

Abstract
The aim of the present study was to investigate the molecular mechanisms underlying the neuroprotective effect of the hydrophilic statin rosuvastatin on cortical neurons exposed to oxygen and glucose deprivation (OGD) followed by reoxygenation. Rosuvastatin (RSV), at concentrations ranging from 10 nM to 1μM, was able to ameliorate the survival of cortical neurons exposed to OGD followed by reoxygenation. This effect was observed either if neurons were pretreated with RSV 24 hrs before OGD/reoxygenation exposure or if RSV was added during the OGD or the reoxygenation phase. Moreover, RSV was also able to improve mitochondrial oxidative capacity in basal conditions, an effect that was already observed at 10 nM either after 24 or after 48 hrs of treatment. These neuroprotective actions were not counteracted by mevalonate, an intermediate of cholesterol biosynthesis that bypasses RSV induced blockade of cholesterol synthesis. Furthermore, the hypothesis that RSV might affect neuronal nitric oxide synthase (nNOS) activity during OGD/reoxygenation was explored. RSV was able to reduce the increase of NO occurring during the reoxygenation phase, an effect prevented by NPLA, the selective inhibitor of nNOS. Finally, the possibility that RSV-induced NO reduction during OGD/reoxygenation might involve ERK1/2 activation was also investigated. The treatment of neurons with PD98059, an ERK1/2 kinase inhibitor, abolished the neuroprotective effect exerted by RSV in cortical neurons exposed to OGD/reoxygenation. In conclusion, these results demonstrated that RSV-induced neuroprotection involves an impairment of constitutive and inducible NOS activity which in turn causes the improvement of mitochondrial function and the stimulation of ERK1/2 via H-Ras activation.
Keywords: Cortical neurons, oxygen glucose deprivation/reoxygenation, nitric oxide, ERK1/2, mitochondria, cell death
Introduction
Statins as inhibitors of HMG-CoA reductase are potent cholesterol lowering drugs. All statins contain an HMG-like component that binds to HMG-CoA reductase. Evidence suggests that statins do more than just lowering cholesterol. Statins enhance NO generation [1], augment the production of the bone morphogenetic protein (BMP)-2 [2], disrupt the function of onco-genic forms of Ras [3], and inhibit the production of proinflammatory cytokines [4] and C-reactive protein [5]. These mechanisms may explain their beneficial actions in many chronic and acute pathological conditions such as Alzheimer's disease [6] and stroke [7]. Indeed, several clinical studies indicate that statins reduce the incidence of stroke in some patients [8,9] and that statin therapy is strongly associated with reduced incidence and severity of stroke and dementia in the central nervous system.
The above mentioned pleiotropic effects of statins are mediated by their ability to block the synthesis of isoprenoids which represent intermediates in the cholesterol biosynthetic pathway. Isoprenoids serve as lipid anchors and activators for a variety of intracellular signalling molecules such as the GTP-binding proteins, Rho, Ras, and Rac, which play a fundamental role in the control of cell growth, signal transduction, and mitogenesis. Particularly, Ras plays a crucial role in the regulation of cell response to oxidative stress, since its isoform Ki-Ras activates the antioxidant mitochondrial Mn-SOD enzyme, whereas Ha-Ras causes NADPH-oxidase stimulation [10]. Therefore, the reduction of isoprenoid levels by statins might result in an accumulation of inactive small G-proteins thus exerting a neuroprotective effect. This hypothesis is supported by the recently described findings that statins are able to restore superoxide dismutase (SOD) 1 expression and to exert antioxidant activity [11]. Moreover, Sironi and colleagues [12] demonstrated that simvastatin exerts a neuroprotective effect in ischemic brain damage by modulating ERK1/2 and NF-kB pathways.
Among the several members of the statin family, rosuvastatin (RSV) is one of the most potent statin and is able to form multiple polar bonds with the HMG-CoA reductase enzyme [13]. Rosuvastatin is also relatively hydrophilic and displays a potential protective effect in cerebral ischemia in mice [14], if administered for 10 days before the ischemic insult. Nevertheless, the molecular mechanisms underlying this effect are not completely clear.
In the present study, we used an in vitro model that mimics cerebral ischemia in vivo to investigate the possible neuroprotective effects of RSV and the molecular mechanisms involved in neuroprotection. Rat cortical neurons were exposed to oxygen-glucose deprivation (OGD) and subsequent reoxygenation in the presence of different concentrations of RSV and the effect on neuronal survival was assessed by MTT assay.
Furthermore, in order to understand whether the mechanism of RSV-induced neuroprotection might be due to RSV inhibition of cholesterol biosynthesis or alternatively might be related to mechanisms independent of the control of cholesterol metabolism, the same experiments were performed in the presence of mevalonate, an intermediate of cholesterol biosynthesis that bypasses RSV induced blockade of cholesterol synthesis.
Materials and methods
Cell Cultures
Postnatal neurons: Mixed cultures of cortical neurons from Wistar rat pups, 2–4 days old, were prepared by modifying a previously described method [15]. The tissue was minced and trypsinized (0.1% for 15 min at 37°C), triturated, plated on poly-D-lysine-coated coverslips, and cultured in Neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen) and 2 mM L-glutamine. Cells were plated at concentration of 1.8×106 on 25-mm glass coverslips and at a concentration of 0,8-1×106 on twelve plastic multiwells both pre-coated with poly-D-lysine (10 μg/ml). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air, fed twice a week, and maintained for a minimum of 10 days before experimental use.
Embryonic neurons: Pure Cortical neurons were prepared from brains of 16-day-old Wistar rat embryos [16]. Briefly, the rats were first anesthetized and then decapitated. Dissection and dissociation of brains were performed in Ca2+/Mg2+-free phosphate-buffered saline (PBS) containing glucose (30 mM). Tissues were incubated with papain for 10 min at 37° C and dissociated by trituration in EBSS containing DNAse, bovine serum albumine (BSA), and ovomucoid. Cells were plated at a concentration of 0,8-1×106 in twelve plastic multiwells pre-coated with poly-D-lysine (20 μg/ml) in MEM/F12 (Life Technology) - containing glucose, 5% deactivated Fetal Calf Serum (FCS) and 5% Horse Serum (HS) (Life Technology), glutamine, and antibiotics - at a concentration of 5×106. Ara-C (10 μM) was added within 48 hrs of plating to prevent the growth of non-neuronal cells. Neurons were cultured at 37°C in a humidified 5% CO2 atmosphere and used after 12 days of culture.
Combined oxygen and glucose deprivation and reoxygenation
Cortical neurons were exposed to OGD for 3 hrs followed by 24 hrs reoxygenation according to a previously reported protocol [17]. Briefly, the culture medium was replaced with a hypoxia medium previously saturated for 20 min with 95% N2 and 5% CO2 and containing NaCl 116 mM, KCl 5.4 mM, MgSO4 0.8 mM, NaHCO3 26.2 mM, NaH2PO4 1 mM, CaCl2 1.8 mM, glycine 0.01 mM and 0.001 w/v phenol red. Hypoxic conditions were maintained using a hypoxia chamber (temperature 37°C, atmosphere 95% N2 and 5% CO2). These experimental conditions induced 30% decrease of pO2 in the medium.
Deprivation of oxygen and glucose was stopped by placing the cells in the regular culture medium saturated with a mixture of 95% O2 and 5% CO2 for 10 min. Reoxygenation was achieved by returning neurons to normoxic conditions (37°C in a humidified 5% CO2 atmosphere) for 24 hrs.
MTT assay
Mitochondrial activity was assessed by measuring the level of mitochondrial dehydrogenase activity using 3-(4,5-dimethylthiazol-2-yl)-2,5, dipheniltetrazolium bromide (MTT) as substrate [18, 19]. The assay was based on the redox ability of living mitochondria to convert dissolved MTT into insoluble formazan. Briefly, after treatments, the medium was removed and cells were incubated in 1 ml of MTT solution (0.5 mg/ml) for 1 hr in a humidified 5% CO2 incubator at 37°C. To stop incubation, MTT solution was removed and 1 ml DMSO was added to solubilize the formazan product. The absorbance was monitored at 540 nm with a Perkin Elmer LS55 Luminescence Spectrometer. The data were expressed as a percentage of cell viability compared to sham-treated cultures.
Imaging mitochondrial membrane potential and calcium concentrations
Mitochondrial membrane potential was assessed using the fluorescent dye tetramethyl rhodamine ethyl ester (TMRE) in the “redistribution mode” [20]. Cortical neurons were loaded with TMRE 20 nM for 30 min in a medium containing in mM: 156 NaCl, 3 KCl, 2 MgSO4, 1.25 KH2PO4, 2 CaCl2, 10 glucose, and 10 Hepes, pH adjusted to 7.35 with NaOH [21]. At the end of the incubation, cells were washed in the same medium in which the dye was still present at 20 nM and allowed to equilibrate. A decline of mitochondrial localized intensity of fluorescence was indicative of mitochondrial membrane depolarization. Mitochondrial calcium concentration was assessed by incubating the cells with the fluorescent dye X-Rhod-1-AM at a concentration of 5 μM in the above described medium for 15 min. Calcium increase within mitochondria was expressed by an improvement of fluorescence intensity. Confocal images were obtained using a Ziess inverted 510 confocal laser scanning microscopy and a 63X oil immersion objective lens. The illumination intensity of 543 Xenon laser, used to excite TMRE and X-Rhod-1-AM fluorescence, was kept to a minimum of 0.5% of laser output in order to avoid phototoxicity.
NO detection
NO production was assayed by the Griess method as previously described [22].
Treatments
Rosuvastatin (AstraZeneca) was dissolved in water to a concentration of 7mM (Stock solution) and diluted for use at final concentrations ranging from 1 μM to 10 nM. Rosuvastatin has been added to the culture medium as reported below: (1) in the culture medium 24 hrs before OGD exposure, in the hypoxia medium for 3 hrs OGD, in the culture medium for 24 hrs reoxygenation; (2) in the hypoxia medium for 3 hrs OGD; (3) in the culture medium for 24 hrs reoxygenation. Mevalonate was purchased from Sigma-Aldrich (St.Louis, MO) and applied at the final concentration of 76 μM. The specific inhibitor of nNOS activity, NPLA, was added to the culture medium at a concentration of 5 nM. The specific ERK1/2 inhibitor PD98059 was used at the final concentration of 40 μM. Cortical neurons were pre-treated for 24 hrs with NPLA (5 nM) or with PD98059 (40 μM) in the culture medium and than exposed to OGD/ reoxygenation.
Results
The first part of this study was carried out to identify the RSV concentration that would exert a neuroprotective effect in neurons exposed to 3 hr of OGD followed by 24 hrs of reoxygenation. RSV in concentrations ranging from 10 nM to 1μM (10 nM, 100 nM, 1 μM), was added to cortical neurons 24 hrs before OGD until the end of the reoxygenation period (24 hrs). As expected, the exposure of neurons to 3 hrs OGD caused a significant impairment of mitochondrial function. This effect was completely abolished if neurons were pretreated 24 hrs before OGD and during the period of OGD with RSV at the concentrations of 10 nM, 100 nM and 1 μM, (Figure 1A). RSV already exerted its neuroprotective effect at the concentration of 10 nM. RSV, at all three concentrations tested, was also able to prevent the impairment of mitochondrial function occurring in cortical neurons exposed to OGD followed by 24 hrs reoxygenation, a model mimicking ischemia followed by reperfusion (Figure 1B). Interestingly, RSV was also able to improve neuronal survival in basal conditions, an effect that already reached significance at a concentration of 10 nM either after 24 or 48 hrs of treatment (Figure 1A and B).
Figure 1.

Dose-response effect of Rosuvastatin on neuronal survival in cortical neurons exposed to OGD and OGD/reoxygenation. A. Cortical neurons were pre-treated for 24 hrs with RSV at concentrations of 10 nM, 100 nM and 1 μM in culture medium and then exposed to 3 hrs OGD in the presence of RSV. B. Cortical neurons were pre-treated with RSV for 24 hrs and then exposed to OGD followed by 24 hrs reoxygenation in the continuos presence of RSV. At the end of the treatments mitochondrial oxidative capacity was evaluated by MTT assay. Data are expressed as a percentage ± SEM vs. control untreated cells *P<0.05 vs. its respective control group; **P<0.05 vs OGD or OGD/Rx groups. CTL=Control, OGD=Oxygen and Glucose Deprivation, Rx = reoxygenation.
In order to identify the time window in which RSV exerts its neu-roprotective effect in cortical neurons exposed to OGD followed by reoxygenation, the two RSV concentrations of 10 nM and 1 μM, that previously displayed a significant protection, were added: (a) just before the exposure of neurons to OGD, and (b) at the beginning of the reoxygenation time. The effects of these treatments were compared to those observed by exposing neurons to RSV 24 hr before OGD, during the OGD and after the reoxygenation phase. The results obtained showed that the exposure of neurons to RSV (10 nM and 1 μM) just before the induction of OGD prevented the detrimental effect exerted by OGD alone (Figure 2A). Interestingly, the impairment of neuronal survival induced by OGD/reoxygenation was also abolished by RSV when it was administered to neurons only at the beginning of the reoxygenation phase (Figure 2B).
Figure 2.

Time window of the effect of Rosuvastatin on neuronal survival in cortical neurons exposed to OGD or to OGD/reoxygenation. A. Cortical neurons were treated with RSV at the concentrations of 10 nM and 1 μM in the OGD phase. At the end of the treatment mitochondrial oxidative capacity was evaluated by MTT assay. Data are expressed as a percentage + SEM vs. control untreated cells. *P<0.05 vs. respective control; **P<0.05 vs. OGD group. B. Cortical neurons were exposed to 3 hr OGD in the absence of RSV and subsequently to 24 hrs reoxygenation in the presence of 10 nM and 1 μM RSV concentration. At the end of the treatments mitochondrial oxidative capacity was evaluated by MTT assay. Data are expressed as a percentage + SEM vs. control untreated cells *P<0.05 vs its respective control group; **P<0.05 vs OGD/Rx group. CTL=Control, OGD=Oxygen and Glucose Deprivation, RX= reoxygenation.
Since we observed that the treatment with RSV at the concentrations of 10 nM and 1 μM induced a time-dependent increase in MTT levels in basal conditions, an index of mitochondrial oxidative capacity, the effect of 1 μM RSV on mitochondrial membrane potential and mitochondrial calcium concentrations was explored by confocal microscopy analysis. The results obtained showed that treatment of cortical neurons with RSV for 48 hrs in basal conditions induced mitochondrial membrane hyperpolarization. Similarly, RSV was able to lower mitochondrial calcium concentrations, thus indicating that RSV may improve mitochondrial function (Figure 3).
Figure 3.
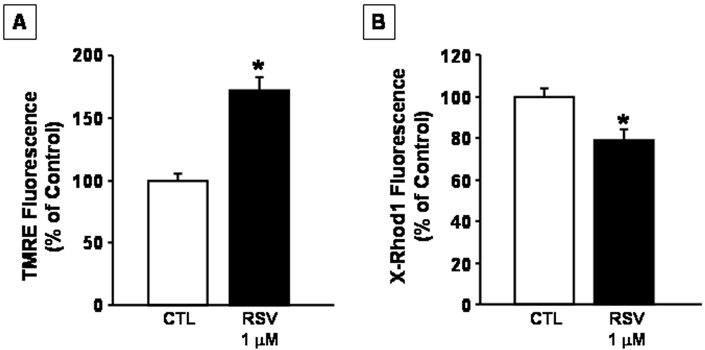
Effect of Rosuvastatin on mitochondrial functions. A. Effect of RSV on mitochondrial membrane potential. Cortical neurons were treated with Rosuvastatin 1μM added to the culture medium for 48 hrs and than loaded with TMRE and examined by confocal microscopy. B. Effect of RSV on mitochondrial calcium concentration. Cortical neurons were treated for 48 hrs with RSV 1μM added to the culture medium and then loaded with X -Rhod 1-AM and examined by confocal microscopy. Data are expressed as a percentage of fluorescence + SEM vs control untreated cells. *P<0.05 vs control group. CTL=Control, RSV =Rosuvastatin.
To establish whether the mechanism of RSV-induced neuroprotection was due to the inhibition of HMG-CoA reductase, neurons subjected to OGD/reoxygenation were incubated with both mevalonate, an intermediate of cholesterol biosynthesis that bypasses RSV induced blockade of cholesterol synthesis, and RSV (10nM). The results obtained demonstrated that neuroprotection induced by RSV was still observed in the presence of mevalonate (Figure 4) thus suggesting that the statin effect was not related to the interference with cholesterol metabolism. Furthermore, to investigate whether RSV-induced neuroprotection might be mediated by a reduction in NO production, further experiments were performed in cortical neurons treated with RSV and then exposed to OGD/ reoxygenation in the presence of the selective inhibitor of nNOS, NPLA [23]. The results obtained demonstrated that RSV was able to reduce the increase in NO occurring during the reoxygenation phase (Figure 5A). Interestingly, NPLA was able to prevent the neuro-protective effect played by RSV on survival of cortical neurons exposed to OGD/ reoxygenation. Moreover, the possibility that RSV-induced NO impairment during OGD/ reoxygenation might involve ERK1/2 pathway was also investigated. To this aim cortical neurons were treated with RSV in the presence of PD98059, an ERK1/2 kinase inhibitor [22]. As observed with NPLA, PD98059 was also able to counteract the neuroprotective effect exerted by RSV in cortical neurons exposed to OGD/ reoxygenation (Figure 5B).
Figure 4.

Effect of mevalonate on rosuvastatin-induced neuroprotection in cortical neurons exposed to OGD/reoxygenation. Cortical neurons were pre-treated for 24 hrs with RSV (10 nM) and with mevalonate (76 μM) in the culture medium and then exposed to OGD/reoxygenation. At the end of the treatments mitochondrial oxidative capacity was evaluated by MTT assay. Data are expressed as a percentage ± SEM vs. control untreated cells. *P<0.05 vs its respective control group; **P<0.05 vs OGD/Rx group.
Figure 5.

Effect of Rosuvastatin (10 nM) on NO production in cortical neurons exposed to OGD/reoxygenation. The cells were pre-treated with RSV for 24 hrs and, after OGD/reoxygenation, NO production was assayed by the Griess method. Data are expressed as a percentage + SEM vs control untreated cells. *P<0.05 vs its respective control group; **P<0.05 vs OGD/Rx group. B. Effect of NPLA and PD98059 on survival of cortical neurons exposed to OGD/reoxygenation in the presence of Rosuvastatin. Cortical neurons were pre-treated for 24 hrs with RSV (10 nM) and with NPLA (5 nM) or with PD98059 (40 μM) in the culture medium and then exposed to OGD/reoxygenation. At the end of the treatments mitochondrial oxidative capacity was evaluated by MTT assay. Data are expressed as a percentage +SEM vs. control untreated cells. *P<0.05 vs its respective control group; **P<0.05 vs RSV treated group; #P<0.05 vs OGD/Rx group; ##P*<0.05 vs OGD/Rx + RSV group
Discussion
The results described in the present study demonstrated that RSV exerts neuroprotective effects in neurons exposed to OGD and OGD/ reoxygenation both in a short and in a long time window since neuroprotection still occurred in neurons treated with RSV just in the OGD phase (3 hrs exposure), or in the reoxygenation period (24 hrs exposure). Experiments performed in the presence of mevalonate, an intermediate of cholesterol biosynthesis that bypasses RSV induced blockade of cholesterol synthesis, suggest that the beneficial effect of RSV was continued. Mevalonate is in fact produced downstream of HMG-CoA reductase and it represents an intermediate in the common pathway of cholesterol and nonsterol biosynthesis. It has been hypothesized that the inhibition of HMG-CoA reductase by statins through the impairment of mevalonate formation might deplete the synthesis of intermediate isoprenoids thereby preventing isoprenylation of small GTPases [24]. This effect results in the blockade of the activity of these signaling molecules which could explain the neuroprotective effects of statins observed in in vitro or in vivo models of ischemia [25]. In this regard, it has been reported that RSV may up-regulate eNOS through Rho GTPase inhibition, may inhibit iNOS by inhibiting NF-kB, but it does not affect nNOS activity [26]. All these effects seem to be reverted by mevalonate, thus confirming that the blockade of intermediate isoprenoids is a possible mechanism to explain RSV-induced neuroprotection in ischemia as well as in other neurodegenerative diseases [6].
The results obtained in the present study let to the hypothesis that RSV-induced neuroprotection might be related to: (1) an improvement of mitochondrial oxidative capacity, (2) an impairment of NO production and (3) a reduction of ERK1/2 activation. Several considerations further support this hypothesis. First of all, the finding that neuroprotection in cortical neurons also occurred when RSV was added to the cells in the reoxygenation phase, during which mitochondria are more sensitive to the oxidative stress, suggests that mitochondria represent a target of RSV. Moreover, the finding that NPLA, a selective inhibitor of nNOS, was able to prevent the beneficial effect exerted by RSV both in basal conditions and after OGD/reoxygenation, strongly suggests that an interference with the pathway responsible for NO production represents the key mechanism leading to RSV-induced neuroprotection. These results are in line with those previously obtained in cerebellar neurons exposed to OGD and OGD/Rx [27]. Moreover, we also demonstrated that the beneficial effects of RSV on neuronal survival might involve the activation of ERK1/2, a member of the family of MAP Kinases, since the treatment with the selective inhibitor PD98059 was able to completely abolish RSV-induced neuroprotection in cortical neurons exposed to OGD/ reoxygenation. This effect might be correlated to RSV-induced NO inhibition during OGD/ reoxygenation. Indeed, it has recently been demonstrated that NO was able to induce H-Ras S-nitrosylation and in turn to inhibit ERK1/2 activity in HEK-293-nNOS transfected cells [28]. Therefore it is possible that RSV, by reducing NO production during OGD/reoxygenation, indirectly stimulates ERK1/2 activity thereby preventing H -RAS-nytrosilation, and thus inducing neuroprotection. Nevertheless, the finding that the effects of RSV in basal conditions were not affected by PD98059, but were prevented by NPLA, led to the hypothesis that the beneficial activity of RSV in basal conditions might be related to a reduction of NO production and not to an involvement of ERK1/2 activation. The increase in mitochondrial membrane potential (hyperpolarization) and the lowering of mitochondrial calcium concentration obtained with RSV treatment in basal conditions support this hypothesis. In fact, it is well known that mitochondria are a target for NO, which can compete with O2 and inhibit mitochondrial cytochrome oxidase [29]. Moreover, peroxynitrite, a product of NO interaction with reactive oxygen species (ROS), may also inhibit NADH-ubiquinone oxidoreductase thus impairing mitochondrial respiration [30]. Finally, it has also been reported that NO dissipates mitochondrial membrane potential and induces neuronal damage [31]. All these above mentioned actions of RSV might render mitochondria more resistant to oxidative stress occurring during OGD/reoxygenation.
In conclusion, the results of the present study demonstrated that the molecular mechanisms underlying the neuroprotective effect of RSV are different from those involved in the hypocholesterolemic actions of statins. This mechanism involves an impairment of constitutive and inducible NOS activity which in turn causes the improvement of mitochondrial function and the stimulation of ERK1/2 via H-Ras activation.
Acknowledgments
The authors of the present work are truly grateful to Mr Vincenzo Grillo in setting up the primary neuronal cultures, Dr. Paola Merolla for editorial revision, and to Astrazeneca in providing Rosuvastatin. This work was supported by an AstraZeneca grant to LA.
References
- 1.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mundy G, Garrett R, Harris S, Chan J, Chen D, Rossini G, Boyce B, Zhao M, Gutierrez G. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999;286:1946–1949. doi: 10.1126/science.286.5446.1946. [DOI] [PubMed] [Google Scholar]
- 3.Chan KK, Oza AM, Siu LL. The statins as anticancer agents. Clin Cancer Res. 2003;9:10–19. [PubMed] [Google Scholar]
- 4.Rosenson RS, Tangney CC, Casey LC. Inhibition of proinflammatory cytokine production by pravastatin. Lancet. 1999;353:983–984. doi: 10.1016/S0140-6736(98)05917-0. [DOI] [PubMed] [Google Scholar]
- 5.Strandberg TE, Vanhanen H, Tikkanen MJ. Effect of statins on C-reactive protein in patients with coronary artery disease. Lancet. 1999;353:118–119. doi: 10.1016/S0140-6736(05)76154-7. [DOI] [PubMed] [Google Scholar]
- 6.Frears ER, Stephens DJ, Walters CE, Davies H, Austen BM. The role of cholesterol in the biosynthesis of beta-amyloid. Neuroreport. 1999;10:1699–1705. doi: 10.1097/00001756-199906030-00014. [DOI] [PubMed] [Google Scholar]
- 7.Rosendorff C. Statins for prevention of stroke. Lancet. 1998;351:1002–1003. doi: 10.1016/S0140-6736(05)78991-1. [DOI] [PubMed] [Google Scholar]
- 8.Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–1307. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- 9.Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 10.Santillo M, Mondola P, Seru R, Annella T, Cassano S, Ciullo I, Tecce MF, Iacomino G, Damiano S, Cuda G, Paterno R, Martignetti V, Mele E, Feliciello A, Avvedimento EV. Opposing functions of Ki- and Ha-Ras genes in the regulation of redox signals. Curr Biol. 2001;11:614–619. doi: 10.1016/s0960-9822(01)00159-2. [DOI] [PubMed] [Google Scholar]
- 11.Verret L, Trouche S, Zerwas M, Rampon C. Hippocampal neurogenesis during normal and pathological aging. Psychoneuroendocrinology. 2007;32(Suppl 1):S26–30. doi: 10.1016/j.psyneuen.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 12.Sironi L, Banfi C, Brioschi M, Gelosa P, Guerrini U, Nobili E, Gianella A, Paoletti R, Tremoli E, Cimino M. Activation of NF-kB and ERK1/2 after permanent focal ischemia is abolished by simvastatin treatment. Neurobiol Dis. 2006;22:445–451. doi: 10.1016/j.nbd.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 14.Laufs U, Gertz K, Dirnagl U, Bohm M, Nickenig G, Endres M. Rosuvastatin, a new HMG-CoA reductase inhibitor, upregulates endothelial nitric oxide synthase and protects from ischemic stroke in mice. Brain Res. 2002;942:23–30. doi: 10.1016/s0006-8993(02)02649-5. [DOI] [PubMed] [Google Scholar]
- 15.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scorziello A, Pellegrini C, Forte L, Tortiglione A, Gioielli A, Iossa S, Amoroso S, Tufano R, Di Renzo G, Annunziato L. Differential vulnerability of cortical and cerebellar neurons in primary culture to oxygen glucose deprivation followed by reoxygenation. J Neurosci Res. 2001;63:20–26. doi: 10.1002/1097-4547(20010101)63:1<20::AID-JNR3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 17.Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 19.Amoroso S, Gioielli A, Cataldi M, Di Renzo G, Annunziato L. In the neuronal cell line SH-SY5Y, oxidative stress-induced free radical overproduction causes cell death without any participation of intracellular Ca(2+) increase. Biochim BiophysActa. 1999;1452:151–160. doi: 10.1016/s0167-4889(99)00110-x. [DOI] [PubMed] [Google Scholar]
- 20.Livigni A, Scorziello A, Agnese S, Adornetto A, Carlucci A, Garbi C, Castaldo I, Annunziato L, Avvedimento EV, Feliciello A. Mitochondrial AKAP121 links cAMP and src signaling to oxidative metabolism. Mol Biol Cell. 2006;17:263–271. doi: 10.1091/mbc.E05-09-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abramov AY, Canevari L, Duchen MR. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci. 2004;24:565–575. doi: 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scorziello A, Santillo M, Adornetto A, Dell'aversano C, Sirabella R, Damiano S, Canzoniero LM, Renzo GF, Annunziato L. NO-induced neuroprotection in ischemic preconditioning stimulates mitochondrial Mn-SOD activity and expression via Ras/ERK1/2 pathway. J Neurochem. 2007;103:1472–1480. doi: 10.1111/j.1471-4159.2007.04845.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhang HQ, Fast W, Marletta MA, Martasek P, Silverman RB. Potent and selective inhibition of neuronal nitric oxide synthase by N omega-propyl-L-arginine. J Med Chem. 1997;40:3869–3870. doi: 10.1021/jm970550g. [DOI] [PubMed] [Google Scholar]
- 24.Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res. 2005;97:1232–1235. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Die J, Wang K, Fan L, Jiang Y, Shi Z. Rosuvastatin preconditioning provides neuroprotection against spinal cord ischemia in rats through modulating nitric oxide synthase expressions. Brain Res. 2010;1346:251–261. doi: 10.1016/j.brainres.2010.05.068. [DOI] [PubMed] [Google Scholar]
- 26.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 27.Scorziello A, Pellegrini C, Secondo A, Sirabella R, Formisano L, Sibaud L, Amoroso S, Canzoniero LM, Annunziato L, Di Renzo GF. Neuronal NOS activation during oxygen and glucose deprivation triggers cerebellar granule cell death in the later reoxygenation phase. J Neurosci Res. 2004;76:812–821. doi: 10.1002/jnr.20096. [DOI] [PubMed] [Google Scholar]
- 28.Raines KW, Cao GL, Lee EK, Rosen GM, Shapiro P. Neuronal nitric oxide synthase-induced S-nitrosylation of H-Ras inhibits calcium ionophore-mediated extracellular-signal-regulated kinase activity. Biochem J. 2006;397:329–336. doi: 10.1042/BJ20052002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- 30.Brorson JR, Schumacker PT, Zhang H. Nitric oxide acutely inhibits neuronal energy production. The Committees on Neurobiology and Cell Physiology. J Neurosci. 1999;19:147–158. doi: 10.1523/JNEUROSCI.19-01-00147.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rintoul GL, Bennett VJ, Papaconstandinou NA, Reynolds IJ. Nitric oxide inhibits mitochondrial movement in forebrain neurons associated with disruption of mitochondrial membrane potential. J Neurochem. 2006;97:800–806. doi: 10.1111/j.1471-4159.2006.03788.x. [DOI] [PubMed] [Google Scholar]