

Abstract
Macrophage recruitment to sites of inflammation is an essential step in host defense. However, the mechanisms preventing excessive accumulation of macrophages remain relatively unknown. The lysophospholipid sphingosine 1-phosphate (S1P) promotes T and B cell egress from lymphoid organs by acting on S1P receptor 1 (S1P1R). More recently, S1P5R was shown to regulate NK cell mobilization during inflammation, raising the possibility that S1P regulates the trafficking of other leukocyte lineages. In this study, we show that S1P2R inhibits macrophage migration in vitro and that S1P2R-deficient mice have enhanced macrophage recruitment during thioglycollate peritonitis. We identify the signaling mechanisms used by S1P2R in macrophages, involving the second messenger cAMP and inhibition of Akt phosphorylation. In addition, we show that the phosphoinositide phosphatase and tensin homolog deleted on chromosome 10, which has been suggested to mediate S1P2R effects in other cell types, does not mediate S1P2R inhibition in macrophages. Our results suggest that S1P serves as a negative regulator of macrophage recruitment by inhibiting migration in these cells and identify an additional facet to the regulation of leukocyte trafficking by S1P.
Sphingosine 1-phosphate (S1P) is a bioactive lipid with roles in diverse physiologic processes including angiogenesis, lymphocyte recirculation, and vascular permeability (1–3). S1P exerts its biological effects by signaling through five G protein-coupled receptors (S1P receptors 1–5 [S1P S1P1–5R]). At the cellular level, S1PRs regulate the migration of a variety of cell types, including endothelial cells, smooth muscle cells, lymphocytes, and cancer cells. S1P1R and S1P2R have opposing effects on cell migration. S1P1R is a Gi-coupled receptor that stimulates migration in endothelial cells and lymphocytes (4, 5). In contrast, S1P2R is a G12/13-coupled receptor known to inhibit migration in a variety of cell types, including cancer cells, smooth muscle cells, and fibroblasts (6–10). Importantly, S1P also inhibits the trans-endothelial migration of neutrophils (6). The S1P2R-mediated inhibition of migration involves altered Rho GTPase activity and the inositol phosphatase and tensin homolog (PTEN) deleted on chromosome 10 (8–13).
S1P is concentrated in blood (and to a lesser extent in lymph), whereas S1P levels in interstitial fluids are much lower (14). Thus, a gradient of S1P normally exists between the vascular space and interstitial fluid, forming the vascular S1P gradient. The existence of this gradient is particularly relevant to leukocytes, which rely on chemotactic gradients for homing to lymphoid organs and sites of inflammation. Indeed, the vascular S1P gradient plays a central role in lymphocyte egress from lymphoid organs, where S1P1R has been shown to direct the chemotaxis of lymphocytes out of lymphoid tissue and toward S1P in blood and lymph (4). S1P has also been shown to be chemotactic for dendritic cells (DCs) (15–17). Consistent with a role for S1P1R, the immunosuppressant FTY720, a functional antagonist of S1P1R, inhibits the migration of DCs toward S1P in vitro and to lymph nodes (LNs) in vivo (15, 18, 19). More recently, osteoclasts and NK cells have been shown to use S1P for trafficking (20, 21).
Macrophages are monocyte DCs of the innate immune system important in combating infection, wound repair, and adaptive immunity (22). Macrophages modulate immune responses, performing proinflammatory and anti-inflammatory roles by secreting cytokines and lipid mediators. They are proficient in the phagocytosis of pathogens and cellular debris, enabling them to clear infections and restore tissue homeostasis. Inherent in their role as immune regulators is the ability of the macrophage lineage to respond to immune challenge by migrating to sites of infection and inflammation. Together with varying cellular phenotypes, macrophages display a wide array of migratory behaviors. For instance, subtypes of macrophages are long-term residents in tissues, whereas others are highly motile cells generated from monocytes during inflammation. Once recruited to sites of inflammation, these motile macrophages actively drain to LNs (23). Macrophage chemotaxis is stimulated by microbial products, complement, and chemokines, allowing them to seek out pathogenic, inflammatory, and non-inflammatory foci. The chemoattractants responsible for recruiting macrophage during inflammation include the CC-motif family of chemokines. Yet, the negative regulation of macrophage migration, and the negative regulation of leukocyte chemotaxis in general, remain poorly understood.
Although S1P is known to regulate the migration of lymphocytes and DCs, the role of S1P in macrophage migration remains unknown. In this study, we characterize the role of S1PRs on macrophage migration. Using the thioglycollate (TG) peritonitis model of acute inflammation and S1p2r knockout mice, we show that S1P2R negatively regulates the accumulation of macrophage during inflammation. In vitro, we show that S1P inhibits macrophage migration through S1P2R ligation. S1P2R signaling stimulates cAMP production and inhibits chemoattractant-induced Akt phosphorylation. However, chemoattractant-induced receptor signaling remains intact, including calcium flux and MAPK phosphorylation. In addition, protein kinase A (PKA)-selective cAMP analogs mimic the effects of S1P2R signaling, inhibiting migration, and Akt phosphorylation. Our results demonstrate that S1P2R can influence macrophage recruitment during inflammation, and we identify a novel mechanism for S1P2R in regulating leukocyte migration in vivo.
Materials and Methods
Mice
S1p2r−/− mice were obtained from Dr. Richard Proia (National Institutes of Health, Bethesda, MD), Ptenlox/loxP mice were obtained from The Jackson Laboratory (Bar Harbor, ME), and Rosa26 CreERT2 mice were obtained from Dr. Guo-Hua Fong (University of Connecticut Health Center, Farmington, CT) (24). All mice were fertile and had developed normally. Mice were genotyped as described (25, 26) and housed under specific pathogen-free conditions. Experimental procedures were approved by the University of Connecticut Animal Care and Use Committee.
Reagents and Abs
The following Abs were purchased from eBioscience (San Diego, CA): APC anti-CD11b, PE anti-F4/80, PE-Cy7 anti-CD11c, and Pacific Blue anti-CD45. HBSS and DMEM were from Invitrogen (San Diego, CA), 8-Br-cAMP and fura 2 were from Calbiochem (San Diego, CA), and 8-pCPT-2′-O-Me-cAMP and 6-Phe-cAMP were from Biolog (Bremen, Germany). Recombinant mouse C5a and CXCL12 were from R&D Systems (Minneapolis, MN). Tamoxifen and fatty acid-free BSA were from Sigma-Aldrich (St. Louis, MO). TG was from Fisher (Pittsburgh, PA). S1P was from Biomol (Plymouth Meeting, PA).
TG peritonitis
Twelve-week-old mice were injected i.p. with 1 ml sterile TG (3% w/v). At indicated times after challenge, mice were euthanized by using CO2, peritoneum lavaged with 5 ml cold HBSS, and draining para-thymic and nondraining ileal LNs collected. LN cell suspensions were obtained by brief dissection, collagenase digestion, and filtering through 40-μm nylon mesh. Total leukocyte counts were determined using a hemocytometer and cell suspensions were analyzed by flow cytometry. CD45+ macrophage, polymorphonuclear neutrophils, and lymphocytes were delineated using forward and side scatter characteristics, CD11b, F4/80, and CD11c.
Generation of bone marrow macrophage
Bone marrow was collected by aseptically removing femurs and tibias of S1p2r+/+ and S1p2r−/− mice and flushing with cold HBSS using a 25G needle. Cell suspensions were then centrifuged, lysed of red cells using ammonium chloride, and cultured in sterile bacterial dishes for 7 d in DMEM supplemented with 10% FBS and 20% L cell conditioned media, as a source of M-CSF. The resulting cultures were analyzed by flow cytometry for CD11b and F4/80 expression. To generate PTEN-deficient bone marrow-derived macrophages (BMDM), PtenloxP/loxP. Rosa26 CreERT2, and PtenloxP/loxP mice were fed 200 mg/kg tamoxifen in corn oil by oral gavage for 5 consecutive days. One day later BMDM were cultured from total bone marrow. PTEN- and S1P2R-deficiency had no effect on the efficiency of BMDM generation, which typically yielded ~95% CD11b+ F4/80+ cells.
Peritonitis in S1P1R−/− fetal liver chimeras
Livers from CD45.2 S1P1R−/− or S1P1R+/+ mice were collected at day 13.5, and used to reconstitute CD45.1 mice. Recipient mice were subjected to whole body irradiation from a [137Cs] radiation source (Gamma cell-40, MDS Nordion, Kanata, Ontario, Canada) in two fractions 4 h apart at total doses of 10 Gy. For bone marrow transplant experiments, recipient wild-type mice (WT CD45.1) were reconstituted with 1 × 106 fetal liver cells via the lateral tail vein immediately after whole body irradiation. Engraftments were confirmed by flow cytometry analysis of peripheral blood CD45.1 cell numbers. Ten weeks after engraftment, chimeric mice were given TG peritonitis and peritoneal cell numbers analyzed by flow cytometry.
In vitro macrophage chemotaxis
Chemotaxis assays were performed using a modified Boyden chamber (NeuroProbe, Gaithersburg, MD). BMDM were washed in PBS, and serum starved for 2 h in DMEM. For cAMP studies, cells were pretreated for 30 min prior to migration. Cells were removed from dishes using 5 mM EDTA in PBS and resuspended in DMEM. The 3 × 105 cells were placed in the top well of the migration chamber and separated from chemoattractants in the bottom well by a filter with 5-μm pores. Cells were allowed to migrate at 37° C, 5% CO2 for 2 h, filters were fixed in 4% paraformaldehyde, the top surface wiped clean of nonmigrating cells, and the filters were stained in 0.1% crystal violet. Migration was quantified by eluting the crystal violet in 10% acetic acid and measuring the OD at 495 nm.
Live cell chemotaxis was performed using a Dunn chamber (Hawksley, Lansing, U.K.) (27). BMDM were washed in PBS and starved for 2 h in DMEM. Cells removed from petri dishes using 5 mM EDTA in PBS and resuspended in DMEM with 2% charcoal-stripped FBS and 10 mM HEPES. The 100 × 105 cells were allowed to adhere to precleaned glass coverslips and placed inverted on top of the Dunn chamber. Chemoattractants were placed in the central well and cell chemotaxis was recorded at 37°C, 5% CO2 for 2 h using an Axiovert 200M 510 Meta confocal microscope (Zeiss, Jena, Germany). Image analysis was performed using MetaMorph (Molecular Devices, Sunnyvale, CA).
Western blot and mRNA analysis
BMDM were grown in 10-cm sterile bacterial dishes, stimulated, washed in PBS, and lysed with RIPA buffer (10 mM Tris-Cl pH 8.0, 0.5 mM EGTA, 1% Triton X-100, 0.1% Na deoxycholate, 0.1% SDS, 140 mM NaCl, 1 mM Na orthovanadate, and 1 mM NaF). Equal amounts of protein were separated by 10% SDS-PAGE and blotted onto a nitrocellulose membrane. Immunoblot analyses were performed using phosphospecific Abs for p42/44 MAPK, AKT (Cell Signaling, Danvers, MA), and α p21-activated kinase (PAK, Santa Cruz Biotechnology, Santa Cruz, CA). mRNA expression analysis, RNA extraction, cDNA synthesis, and real-time PCR analysis were performed as described (7). Briefly, total RNA was extracted from elicited neutrophils using RNA-STAT-60 (Tel-Test, Friendswood, TX) according to the manufacturer’s instructions. RNA was treated with DNase I, followed by reverse transcription. Equal amounts of cDNA were run in duplicate SYBR green PCR reactions using an ABI 7900HT sequence detection system (Applied Biosystems, Foster City, CA). Data were analyzed to account for reaction efficiency (28) and results expressed relative to β-actin expression.
Measurement of intracellular Ca2+ concentration
Cells were sedimented, resuspended in HEPES-buffered medium, containing 20 mM HEPES (pH 7.4), 103 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 0.5 mM CaCl2, 25 mM NaHCO3, 15 mM glucose, and 0.1% BSA (fatty acid-free), and then incubated for 40 min with 5 μM fura 2-AM. Fluorescence emission at 510 nm after excitation at 340 nm wavelength was measured every 0.1 s using a FluoroLog fluorescent spectrophotometer (HORIBA Jobin Yvon, Edison, NJ) and the amount of [Ca2+]i was estimated from the change in the fluorescence of fura 2-loaded cells as described previously (29).
cAMP measurement
cAMP was measured using a cAMP ELISA according to the manufacturer’s instructions (Assay Designs, Ann Arbor, MI). Briefly, BMDM were pre-treated for 10 min with 200 μM 3-isobutyl-1-methylxanthine in HBSS, stimulated with S1P for 15 min, and cAMP was extracted with HCl.
Statistical analysis
Results are representative of three to five independent experiments, including in vivo experiments comparing independent groups of mice, and in vitro cell cultures obtained from separate mice. Statistical significance was determined by unpaired Student t test, or χ2 test (where appropriate), using p < 0.05 as significant. Statistical results are represented as: *p < 0.05; **p < 0.01; and ***p < 0.001.
Results
The S1P2 receptor regulates macrophage recruitment during peritonitis
Macrophages are recruited to sites of inflammation as peripheral blood monocytes, which mature into macrophages in the extravascular space. To examine the possible role of S1P2R in macrophage recruitment, we analyzed the response of WT (S1p2r+/+) and S1p2r-null (S1p2r−/−) mice during TG-induced peritonitis. As shown in Fig. 1A, CD11b+F4/80+ macrophages were the major population of cells recruited to the peritoneum 5 d after challenging mice with TG. Many of the recruited macrophages also expressed an intermediate level of the murine DC marker CD11c (Fig. 1A). This CD11b+F4/80+CD11cint macrophage population was efficiently recruited to the peritoneum in both S1p2r+/+ and S1p2r−/− mice (Fig. 1A). However, on day 5 of peritonitis S1p2r−/− mice showed a 44% increase in the absolute number of macrophages when compared with S1p2r+/+ mice (Fig. 1B). No significant differences in neutrophil and lymphocyte counts were observed (Fig. 1B). Together, these data suggest that S1P2R restrains macrophage migration into the inflamed peritoneum.
FIGURE 1.

The S1P2 receptor regulates macrophage recruitment. S1p2r+/+ and S1p2r−/− mice were injected i.p. with TG. Five or 15 d later, cells were collected by peritoneal lavage and analyzed for macrophage content. A, Flow cytometric analysis of peritoneal cells on days 5 and 15, showing percentage of cells that are CD11b+F480+, and CD11c expression in this population; (day 5 S1p2r+/+ n = 14, S1p2r−/− n = 11) (day 15 S1p2r+/+ n = 10, S1p2r−/− n = 9). B, Absolute numbers of peritoneal macrophages, polymorphonuclear neutrophils, and lymphocytes recovered from the peritoneum on day 5 and 15. C, Flow cytometric analysis of draining LNs and nondraining LNs on day 15, showing percentage of cells that are CD11b+F480+.
Macrophages recruited to inflamed tissues actively migrate to draining LNs (23), and their egress is an essential part of resolution. This raised the possibility that macrophage numbers in the peritoneum of S1p2r−/− mice were increased because of impaired migration to draining LNs. Fifteen days after TG challenge, macrophages remained the major cell type in the peritoneum of S1p2r+/+ and S1p2r+/+ mice (Fig. 1A). However, inflammation had resolved, as less macrophages remained in the peritoneum on day 15 when compared with day 5 (Fig. 1B). Loss of S1P2R did not appear to alter resolution, as equivalent number of macrophages remained in the peritoneum of S1p2r+/+ and S1p2r−/− mice (Fig. 1B). The egress of macrophages in this setting was confirmed by an increase in macrophage content in draining LNs. As shown in Fig. 1C, both S1p2r+/+ and S1p2r−/− mice had equivalent numbers of CD11b+F4/80+ macrophages in their LNs on day 15. These cells were notably absent in non-draining LNs (Fig. 1C), and were also absent in draining LNs on day 5 (data not shown). To assess the distribution of myeloid cells during peritonitis, we measured peripheral monocyte and splenic macrophage numbers. No significant differences were found between S1p2r+/+ and S1p2r−/− mice (Supplemental Fig. 1). These results suggest that S1P2R inhibits the recruitment of macrophages into the inflamed peritoneum, whereby loss of S1P2R results in enhanced macrophage accumulation during peritonitis. In addition, we show that S1P2R does not influence the egress of macrophages into LNs.
The S1P2 receptor inhibits macrophage chemotaxis
The enhanced macrophage influx during peritonitis in S1p2r-null mice led us to the hypothesis that S1P serves as a negative regulator of migration in the macrophage lineage. To study the effects of S1P in more detail, we sought to use in vitro models of migration. Although monocytes are the initial cell type recruited during inflammation, this cell type is difficult to isolate from mice for in vitro studies, whereas primary BMDM have been used widely to study myeloid cell migration in vitro (30, 31). Primary mouse macrophages express mainly S1P1R and S1P2R, as determined by real-time RT-PCR (Fig. 2A). This S1P receptor expression profile gives S1P the potential to both stimulate and inhibit the migration of macrophages. To test the effects of S1P on macrophage migration, we generated BMDM and exposed them to chemoattractant gradients using a modified Boyden chamber. S1P was not chemotactic for BMDM over a wide concentration range (1 nM–10 μM) (Supplemental Fig. 2). As expected, BMDM migrated efficiently to both C5a and CXCL12 (SDF-1) (Fig. 2B, 2C). When BMDM were exposed to S1P in combination with C5a or CXCL12, S1P inhibited migration toward both chemoattractants (Fig. 2B, 2C). To examine the role of S1P2R in S1Ps inhibition of macrophage migration we generated BMDM from S1p2r knockout mice (25). Although S1P inhibited migration in S1p2r+/+ BMDM, S1P was not inhibitory in S1p2r−/− cells (Fig. 2B, 2C). These results demonstrate that S1P2R signaling inhibits chemotaxis in BMDM. They also suggest that loss of S1P2R in vivo may lead to enhance migration in the macrophage lineage, and may explain the increased accumulation of macrophages during peritonitis (Fig. 1B).
FIGURE 2.

The S1P2 receptor inhibits macrophage chemotaxis. A, Real-time PCR analysis of macrophage S1P receptor expression. B and C, In vitro chemotaxis of BMDM analyzed using a modified Boyden chamber. Migration of S1p2r+/+ and S1p2r−/− BMDM toward 10 nM C5a (B) or 10 nM CXCL12 (C), with or without 10 nM S1P.
S1P reduces macrophage migration speed but does not alter directionality
To examine the cellular effects of S1P in more detail, we performed live cell chemotaxis assays using a direct-viewing Dunn chemotaxis chamber (27). BMDM were exposed to gradients of C5a, with or without S1P, and migration patterns of individual cells were recorded to determine cell migration speed and directionality, as described in Materials and Methods. As shown in Fig. 3A, C5a induced the directional migration of WT BMDM away from their starting position (at X = 0, Y = 0) and up the concentration gradient of C5a (upward on the y-axis). However, when exposed to C5a and S1P together, WT BMDM migrated less, as evident by their clustering around their starting position (at X = 0, Y = 0) (Fig. 3B). These effects were absent in S1p2r−/− cells (Fig. 3C, 3D), confirming the inhibitory role of S1P2R in regulating macrophage migration. We next quantified the effects of S1P on directionality, an important component of efficient chemotaxis. As shown in Fig. 3E and 3F, BMDM migrated up the concentration gradient of C5a (arrows mark the mean direction), and S1P did not alter this directionality. In addition, we found no evidence that S1P alters C5a-induced cell polarization in BMDM. In the presence of a C5a gradient, the percentage of BMDM with lamellipods was unaltered by S1P (data not shown). Likewise, membrane translocation of PH-Akt-cerulean in RAW263.7 cells in response to C5a was unaltered by S1P (data not shown). We also quantified the effects of S1P on migration speed and distance (see Materials and Methods). S1P2R inhibited the average velocity of cells migrating toward C5a (Fig. 3G). This resulted in a corresponding decrease in the average distance that BMDM traveled (Fig. 3H) and also greatly inhibited the ability of BMDM to travel distances of >100 μm (Fig. 3I). These results confirm that S1P, signaling through S1P2R, inhibits macrophage migration. In addition, they show that S1P inhibits cell migration speed without altering directionality.
FIGURE 3.

S1P reduces macrophage migration speed but does not alter directionality. A Dunn chamber was used to analyze live cell chemotaxis of S1p2r+/+ and S1p2r−/− BMDM migrating toward 10 nM C5a, with or without 10 nM S1P. A–D, Scatter plots indicating final cell positions. E and F, Direction of migration of S1p2r+/+ (E) or S1p2r−/− (F) BMDM, represented as mean direction (arrow) and 95% CI (shaded area). G–I, Quantification of velocity (G), distance from origin (H), and the percentage of cells reaching 100 μm from origin (I).
S1P2R selectively inhibits chemoattractant-induced Akt phosphorylation
To determine whether S1P-mediated inhibition of chemotaxis resulted from alterations in chemoattractant receptor signaling, we analyzed the effects of S1P on C5a receptor (C5aR) signaling in BMDM. As shown in Fig. 4A, C5a induced rapid phosphorylation of Akt and p42/44 MAPK. Akt is a serine/threonine kinase whose activity and phosphorylation are controlled by the PI3K product phosphatylinositol (3, 4, 5)-trisphosphate (PIP3). Thus, Akt phosphorylation is one indicator of PIP3 production. PIP3 species are vital to cell migration, regulating polarity in eukaryotic cells, including neutrophils and macrophages (32). In BMDM that stimulated simultaneously with S1P and C5a, S1P inhibited Akt phosphorylation, but not the phosphorylation of p42/44 MAPK (Fig. 4A). S1P stimulation also selectively inhibited CXCL12-induced Akt phosphorylation (data not shown). S1P stimulation in S1p2r−/− BMDM failed to inhibit C5a-induced Akt phosphorylation (Fig. 4A). S1P2R has previously been shown to inhibit Akt phosphorylation and inhibit the small GTPase Rac (9, 10). Next, we examined the phosphorylation of the PAK, a common mediator of Rac function in leukocytes, including chemotaxis (33, 34). As shown in Fig. 4B, C5a-induced PAK phosphorylation was not altered by S1P, at any of the time points tested. Thus, S1P2R signaling selectively alters chemoattractant-stimulated Akt phosphorylation in BMDM. These results suggest that S1P2R disrupts PI3K signaling, leading to inhibition of migration.
FIGURE 4.

S1P2R selectively inhibits chemoattractant-induced Akt phosphorylation. S1p2r+/+ and S1p2r−/− BMDM were analyzed by Western blot for protein phosphorylation. A, Phospho-Akt and phospho-ERK analysis after 5-min stimulation with 10 nM C5a, with or without 100 nM S1P. B, Time-course of phospho-PAK1 analysis in S1p2r+/+ BMDM stimulated with 10 nM C5a, with or without 100 nM S1P.
Like C5aR, S1P1R is known to activate Akt and p42/44 MAPK (35). However, only minimal phosphorylation of Akt and p42/44 MAPK was observed with S1P stimulation (Fig. 4A), consistent with the lack of migration observed toward S1P in BMDM (Supplemental Fig. 2A). Thus, we have demonstrated that the S1P1 receptor is expressed in BMDM at the mRNA level (Fig. 2A), and that the receptor can signal to downstream intermediates, but S1P2R signaling appears to have greater functional significance in both signaling regulation and migration. In support of the lack of S1P1R function in macrophage migration, we also failed to detect any differences in peritonitis in chimeric mice lacking hematopoetic S1P1R (Supplemental Fig. 2B).
Attenuation of chemoattractant receptor signaling can occur as a result of receptor desensitization, where repeated or continuous stimulation of G protein coupled receptors results in diminished responsiveness. Originally identified in β-adrenergic receptors, receptor desensitization can result from sequential stimulation of cells with ligands for the same receptor (homologous desensitization) or with ligands of different receptors (heterologous desensitization) (36). C5aR is known to undergo both homologous and heterologous desensitization (37–39). Because costimulation of macrophages with S1P and C5a, resulted in decreased migration and Akt phosphorylation compared with C5a alone, we considered the possibility that S1P2R ligation heterologously desensitizes C5aR responsiveness. We measured intracellular Ca2+ concentration to examine C5aR signaling in real time. As shown in Fig. 5A, sequential stimulation of macrophages with C5a resulted in a diminished Ca2+ response, indicating homologous desensitization of the C5aR. However, S1P stimulation did not inhibit the Ca2+ response to C5a, or did S1P induce an increase in intracellular Ca2+ (Fig. 5B). The lack of Ca2+ response after S1P stimulation is surprising, given that S1P2R has been shown to couple to Gq and phospholipase C, and to induce Ca2+ flux in other cell types (40). Our results indicate that S1P does not desensitize C5aR, and implicate an inhibitory pathway downstream from C5aR.
FIGURE 5.

S1P2R does not desensitize the C5aR. Intracellular calcium was measured in cell suspensions of WT macrophages loaded with fura 2, as described in Materials and Methods. Cells were sequentially stimulated with 1 nM C5a (A) or stimulated with 1 μM S1P, followed by 1 nM C5a (B), and intracellular calcium content measured in real time.
PTEN is not required for S1P2R-mediated inhibition
The phosphatase PTEN has been shown to be important for efficient cell migration by localizing and balancing PI3K signaling (41–43). PTEN serves as a negative regulator of the PI3K pathway by dephosphorylating PIP3 at the 5′ position. PTEN has also been postulated to mediate the inhibitory effects of S1P2R on migration and Akt phosphorylation in endothelial cells and fibroblasts but not in glioblastoma cells (7, 8, 10). Global deletion of Pten is embryonic lethal (44, 45). To investigate whether PTEN is required for S1P2R inhibition in macrophages, we generated inducible Pten knockout mice by crossing floxed Pten mice with mice expressing inducible Cre recombinase (Pten loxP/loxP;CreERT2 mice). As described in Materials and Methods, BMDM lacking PTEN (PtenΔ/Δ) were generated after Pten deletion in vivo. PtenΔ/Δ BMDM essentially lacked the PTEN protein, and had a corresponding enhancement of basal Akt phosphorylation (Fig. 6A). Pten+/+ and PtenΔ/Δ BMDM migrated equally well toward C5a, and despite the loss of PTEN, PtenΔ/Δ BMDM migration was equally inhibited by S1P (Fig. 6B). In addition, loss of PTEN did not alter the ability of S1P to inhibit Akt phosphorylation. As shown in Fig. 6C, S1P-inhibited C5a induced Akt phosphorylation in both Pten+/+ and PtenΔ/Δ BMDM, whereas p42/44 MAPK phosphorylation remained unaltered by S1P. Our results clearly indicate that PTEN is not required for S1P2R to inhibit migration in macrophages. Also, lack of PTEN does not alter the ability of S1P2R to reduce chemoattractant-stimulated Akt phosphorylation. Our results in macrophages confirm the PTEN-independent effect of S1P2R recently reported in glioma cells (7, 8, 46).
FIGURE 6.

PTEN is not required for S1P2R-mediated inhibition. A, PtenloxP/loxP, Cre ERT2, and PtenloxP/loxP mice were fed tamoxifen for 5 d and bone marrow was cultured to generate PtenΔ/Δ and Pten+/+ BMDM, respectively. Inset shows immunoblot analysis of BMDM extracts for respective Abs. B, In vitro migration of PtenΔ/Δ and Pten+/+ BMDM toward 10 nM C5a, with or without 10 nM S1P, was measured using a modified Boyden chamber. C, Western blot analysis of phospho-Akt in PtenΔ/Δ and Pten+/+ BMDM stimulated with 10 nM C5a, with or without 100 nM S1P.
S1P2R stimulates cAMP production to inhibit macrophage migration
The second messenger cAMP has been shown to suppress multiple leukocyte functions, inhibiting cytokine production, phagocytosis, and chemotaxis (47–49). cAMP is synthesized from ATP by adenylate cyclase, which is typically activated by ligand binding to Gs coupled receptors (50). More recently, Jiang and colleagues demonstrated a novel mechanism for cAMP production, whereby S1P2R activates adenylate cyclase by coupling to G12/13 (51, 52). The actions of cAMP are primarily mediated by two intracellular cAMP effectors: PKA and exchange proteins activated by cAMP (Epac). PKA is a heterotetrameric serine/threonine kinase that phosphorylates a wide range of proteins, to mediate the majority of cAMP effector functions. Epac is a guanine nucleotide exchange factor for the small GTPase Rap (53). Although Epac is less well studied than PKA, it is likely that PKA and Epac can act both alone and in concert to inhibit leukocyte functions (48, 54, 55).
Because the role of S1P2R in negatively regulating migration parallels the inhibitory actions of cAMP, we hypothesized that cAMP mediates S1P2Rs inhibition of macrophage migration. We first sought to confirm that S1P induces cAMP production in macrophages. As shown in Fig. 7A, S1P stimulation of WT BMDM led to a significant increase in cAMP. The increase in cAMP was mediated by S1P2R, as S1p2r−/− BMDM did not generate cAMP in response to S1P stimulation (Fig. 7A). The increase in cAMP in BMDM is likely mediated by G12/13, as S1P receptors, including S1P2R, do not couple to Gs receptors (56). Also, it is likely that S1P2R increases cAMP by activating adenylate cyclase, and not by inhibiting phosphodiesterases, because the increase in cAMP occurred even in the presence of phosphodiesterase inhibitors, as described in Materials and Methods. As shown in Fig. 7B, the cell-permeable cAMP analog 8-Br-cAMP inhibited BMDM migration toward C5a. The inhibitory actions of cAMP on leukocytes have primarily been attributed to the effects of PKA. To better define the pathway of cAMP inhibition, we used cAMP analogs with specificity for activating PKA (6-Bnz-cAMP) and Epac (8-pCPT-2′-O-Me-cAMP) (57, 58). The PKA-specific analog 6-Bnz-cAMP exhibited identical inhibition of macrophage migration, whereas the Epac-specific analog 8-pCPT-2′-O-Me-cAMP did not inhibit migration (Fig. 7B). Next, we tested the effects of 8-Br-cAMP on Akt phosphorylation. As shown in Fig. 7C, 8-Br-cAMP treatment of BMDM blocked the ability of C5a to induce Akt phosphorylation, in a similar fashion to S1P. Thus, S1P2R stimulation causes cAMP generation in macrophages, and elevated levels of intracellular cAMP inhibit macrophage migration and Akt phosphorylation. Taken together, these results clearly suggest that S1P2R stimulates the production of cAMP to inhibit macrophage migration. In addition our results suggest that PKA is the primary effector for cAMP inhibition of migration in macrophages. To our knowledge, this is the first report examining the effects of PKA and Epac selective cAMP analogs on macrophage migration.
FIGURE 7.
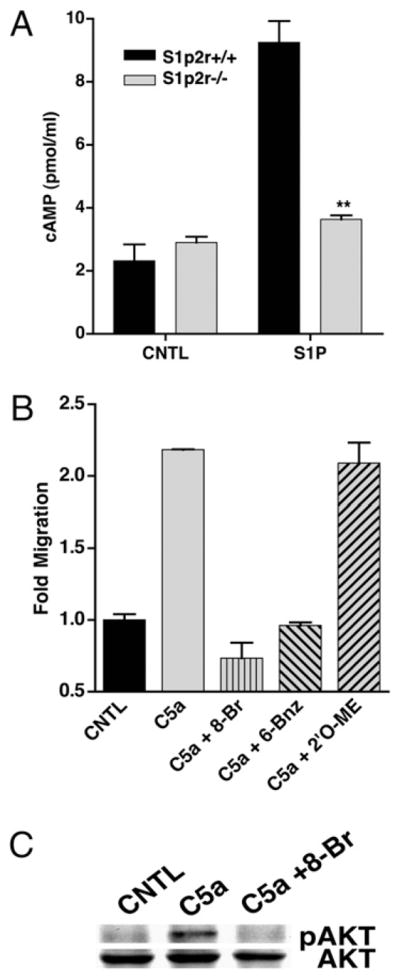
S1P2R stimulates cAMP production to inhibit BMDM migration. A, Intracellular cAMP levels were measured by ELISA in S1p2r+/+ and S1p2r−/− BMDM after stimulation with 10 nM S1P. B, BMDM were pretreated for 30 min with 500 μM cAMP analogs and then migrated toward 10 nM C5a in a modified Boyden chamber, in the presence of cAMP analogs. C, Western blot analysis of phospho-Akt in BMDM pretreated with 500 μM 8-Br-cAMP for 30 min.
Discussion
The mobilization of leukocytes to sites of inflammation is an essential step in host defense; however, excessive or prolonged influx of effector cells can lead to tissue destruction and dysfunction. With the potential for both benefit and harm, it is likely that leukocyte mobilization is tightly regulated. Although there has been considerable progress in identifying positive regulators of leukocyte recruitment, negative regulators of this process remain poorly understood. In this report, we have identified S1P as a negative regulator of macrophage recruitment during peritonitis. The negative effects of S1P are mediated by S1P2R, as loss of S1P2R resulted in increased accumulation of macrophages after TG challenge. This suggests that pharmacologic agonists of S1P2R may reduce macrophage accumulation at sites of inflammation. These results contrast the known functions of S1P on the trafficking of other leukocytes. The best-studied role for S1P is in lymphocyte recirculation, where S1P1R is a positive regulator of lymphocyte egress from lymphoid organs (4). This process is thought to rely on the vascular S1P gradient high levels of S1P in blood and lymph, and low levels of S1P in interstitial fluid (14). Our laboratory has found biochemical data for an S1P gradient between interstitial fluid, plasma, and lymph. We found that plasma contains ~400 nM S1P and lymph ~80 nM S1P (59), whereas peritoneal exudate during peritonitis contained only 20 nM S1P(60). We propose that macrophage recruitment may rely on S1P within the vasculature to dampen the influx of myeloid cells to sites of inflammation. Our results raise the interesting evolutionary concept of opposing roles for S1P in the trafficking of the lymphocyte and macrophage lineages. This also raises the possibility that disrupting the vascular S1P gradient may modulate macrophage trafficking.
Although macrophages can be long-term residents of tissues, they actively migrate to draining LNs during inflammation (23). Their egress is thought to be important in resolution, by clearing debris and potentially harmful effector cells. We examined the effects of S1P2R on macrophage egress to LNs and found that S1p2r-null macrophages still migrated out of the peritoneum to draining LNs. Although S1P2R did not effect emigration in this setting, S1P may regulate the migratory behavior of macrophages in other settings. For example, macrophage egress from athero-sclerotic plaques has been suggested as a potentially disease modifying event and hence an important therapeutic target (61). In this setting, S1P2R could function as a retention signal by inhibiting the migration of macrophage foam cells out of plaques.
In an effort to understand the phenotype of S1P2R-null mice during inflammation, we have demonstrated that S1P inhibits macrophage migration in vitro in an S1P2R-dependent manner. S1P2R is known to inhibit migration in variety of cell types (6–10) and we have expanded on previous observations of S1P2R function. Using live cell imaging, we found that S1P inhibits migratory speed, but does not affect the ability of macrophages to sense the direction of a chemoattractant gradient. S1P2R signaling selectively inhibited Akt phosphorylation, suggesting that S1P2R alters the PI3K pathway in macrophages. Together, these findings support the previous observation that altering the PI3K pathway in primary macrophages reduces migratory speed but not directionality (62). S1P2R-mediated inhibition of Akt has previously been thought to rely on degradation of the Akt activator PIP3 by the inositide phosphatase PTEN (10). Using PTEN-deficient BMDM, we demonstrated that PTEN is dispensable for the effects of S1P on macrophage migration. The lack of involvement of PTEN in macrophages is not surprising given the conflicting reports on the role of PTEN in the migration of other leukocytes. PTEN has been shown to be dispensable for neutrophil and macrophage migration (62). PTEN was also reported to both inhibit (63, 64) and stimulate (65) B cell chemotaxis.
Elevated levels of intracellular cAMP were found to inhibit neutrophil and macrophage migration >30 y ago (66–69). This long-standing inhibitory role for cAMP and the identification of S1P2R-mediated cAMP signaling in macrophage (51, 52) led us to examine the role of cAMP in macrophage migration. S1P stimulation of macrophages led to a significant increase in intracellular cAMP, in an S1P2R-dependent manner. In addition, treatment of macrophages with cell-permeable cAMP analogs mimicked both the inhibitory effect of S1P on migration and the inhibition of C5a-induced Akt phosphorylation. PKA-selective cAMP analogs also demonstrated that the antimigratory effect of cAMP appeared to be mediated by PKA, and not by the alternate cAMP effector Epac. These results highlight the divergent and independent roles for PKA and Epac in cellular responses to cAMP. This raises questions about the way PKA and Epac responses to intracellular cAMP are separated. Like other intracellular mediators, we suspect that cAMP signaling during migration is localized based on the activator of adenylate cyclase. A-kinase anchoring proteins, which are known to localize PKA, are likely to be involved. Of interest to S1P2R signaling, G12/13 has been shown to interact with PKA and/or a-kinase anchoring protein 110 (70), but the significance of this interaction remains to be determined.
In summary, we have identified S1P as a regulator of macrophage recruitment to sites of inflammation. S1P2R-mediated inhibition of macrophage migration likely limits excessive macrophage accumulation at inflammatory sites. Thus, S1P2R represents a potential pharmacologic target to control the damaging effects of excessive macrophage recruitment. In vitro, we have identified cAMP as a novel pathway in the antimigratory signaling of S1P2R. This raises the possibility that S1P2R-induced cAMP may modulate macrophage functions in disease states such as atherosclerosis.
Supplementary Material
Acknowledgments
We thank Dr. Richard Proia for S1P2R-null mice, Dr. Zihai Li for advice in culturing BMDM, Dr. Guo-Hua Fong for Rosa26 CreERT2 mice, Dr. Kimberly Dodge-Kafka for helpful suggestions on cAMP sgnaling, Dr. James Watras for PH-Akt-cerulean constructs, and the Center for Cell Analysis and Modeling at the University of Connecticut for help with microscopy. J. M. also thanks the University of Connecticut M.D./Ph.D. Program.
This work was supported by National Institutes of Health Grants HL67330 and HL89934 (to T.H.).
Abbreviations used in this paper
- BMDM
bone marrow-derived macrophage
- C5aR
C5a receptor
- DC
dendritic cell
- Epac
exchange proteins activated by cAMP
- LN
lymph node
- PAK
p21-activated kinase
- PIP3
phosphatidylinositol (3, 4, 5)-triphosphate
- PKA
protein kinase A
- PTEN
phosphatase and tensin homolog
- S1P
sphingosine 1-phosphate
- S1P1–5R
S1P receptor 1–5
- TG
thioglycollate
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Goetzl EJ, Rosen H. Regulation of immunity by lysosphingolipids and their G protein-coupled receptors. J Clin Invest. 2004;114:1531–1537. doi: 10.1172/JCI23704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hla T. Physiological and pathological actions of sphingosine 1-phosphate. Semin Cell Dev Biol. 2004;15:513–520. doi: 10.1016/j.semcdb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 3.McVerry BJ, Garcia JG. In vitro and in vivo modulation of vascular barrier integrity by sphingosine 1-phosphate: mechanistic insights. Cell Signal. 2005;17:131–139. doi: 10.1016/j.cellsig.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 5.Paik JH, Chae Ss, Lee MJ, Thangada S, Hla T. Sphingosine 1-phosphate-induced endothelial cell migration requires the expression of EDG-1 and EDG-3 receptors and Rho-dependent activation of α vβ3- and β1-containing integrins. J Biol Chem. 2001;276:11830–11837. doi: 10.1074/jbc.M009422200. [DOI] [PubMed] [Google Scholar]
- 6.Kawa S, Kimura S, Hakomori S, Igarashi Y. Inhibition of chemotactic motility and trans-endothelial migration of human neutrophils by sphingosine 1-phosphate. FEBS Lett. 1997;420:196–200. doi: 10.1016/s0014-5793(97)01516-0. [DOI] [PubMed] [Google Scholar]
- 7.Lepley D, Paik JH, Hla T, Ferrer F. The G protein-coupled receptor S1P2 regulates Rho/Rho kinase pathway to inhibit tumor cell migration. Cancer Res. 2005;65:3788–3795. doi: 10.1158/0008-5472.CAN-04-2311. [DOI] [PubMed] [Google Scholar]
- 8.Malchinkhuu E, Sato K, Maehama T, Mogi C, Tomura H, Ishiuchi S, Yoshimoto Y, Kurose H, Okajima F. S1P(2) receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem Biophys Res Commun. 2008;366:963–968. doi: 10.1016/j.bbrc.2007.12.054. [DOI] [PubMed] [Google Scholar]
- 9.Ryu Y, Takuwa N, Sugimoto N, Sakurada S, Usui S, Okamoto H, Matsui O, Takuwa Y. Sphingosine-1-phosphate, a platelet-derived lysophospholipid mediator, negatively regulates cellular Rac activity and cell migration in vascular smooth muscle cells. Circ Res. 2002;90:325–332. doi: 10.1161/hh0302.104455. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez T, Thangada S, Wu MT, Kontos CD, Wu D, Wu H, Hla T. PTEN as an effector in the signaling of antimigratory G protein-coupled receptor. Proc Natl Acad Sci USA. 2005;102:4312–4317. doi: 10.1073/pnas.0409784102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arikawa K, Takuwa N, Yamaguchi H, Sugimoto N, Kitayama J, Nagawa H, Takehara K, Takuwa Y. Ligand-dependent inhibition of B16 melanoma cell migration and invasion via endogenous S1P2 G protein-coupled receptor. Requirement of inhibition of cellular RAC activity. J Biol Chem. 2003;278:32841–32851. doi: 10.1074/jbc.M305024200. [DOI] [PubMed] [Google Scholar]
- 12.Goparaju SK, Jolly PS, Watterson KR, Bektas M, Alvarez S, Sarkar S, Mel L, Ishii I, Chun J, Milstien S, Spiegel S. The S1P2 receptor negatively regulates platelet-derived growth factor-induced motility and proliferation. Mol Cell Biol. 2005;25:4237–4249. doi: 10.1128/MCB.25.10.4237-4249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugimoto N, Takuwa N, Okamoto H, Sakurada S, Takuwa Y. Inhibitory and stimulatory regulation of Rac and cell motility by the G12/13-Rho and Gi pathways integrated downstream of a single G protein-coupled sphingosine-1-phosphate receptor isoform. Mol Cell Biol. 2003;23:1534–1545. doi: 10.1128/MCB.23.5.1534-1545.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hla T, Venkataraman K, Michaud J. The vascular S1P gradient-cellular sources and biological significance. Biochim Biophys Acta. 2008;1781:477–482. doi: 10.1016/j.bbalip.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Czeloth N, Bernhardt G, Hofmann F, Genth H, Förster R. Sphingosine-1-phosphate mediates migration of mature dendritic cells. J Immunol. 2005;175:2960–2967. doi: 10.4049/jimmunol.175.5.2960. [DOI] [PubMed] [Google Scholar]
- 16.Idzko M, Panther E, Corinti S, Morelli A, Ferrari D, Herouy Y, Dichmann S, Mockenhaupt M, Gebicke-Haerter P, Di Virgilio F, et al. Sphingosine 1-phosphate induces chemotaxis of immature and modulates cytokine-release in mature human dendritic cells for emergence of Th2 immune responses. FASEB J. 2002;16:625–627. doi: 10.1096/fj.01-0625fje. [DOI] [PubMed] [Google Scholar]
- 17.Maeda Y, Matsuyuki H, Shimano K, Kataoka H, Sugahara K, Chiba K. Migration of CD4 T cells and dendritic cells toward sphingosine 1-phosphate (S1P) is mediated by different receptor subtypes: S1P regulates the functions of murine mature dendritic cells via S1P receptor type 3. J Immunol. 2007;178:3437–3446. doi: 10.4049/jimmunol.178.6.3437. [DOI] [PubMed] [Google Scholar]
- 18.Lan YY, De Creus A, Colvin BL, Abe M, Brinkmann V, Coates PT, Thomson AW. The sphingosine-1-phosphate receptor agonist FTY720 modulates dendritic cell trafficking in vivo. Am J Transplant. 2005;5:2649–2659. doi: 10.1111/j.1600-6143.2005.01085.x. [DOI] [PubMed] [Google Scholar]
- 19.Idzko M, Hammad H, van Nimwegen M, Kool M, Müller T, Soullié T, Willart MA, Hijdra D, Hoogsteden HC, Lambrecht BN. Local application of FTY720 to the lung abrogates experimental asthma by altering dendritic cell function. J Clin Invest. 2006;116:2935–2944. doi: 10.1172/JCI28295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishii M, Egen JG, Klauschen F, Meier-Schellersheim M, Saeki Y, Vacher J, Proia RL, Germain RN. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature. 2009;458:524–528. doi: 10.1038/nature07713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walzer T, Chiossone L, Chaix J, Calver A, Carozzo C, Garrigue-Antar L, Jacques Y, Baratin M, Tomasello E, Vivier E. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat Immunol. 2007;8:1337–1344. doi: 10.1038/ni1523. [DOI] [PubMed] [Google Scholar]
- 22.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bellingan GJ, Caldwell H, Howie SE, Dransfield I, Haslett C. In vivo fate of the inflammatory macrophage during the resolution of inflammation: inflammatory macrophages do not die locally, but emigrate to the draining lymph nodes. J Immunol. 1996;157:2577–2585. [PubMed] [Google Scholar]
- 24.Takeda K, Cowan A, Fong GH. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. 2007;116:774–781. doi: 10.1161/CIRCULATIONAHA.107.701516. [DOI] [PubMed] [Google Scholar]
- 25.Kono M, Mi Y, Liu Y, Sasaki T, Allende ML, Wu YP, Yamashita T, Proia RL. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem. 2004;279:29367–29373. doi: 10.1074/jbc.M403937200. [DOI] [PubMed] [Google Scholar]
- 26.Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 27.Zicha D, Dunn GA, Brown AF. A new direct-viewing chemotaxis chamber. J Cell Sci. 1991;99:769–775. doi: 10.1242/jcs.99.4.769. [DOI] [PubMed] [Google Scholar]
- 28.Liu W, Saint DA. Validation of a quantitative method for real time PCR kinetics. Biochem Biophys Res Commun. 2002;294:347–353. doi: 10.1016/S0006-291X(02)00478-3. [DOI] [PubMed] [Google Scholar]
- 29.Im DS, Fujioka T, Katada T, Kondo Y, Ui M, Okajima F. Characterization of sphingosine 1-phosphate-induced actions and its signaling pathways in rat hepatocytes. Am J Physiol. 1997;272:G1091–G1099. doi: 10.1152/ajpgi.1997.272.5.G1091. [DOI] [PubMed] [Google Scholar]
- 30.Costa C, Barberis L, Ambrogio C, Manazza AD, Patrucco E, Azzolino O, Neilsen PO, Ciraolo E, Altruda F, Prestwich GD, et al. Negative feedback regulation of Rac in leukocytes from mice expressing a constitutively active phosphatidylinositol 3-kinase γ. Proc Natl Acad Sci USA. 2007;104:14354–14359. doi: 10.1073/pnas.0703175104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiss-Haljiti C, Pasquali C, Ji H, Gillieron C, Chabert C, Curchod ML, Hirsch E, Ridley AJ, Hooft van Huijsduijnen R, Camps M, Rommel C. Involvement of phosphoinositide 3-kinase γ, Rac, and PAK signaling in chemokine-induced macrophage migration. J Biol Chem. 2004;279:43273–43284. doi: 10.1074/jbc.M402924200. [DOI] [PubMed] [Google Scholar]
- 32.Cain RJ, Ridley AJ. Phosphoinositide 3-kinases in cell migration. Biol Cell. 2009;101:13–29. doi: 10.1042/BC20080079. [DOI] [PubMed] [Google Scholar]
- 33.Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- 34.Li Z, Hannigan M, Mo Z, Liu B, Lu W, Wu Y, Smrcka AV, Wu G, Li L, Liu M, et al. Directional sensing requires G β γ-mediated PAK1 and PIX α-dependent activation of Cdc42. Cell. 2003;114:215–227. doi: 10.1016/s0092-8674(03)00559-2. [DOI] [PubMed] [Google Scholar]
- 35.Lee MJ, Thangada S, Paik JH, Sapkota GP, Ancellin N, Chae SS, Wu M, Morales-Ruiz M, Sessa WC, Alessi DR, Hla T. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol Cell. 2001;8:693–704. doi: 10.1016/s1097-2765(01)00324-0. [DOI] [PubMed] [Google Scholar]
- 36.Böhm SK, Grady EF, Bunnett NW. Regulatory mechanisms that modulate signalling by G-protein-coupled receptors. Biochem J. 1997;322:1–18. doi: 10.1042/bj3220001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blackwood RA, Hartiala KT, Kwoh EE, Transue AT, Brower RC. Unidirectional heterologous receptor desensitization between both the fMLP and C5a receptor and the IL-8 receptor. J Leukoc Biol. 1996;60:88–93. doi: 10.1002/jlb.60.1.88. [DOI] [PubMed] [Google Scholar]
- 38.Christophe T, Rabiet MJ, Tardif M, Milcent MD, Boulay F. Human complement 5a (C5a) anaphylatoxin receptor (CD88) phosphorylation sites and their specific role in receptor phosphorylation and attenuation of G protein-mediated responses. Desensitization of C5a receptor controls superoxide production but not receptor sequestration in HL-60 cells. J Biol Chem. 2000;275:1656–1664. doi: 10.1074/jbc.275.3.1656. [DOI] [PubMed] [Google Scholar]
- 39.Didsbury JR, Uhing RJ, Tomhave E, Gerard C, Gerard N, Snyderman R. Receptor class desensitization of leukocyte chemoattractant receptors. Proc Natl Acad Sci USA. 1991;88:11564–11568. doi: 10.1073/pnas.88.24.11564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An S, Bleu T, Zheng Y. Transduction of intracellular calcium signals through G protein-mediated activation of phospholipase C by recombinant sphingosine 1-phosphate receptors. Mol Pharmacol. 1999;55:787–794. [PubMed] [Google Scholar]
- 41.Iijima M, Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- 42.Leslie NR, Yang X, Downes CP, Weijer CJ. The regulation of cell migration by PTEN. Biochem Soc Trans. 2005;33:1507–1508. doi: 10.1042/BST0331507. [DOI] [PubMed] [Google Scholar]
- 43.Subramanian KK, Jia Y, Zhu D, Simms BT, Jo H, Hattori H, You J, Mizgerd JP, Luo HR. Tumor suppressor PTEN is a physiologic suppressor of chemoattractant-mediated neutrophil functions. Blood. 2007;109:4028–4037. doi: 10.1182/blood-2006-10-055319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 45.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez T, Hla T. Structural and functional characteristics of S1P receptors. J Cell Biochem. 2004;92:913–922. doi: 10.1002/jcb.20127. [DOI] [PubMed] [Google Scholar]
- 47.Elferink JG, VanUffelen BE. The role of cyclic nucleotides in neutrophil migration. Gen Pharmacol. 1996;27:387–393. doi: 10.1016/0306-3623(95)00070-4. [DOI] [PubMed] [Google Scholar]
- 48.Serezani CH, Ballinger MN, Aronoff DM, Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol. 2008;39:127–132. doi: 10.1165/rcmb.2008-0091TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torgersen KM, Vang T, Abrahamsen H, Yaqub S, Taskén K. Molecular mechanisms for protein kinase A-mediated modulation of immune function. Cell Signal. 2002;14:1–9. doi: 10.1016/s0898-6568(01)00214-5. [DOI] [PubMed] [Google Scholar]
- 50.Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv. 2002;2:168–184. doi: 10.1124/mi.2.3.168. [DOI] [PubMed] [Google Scholar]
- 51.Jiang LI, Collins J, Davis R, Fraser ID, Sternweis PC. Regulation of cAMP responses by the G12/13 pathway converges on adenylyl cyclase VII. J Biol Chem. 2008;283:23429–23439. doi: 10.1074/jbc.M803281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang LI, Collins J, Davis R, Lin KM, Decamp D, Roach T, Hsueh R, Rebres RA, Ross EM, Taussig R, Fraser I, et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine-1-phosphate/G13 pathway. J Biol Chem. 2007;282:10576–10584. doi: 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 54.Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol. 2005;174:595–599. doi: 10.4049/jimmunol.174.2.595. [DOI] [PubMed] [Google Scholar]
- 55.Bryn T, Mahic M, Enserink JM, Schwede F, Aandahl EM, Taskén K. The cyclic AMP-Epac1-Rap1 pathway is dissociated from regulation of effector functions in monocytes but acquires immunoregulatory function in mature macrophages. J Immunol. 2006;176:7361–7370. doi: 10.4049/jimmunol.176.12.7361. [DOI] [PubMed] [Google Scholar]
- 56.Windh RT, Lee MJ, Hla T, An S, Barr AJ, Manning DR. Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the G(i), G(q), and G(12) families of heterotrimeric G proteins. J Biol Chem. 1999;274:27351–27358. doi: 10.1074/jbc.274.39.27351. [DOI] [PubMed] [Google Scholar]
- 57.Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, Døskeland SO. cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem. 2003;278:35394–35402. doi: 10.1074/jbc.M302179200. [DOI] [PubMed] [Google Scholar]
- 58.Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- 59.Lee YM, Venkataraman K, Hwang SI, Han DK, Hla T. A novel method to quantify sphingosine 1-phosphate by immobilized metal affinity chromatography (IMAC) Prostaglandins Other Lipid Mediat. 2007;84:154–162. doi: 10.1016/j.prostaglandins.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michaud J, Kohno M, Proia RL, Hla T. Normal acute and chronic inflammatory responses in sphingosine kinase 1 knockout mice. FEBS Lett. 2006;580:4607–4612. doi: 10.1016/j.febslet.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 61.Randolph GJ. Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr Opin Lipidol. 2008;19:462–468. doi: 10.1097/MOL.0b013e32830d5f09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishio M, Watanabe K, Sasaki J, Taya C, Takasuga S, Iizuka R, Balla T, Yamazaki M, Watanabe H, Itoh R, et al. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat Cell Biol. 2007;9:36–44. doi: 10.1038/ncb1515. [DOI] [PubMed] [Google Scholar]
- 63.Fox JA, Ung K, Tanlimco SG, Jirik FR. Disruption of a single Pten allele augments the chemotactic response of B lymphocytes to stromal cell-derived factor-1. J Immunol. 2002;169:49–54. doi: 10.4049/jimmunol.169.1.49. [DOI] [PubMed] [Google Scholar]
- 64.Suzuki A, Kaisho T, Ohishi M, Tsukio-Yamaguchi M, Tsubata T, Koni PA, Sasaki T, Mak TW, Nakano T. Critical roles of Pten in B cell homeostasis and immunoglobulin class switch recombination. J Exp Med. 2003;197:657–667. doi: 10.1084/jem.20021101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anzelon AN, Wu H, Rickert RC. Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol. 2003;4:287–294. doi: 10.1038/ni892. [DOI] [PubMed] [Google Scholar]
- 66.Hill HR, Estensen RD, Quie PG, Hogan NA, Goldberg ND. Modulation of human neutrophil chemotactic responses by cyclic 3′,5′-guanosine monophosphate and cyclic 3′,5′-adenosine monophosphate. Metabolism. 1975;24:447–456. doi: 10.1016/0026-0495(75)90124-9. [DOI] [PubMed] [Google Scholar]
- 67.Pick E. Cyclic AMP affects macrophage migration. Nat New Biol. 1972;238:176–177. doi: 10.1038/newbio238176a0. [DOI] [PubMed] [Google Scholar]
- 68.Rivkin I, Neutze JA. Influence of cyclic nucleotides and a phosphodiesterase inhibitor on in vitro human blood neutrophil chemotaxis. Arch Int Pharmacodyn Ther. 1977;228:196–204. [PubMed] [Google Scholar]
- 69.Tse RL, Phelps P, Urban D. Polymorphonuclear leukocyte motility in vitro. VI. Effect of purine and pyrimidine analogues: possible role of cyclic AMP. J Lab Clin Med. 1972;80:264–274. [PubMed] [Google Scholar]
- 70.Niu J, Vaiskunaite R, Suzuki N, Kozasa T, Carr DW, Dulin N, Voyno-Yasenetskaya TA. Interaction of heterotrimeric G13 protein with an A-kinase-anchoring protein 110 (AKAP110) mediates cAMP-independent PKA activation. Curr Biol. 2001;11:1686–1690. doi: 10.1016/s0960-9822(01)00530-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.