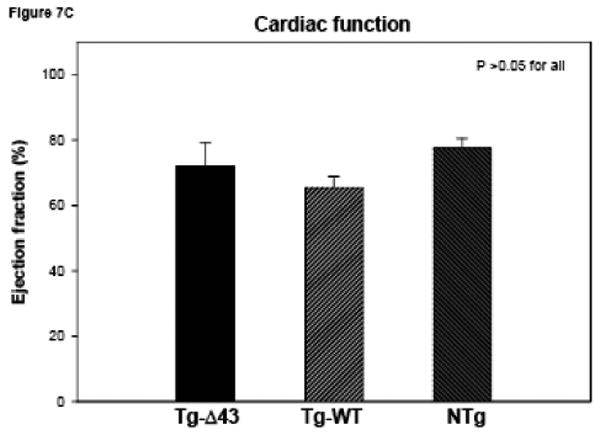
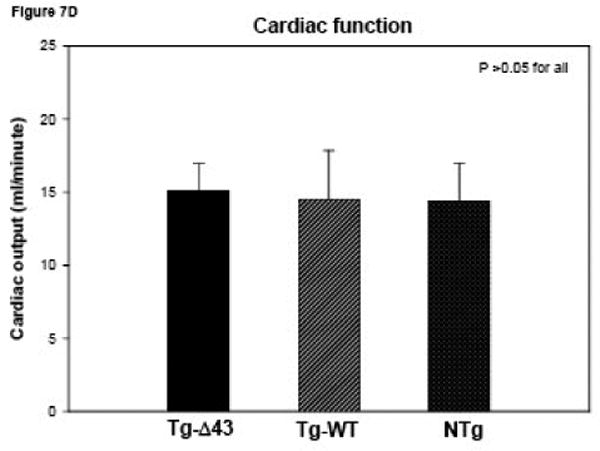
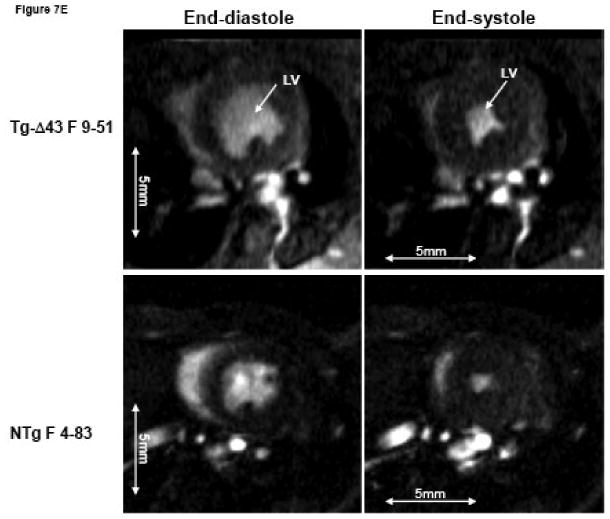
The MRI study of Tg-Δ43, Tg-WT and NTg mice. (A, B) Inner wall thickness in systole and in diastole. Three hearts from Tg-Δ43 mice and four from control WT and NTg mice were examined. Three slices from each heart were subjected to interventricular septal thickenss measurements and three measurements per slice were made. Bar graphs represent the average ± SD. (C, D) The effect of the Δ43 truncation in myosin ELC on ejection fraction (%) and cardiac output (ml/minute) in Tg-Δ43 mice compared to Tg-WT and NTg mice. Bar graphs represent the mean values derived for three Tg-Δ43 mice and four control WT and NTg mice ± SD. (E) Representative images of the mutant (upper panel) vs. control (bottom panel) mouse hearts at the end-diastole (left panel) and end-systole (right panel). Abbreviations: LV –left ventricle, Tg-Δ43 F 9-51 – mutant female mouse L9-51, NTg F 4-83 non-transgenic female mouse L4-83.
Figs. 7A and B illustrate inner wall thickness in systole and diastole. As demonstrated there is a significant increase in interventricular septal wall thickness in systole between Tg-Δ43 mice and control Tg-WT and NTg mice (Fig. 7A). Likewise, a significant increase in wall thickness was observed between Tg-Δ43 and Tg-WT mice in diastole (Fig. 7B). Functional MRI data derived by three independent investigators using two different programs are presented in Figs. 7C and D. No effects of Δ43 truncation in ELC on the ejection fraction (in %) and cardiac output (ml/minute) in all tested Tg-Δ43 mice compared to Tg-WT and NTg mice were observed (Fig. 7C). The same was true for cardiac output and no statistically significant changes between the groups were monitored (Fig. 7D). The average heart rates (in beats per minute) in Tg-Δ43, Tg-WT and NTg mice were respectively 399 ± 23, 467 ± 33 and 368 ± 35. Fig. 7E demonstrates the representative images of end-diastole and end-systole for Tg-Δ43 F (female) 9-51 mouse compared to control NTg F 4-83 mouse. Septal wall thickness measurements in diastole for these two representative mice were: 1.49 ± 0.19 mm (Tg-Δ43) and 1.11± 0.09 mm (NTg). In systole the values were: 1.83 ± 0.19 mm (Tg-Δ43) and 1.42± 0.14 mm (NTg). Our MRI results suggest that as the mice grow older, the hearts of Tg-Δ43 mice hypertrophy but do not manifest functional abnormalities and present unchanged cardiac performance.
Histopathology
To gain further insight into the nature of the skinned muscle fiber and MRI phenotypes observed in maturing Tg-Δ43 mice, the hearts of the mice subjected to the MRI examination were assessed for overall morphology and histopathology. For comparison, a histologic evaluation of hearts from young (∼ 2 months of age) Tg-Δ43, Tg-WT and NTg mice was also performed (Fig. 8). Consistent with the hypertrophic phenotype observed in older Tg-Δ43 mice (Figs. 7A and B), the gross morphological assessment of the hearts evaluated by MRI shows a large increase in heart size in Tg-Δ43 mice (males and females) compared to the age and gender matched control hearts from Tg-WT and NTg mice (Fig. 8A). However, the subsequent histological analysis of hematoxylin and eosin (H&E) and Masson's trichrome (Masson) stained left ventricular (LV) sections from these hearts demonstrated no tissue abnormalities, myofilament disarray or fibrosis (Fig. 8B). Likewise, there was no difference in morphology of LV tissue between ∼2 month old and ∼7 month old mice from all groups (Fig. 8B). In summary, we show that as the mutant mice mature the hearts of Tg-Δ43 mice grow larger, most likely due to activation of some compensatory mechanism to balance the reduced contractile force in the mutated myocardium. However, the ventricles of Tg-Δ43 mice do not show any pathologic phenotype or changes in the MRI assessed cardiac function. Perhaps, the structural changes in the Δ43 truncated myofilaments trigger an ELC mediated cardiac remodeling resulting in unchanged cardiac performance in Tg-Δ43 mice.
Figure 8.
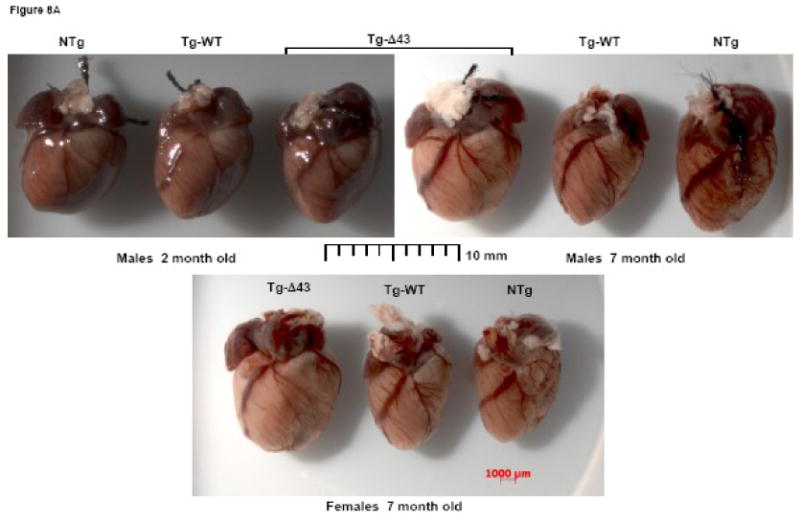
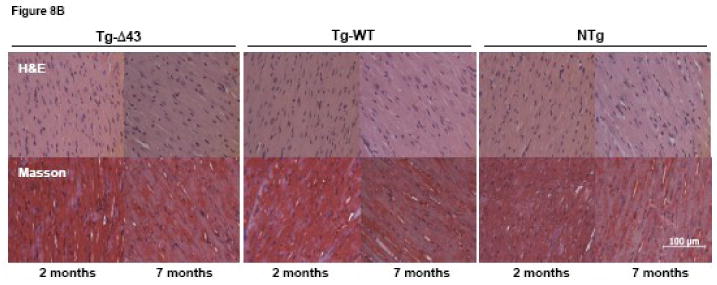
Gross morphology (A) and histological (B) assessments of the hearts from ∼2 and ∼7 month old NTg, Tg-WT and Tg-Δ43 mice. Note, that the hearts of ∼7 month old Tg-Δ43 mice are larger than those of ∼7 month old Tg-WT and NTg mice. The hearts shown in (A) are from the mice examined by MRI. (B) Hematoxylin and eosin (H&E) and Masson's trichrome stained longitudinal sections from the left ventricles of the hearts shown in (A). The scale bar, 100 μm.
Discussion
To assess the role of the myosin essential light chain N-terminal extension in cardiac muscle contraction, we have examined the physiological, biochemical and histological properties of transgenic hearts expressing a 43 amino acid truncated form of the human ventricular ELC (Tg-Δ43) compared to a full-length human ventricular wild-type ELC (Tg-WT) in mice (Fig. 1). Our long term objective to fully understand the ELC physiology of the heart, including the effects of familial hypertrophic cardiomyopathy mutations in ELC on cardiac muscle function, prompted us to the use the human rather than the mouse ELC isoform. The results show that the genetic ablation of the N-terminal ELC extension induces reduced force per cross-sectional area of muscle and leads to age dependent compensatory cardiac hypertrophy. We also demonstrate that the removal of the N-terminus of ELC produces an approximately 30% deficit in myosin cross-bridge content in the myocardium of aging Tg-Δ43 mice (Fig. 5) and that the force/cross-sectional area further decreases in the hearts of older mutant mice (Table II). However, the myocardium of Tg-Δ43 animals does not develop an age dependent pathologic phenotype and maintains unchanged cardiac performance (Figs. 7 and 8).
Here we address the molecular mechanisms underlying the N-terminal ELC extension-dependent regulation of force generation in muscle and discuss our findings along with the data and the mechanisms put forward by others 21; 36; 37; 38. In addition to the major actin binding site located in the 50 kDa domain of the MHC 6, it has been shown that the positively charged N-terminus of the long ELC variant, present in skeletal and cardiac muscles, can interact with the negatively charged C-terminus of actin (reviewed in Hermandez et al. 4). Since the atomic information of the “free-state” ELC (not bound to MHC) or the structural details regarding the N-terminal ELC extension are not currently available, these direct ELC-actin molecular contacts proposed previously 9; 11; 16 and in the recent studies 19; 20 are still under debate. Contrary to the fast skeletal muscle containing a combination of the long (MLC1) and short (MLC3) skeletal ELC variants, the cardiac myosin exclusively contain the long ELC isoform that is similar in sequence to the fast skeletal MLC1 (Fig. 1). The additional question that we address in our Tg-Δ43 study is the role of the short MLC3 in skeletal muscle contraction and its lack of expression in cardiac muscle. Interestingly, the resultant cardiac ELC distribution in Tg-Δ43 hearts, containing a mixture of a long (endogenous) and short (transgenic) ELC variants (Fig. 2B), resembles that present in fast skeletal muscle 24 (Fig. 2C). We hypothesize that the presence of the short transgenic Δ43 protein, partially replacing the long endogenous ELC in the murine cardiac myosin leads to altered interaction of myosin with actin through one or more of the following pathways: 1) ablation of the N-terminal myosin ELC induced cooperative activation of the thin filament; 2) lack of an N-terminal myosin ELC “tether” between the thick and thin filaments; 3) increased compliance of the neck region of the myosin head and its reduced ability to promote and/or support the strong cross-bridge formation. We further hypothesize that one or more of these mechanisms is responsible for a reduced force per cross-sectional area of muscle observed in the Δ43 ablated myocardium that is most likely caused by a decreased force per cross-bridge and/or a reduced number of force generating myosin cross-bridges.
Regarding hypothesis #1, it has been suggested that the N-termini of the long cardiac myosin ELC proteins activate the Tm-Tn regulated thin filaments in a cooperative manner and thus the genetic ablation of the N-terminal ELC extension would result in a lower myofilament cooperativity and decreased calcium sensitivity. As shown in Table II, the N-terminal Δ43 truncation led to a slight decrease in myofilament calcium sensitivity in Tg-Δ43 myocardium compared to controls. The mutant myofibrils (Fig. 4) and fibers (Table II) also demonstrated lower myofilament cooperativity compared to Tg-WT and NTg preparations. An N-terminal ELC extension induced cooperative activation was proposed by Rarick et al. 37. The authors studied the regulation of ATPase activity by the peptides derived from the sequence of the N-terminal myosin ELC actin binding domain (residues 5–14 of ELC) added in rat cardiac myofibrils. The results showed that the presence of nano-molar concentrations of peptides was able to induce a supra-maximal ATPase activation at submaximal Ca2+ levels suggesting a peptide induced cooperative interaction of the thin filament 37. An increase in isometric tension generation at both sub-maximal and maximal Ca2+ concentrations was also observed in human cardiac muscle fibers incubated with N-terminal ELC peptides spanning amino acids 5-14, 5-10 and 5-8 and the strongest effect was observed with 5-14 peptide 36. In addition, study by Haase et al. showed that the expression of the N-terminal human ELC peptides in transgenic rats positively correlate with improvements in the intrinsic contractile state of isolated perfused hearts 39. Although the Δ43 induced changes in myofilament calcium sensitivity and cooperativity measured in Tg-Δ43 preparations are small, our results are consistent with mechanism #1 suggesting that the N-terminal ELC extension could exert its action through a cooperative activation of the thin filament.
Furthermore, lower myofilament sensitivity to calcium could be due to faster kinetics of the cycling myosin cross-bridges and/or increased calcium dissociation rates from troponin C (TnC), the major myofilament calcium buffer 40. While the latter was not tested in this study, our results shown in Figs. 3, 4 and 6 suggest that the Δ43-ELC truncation only slightly decreased Vmax and the rate of cross-bridge dissociation (g) in Tg-Δ43 cardiac muscle preparations compared to Tg-WT and NTg controls. Using a transgenic mouse model similar to ours expressing the N-terminal ELC truncation mutant (ELCΔ5–14), Miller et al. showed significant kinetic differences in skinned cardiac muscle strips osmotically compressed to the intact lattice spacing 38. The authors proposed that the deletion of these N-terminal ELC residues most likely increases the probability of the cross-bridges to undergo a backwards power stroke suggesting that the role of the N-terminus of ELC is to inhibit the reversal of the power stroke 38. Strong support for the long ELC mediated changes in kinetics of cross-bridges comes from the previous in vitro studies on MLC1 or MLC3 reconstituted skeletal muscle fibers 41; 42; 43. The authors demonstrated that the long ELC (MLC1) may contribute to a slower shortening velocity suggesting that the removal of the N-terminal ELC extension would result in a faster cross-bridge kinetics. A recent study by Andruchov et al. 44 however showed no effect of the MLC1/MLC3 ratio on the kinetics of force transients. Our data are in line with the latter study 45. Nevertheless, the regulation of the myosin cross-bridge kinetics by the N-terminal ELC extension in cardiac muscle contraction is still an open question and perhaps the employment of a single molecule approach, e.g. in vitro motility assays, would help to determine the effect of the Δ43 mutation in ELC on myosin cross-bridge kinetics. Likewise, a decrease in calcium sensitivity of force/ATPase caused by a faster release of Ca2+ from TnC could be further tested in intact papillary muscle fibers to conclude whether the Δ43 mutation in ELC does or does not affect the kinetics of calcium release from TnC.
Considering the N-terminal ELC extension as a regulator of the myosin cross-bridge kinetics is also in line with mechanism #2. One can hypothesize that the N-terminus of ELC functions as a “tether” between the myosin head and actin thereby restricting (partially blocking) binding of myosin to actin. The removal of the N-terminal ELC tether (Δ43) would then lead to faster myosin cross-bridge kinetics. Again, our data at present do not support the faster kinetics hypothesis but as mentioned above, future experiments with Tg-Δ43 myocardial preparations performed at the single molecule level may bring a different perspective to these questions. Following the “tether” hypothesis one can anticipate that the removal of the tether (interference with binding to actin) would lead to tighter binding of myosin cross-bridges to actin. The results presented in Fig. 3 demonstrate an age dependent decrease in Km (higher affinity) of Tg-Δ43 myosin for actin compared to control Tg-WT and NTg myosins. Myosin from ∼8 month Tg-Δ43 mice had about 40% lower Km but the differences for both age groups were not statistically significant (Fig. 3). The tether hypothesis would still allow for the N-terminus of ELC to modulate force generation by binding to actin and possibly regulating force by increasing the number of force generating myosin cross-bridges. The work by Miller at al. also suggests that the role for the N-terminal ELC extension is to inhibit the reversal of the power stroke, thereby increasing the number of myosin heads in the force-producing and force-maintaining states 38. This mechanism is also supported by the work of others suggesting that the role of the N-terminus of ELC might be to tune the myosin motor function 22; 46.
Finally, according to hypothesis #3, the role of the N-terminal ELC extension would be to support myosin neck region compliance as this region of the myosin head transmits strain (load) from the thick filament to the active site of the myosin head bound to the actin filament. Our results are in agreement with this hypothesis. We propose that the genetic ablation of the N-terminus of ELC most likely leads to alterations in cross-bridge compliance resulting in a lower force per myosin cross-bridge. Therefore, the short form of cardiac ELC in the background of the long ELC form is detrimental to cardiac muscle contraction and results in reduced force per cross-section of muscle most likely through affecting the force per myosin cross-bridge. Since the skeletal muscle contains the combination of both ELC forms, one can only speculate that a background of only the long form of ELC in skeletal muscle would result in more force per cross-bridge. If so, then perhaps the short form of ELC in the skeletal muscle tunes the force per cross-bridge to the physiologically optimal level.
Clinical studies have revealed that several mutations in the human ventricular ELC gene (MYL3) lead to Familial Hyperthrophic Cardiomyopathy (FHC) with phenotypes of asymmetric left ventricular hypertrophy, mid-cavity obstruction and in restricted ventricular filling (reviewed in 4). Interestingly, the results from this study show that the hearts of ∼7 month old Tg-Δ43 mice (both females and males) resemble the phenotype of hypertrophic cardiomyopathy whereas the hearts of young mice (∼2 months of age) from all groups were similar in size (Fig. 8A). Interestingly, histologic assessments of the hearts from all groups of mice did not reveal any abnormalities in the ventricular tissues of young or older animals (Fig. 8B). The MRI experiments show increased interventricular wall thickness (hypertrophy) in the mutated hearts (Figs. 7A, B) but at the same time no abnormalities in either ejection fraction or cardiac output in the ∼7 month old Tg-Δ43 mice compared to controls (Figs. 7C, D). One can conclude that the N-terminal ELC truncation does not result in a functionally compromised Tg-Δ43 myocardium and that the hearts of Tg-Δ43 mice are fully capable of adapting to the ELC structural myofilament changes. The lower force in the mutated myocardium, as explained above, was primarily triggering compensatory growth in Tg-Δ43 mice but the change was not of a pathologic nature. The gene expression profile in Tg-Δ43 mice supports this notion and shows normal expression of the key genes that could possibly be altered by the Δ43 truncation (Table I). These include sarcomeric mouse cardiac α-actin, α- and β-myosin heavy chains, ventricular myosin regulatory light chain, fast skeletal and atrial ELC. No change was also observed in gene expression of the Ca2+-ATPase protein present in sarcoplasmic reticulum (Table I). Furthermore, expression of the truncated ELC mutant in the mouse heart did not affect expression of the fast skeletal ELC in the fast skeletal muscle of Tg-Δ43 mice at either the gene (Table I) or protein (Fig. 2) levels.
Taken together, we have demonstrated that the partial replacement of the long ventricular ELC with the short transgenic huELCv-Δ43 results in reduced contractile force in Tg-Δ43 myocardium most likely due to a lower force per myosin cross-bridge and/or lower number of force generating myosin cross-bridges. To compensate and to meet the hemodynamic demands, the mutated myocardium increases in size but the hypertrophied hearts do not manifest pathologic phenotypes and are able to maintain normal cardiac performance. We conclude that the N-terminus of ELC may modulate force generation in muscle by altering a cross-bridge mechanics (decreasing myosin neck region compliance, increasing force per cross-bridge) and/or enhancing myosin attachment to actin and increasing the number of force generating myosin cross-bridges.
Material and Methods
Generation of transgenic mice
All animal studies were conducted in accordance with institutional guidelines (#A3711-01). The cDNA clones expressing the wild type human ventricular ELC (NCBI accession # NP_000249) and the 43 amino acid N-terminal ELC deletion mutant huELCv-Δ43 were cloned into the unique SalI site of the plasmid, α-myosin heavy-chain clone 26 (a generous gift from Jeffrey Robbins, Cincinnati Children's Hospital Medical Center, Cincinnati, OH). The resulting constructs contained about 5.5 kb of the mouse α-myosin heavy-chain promoter, including the first two exons and part of the third, followed by the WT/Δ43 cDNAs and a 600 bp 3′ untranslated region from the human growth hormone transcript. Founder mice were bred with B6/SJL mice.
Determination of transgenic ELC protein expression
Protein concentrations from ventricular tissue extracts or myofibrils was determined in 1% SDS, 10% β-mercaptoethanol, 1 mM EDTA, 1 mM PMSF using Coomassie Plus reagent (Pierce). Tissues were processed on ice. Proteins were transferred from the gel to nitrocellulose membrane over 1 hour on ice at a constant voltage of 95 V and 260-500 mA. Membranes were washed 5 minutes at room temperature in TBS, blocked 1 hour in a 1:1 mixture of TBS/Odyssey blocking buffer. All subsequent washes and antibody incubations were done in a 1:1 mixture of 0.05% Tween 20-TBS/Odyssey blocking buffer. Specific detection of ELC proteins was accomplished using 1:2000 mouse monoclonal antibody, clone H22.1, (Accurate Chemical and Scientific Corporation). Myosin RLC was utilized as a loading control and visualized with 1:5000 rabbit polyclonal antibody against a C-terminal RLC peptide, CT-1 developed in this lab 27. Differential detection of primary antibody bound to ELC and RLC was done by use of affinity purified polyclonal Infrared CY5.5 (700 nm) labeled goat anti mouse IgG H&L, (Rockland Antibodies) and a parallel polyclonal Infrared IR 800 Red Dye (800 nm), both at 1:4000. Percent ELC transgene expression was defined as: % protein expression = ELCtransgenic signal / (ELCendogenous signal + ELCtransgenic signal) × 100.
Gene expression analysis using real- time PCR
Tissue processing and total RNA isolation
Left ventricles and extensor digitorum longus (EDL) muscles from ∼3 month old Tg-Δ43, Tg-WT and NTg mice were rapidly harvested, submerged immediately in 25 volumes of room temperature RNAlater RNA stabilization reagent (Qiagen) and incubated overnight at 4°C. The samples were stored frozen at -20°C until used. Total RNA was isolated from RNAlater stabilized tissues using an RNeasy Fibrous Tissue Mini Kit (Qiagen, Valencia, CA) after being homogenized in a Mixer-Mill MM301 (Retsch) according to the manufacturer's protocol 26.
Real-time PCR
RNA concentration and integrity was assessed using spectrophotometric analysis and non-denaturing agarose gel electophoresis prior to cDNA synthesis. 1.5 μg of total RNA from each sample was converted to double stranded cDNA using Random Primers and a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and reverse transcription was performed in an MJ Research PTC-200 according to the manufacturer's protocols. Reverse transcription reactions were stored at -80°C until needed for quantitative PCR. Quantitative PCR was conducted using SYBR Green I chemistry with gene specific Quantitect Primer Sets (Qiagen) for murine: Myl1 (myosin essential light chain, fast skeletal; NCBI accession # NM_021285), Myl2 (myosin regulatory light chain, ventricular; NCBI accession # NM_010861), Myl4 (myosin essential light chain, atrial; NCBI accession # NM_010858), Myh6 (alpha-myosin heavy chain, cardiac; NCBI accession # NM_010856), Myh7 (beta-myosin heavy chain, cardiac; NCBI accession # NM_080728), Atp2a2 (Ca2+-ATPase (SERCA2), cardiac; NCBI accession # NM_009722), Actc1 (alpha-actin, cardiac; NCBI accession # NM_009608), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; NCBI accession # NM_008084), QuantumRNA 18S Internal standards (Ambion) and Power Sybr Green PCR Master-Mix (Applied Biosystems) using the manufacturer's protocol. Following optimization using a Standard Curve (AQ) assay, 50 ng of cDNA was used in a 25 μl PCR reaction containing 12.5 μl of Master Mix, 2.5 μl of 10× Quantitect primers or 0.4 μM final concentration of 4:6 ratio of 18S primer:competimer and RNAase/DNAase free water. All reactions were performed in duplicates and were run in an ABI 7900 HT Fast Real-Time PCR System with the following cycle parameters: 1 cycle of 50°C (2 minutes) followed by 95°C (10 minutes), 40 cycles of 95°C (15 seconds) followed by 60°C (1 minute). Raw data were analyzed using the Sequence Detection Software (ABI), and fold change in expression of each gene was calculated using the relative quantification (RQ) ΔΔCt method with the levels of 18S rRNA as the normalizer gene.
Isolation and purification of cardiac muscle myosin, actin and cardiac myofibrils
Transgenic cardiac myosin
Cardiac myosin was isolated from mouse hearts from age matched groups of Tg-Δ43, Tg-WT and NTg, mice according to Szczesna-Cordary et al. 26. One myosin preparation was obtained from a pool of 5-6 hearts for each group of mice. Briefly, after euthanasia, whole hearts were isolated and the atria were removed. Left and right ventricles varying from 0.1-0.2 g were flash frozen and stored at -80°C until processed. The ventricular tissue was later thawed in an ice cold Guba Straub-type buffer, pH 6.5 consisting of 300 mM NaCl, 100 mM NaH2PO4, 50 mM Na2HPO4, 1mM MgCl2, 10mM EDTA, 0.1% NaN3, 10 mM Na4P2O7, 1 mM DTT and protease inhibitor cocktail in a volume of 0.75 ml buffer per 0.2 g tissue. Ventricles kept on ice were first minced by hand and then homogenized for 2 minutes at a frequency of 30 Hz in a Mixer-Mill MM301 (Retsch). The homogenate was then incubated on ice for 40 minutes before centrifugation at 200,000 × g for 1 hour. The supernatant was then diluted 60-fold (by volume) with 2 mM DTT and incubated on ice 30 minutes with stirring and left standing without stirring for an additional 30 minutes. The samples were centrifuged again at 8,000 × g for 10 minutes and resultant pellets were then re-suspended in a minimal volume of buffer containing 0.4 M KCl, 10 mM MOPS, pH 7.0, 5 mM DTT and protease inhibitor cocktail. Samples were then diluted 1:1 with glycerol mixed gently and stored at -20°C for 2-5 days. For ATPase assays, myosins were precipitated with 14 volumes of ice cold 2 mM DTT and collected by centrifugation at 8,000 × g for 10 minutes. The myosin pellets were then resuspended in a minimal volume of ATPase buffer consisting of 0.4 M KCl, 10 mM MOPS, pH 7.0, and 1mM DTT and dialyzed overnight against the same buffer. The concentration of myosin was determined using a Coomassie Plus Assay (Pierce) and all myosins were diluted to a concentration of 2 mg/ml for the assay.
Muscle F-actin
Rabbit skeletal muscle actin was prepared according to Pardee and Spudich 47 with modifications. Briefly, rabbit skeletal acetone powder was extracted with a buffer consisting of 2 mM MOPS, pH 8.0, 0.2 mM Na2ATP, 0.5 mM β–mercaptoethanol, 0.2 mM CaCl2, and 0.0005% NaN3 at a ratio of 20 ml/g for 30 minutes with stirring on ice. The extract was centrifuged at 7,000 × g at 4°C for 1 hour to clarify and the tissue pellet was discarded. The supernatant was removed and adjusted to a final concentration of 0.05 M KCl, 2 mM MgCl2, and 1 mM Na2ATP, pH 8.0. The F-actin was allowed to polymerize for 2 hours at 4°C. The KCl concentration was then increased again very slowly to a final concentration of 0.6 M and the solution was stirred slowly on ice for 30 minutes. The F-actin was then collected by ultracentrifugation at 107,000 × g at 4°C for 1.5 hours. The supernatant was discarded and the F-actin pellets were re-dissolved in a buffer consisting of 10 mM MOPS, pH 7.0 and 40 mM KCl.
Isolation and purification of transgenic cardiac myofibrils
Myofibrils (MF) were prepared from left and right ventricular walls, septa and papillary muscles of transgenic mice according to Solaro et al. 48. One MF preparation was obtained from a pool of 4-5 hearts from each group of mice. Unless stated otherwise, standard MF buffer containing 30 mM imidazole pH 7.0, 60 mM KCl, 2 mM MgCl2, 1 mM DTT, 1 mM PMSF and protease inhibitor cocktail was used for MF purification. Briefly, 100-200 μg of ventricular tissue was minced in 600 μl of 300 mM sucrose and 10 mM imidazole, pH 7, and homogenized in a Mixer-Mill MM301 (Retsch) for 2 minutes at a frequency of 20 Hz. Samples were then centrifuged for 1 minute at 1,500 × g and pellets treated with 300 mM sucrose, homogenized for 30 seconds at 20 Hz and centrifuged. Subsequently, samples were washed with 2 mM EGTA in standard MF buffer, centrifuged and washed 3 times in standard MF buffer containing 1% Triton X-100. After each wash with Triton the samples were left on ice for 30 minutes. Then, samples were washed 4 times with MF buffer mixed with glycerol. The first wash was done in 25% glycerol and the following 3 washes in 50% glycerol. Purified myofibrils were stored in standard MF buffer and 50% glycerol at -20°C until needed.
Actin-activated myosin ATPase activity
Actin activated myosin ATPase activity was measured as a function of actin concentration using myosin purified from ∼4 and ∼8 month old Tg-Δ43, Tg-WT and NTg mice. The ATPase assays were performed with at least 2 myosin preparations, and a pool of 5-6 hearts from each group of mice were used to yield one myosin preparation. Myosins at a concentration of 1 μM were titrated (in triplicates onto 96-well microplates) with rabbit skeletal actin in increasing concentrations of (in μM): 0.1, 3, 6, 9, 12, 15, and 20. ATPases assays were performed in a 120 μl reaction volume in a buffer consisting of 25 mM imidazole, pH 7.0, 4 mM MgCl2, 1 mM EGTA, and 1 mM DTT. The final salt KCl concentration was 0.107 mM. The reactions were initiated with the addition of 2.5 mM ATP with mixing in a Jitterbug incubator shaker (Boekel) and allowed to proceed for 15 minutes at 30°C and then terminated by the addition of 5% trichloroacetic acid (TCA). Samples were then centrifuged at 18,000 × g for 20 minutes and 50 μl of supernatant transferred to a 96-well microplate for determination of inorganic phosphate (Fiske Subbarow method) 49. Data were analyzed using the Michaelis-Menten equation yielding the Vmax and Km parameters 50; 51.
Myofibrillar ATPase activity
Myofibrillar ATPase activity was determined using mouse cardiac myofibrils isolated and purified from the ventricular tissue of ∼4 month old NTg, Tg-WT, Tg-Δ43 mice. To determine the ATPase activity at increasing concentrations of myofibrils in ±Ca2+ conditions increasing amounts of myofibrils (5, 10, 15, 20 and 25 μg) were assayed in the solution of 70 mM KCl, 1 mM MgCl2, 20 mM imidazole, pH 7, 1 mM DTT and 0.5 mM CaCl2 for +Ca2+ or 1 mM EGTA for -Ca2+ conditions. To determine the ATPase-pCa relationship, 20 μg myofibrillar samples were suspended in a solution of 20 mM MOPS pH 7.0, 40 mM KCl, 3.5 mM MgCl2, 2 mM EGTA and increasing concentrations of Ca2+ from pCa 8 to pCa 4.5. The final ionic strength was 73 mM and the free concentration of Mg2+ was 1 mM 25. The assays were performed using 3-4 individual myofibrillar preparations from all groups of mice. After 5 minutes incubation at 30°C, the ATPase reaction was initiated with addition of 2.5 mM ATP and terminated after 10 minutes with 5% trichloroacetic acid (TCA). Released inorganic phosphate was measured according to Fiske and Subbarow 49. The ATPase-pCa data were analyzed with a Hill equation yielding the pCa value which gives 50% of the ATPase activity (pCa50) and the Hill coefficient, nH 52.
Papillary muscle fiber studies
Two series of experiments in skinned muscle fibers were performed, one utilizing ∼3 and the second ∼ 7 month old Tg-Δ43 mice matched by age with Tg-WT and NTg control mice. Simultaneous ATPase and force measurements were performed using the Guth Muscle Research System (Scientific Instruments, Heidelberg, Germany), as described in detail in Szczesna-Cordary et al. 26. The mechanical parts of the apparatus consisted of a force transducer for measuring force. The optics consisted of a microscope photometer unit for monitoring emission light from the muscle fiber. The light was focused by an Olympus Quartz condenser onto the muscle preparation after passing through filters appropriate for performing [Ca2+] transient and ATPase measurements.
Simultaneous ATPase and force measurements
After euthanasia, the hearts of mice were excised and strips of mouse papillary muscle fibers (diameter: 60-70 μm) were dissected in ice-cold relaxing solution (85 mM K+, 2 mM MgATP2-, 1 mM Mg2+, 7 mM EGTA, pH 7.0 and propionate as the major anion). The fibers were treated with 1% Triton X-100 for 30 minutes and processed immediately without glycerination 26. The skinned fiber was then mounted in the Guth apparatus and the sarcomere length adjusted to 2.2 μm by use of a laser diffraction pattern. The cross-sectional area was calculated based on measurement of the fiber width by microscope and the assumption that the fiber is circular in diameter. The ATPase rate was measured using the NADH fluorescence method 53 as described earlier 26. The fiber was subjected to an increasing Ca2+ gradient assessed by mixing the relaxing (pCa 9) and contracting (pCa 3.4) solutions. Both solutions contained: 85 mM K+, 2 mM MgATP2-, 1 mM Mg2+, 7 mM EGTA, 5 mM PEP, 100 units/ml PK, 0.4 mM NADH, 140 U/ml LDH, ionic strength 150 mM, pH 7.0 and propionate as the major anion. Fresh, unoxidized NADH solution was introduced into the cuvette every 20 seconds. The decrease in NADH concentration was determined by a decrease in the fluorescence signal detected at 450 nm. The slope of the linear decrease in NADH concentration was used to calculate the ATPase rate. The Ca2+ concentration gradient was calibrated by use of the fluorescent Ca2+ indicator calcium green-2 (Molecular Probes) 54. Force development was monitored simultaneously with ATPase measurements utilizing the force transducer of the Guth apparatus. The ratio of ATPase/force expresses the energy cost per unit of isometric force.
Magnetic Resonance Imaging
In vivo cardiac MRI (magnetic resonance imaging) datasets were collected on a 4.7-Tesla (200 MHz) 40-cm bore magnet interfaced with a Bruker Avance™ console using an actively shielded gradient set with an inner diameter of 120-mm and a maximum gradient strength of 400 mT/m (MRI Core Facility, University of Miami, Miller School of Medicine, Miami FL). About 7 month old male and female mice from all experimental groups were used for imaging. After induction the subject was placed in an MRI compatible cradle equipped with a nose cone to deliver gaseous anesthesia, head restraints to reduce motion, a respiratory sensor, an ECG sensor and a body temperature regulating system. For anesthesia approximately 0.8% to 1.5% isoflurane with 0.8 l/minute of nitrous oxide and 0.4 l/minute oxygen was used. The concentration of isoflurane was adjusted to maintain a respiratory rate of approximately 125 breaths per minute. Following subject placement in an MRI compatible cradle a quadrature saddle surface RF coil with an inner diameter of 30 mm was attached to the thorax for transmission and reception of NMR signal. Pilot images were acquired in both transverse and coronal planes to ensure proper slice positioning. Short-axis slices were prescribed based on these pilot images and were positioned perpendicular to the interventricular septum. Typically, 8, 9 or 10 slices were collected to achieve complete heart coverage. MRI acquisitions employed an electrocardiogram (ECG) triggered Fast Low Angle SHot (FLASH) gradient echo cine sequence. The following parameters were used: echo time of 1.3 ms, a time of repetition of approximately 10 ms; a field of view of 1.5 cm by 1.5 cm; a matrix size of 100 × 100; an in-plane resolution of 150 μm by 150 μm; and a slice thickness of 1.0 mm. Thirteen frames per cardiac cycle were collected using a Time of Repetition (TR) period that varied to ensure that all thirteen frames were contained within one cardiac cycle. A heart rate of 360 to 500 beats per minute was typically achieved which gave a range of TR times of 9.23 ms to 10.25 ms. Image analysis was performed off-line by 3 independent investigators using two different methods: 1) The freely available Segment v1 software (http://segment.heiberg.se) 55; 56, and 2) The Paravision™ software package (Bruker, Biospin), employing semiautomatic and manual routines to derive the endocardial borders. Determination of the left ventricular volumes in diastole (end-diastolic volume, EDV) and systole (end-systolic volume, ESV) were assessed by manual delineation of endocardial borders in the left ventricle of the heart at the end-diastole (when the heart is the biggest) and end-systole (when the heart is the smallest), which demonstrated a higher reliability than the semi-automatic methodology. Total ventricular volumes were calculated according to Simpson's rule in short-axis views 35. Determination of inner wall thickness in systole and diastole was performed on three short-axis slices for each heart and three measurements of interventricular septal wall thickenss were performed for each slice.
Histopathology
After euthanasia, the hearts from ∼2 and ∼7 month old Tg-Δ43, Tg-WT and NTg mice were excised, weighed and immersed in 10% buffered formalin. Pictures of whole hearts were taken using a Stereoscope Discovery.V12 Zeiss and 0.63×/5× PlanApo S objective and AxioCam HRc (Zeiss). Slides of whole mouse hearts were prepared by American Histolabs, Inc. (Gaithersburg, MD). The paraffin embedded longitudinal sections of whole mouse hearts stained with hematoxylin and eosin (H&E) and Masson's trichrome were examined for overall morphology and fibrosis using a Dialux 20 microscope, 40×/0.65 Leitz Wetzlar objective and AxioCam HRc (Zeiss).
Statistical Analysis
Data are expressed as the average of n experiments ± SE (standard error). For MRI experiments due to a relatively low number of available animals the error bars represent standard deviation (SD). Statistically significant differences were determined utilizing an unpaired Student's t-test with significance defined as p< 0.05 (Sigma Plot 11).
Acknowledgments
The authors thank Dr. Kyle Padgett, Joshi Vivek and Roth Nathan for their work with MRI and Zoraida Diaz-Perez for her excellent technical assistance regarding transgenic mice. This work was supported by NIH-HL071778 (D.S-C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lowey S, Waller GS, Trybus KM. Function of skeletal muscle myosin heavy and light chain isoforms by an in vitro motility assay. J Biol Chem. 1993;268:20414–20418. [PubMed] [Google Scholar]
- 2.Trybus KM. Role of myosin light chains. J Muscle Res Cell Motil. 1994;15:587–94. doi: 10.1007/BF00121066. [DOI] [PubMed] [Google Scholar]
- 3.Szczesna D. Regulatory light chains of striated muscle myosin. Structure, function and malfunction. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:187–97. doi: 10.2174/1568006033481474. [DOI] [PubMed] [Google Scholar]
- 4.Hernandez OM, Jones M, Guzman G, Szczesna-Cordary D. Myosin essential light chain in health and disease. Am J Physiol Heart Circ Physiol. 2007;292:H1643–1654. doi: 10.1152/ajpheart.00931.2006. [DOI] [PubMed] [Google Scholar]
- 5.Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–8. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 6.Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 7.Geeves MA, Holmes KC. The molecular mechanism of muscle contraction. Adv Protein Chem. 2005;71:161–93. doi: 10.1016/S0065-3233(04)71005-0. [DOI] [PubMed] [Google Scholar]
- 8.Sutoh K. An actin-binding site on the 20K fragment of myosin subfragment 1. Biochemistry. 1982;21:4800–4. doi: 10.1021/bi00262a043. [DOI] [PubMed] [Google Scholar]
- 9.Henry GD, Winstanley MA, Dalgarno DC, Scott GM, Levine BA, Trayer IP. Characterization of the actin-binding site on the alkali light chain of myosin. Biochim Biophys Acta. 1985;830:233–43. doi: 10.1016/0167-4838(85)90279-1. [DOI] [PubMed] [Google Scholar]
- 10.Trayer IP, Trayer HR, Levine BA. Evidence that the N-terminal region of A1-light chain of myosin interacts directly with the C-terminal region of actin. A proton magnetic resonance study. Eur J Biochem. 1987;164:259–66. doi: 10.1111/j.1432-1033.1987.tb11019.x. [DOI] [PubMed] [Google Scholar]
- 11.Milligan RA, Whittaker M, Safer D. Molecular structure of F-actin and location of surface binding sites. Nature. 1990;348:217–21. doi: 10.1038/348217a0. [DOI] [PubMed] [Google Scholar]
- 12.Morano M, Zacharzowski U, Maier M, Lange PE, Alexi-Meskishvili V, Haase H, Morano I. Regulation of Human Heart Contractility by Essential Myosin Light Chain Isoforms. J Clin Invest. 1996;98:467–473. doi: 10.1172/JCI118813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Timson DJ, Trayer HR, Trayer IP. The N-terminus of A1-type myosin essential light chains binds actin and modulates myosin motor function. Eur J Biochem. 1998;25:654–662. doi: 10.1046/j.1432-1327.1998.2550654.x. [DOI] [PubMed] [Google Scholar]
- 14.Timson DJ, Trayer HR, Smith KJ, Trayer IP. Size and charge requirements for kinetic modulation and actin binding by alkali 1-type myosin essential light chains. J Biol Chem. 1999;274:18271–7. doi: 10.1074/jbc.274.26.18271. [DOI] [PubMed] [Google Scholar]
- 15.Andreev OA, Saraswat LD, Lowey S, Slaughter C, Borejdo J. Interaction of the N-terminus of chicken skeletal essential light chain 1 with F-actin. Biochemistry. 1999;38:2480–5. doi: 10.1021/bi981706x. [DOI] [PubMed] [Google Scholar]
- 16.Miyanishi T, Ishikawa T, Hayashibara T, Maita T, Wakabayashi T. The Two Actin-Binding Regions on the Myosin Heads of Cardiac Muscle. Biochemistry. 2002;41:5429–5438. doi: 10.1021/bi0118355. [DOI] [PubMed] [Google Scholar]
- 17.Nieznanska H, Nieznanski K, Stepkowski D. The effects of the interaction of myosin essential light chain isoforms with actin in skeletal muscles. Acta Biochim Pol. 2002;49:709–19. [PubMed] [Google Scholar]
- 18.Houdusse A, Cohen C. Structure of the regulatory domain of scallop myosin at 2 A resolution: implications for regulation. Structure. 1996;4:21–32. doi: 10.1016/s0969-2126(96)00006-8. [DOI] [PubMed] [Google Scholar]
- 19.Aydt EM, Wolff G, Morano I. Molecular modeling of the myosin-S1(A1) isoform. J Struct Biol. 2007;159:158–163. doi: 10.1016/j.jsb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Lowey S, Saraswat LD, Liu H, Volkmann N, Hanein D. Evidence for an Interaction between the SH3 Domain and the N-terminal Extension of the Essential Light Chain in Class II Myosins. J Mol Biol. 2007;371:902–913. doi: 10.1016/j.jmb.2007.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morano I. Tuning the human heart molecular motors by myosin light chains. J Mol Med. 1999;77:544–55. doi: 10.1007/s001099900031. [DOI] [PubMed] [Google Scholar]
- 22.Timson DJ. Fine tuning the myosin motor: the role of the essential light chain in striated muscle myosin. Biochimie. 2003;85:639–45. doi: 10.1016/s0300-9084(03)00131-7. [DOI] [PubMed] [Google Scholar]
- 23.Arad M, Penas-Lado M, Monserrat L, Maron BJ, Sherrid M, Ho CY, Barr S, Karim A, Olson TM, Kamisago M, Seidman JG, Seidman CE. Gene Mutations in Apical Hypertrophic Cardiomyopathy. Circulation. 2005;112:2805–2811. doi: 10.1161/CIRCULATIONAHA.105.547448. [DOI] [PubMed] [Google Scholar]
- 24.Lowey S, Risby D. Light Chains from Fast and Slow Muscle Myosins. Nature. 1971;234:81–85. doi: 10.1038/234081a0. [DOI] [PubMed] [Google Scholar]
- 25.Dweck D, Reyes-Alfonso A, Jr, Potter JD. Expanding the range of free calcium regulation in biological solutions. Anal Biochem. 2005;347:303–15. doi: 10.1016/j.ab.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 26.Szczesna-Cordary D, Jones M, Moore JR, Watt J, Kerrick WGL, Xu Y, Wang Y, Wagg C, Lopaschuk GD. Myosin regulatory light chain E22K mutation results in decreased cardiac intracellular calcium and force transients. FASEB J. 2007;21:3974–3985. doi: 10.1096/fj.07-8630com. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Xu Y, Kerrick WGL, Wang Y, Guzman G, Diaz-Perez Z, Szczesna-Cordary D. Prolonged Ca2+ and Force Transients in Myosin RLC Transgenic Mouse Fibers Expressing Malignant and Benign FHC Mutations. J Mol Biol. 2006;361:286–299. doi: 10.1016/j.jmb.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 28.Kerrick WG, Xu Y. Inorganic phosphate affects the pCa-force relationship more than the pCa-ATPase by increasing the rate of dissociation of force generating cross-bridges in skinned fibers from both EDL and soleus muscles of the rat. J Muscle Res Cell Motil. 2004;25:107–17. doi: 10.1023/b:jure.0000035841.04314.16. [DOI] [PubMed] [Google Scholar]
- 29.Wen Y, Pinto JR, Gomes AV, Xu Y, Wang Y, Wang Y, Potter JD, Kerrick WGL. Functional Consequences of the Human Cardiac Troponin I Hypertrophic Cardiomyopathy Mutation R145G in Transgenic Mice. J Biol Chem. 2008;283:20484–20494. doi: 10.1074/jbc.M801661200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muthu P, Talent JM, Gryczynski I, Borejdo J. Cross-Bridge Duty Cycle in Isometric Contraction of Skeletal Myofibrils. Biochemistry. 2008;47:5657–5667. doi: 10.1021/bi7023223. [DOI] [PubMed] [Google Scholar]
- 31.Huxley AF. A hypothesis for the mechanism of contraction of muscle. Prog Biophys Biophys Chem. 1957;7:255–318. [PubMed] [Google Scholar]
- 32.Brenner B. Effect of Ca2+ on Cross-Bridge Turnover Kinetics in Skinned Single Rabbit Psoas Fibers: Implications for Regulation of Muscle Contraction. Proc Natl Acad Sci USA. 1988;85:3265–3269. doi: 10.1073/pnas.85.9.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huxley AF. Cross-bridge action: present views, prospects, and unknowns. J Biomech. 2000;33:1189–1195. doi: 10.1016/s0021-9290(00)00060-9. [DOI] [PubMed] [Google Scholar]
- 34.Ferenczi MA, Homsher E, Trentham DR. The kinetics of magnesium adenosine triphosphate cleavage in skinned muscle fibres of the rabbit. J Physiol. 1984;352:575–99. doi: 10.1113/jphysiol.1984.sp015311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiesmann F, Frydrychowicz A, Rautenberg J, Illinger R, Rommel E, Haase A, Neubauer S. Analysis of right ventricular function in healthy mice and a murine model of heart failure by in vivo MRI. Am J Physiol Heart Circ Physiol. 2002;283:H1065–71. doi: 10.1152/ajpheart.00802.2001. [DOI] [PubMed] [Google Scholar]
- 36.Morano I, Ritter O, Bonz A, Timek T, Vahl CF, Michel G. Myosin light chain-actin interaction regulates cardiac contractility. Circ Res. 1995;76:720–5. doi: 10.1161/01.res.76.5.720. [DOI] [PubMed] [Google Scholar]
- 37.Rarick HM, Opgenorth TJ, von Geldern TW, Wu-Wong JR, Solaro RJ. An essential myosin light chain peptide induces supramaximal stimulation of cardiac myofibrillar ATPase activity. J Biol Chem. 1996;271:27039–43. doi: 10.1074/jbc.271.43.27039. [DOI] [PubMed] [Google Scholar]
- 38.Miller MS, Palmer BM, Ruch S, Martin LA, Farman GP, Wang Y, Robbins J, Irving TC, Maughan DW. The Essential Light Chain N-terminal Extension Alters Force and Fiber Kinetics in Mouse Cardiac Muscle. J Biol Chem. 2005;280:34427–34434. doi: 10.1074/jbc.M508430200. [DOI] [PubMed] [Google Scholar]
- 39.Haase H, Dobbernack G, Tunnemann G, Karczewski P, Cardoso C, Petzhold D, Schlegel WP, Lutter S, Pierschalek P, Behlke J, Morano I. Minigenes encoding N-terminal domains of human cardiac myosin light chain-1 improve heart function of transgenic rats. FASEB J. 2006;20:865–873. doi: 10.1096/fj.05-5414com. [DOI] [PubMed] [Google Scholar]
- 40.Robinson JM, Wang Y, Kerrick WG, Kawai R, Cheung HC. Activation of striated muscle: nearest-neighbor regulatory-unit and cross-bridge influence on myofilament kinetics. J Mol Biol. 2002;322:1065–88. doi: 10.1016/s0022-2836(02)00855-0. [DOI] [PubMed] [Google Scholar]
- 41.Sweeney HL. Function of the N terminus of the myosin essential light chain of vertebrate striated muscle. Biophys J. 1995;68:112S–118S. discussion 118S-119S. [PMC free article] [PubMed] [Google Scholar]
- 42.Bottinelli R, Betto R, Schiaffino S, Reggiani C. Unloaded shortening velocity and myosin heavy chain and alkali light chain isoform composition in rat skeletal muscle fibres. J Physiol. 1994;478:341–349. doi: 10.1113/jphysiol.1994.sp020254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowey S, Waller GS, Trybus KM. Skeletal muscle myosin light chains are essential for physiological speeds of shortening. Nature. 1993;365:454–6. doi: 10.1038/365454a0. [DOI] [PubMed] [Google Scholar]
- 44.Andruchov O, Galler S. Influence of fast and slow alkali myosin light chain isoforms on the kinetics of stretch-induced force transients of fast-twitch type IIA fibres of rat. Pflugers Arch. 2007 doi: 10.1007/s00424-007-0369-1. [DOI] [PubMed] [Google Scholar]
- 45.Andruchov O, Galler S. Influence of fast and slow alkali myosin light chain isoforms on the kinetics of stretch-induced force transients of fast-twitch type IIA fibres of rat. Pflügers Archiv Eur J Physiol. 2008;455:1165–1172. doi: 10.1007/s00424-007-0369-1. [DOI] [PubMed] [Google Scholar]
- 46.Andruchov O, Andruchova O, Galler S. Fine-tuning of cross-bridge kinetics in cardiac muscle of rat and mouse by myosin light chain isoforms. Pflugers Arch. 2006;452:667–73. doi: 10.1007/s00424-006-0080-7. [DOI] [PubMed] [Google Scholar]
- 47.Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85:164–81. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 48.Solaro RJ, Pang DC, Briggs FN. The purification of cardiac myofibrils with Triton X-100. Biochim Biophys Acta. 1971;245:259–62. doi: 10.1016/0005-2728(71)90033-8. [DOI] [PubMed] [Google Scholar]
- 49.Fiske CH, Subbarow Y. The Colorimetric Determination of Phosphorus. J Biol Chem. 1925;66:375–400. [Google Scholar]
- 50.Hanson KR, Ling R, Havir E. A computer program for fitting data to the Michaelis-Menten equation. Bioch Biophys Res Com. 1967;29:194–197. doi: 10.1016/0006-291x(67)90586-4. [DOI] [PubMed] [Google Scholar]
- 51.Trybus KM. Biochemical studies of myosin. Methods. 2000;22:327–35. doi: 10.1006/meth.2000.1085. [DOI] [PubMed] [Google Scholar]
- 52.Hill TL, Eisenberg E, Chen YD, Podolsky RJ. Some self-consistent two-state sliding filament models of muscle contraction. Biophys J. 1975;15:335–72. doi: 10.1016/S0006-3495(75)85823-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guth K, Wojciechowski R. Perfusion cuvette for the simultaneous measurement of mechanical, optical and energetic parameters of skinned muscle fibres. Pflugers Arch. 1986;407:552–7. doi: 10.1007/BF00657515. [DOI] [PubMed] [Google Scholar]
- 54.Allen K, Xu YY, Kerrick WG. Ca2+ measurements in skinned cardiac fibers: effects of Mg2+ on Ca2+ activation of force and fiber ATPase. J Appl Physiol. 2000;88:180–5. doi: 10.1152/jappl.2000.88.1.180. [DOI] [PubMed] [Google Scholar]
- 55.Heiberg E, Engblom H, Engvall J, Hedstrom E, Ugander M, Arheden H. Semi-automatic quantification of myocardial infarction from delayed contrast enhanced magnetic resonance imaging. Scand Cardiovasc J. 2005;39:267–75. doi: 10.1080/14017430500340543. [DOI] [PubMed] [Google Scholar]
- 56.Rosendahl L, Blomstrand P, Heiberg E, Ohlsson J, Bjorklund PG, Ahlander BM, Engvall J. Computer-assisted calculation of myocardial infarct size shortens the evaluation time of contrast-enhanced cardiac MRI. Clin Physiol Funct Imaging. 2008;28:1–7. doi: 10.1111/j.1475-097X.2007.00765.x. [DOI] [PubMed] [Google Scholar]