

Abstract
Enzyme activation by monovalent cations is widely documented in plants and the animal world. In type II enzymes, activation entails two steps: binding of the monovalent cation to its allosteric site and transduction of this event into enhanced catalytic activity. The effect has exquisite specificity for either Na+ or K+, the most abundant cations present in physiological environments. Enzymes requiring K+ such as kinases and molecular chaperones are not activated as well or at all by the larger cation Cs+ or the smaller cations Na+ and Li+. Enzymes requiring Na+ such as β-galactosidase and clotting proteases are not activated as well by Li+, or the larger cations K+, Rb+, and Cs+. Efforts to switch specificity between Na+ and K+ in this large class of enzymes and completely redesign the mechanism of allosteric transduction leading to enhanced catalytic activity have so far been unsuccessful. Here we show how mutagenesis of two loops defining the Na+ binding site of thrombin, a Na+-activated clotting protease, generates a construct that is most active in the presence of K+ toward synthetic and physiological substrates. The effect is the result of a higher binding affinity and more efficient allosteric transduction of binding into enhanced catalytic activity for K+ compared to Na+, which represents a complete reversal of the properties of wild type. In addition, the construct features altered specificity toward physiological substrates resulting in a significant anticoagulant profile. The findings are relevant to all Na+-activated proteases involved in blood coagulation and the complement system.
Keywords: activated protein C, allosteric enzyme, enzyme specificity
Enzymes activated by monovalent cations are abundantly represented in plants and the animal world (1, 2). Following the original observations by Boyer et al. (3) on the absolute requirement of K+ by pyruvate kinase (4) and by Cohn and Monod on the Na+-dependent activation of β-galactosidase (5), hundreds of enzymes have been reported to display increased activity in the presence of monovalent cations (2). The effect has exquisite specificity, with Na+ or K+ being the preferred activators (6). In general, enzymes requiring K+ such as kinases and molecular chaperones are also activated by 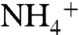 and Rb+, but are not activated as well or at all by the larger cation Cs+ or the smaller cations Na+ and Li+. Enzymes requiring Na+ such as β-galactosidase and clotting proteases are not activated as well by Li+, or the larger cations K+, Rb+, and Cs+. The mechanism of activation is cofactor-like (type I) or allosteric (type II) (1). In the former case, the monovalent cation anchors substrate to the active site of the enzyme, often acting in tandem with a divalent cation like Mg2+, and is absolutely required for catalysis. In the latter, the monovalent cation enhances enzyme activity by binding to an allosteric site and is not absolutely required for catalysis.
and Rb+, but are not activated as well or at all by the larger cation Cs+ or the smaller cations Na+ and Li+. Enzymes requiring Na+ such as β-galactosidase and clotting proteases are not activated as well by Li+, or the larger cations K+, Rb+, and Cs+. The mechanism of activation is cofactor-like (type I) or allosteric (type II) (1). In the former case, the monovalent cation anchors substrate to the active site of the enzyme, often acting in tandem with a divalent cation like Mg2+, and is absolutely required for catalysis. In the latter, the monovalent cation enhances enzyme activity by binding to an allosteric site and is not absolutely required for catalysis.
Type II activation comprises two steps: binding of the monovalent cation to its site followed by transduction of this event into enhanced catalytic activity. The steps are thermodynamically independent because binding obeys detailed balancing but transduction does not (7, 8). Residues responsible for monovalent cation binding need not be the same as those involved in the allosteric transduction of this event into enhanced catalytic activity. Consequently, mutagenesis of the enzyme may succeed in redesigning monovalent cation binding specificity but without bringing about changes in the allosteric mechanism of activation. The clotting protease thrombin (9) can be redesigned to convert its binding specificity from Na+ to K+, but that does not result in a K+-activated enzyme because catalytic activity remains higher in the presence of Na+ (10). Paradoxically, redesigning monovalent cation activation remains an unsolved task in protein engineering, despite the fact that the activation mechanism can be introduced de novo in protein scaffolds devoid of such property (11, 12).
Redesigning allosteric activation in an enzyme requires interference with the mechanism that transduces monovalent cation binding into enhanced catalytic activity. This is equivalent to redesigning signaling for a receptor activated by two different agonists (13), so that the final result is a complete swap of the efficacy between the agonists. In either case, successful engineering must involve components of the activation downstream of the initial binding event, which are intrinsically more challenging to identify than the determinants responsible for ligand binding readily accessible to structural analysis (1, 6). In this study we present a thrombin construct for which monovalent cation specificity has been shifted from Na+ to K+ and the resulting catalytic activity toward synthetic and physiological substrates is also significantly higher in the presence of K+. The results provide insights into the structural determinants of monovalent cation activation in other trypsin-like enzymes involved in blood coagulation and the complement system.
Results
Among all monovalent cations, Na+ binds to thrombin with the highest affinity and elicits the largest enhancement in catalytic activity (10, 14) by ensuring the correct architecture of the oxyanion hole formed by the backbone N atoms of G193 and the catalytic S195 (15). Comparison of the structures of thrombin bound to Na+ (16) and K+ (17) reveals differences in the coordination shell defined by the 186 and 220 loops that explain the different affinities of the two cations. In addition, the C191-C220 disulfide bond is twisted in the K+-bound form and causes the E192-G193 peptide bond downstream to flip, leading to a perturbed conformation of the oxyanion hole. These observations support the conclusion that Na+ activation is controlled upstream by residues of the Na+ coordination shell. Na+ binding and activation are present in all vitamin K-dependent clotting proteases, i.e., thrombin, factors VIIa, IXa, Xa, and activated protein C (18). Activated protein C is peculiar insofar as it shows comparable enhancement of catalytic activity in the presence of Na+ or K+ (18). We therefore tested the hypothesis that replacement of the 186 and 220 loops of thrombin with those of protein C would produce changes in monovalent cation activation. The 186 loop of protein C has a sequence 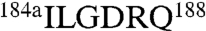 and is three residues shorter than the loop in thrombin with a sequence
and is three residues shorter than the loop in thrombin with a sequence 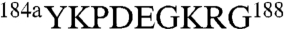 . The 220 loop in the two enzymes has the same length but a very different sequence, i.e.,
. The 220 loop in the two enzymes has the same length but a very different sequence, i.e., 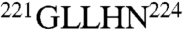 in protein C and
in protein C and 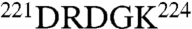 in thrombin.
in thrombin.
Replacement of the 186 and 220 loops in thrombin with those of protein C yields a chimera featuring reduced catalytic activity toward synthetic and physiological substrates (Table 1). The defect reaches 10,000-fold in the case of fibrinogen. The chromogenic substrate H-D-Phe-Pro-Arg-p-nitroanilide (FPR) is affected more than H-D-Phe-Pro-Lys-p-nitroanilide (FPK) and H-D-Phe-Pro-Phe-p-nitroanilide (FPF), supporting a direct perturbation of the primary specificity pocket that preferentially engages Arg at the P1 position. The functional profile observed in the chimera reflects the importance of the 186 and 220 loops in shaping the architecture of the primary specificity pocket (19–22). However, the drastic catalytic impairment caused by the mutation is partially corrected by the presence of Na+ or K+. Although Na+ retains its activating effect, the magnitude of the change in kcat/Km observed in the presence of K+ is more pronounced (Fig. 1). With the exception of the anticoagulant protein C, the substrates analyzed in this study are cleaved more efficiently by the chimera in the presence of K+. The chimera is de facto a K+-activated enzyme. Clotting of fibrinogen directly documents the consequences of this change in monovalent cation specificity (Fig. 2). In the case of wild type, clotting is more rapid in the presence of Na+ relative to K+ and is insignificant in the presence of the inert cation Ch+. In the case of the chimera, clotting is significantly faster in the presence of K+ compared to Na+, and no appreciable activity is observed in the presence of Ch+.
Table 1.
Values of kcat/Km (mM-1 s-1) for thrombin wild type and chimera toward synthetic and physiological substrates
| Enzyme | Cation | FPR* | FPK* | FPF* | FpA† | PAR1† | Protein C† |
| WT | Ch+ | 3,000 ± 200 (1.0) | 440 ± 20 (1.0) | 13 ± 1 (1.0) | 1,500 ± 100 (1.0) | 3,000 ± 200 (1.0) | 310 ± 20 (1.0) |
| WT | Na+ | 89,000 ± 5,000 (30) | 4,600 ± 200 (10) | 400 ± 20 (31) | 15,000 ± 1,000 (10) | 32,000 ± 2,000 (11) | 220 ± 10 (0.71) |
| WT | K+ | 42,000 ± 2,000 (14) | 1,100 ± 100 (2.5) | 300 ± 20 (23) | 6,000 ± 300 (4.0) | 16,000 ± 1,000 (5.3) | 110 ± 10 (0.35) |
| Chimera | Ch+ | 2.9 ± 0.2 (1.0) | 0.29 ± 0.02 (1.0) | 0.069 ± 0.004 (1.0) | 0.30 ± 0.01 (1.0) | 0.45 ± 0.02 (1.0) | 0.097 ± 0.005 (1.0) |
| Chimera | Na+ | 78 ± 4 (27) | 1.1 ± 0.1 (3.8) | 0.19 ± 0.01 (2.8) | 1.5 ± 0.1 (5.0) | 5.0 ± 0.2 (11) | 3.0 ± 0.1 (31) |
| Chimera | K+ | 240 ± 10 (83) | 3.1 ± 0.2 (11) | 0.34 ± 0.02 (4.9) | 2.4 ± 0.1 (8.0) | 15 ± 1 (33) | 2.0 ± 0.1 (21) |
FpA: fibrinopeptide A.
*Experimental conditions are 5 mM Tris, 0.1% PEG 8K, pH 8.0, 25 °C, 200 mM chloride salt as indicated.
†Experimental conditions are 5 mM Tris, 0.1% PEG 8K, pH 7.4, 37 °C, 145 mM chloride salt as indicated. Enhancement relative to Ch+ is given by parentheses.
Fig. 1.

Shift in monovalent cation activation for the thrombin chimera. Values of the specificity constant s = kcat/Km (see also Table 1) in the presence of Na+ or K+ for various thrombin substrates are plotted in units of the values obtained in the presence of the inert cation choline (Ch+). The continuous line in the plot separates a region where activity is higher in the presence of Na+ (below the line) or K+ (above the line). The wild type (closed circles) cleaves chromogenic (FPR, FPF, and FPK) and physiological (fibrinogen, PAR1) substrates with higher specificity in the presence of Na+ and this cation is preferred over K+. Protein C is an exception, because it is cleaved with slightly higher specificity in the absence of Na+ or K+. Monovalent cation specificity is completely reversed in the chimera (open circles) in favor of K+ over Na+ for all substrates tested, excluding protein C. Note the logarithmic scale on both axes.
Fig. 2.

Clotting of fibrinogen by wild-type thrombin and chimera. Clotting curves obtained as changes in turbidity for wild-type thrombin and chimera in the presence of different monovalent cations, as indicated. Experimental conditions are 5 mM Tris, 0.1% PEG 8000, pH 7.4 at 37 °C, and 145 mM chloride salt. The enzyme concentration is 0.1 nM for wild type and 250 nM for the chimera. Clotting requires the presence of Na+ or K+ in the wild type, but the extent of activation by these cations is reversed in the chimera.
The activating effect of K+ in the chimera is linked to a significant switch in the equilibrium components of the interaction. Binding of Na+ or K+ to thrombin can be measured accurately by monitoring the increase in intrinsic fluorescence (10), but no appreciable change could be detected in the case of the chimera. The observation is intriguing because the sequence replaced in thrombin does not contain any known fluorophores of the enzyme (23). Binding of Na+ and K+ to the chimera was therefore measured from the linkage effect on hirudin binding (24). Both Na+ and K+ significantly enhance hirudin binding to the chimera, but K+ exceeds Na+ in the extent of this enhancement (72- vs. 18- fold) as well as in the binding affinity (Fig. 3). The Kd values for K+ and Na+ in the chimera are 59 ± 6 mM and 270 ± 30 mM, respectively, which represents a complete reversal of affinity compared to the values of 260 ± 30 mM for K+ and 50 ± 4 mM for Na+ measured for the wild type under identical solution conditions (10). The chimera has higher binding affinity for K+ relative to Na+ and transduces binding into higher catalytic activity more efficiently in K+. The 186 and 220 loops of activated protein C confer thrombin a complete reversal of monovalent cation specificity from Na+ to K+ involving the equilibrium and catalytic components of the activation mechanism.
Fig. 3.

Linkage between monovalent cation and hirudin binding to the thrombin chimera. Shown is the effect of K+ (closed circles) and Na+ (open circles) on the equilibrium association constant for hirudin binding to the thrombin chimera. Continuous lines were drawn according to the linkage equation 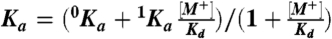 , where
, where 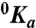 and
and 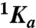 are the values of Ka for hirudin binding in the absence and under saturating [M+] (M+ = Na+ or K+) and Kd is the equilibrium dissociation constant for monovalent cation binding. Best-fit parameter values are (K+)
are the values of Ka for hirudin binding in the absence and under saturating [M+] (M+ = Na+ or K+) and Kd is the equilibrium dissociation constant for monovalent cation binding. Best-fit parameter values are (K+) 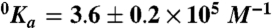 ,
, 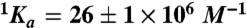 , Kd = 59 ± 9 mM; (Na+)
, Kd = 59 ± 9 mM; (Na+) 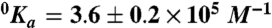 ,
, 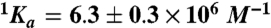 , Kd = 260 ± 30 mM. Experimental conditions are 10 mM Bis-Tris propane, 0.1% PEG 8000, pH 7.4 at 25 °C. The Kd values for K+ and Na+ measured for wild type under identical solution conditions are 260 ± 30 mM and 50 ± 4 mM (10).
, Kd = 260 ± 30 mM. Experimental conditions are 10 mM Bis-Tris propane, 0.1% PEG 8000, pH 7.4 at 25 °C. The Kd values for K+ and Na+ measured for wild type under identical solution conditions are 260 ± 30 mM and 50 ± 4 mM (10).
The procoagulant effect of Na+ on thrombin is well established (25, 26) and is directly illustrated in Fig. 4. The plot contains information on three different substrates. The value of sPAR1 is the specificity constant kcat/Km for the hydrolysis of PAR1 by thrombin and is plotted vs. the analogous value for fibrinogen cleavage sFpA. Both of these values are expressed in units of sPC, the kcat/Km value for hydrolysis of protein C in the presence of thrombomodulin and Ca2+. The origin of the axes divides the plot in four regions, with two of them further divided in two regions by the diagonal dotted line where PAR1 cleavage occurs with the same value of kcat/Km as fibrinogen cleavage. The region to the right of the vertical axis denotes activity toward fibrinogen that exceeds that toward protein C and the reverse is seen to the left of the vertical axis. Likewise, the region above the horizontal axis denotes activity toward PAR1 that exceeds that toward protein C and the reverse is seen below the horizontal axis. Closed circles refer to wild type and open circles refer to the chimera. Under physiological conditions (Na+) thrombin specificity is significantly shifted toward fibrinogen and PAR1. Replacement of Na+ with K+ does not alter this property, but removal of Na+ or K+ from the buffer (Ch+) significantly increases the relative specificity toward protein C. The chimera retains an intriguing anticoagulant profile in the absence of cations (Ch+) that is the result of very similar values of kcat/Km toward fibrinogen, PAR1, and protein C. The effect is even more evident under physiological conditions (Na+) and the chimera maps in the plot near the thrombin mutants Δ146-149e and W215A/E217A that have well established anticoagulant profile in vitro (27, 28) and in vivo (29, 30). Therefore, Na+ is a procoagulant cofactor in the wild type and has no effect on protein C activation (18, 26), but promotes cleavage of all physiological substrates in the chimera. This fundamental change in the response of thrombin to Na+ contributes to turning the chimera into an anticoagulant enzyme. In the presence of K+ there is a further shift in macromolecular substrate specificity and PAR1 becomes the preferred substrate. The change in monovalent cation specificity in the chimera is linked to significant changes in macromolecular substrate specificity, confirming the dual importance of the 186 and 220 loops in both cation and substrate recognition.
Fig. 4.

Multifunctional plot of thrombin activity. Functional properties of wild-type thrombin (closed circles) and chimera (open circles) shown as the values of s = kcat/Km for the hydrolysis of fibrinogen (sFpA), PAR1 (sPAR1), or protein C (sPC) in the presence of 50 nM thrombomodulin and 5 mM Ca2+. Values are plotted as the logarithm of the ratios sPAR1/sPC vs sFpA/sPC. Experimental conditions are 5 mM Tris, 0.1% PEG 8000, 145 mM NaCl, pH 7.4 at 37 °C. The anticoagulant mutants W215A/E217A (WE) and Δ146-149e (del) are also shown for comparison (gray circles). Note how Na+ and K+ binding drastically increase the specificity of thrombin toward fibrinogen and PAR1. In the absence of monovalent cation, specificity shifts toward the anticoagulant protein C, which is a property retained by the chimera. In the presence of Na+, the anticoagulant profile of the chimera is even more pronounced.
Discussion
Binding of monovalent cations such as Na+ or K+ is central to the function and stability of many proteins (1, 6). The molecular origin of the exquisite specificity of the effect that enables proteins to select Na vs. K+ and vice versa has been the subject of extensive investigation for decades. The current view evolved from earlier proposals (31, 32) is that the interplay among ion size, hydration, and electrostatic coupling with protein carbonyls or charged residues selects the correct cation for binding or transport (6, 33–38). Ion channels provide the most striking example of selectivity (39), with discrimination reaching 1,000-fold (37). Even more remarkable is that this selectivity can be reengineered with few amino acid substitutions. Selectivity of a K+ channel in Drosophila can be shifted to cations of larger ionic radius such as Rb+ and 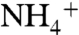 by single amino acid substitutions (40). Similar findings have been reported for a K+ channel in Arabidopsis thaliana (41). Mutagenesis of Na+ channels has shown that selectivity can be shifted to K+ and
by single amino acid substitutions (40). Similar findings have been reported for a K+ channel in Arabidopsis thaliana (41). Mutagenesis of Na+ channels has shown that selectivity can be shifted to K+ and 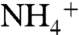 (42, 43) or even to Ca2+ when the net negative charge at the site is increased (44). In ligand-gated ion channels, selectivity can be exchanged between anionic and cationic. Neutralization of glutamate residues converts the nicotinic acetylcholine receptor (45) and the 5-hydroxytryptamine receptor (46) from cationic to anionic, whereas the converse mutations convert the glycine receptor from anionic to cationic (47, 48).
(42, 43) or even to Ca2+ when the net negative charge at the site is increased (44). In ligand-gated ion channels, selectivity can be exchanged between anionic and cationic. Neutralization of glutamate residues converts the nicotinic acetylcholine receptor (45) and the 5-hydroxytryptamine receptor (46) from cationic to anionic, whereas the converse mutations convert the glycine receptor from anionic to cationic (47, 48).
Enzymes activated by monovalent cations share with ion channels remarkable selectivity, but in this case no transport phenomena are involved and specificity is decoupled in terms of binding and transduction into higher catalytic activity. Binding selectivity is an equilibrium property and is accounted for by geometric and thermodynamic features of the coordination shell (37, 38). This component of the activation process has been redesigned by altering the size of access to the monovalent cation binding site (10) or the electrostatic coupling with ligands of the coordination shell (49). However, in no case has a change in the equilibrium binding component of selectivity translated into a change in the activation of the enzyme. Thrombin mutants selective for K+ or Li+ over Na+ remain most active in the presence of Na+ (10, 49). Although this result may seem paradoxical, it is readily explained by the thermodynamic decoupling between binding and transduction inherent to the activation mechanism (8). The results presented here provide evidence that the entire mechanism of activation can be redesigned. A thrombin chimera carrying the 186 and 220 loops of the cognate protease activated protein C not only binds K+ with higher affinity than Na+, but also transduces this event into significantly higher catalytic activity. The 186 and 220 loops are critical to substrate specificity in trypsin-like enzymes (19, 21, 22, 50). Some members of the trypsin family, such as clotting and complement factors, are endowed with Na+ activation made possible by the necessary presence of residue Y225 (18). The Na+ binding site in these enzymes is formed by carbonyl O atoms from residues of the 186 and 220 loops (16, 51–55) and Na+ binding causes significant activation of thrombin (14, 56), activated protein C (57–61), and clotting factors VIIa (18, 62), IXa (18, 63), and Xa (64–70). Activated protein C is unique among these enzymes insofar as it features a comparable degree of activation in Na+ and K+ (18). However, grafting the 186 and 220 loops of activated protein C into thrombin affects the monovalent cation specificity of the enzyme and its activation mechanism in ways that cannot be fully anticipated from the properties of the guest-host pair. The chimera shows higher affinity for K+ relative to Na+, which is a property not present in either thrombin or activated protein C (10, 71). In addition, the chimera features highest activity in K+, as opposed to the preference for Na+ seen in wild-type thrombin and the comparable activity between K+ and Na+ seen for activated protein C. The change in specificity is observed for a variety of chromogenic substrates carrying different residues at the P1 position, as well as for the physiological substrates fibrinogen and PAR1. However, this remarkable switch in cation specificity and activation comes at the expenses of the catalytic activity of the enzyme, which is reduced significantly relative to wild type. This introduces an element of uncertainty in the process of rational engineering enzyme activation, especially because the structural domains involved are critical to direct substrate recognition. We conclude that the 186 and 220 loops contain key determinants for monovalent cation specificity and activation in trypsin-like proteases such as thrombin. Future mutagenesis studies should clarify which residues in these loops control monovalent activation and how these residues interface with the recently established mechanism that transduces Na+ binding into higher catalytic activity by organizing the architecture of the oxyanion hole (15).
The results presented here are also relevant to practical applications. Mutants of thrombin with differential perturbation of fibrinogen and PAR1 cleavage relative to protein C activation have been shown to be effective anticoagulants in vivo (29, 30, 72, 73). The chimera presented in this study has a significant anticoagulant profile that, in the presence of the physiological cation Na+, matches the properties of the mutant Δ146-149e (28) and some mutants of W215 recently identified as significantly improved versions (74) of the potent anticoagulant mutant W215/E217A (27, 29, 30). The 186 and 220 loops offer previously undescribed targets for engineering thrombin into an anticoagulant enzyme thereby expanding and optimizing the existing repertoire dominated by residue 215 (24, 27, 74) and the autolysis loop (28, 75).
Materials and Methods
The thrombin chimera was constructed, expressed, and purified to homogeneity as described (10, 16, 26) using the QuikChange site-directed mutagenesis kit from Stratagene in a HPC4-modified pNUT expression vector containing the human prethrombin-1 gene. The segments 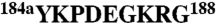 and
and 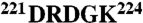 in thrombin were swapped with the segments
in thrombin were swapped with the segments 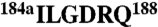 and
and 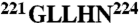 of protein C to construct a thrombin chimera carrying the 186 and 220 loops of protein C.
of protein C to construct a thrombin chimera carrying the 186 and 220 loops of protein C.
Values of the specificity constant kcat/Km for the hydrolysis of the chromogenic substrates FPR, FPK, and FPF were determined from analysis of progress curves and corrected for product inhibition as detailed elsewhere (76, 77) under experimental conditions of 5 mM Tris, 0.1% PEG 8000, pH 8.0 at 25 °C, in the presence of 200 mM NaCl, KCl, or choline chloride (ChCl), with the inert Ch+ used as reference. Values of kcat/Km for release of fibrinopeptide A from fibrinogen, cleavage of the protease activated receptor PAR1, and activation of protein C in the presence of 50 nM thrombomodulin and 5 mM CaCl2 were obtained as reported elsewhere (26, 78, 79) under experimental conditions of 5 mM Tris, 0.1% PEG 8000, 145 mM NaCl, pH 7.4 at 37 °C.
The equilibrium constants for Na+ and K+ binding to the chimera were resolved from the linkage with hirudin binding (24, 80) measured directly by fluorescence spectroscopy. Binding of Na+ or K+ to the chimera did not elicit a significant change in intrinsic fluorescence, as opposed to wild type (10, 14). Experimental conditions were 10 mM Bis-Tris propane, 0.1% PEG 8000, pH 7.4 at 25 °C. The optical density of the solution was always significantly lower than 0.05 units both at λex (280 nm) and λem (340 nm) and no inner filter effect was observed. Fluorescence data were corrected for baseline reading and sample dilution (< 2% at the end of the titration) to obtain the value of equilibrium association constant Ka for the chimera–hirudin interaction as a function of [Na+] or [K+].
Acknowledgments.
This work was supported in part by the National Institutes of Health Research Grants HL49413, HL58141, HL73813, and HL95315 (to E.D.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 5145.
References
- 1.Di Cera E. A structural perspective on enzymes activated by monovalent cations. J Biol Chem. 2006;281:1305–1308. doi: 10.1074/jbc.R500023200. [DOI] [PubMed] [Google Scholar]
- 2.Suelter CH. Enzymes activated by monovalent cations. Science. 1970;168:789–795. doi: 10.1126/science.168.3933.789. [DOI] [PubMed] [Google Scholar]
- 3.Boyer PD, Lardy HA, Phillips PH. The role of potassium in muscle phosphorylations. J Biol Chem. 1942;146:673–681. [Google Scholar]
- 4.Kachmar JF, Boyer PD. Kinetic analysis of enzyme reactions. II. The potassium activation and calcium inhibition of pyruvic phosphoferase. J Biol Chem. 1953;200:669–682. [PubMed] [Google Scholar]
- 5.Cohn M, Monod J. [Purification and properties of the beta-galactosidase (lactase) of Escherichia coli] Biochim Biophys Acta. 1951;7:153–174. doi: 10.1016/0006-3002(51)90013-3. [DOI] [PubMed] [Google Scholar]
- 6.Page MJ, Di Cera E. Role of Na+ and K+ in enzyme function. Physiol Rev. 2006;86:1049–1092. doi: 10.1152/physrev.00008.2006. [DOI] [PubMed] [Google Scholar]
- 7.Botts J, Morales M. Analytical description of the effects of modifiers and of multivalency upon the steady state catalyzed reaction rate. T Faraday Soc. 1953;49:696–707. [Google Scholar]
- 8.Hill TL. Free Energy Transduction in Biology. New York: Academic; 1977. [Google Scholar]
- 9.Di Cera E. Thrombin. Mol Aspects Med. 2008;29:203–254. doi: 10.1016/j.mam.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prasad S, et al. Redesigning the monovalent cation specificity of an enzyme. Proc Natl Acad Sci USA. 2003;100:13785–13790. doi: 10.1073/pnas.2333109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Page MJ, Carrell CJ, Di Cera E. Engineering protein allostery: 1.05 Å resolution structure and enzymatic properties of a Na+-activated trypsin. J Mol Biol. 2008;378:666–672. doi: 10.1016/j.jmb.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Page MJ, Di Cera E. Combinatorial enzyme design probes allostery and cooperativity in the trypsin fold. J Mol Biol. 2010;399:306–319. doi: 10.1016/j.jmb.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wells CM, Di Cera E. Thrombin is a Na(+)-activated enzyme. Biochemistry. 1992;31:11721–11730. doi: 10.1021/bi00162a008. [DOI] [PubMed] [Google Scholar]
- 15.Niu W, et al. Mutant N143P reveals how Na+ activates thrombin. J Biol Chem. 2009;284:36175–36185. doi: 10.1074/jbc.M109.069500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pineda AO, et al. Molecular dissection of Na+ binding to thrombin. J Biol Chem. 2004;279:31842–31853. doi: 10.1074/jbc.M401756200. [DOI] [PubMed] [Google Scholar]
- 17.Carrell CJ, Bush LA, Mathews FS, Di Cera E. High resolution crystal structures of free thrombin in the presence of K+ reveal the basis of monovalent cation selectivity and an inactive slow form. Biophys Chem. 2006;121:177–184. doi: 10.1016/j.bpc.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Dang QD, Di Cera E. Residue 225 determines the Na+-induced allosteric regulation of catalytic activity in serine proteases. Proc Natl Acad Sci USA. 1996;93:10653–10656. doi: 10.1073/pnas.93.20.10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 20.Hedstrom L, Szilagyi L, Rutter WJ. Converting trypsin to chymotrypsin: The role of surface loops. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 21.Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perona JJ, Hedstrom L, Rutter WJ, Fletterick RJ. Structural origins of substrate discrimination in trypsin and chymotrypsin. Biochemistry. 1995;34:1489–1499. doi: 10.1021/bi00005a004. [DOI] [PubMed] [Google Scholar]
- 23.Bah A, Garvey LC, Ge J, Di Cera E. Rapid kinetics of Na+ binding to thrombin. J Biol Chem. 2006;281:40049–40056. doi: 10.1074/jbc.M608600200. [DOI] [PubMed] [Google Scholar]
- 24.Arosio D, Ayala YM, Di Cera E. Mutation of W215 compromises thrombin cleavage of fibrinogen, but not of PAR-1 or protein C. Biochemistry. 2000;39:8095–8101. doi: 10.1021/bi0006215. [DOI] [PubMed] [Google Scholar]
- 25.Dang OD, Vindigni A, Di Cera E. An allosteric switch controls the procoagulant and anticoagulant activities of thrombin. Proc Natl Acad Sci USA. 1995;92:5977–5981. doi: 10.1073/pnas.92.13.5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang QD, Guinto ER, Di Cera E. Rational engineering of activity and specificity in a serine protease. Nat Biotechnol. 1997;15:146–149. doi: 10.1038/nbt0297-146. [DOI] [PubMed] [Google Scholar]
- 27.Cantwell AM, Di Cera E. Rational design of a potent anticoagulant thrombin. J Biol Chem. 2000;275:39827–39830. doi: 10.1074/jbc.C000751200. [DOI] [PubMed] [Google Scholar]
- 28.Bah A, Carrell CJ, Chen Z, Gandhi PS, Di Cera E. Stabilization of the E* form turns thrombin into an anticoagulant. J Biol Chem. 2009;284:20034–20040. doi: 10.1074/jbc.M109.012344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gruber A, Cantwell AM, Di Cera E, Hanson SR. The thrombin mutant W215A/E217A shows safe and potent anticoagulant and antithrombotic effects in vivo. J Biol Chem. 2002;277:27581–27584. doi: 10.1074/jbc.C200237200. [DOI] [PubMed] [Google Scholar]
- 30.Gruber A, et al. Relative antithrombotic and antihemostatic effects of protein C activator versus low molecular weight heparin in primates. Blood. 2007;109:3733–3740. doi: 10.1182/blood-2006-07-035147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eisenman G, Dani JA. An introduction to molecular architecture and permeability of ion channels. Annu Rev Biophys Biophys Chem. 1987;16:205–226. doi: 10.1146/annurev.bb.16.060187.001225. [DOI] [PubMed] [Google Scholar]
- 32.Eisenman G, Horn R. Ionic selectivity revisited: The role of kinetic and equilibrium processes in ion permeation through channels. J Membrane Biol. 1983;76:197–225. doi: 10.1007/BF01870364. [DOI] [PubMed] [Google Scholar]
- 33.Roux B, MacKinnon R. The cavity and pore helices in the KcsA K+ channel: Electrostatic stabilization of monovalent cations. Science. 1999;285:100–102. doi: 10.1126/science.285.5424.100. [DOI] [PubMed] [Google Scholar]
- 34.Berneche S, Roux B. Energetics of ion conduction through the K+ channel. Nature. 2001;414:473–77. doi: 10.1038/35102067. [DOI] [PubMed] [Google Scholar]
- 35.Noskov SY, Berneche S, Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 36.Noskov SY, Roux B. Importance of hydration and dynamics on the selectivity of the KcsA and NaK channels. J Gen Physiol. 2007;129:135–143. doi: 10.1085/jgp.200609633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu H, Noskov SY, Roux B. Two mechanisms of ion selectivity in protein binding sites. Proc Natl Acad Sci USA. 2010;107:20329–20334. doi: 10.1073/pnas.1007150107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nayal M, Di Cera E. Valence screening of water in protein crystals reveals potential Na+ binding sites. J Mol Biol. 1996;256:228–234. doi: 10.1006/jmbi.1996.0081. [DOI] [PubMed] [Google Scholar]
- 39.Doyle DA, et al. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 40.Yool AJ, Schwarz TL. Alteration of ionic selectivity of a K+ channel by mutation of the H5 region. Nature. 1991;349:700–704. doi: 10.1038/349700a0. [DOI] [PubMed] [Google Scholar]
- 41.Uozumi N, Gassmann W, Cao Y, Schroeder JI. Identification of strong modifications in cation selectivity in an Arabidopsis inward rectifying potassium channel by mutant selection in yeast. J Biol Chem. 1995;270:24276–24281. doi: 10.1074/jbc.270.41.24276. [DOI] [PubMed] [Google Scholar]
- 42.Kellenberger S, Gautschi I, Schild L. A single point mutation in the pore region of the epithelial Na+ channel changes ion selectivity by modifying molecular sieving. Proc Natl Acad Sci USA. 1999;96:4170–4175. doi: 10.1073/pnas.96.7.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsushima RG, Li RA, Backx PH. Altered ionic selectivity of the sodium channel revealed by cysteine mutations within the pore. J Gen Physiol. 1997;109:463–475. doi: 10.1085/jgp.109.4.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yue L, Navarro B, Ren D, Ramos A, Clapham DE. The cation selectivity filter of the bacterial sodium channel, NaChBac. J Gen Physiol. 2002;120:845–853. doi: 10.1085/jgp.20028699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galzi JL, et al. Mutations in the channel domain of a neuronal nicotinic receptor convert ion selectivity from cationic to anionic. Nature. 1992;359:500–505. doi: 10.1038/359500a0. [DOI] [PubMed] [Google Scholar]
- 46.Gunthorpe MJ, Lummis SC. Conversion of the ion selectivity of the 5-HT(3a) receptor from cationic to anionic reveals a conserved feature of the ligand-gated ion channel superfamily. J Biol Chem. 2001;276:10977–10983. [PubMed] [Google Scholar]
- 47.Keramidas A, Moorhouse AJ, French CR, Schofield PR, Barry PH. M2 pore mutations convert the glycine receptor channel from being anion- to cation-selective. Biophys J. 2000;79:247–259. doi: 10.1016/S0006-3495(00)76287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keramidas A, Moorhouse AJ, Pierce KD, Schofield PR, Barry PH. Cation-selective mutations in the M2 domain of the inhibitory glycine receptor channel reveal determinants of ion-charge selectivity. J Gen Physiol. 2002;119:393–410. doi: 10.1085/jgp.20028552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prasad S, et al. Residue Asp-189 controls both substrate binding and the monovalent cation specificity of thrombin. J Biol Chem. 2004;279:10103–10108. doi: 10.1074/jbc.M312614200. [DOI] [PubMed] [Google Scholar]
- 50.Hedstrom L, Farr-Jones S, Kettner CA, Rutter WJ. Converting trypsin to chymotrypsin: Ground-state binding does not determine substrate specificity. Biochemistry. 1994;33:8764–8769. doi: 10.1021/bi00195a018. [DOI] [PubMed] [Google Scholar]
- 51.Di Cera E, et al. The Na+ binding site of thrombin. J Biol Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 52.Zhang E, Tulinsky A. The molecular environment of the Na+ binding site of thrombin. Biophys Chem. 1997;63:185–200. doi: 10.1016/s0301-4622(96)02227-2. [DOI] [PubMed] [Google Scholar]
- 53.Scharer K, et al. Quantification of cation-pi interactions in protein-ligand complexes: crystal-structure analysis of Factor Xa bound to a quaternary ammonium ion ligand. Angew Chem Int Edit. 2005;44:4400–4404. doi: 10.1002/anie.200500883. [DOI] [PubMed] [Google Scholar]
- 54.Bajaj SP, Schmidt AE, Agah S, Bajaj MS, Padmanabhan K. High resolution structures of p-aminobenzamidine- and benzamidine-VIIa/soluble tissue factor: Unpredicted conformation of the 192-193 peptide bond and mapping of Ca2+, Mg2+, Na+, and Zn2+ sites in factor VIIa. J Biol Chem. 2006;281:24873–24888. doi: 10.1074/jbc.M509971200. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt AE, et al. Thermodynamic linkage between the S1 site, the Na+ site, and the Ca2+ site in the protease domain of human activated protein C (APC). Sodium ion in the APC crystal structure is coordinated to four carbonyl groups from two separate loops. J Biol Chem. 2002;277:28987–28995. doi: 10.1074/jbc.M201892200. [DOI] [PubMed] [Google Scholar]
- 56.Orthner CL, Kosow DP. Evidence that human alpha-thrombin is a monovalent cation-activated enzyme. Arch Biochem Biophys. 1980;202:63–75. doi: 10.1016/0003-9861(80)90406-3. [DOI] [PubMed] [Google Scholar]
- 57.He X, Rezaie AR. Identification and characterization of the sodium-binding site of activated protein C. J Biol Chem. 1999;274:4970–4976. doi: 10.1074/jbc.274.8.4970. [DOI] [PubMed] [Google Scholar]
- 58.Steiner SA, Amphlett GW, Castellino FJ. Stimulation of the amidase and esterase activity of activated bovine plasma protein C by monovalent cations. Biochem Biophys Res Commun. 1980;94:340–347. doi: 10.1016/s0006-291x(80)80226-9. [DOI] [PubMed] [Google Scholar]
- 59.Steiner SA, Castellino FJ. Kinetic studies of the role of monovalent cations in the amidolytic activity of activated bovine plasma protein C. Biochemistry. 1982;21:4609–4614. doi: 10.1021/bi00262a015. [DOI] [PubMed] [Google Scholar]
- 60.Steiner SA, Castellino FJ. Effect of monovalent cations on the pre-steady-state kinetic parameters of the plasma protease bovine activated protein C. Biochemistry. 1985;24:1136–1141. doi: 10.1021/bi00326a011. [DOI] [PubMed] [Google Scholar]
- 61.Steiner SA, Castellino FJ. Kinetic mechanism for stimulation by monovalent cations of the amidase activity of the plasma protease bovine activated protein C. Biochemistry. 1985;24:609–617. doi: 10.1021/bi00324a011. [DOI] [PubMed] [Google Scholar]
- 62.Petrovan RJ, Ruf W. Role of residue Phe225 in the cofactor-mediated, allosteric regulation of the serine protease coagulation factor VIIa. Biochemistry. 2000;39:14457–14463. doi: 10.1021/bi0009486. [DOI] [PubMed] [Google Scholar]
- 63.Schmidt AE, Stewart JE, Mathur A, Krishnaswamy S, Bajaj SP. Na+ site in blood coagulation factor IXa: Effect on catalysis and factor VIIIa binding. J Mol Biol. 2005;350:78–91. doi: 10.1016/j.jmb.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 64.Rezaie AR, He X. Sodium binding site of factor Xa: Role of sodium in the prothrombinase complex. Biochemistry. 2000;39:1817–1825. doi: 10.1021/bi992006a. [DOI] [PubMed] [Google Scholar]
- 65.Rezaie AR, Kittur FS. The critical role of the 185-189-loop in the factor Xa interaction with Na+ and factor Va in the prothrombinase complex. J Biol Chem. 2004;279:48262–48269. doi: 10.1074/jbc.M409964200. [DOI] [PubMed] [Google Scholar]
- 66.Orthner CL, Kosow DP. The effect of metal ions on the amidolytic acitivity of human factor Xa (activated Stuart-Prower factor) Arch Biochem Biophys. 1978;185:400–406. doi: 10.1016/0003-9861(78)90182-0. [DOI] [PubMed] [Google Scholar]
- 67.Monnaie D, et al. Identification of a binding site for quaternary amines in factor Xa. Biochemistry. 2000;39:5349–5354. doi: 10.1021/bi9926781. [DOI] [PubMed] [Google Scholar]
- 68.Underwood MC, Zhong D, Mathur A, Heyduk T, Bajaj SP. Thermodynamic linkage between the S1 site, the Na+ site, and the Ca2+ site in the protease domain of human coagulation factor Xa. Studies on catalytic efficiency and inhibitor binding. J Biol Chem. 2000;275:36876–36884. doi: 10.1074/jbc.M001386200. [DOI] [PubMed] [Google Scholar]
- 69.Camire RM. Prothrombinase assembly and S1 site occupation restore the catalytic activity of FXa impaired by mutation at the sodium-binding site. J Biol Chem. 2002;277:37863–37870. doi: 10.1074/jbc.M203692200. [DOI] [PubMed] [Google Scholar]
- 70.Levigne S, et al. Role of the alpha-helix 163-170 in factor Xa catalytic activity. J Biol Chem. 2007;282:31569–31579. doi: 10.1074/jbc.M704837200. [DOI] [PubMed] [Google Scholar]
- 71.Griffon N, Di Stasio E. Thermodynamics of Na+ binding to coagulation serine proteases. Biophys Chem. 2001;90:89–96. doi: 10.1016/s0301-4622(01)00129-6. [DOI] [PubMed] [Google Scholar]
- 72.Gibbs CS, et al. Conversion of thrombin into an anticoagulant by protein engineering. Nature. 1995;378:413–416. doi: 10.1038/378413a0. [DOI] [PubMed] [Google Scholar]
- 73.Tsiang M, et al. Protein engineering thrombin for optimal specificity and potency of anticoagulant activity in vivo. Biochemistry. 1996;35:16449–16457. doi: 10.1021/bi9616108. [DOI] [PubMed] [Google Scholar]
- 74.Marino F, Pelc LA, Vogt A, Gandhi PS, Di Cera E. Engineering thrombin for selective specificity toward protein C and PAR1. J Biol Chem. 2010;285:19145–19152. doi: 10.1074/jbc.M110.119875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dang QD, Sabetta M, Di Cera E. Selective loss of fibrinogen clotting in a loop-less thrombin. J Biol Chem. 1997;272:19649–19651. doi: 10.1074/jbc.272.32.19649. [DOI] [PubMed] [Google Scholar]
- 76.Krem MM, Di Cera E. Dissecting substrate recognition by thrombin using the inactive mutant S195A. Biophys Chem. 2003;100:315–323. doi: 10.1016/s0301-4622(02)00289-2. [DOI] [PubMed] [Google Scholar]
- 77.Vindigni A, Dang QD, Di Cera E. Site-specific dissection of substrate recognition by thrombin. Nat Biotechnol. 1997;15:891–895. doi: 10.1038/nbt0997-891. [DOI] [PubMed] [Google Scholar]
- 78.Ayala YM, et al. Molecular mapping of thrombin-receptor interactions. Proteins. 2001;45:107–116. doi: 10.1002/prot.1130. [DOI] [PubMed] [Google Scholar]
- 79.Vindigni A, Di Cera E. Release of fibrinopeptides by the slow and fast forms of thrombin. Biochemistry. 1996;35:4417–4426. doi: 10.1021/bi952834d. [DOI] [PubMed] [Google Scholar]
- 80.Ayala YM, et al. Thermodynamic investigation of hirudin binding to the slow and fast forms of thrombin: Evidence for folding transitions in the inhibitor and protease coupled to binding. J Mol Biol. 1995;253:787–798. doi: 10.1006/jmbi.1995.0591. [DOI] [PubMed] [Google Scholar]