

Abstract
Mutations in voltage-gated ion channels are responsible for several types of epilepsy. Genetic epilepsies often exhibit variable severity in individuals with the same mutation, which may be due to variation in genetic modifiers. The Scn2aQ54 transgenic mouse model has a sodium channel mutation and exhibits epilepsy with strain-dependent severity. We previously mapped modifier loci that influence Scn2aQ54 phenotype severity and identified Kcnv2, encoding the voltage-gated potassium channel subunit Kv8.2, as a candidate modifier. In this study, we demonstrate a threefold increase in hippocampal Kcnv2 expression associated with more severe epilepsy. In vivo exacerbation of the phenotype by Kcnv2 transgenes supports its identification as an epilepsy modifier. The contribution of KCNV2 to human epilepsy susceptibility is supported by identification of two nonsynonymous variants in epilepsy patients that alter function of Kv2.1/Kv8.2 heterotetrameric potassium channels. Our results demonstrate that altered potassium subunit function influences epilepsy susceptibility and implicate Kcnv2 as an epilepsy gene.
Epilepsy is a common neurological disorder affecting approximately 1% of the population worldwide (1–3). Over the last 15 y, a number of genes responsible for monogenic epilepsy have been identified. Most of the responsible genes are components of neuronal signaling, including voltage-gated ion channels (4). Mutations in the voltage-gated sodium channel genes SCN1A and SCN2A are responsible for several types of human epilepsy, including genetic (generalized) epilepsy with febrile seizures plus (GEFS+), Dravet syndrome, and benign familial neonatal–infantile seizures (BFNIS) (5). More than 700 SCN1A mutations have been reported to date, making this gene the most frequently identified cause of monogenic epilepsy. Approximately 20 SCN2A mutations have been reported in patients with BFNIS, GEFS+, and Dravet syndrome (5). Variable expressivity is a common feature of monogenic epilepsy caused by ion channel mutations, suggesting that genetic modifiers may influence clinical severity (6).
Scn2aQ54 transgenic mice are a model of sodium channel-dependent epilepsy. These mice express a gain-of-function mutation in Scn2a that results in progressive epilepsy that begins with brief, spontaneous partial motor seizures (7). As they age, Scn2aQ54 mice exhibit more frequent partial seizures, as well as secondarily generalized seizures, and have a reduced lifespan. Excitatory hippocampal pyramidal neurons from Scn2aQ54 mice display elevated persistent current that is predicted to increase neuronal excitability and contribute to seizure generation (7). Severity of the epilepsy phenotype in Scn2aQ54 is highly dependent on genetic background. The phenotype is less severe on the resistant strain C57BL/6J (B6), with delayed seizure onset and improved survival, compared with the susceptible strain SJL/J (SJL) (8). We previously mapped two modifier loci that influence the difference in severity between these two strains: Moe1 (Modifier of Epilepsy 1) on chromosome 11 and Moe2 on chromosome 19 (8). Previous linkage analysis identified Kcnv2 as a strong candidate for the Moe2 locus (9). Kcnv2 encodes the voltage-gated potassium channel Kv8.2, which is a silent subunit as a homotetramer. Kv8.2 can form functional heterotetramers with Kv2 subunits and influence membrane translocation and channel properties (10, 11). Members of the Kv2 family are expressed in the nervous system and underlie the neuronal delayed rectifier K+ current, which is important for limiting membrane excitability, particularly under conditions of repetitive stimulation (12). In the hippocampus, which is a region of particular importance for seizure generation, Kv2.1 is the major contributor to the delayed rectifier potassium current (13) and colocalizes with Kv8.2 in hippocampal pyramidal neurons (14).
To test the contribution of strain-dependent variation in Kcnv2 to epilepsy susceptibility, we characterized the functional effects of previously identified nonsynonymous coding variants and compared transcript levels between strains. We also evaluated the in vivo effect of Kcnv2 variants in transgenic mice. To determine whether the human ortholog contributes to human epilepsy, we screened epilepsy patients for genetic variation in KCNV2. Our studies demonstrate Kcnv2 transgene-mediated transfer of seizure susceptibility in mice, as well as human variants that alter delayed rectifier K+ currents. These results implicate KCNV2 as an epilepsy gene in mice and humans.
Results
Functional Analysis of Kcnv2 Variants in the Mouse.
We previously identified two nonsynonymous coding variants in Kcnv2 that differ between the resistant B6 and susceptible SJL strains and hypothesized that functional effects of these variants may explain the differences in seizure susceptibility. Strain SJL carries the Kv8.2 amino acid substitutions R205H and Q252R (9). We compared the functional effects of the B6 and SJL isoforms of Kv8.2 on potassium currents by coexpression with mouse Kv2.1 in a heterologous expression system. Channel cDNAs were cotransfected into CHO cells, and whole-cell currents were measured in the whole-cell configuration of the patch clamp technique (15). Expression of mouse Kv2.1 (mKv2.1) alone produced large voltage-dependent potassium currents, and coexpression with the B6 or SJL isoforms of Kv8.2 (B6-Kv8.2, SJL-Kv8.2) resulted in significant suppression of K+ current density (mKv2.1 alone, 326.1 ± 30.8 pA/pF, n = 8; mKv2.1 + B6-Kv8.2, 84.5 ± 5.3 pA/pF, n = 7, P < 0.001; mKv2.1 + SJL-Kv8.2, 157.1 ± 29.1 pA/pF, n = 7, P < 0.005) (Fig. 1). Coexpression of mKv2.1 with B6-Kv8.2 produced significantly greater suppression than SJL-Kv8.2 for voltage steps between 0 mV and +60 mV (Fig. 1). Further, B6-Kv8.2, but not SJL-Kv8.2, had a significant effect on the kinetics of activation that was limited to a single test potential (0 mV) without a corresponding change in voltage dependence (Fig. S1). Neither Kv8.2 isoform differed in the magnitude of cumulative inactivation evoked by moderate-frequency pulse trains (Fig. S2), but SJL-Kv8.2 did evoke a greater time-dependent decay in whole-cell current (Fig. 1C). However, given their colocalization in excitatory neurons, the differences in biophysical properties of Kv2.1/B6-Kv8.2 and Kv2.1/SJL-Kv8.2 heterotetrameric channels do not provide a clear and compelling explanation for strain differences in epilepsy severity in Scn2aQ54 mice.
Fig. 1.

Functional consequences of amino acid sequence variants in mouse Kcnv2. (A) Averaged whole-cell current traces normalized to membrane capacitance recorded from CHO cells expressing mKv2.1 in combination with either B6-Kv8.2 or SJL-Kv8.2 (n = 7 for each condition). (B) Current density–voltage relationships for mKv2.1 coexpressed with the two Kv8.2 isoforms. Current was measured at 1,990 ms after start of the test pulse then normalized to membrane capacitance. (C) Extent of time-dependent whole-cell current decay illustrated by the ratio of steady-state current (measured at 1,990 ms) to instantaneous current (measured at 100 ms).
Relative Expression of Kcnv2 in Hippocampus.
Whole-brain expression of Kcnv2 does not differ between strains B6 and SJL (9). We further investigated strain-dependent expression in the hippocampus for three reasons: first, previous EEG studies showed that seizures in Scn2aQ54 likely originate in the hippocampus (7); second, Kcnv2 transcripts are enriched in the hippocampus (14); and, third, the biophysical differences described above were counterintuitive and suggested more complex physiology. RNA was isolated from dissected hippocampi of 8-wk-old B6 and SJL mice for quantitative RT-PCR (qRT-PCR) analysis. Relative Kcnv2 expression was assessed by the 2-ΔΔCt method normalizing to TATA binding protein using the same assay previously used for whole-brain analysis (16). Strain SJL exhibited threefold greater expression of hippocampal Kcnv2 transcript relative to B6 (Fig. 2). The finding indicates that higher Kcnv2 expression in hippocampus correlates with greater epilepsy severity.
Fig. 2.
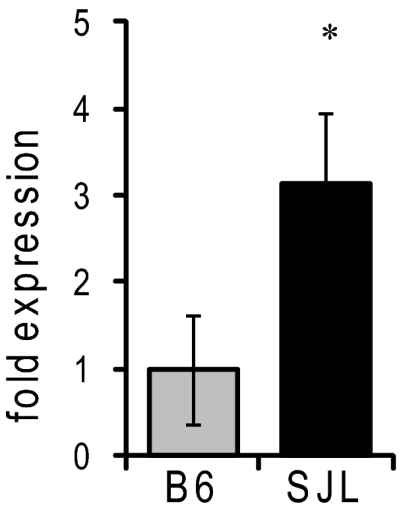
Relative hippocampal expression of Kcnv2. Transcript levels were measured by qRT-PCR (n = 7 per strain; *P = 0.012 compared with B6, pairwise fixed reallocation randomization test).
Transgenic Transfer of the Modified Phenotype.
We evaluated the in vivo modifier effects of Kcnv2 using a transgenic transfer approach, based on the prior observation that the modifier effect is a dominant trait (8). We constructed transgenes containing the B6 or SJL Kcnv2 coding sequence under the control of the neuron-specific enolase (NSE) promoter. This promoter drives panneuronal expression in transgenic mice and is the same promoter that drives expression of the Scn2aQ54 transgene (7, 17). To determine the contribution of the Kcnv2 amino acid variants, we generated transgenic lines expressing the SJL-derived Kcnv2 transgenes (Kcnv2-S1, Kcnv2-S2). To determine whether higher expression alone is sufficient for the severe phenotype, we generated lines carrying the B6-derived Kcnv2 transgenes (Kcnv2-B1, Kcnv2-B2).
Transgenes were microinjected into B6 oocytes and maintained on the B6 background, ensuring a pure genetic background for experiments. The transgenic lines expressed varying amounts of Kcnv2 transgene transcript as determined by qRT-PCR (Fig. 3A). Kv8.2 protein levels exhibited a similar trend in the transgenic lines, as determined by immunoblotting (Fig. S3). Kcnv2 transgenic hemizygotes were crossed with B6.Q54 mice to generate double-transgenic Kcnv2;Q54 offspring and single-transgenic littermate controls that were phenotyped for spontaneous seizure onset and survival. For all Kcnv2 transgenic lines, single-transgenic mice displayed no spontaneous seizures and had a normal lifespan.
Fig. 3.

Transgenic transfer of the modified phenotype in Kcnv2;Q54 double transgenic mice. (A) Relative whole-brain expression of Kcnv2 transcript measured by qRT-PCR (n = 5 per genotype; *P ≤ 0.002 compared with nontransgenic, pairwise fixed reallocation randomization test). (B) Percentage of mice exhibiting more than 1 seizure per 30 min by 6 wk of age (*P < 0.03 compared with B6.Q54, Fisher's exact test). (C) Survival of Q54 mice carrying Kcnv2 transgenes and single transgenic B6.Q54 littermates (dashed black line).
Lines Kcnv2-S2 and Kcnv2-B2, with the highest levels of transgene expression, generated an increased number of Kcnv2;Q54 double-transgenic mice with seizures by 6 wk of age (Fig. 3 A and B). Double-transgenic mice with either the Kcnv2-S2 or Kcnv2-B2 transgenes also exhibited accelerated mortality, with 50% mortality by 10 wk and 14 wk of age, respectively, compared with B6.Q54 single-transgenic mice with 50% survival at 20 wk of age (Fig. 3C). Lines Kcnv2-S1 and Kcnv2-B1, with lower levels of transgene expression, did not influence the Scn2aQ54 phenotype (Fig. 3). The level of Kcnv2 transgene expression was strongly correlated with seizure phenotype (R2 = 0.85) and 24-wk survival (R2 = 0.79), whereas amino acid sequence was not correlated with seizure phenotype (R2 = 0.008) or survival (R2 = 0.04). Although the precise contribution of the amino acid variants in vivo is unclear, elevated expression of Kcnv2 seems to be sufficient to exacerbate the Scn2aQ54 epilepsy phenotype.
Human KCNV2 Screening.
To determine whether KCNV2 contributes to human epilepsy, we screened 209 pediatric epilepsy subjects for variants in KCNV2 by exon amplification, conformation-sensitive gel electrophoresis (CSGE), and sequencing (18). A number of known polymorphic coding SNPs were present in the patients, including seven synonymous SNPs and a common nonsynonymous SNP with a population minor allele frequency of 0.12 (Table S1). Additionally, we identified two unique nonsynonymous coding variants: R7K and M285R. These variants were not detected in 368 neurologically normal control chromosomes [National Institute of Neurological Disorders and Stroke (NINDS) Neurologically Normal Control Panels NDPT079, NDPT093]. Additionally, these variants are not present in dbSNP, nor have they been reported in resequencing projects, including the 1000 Genomes Project (http://browser.1000genomes.org/index.html) and the Baylor Ion Channel Sequencing Project (http://www.hgsc.bcm.tmc.edu/ionchannel-snpList.xsp).
The KCNV2-R7K variant was identified in a patient with febrile and afebrile partial seizures starting at age 2 y (Table S2, patient 1). The variant was inherited from his unaffected mother. However, there is a positive family history of two paternal uncles with childhood epilepsy. R7K substitutes lysine for an evolutionarily conserved arginine in the cytoplasmic amino terminus of the Kv8.2 channel (Fig. S4A).
The KCNV2-M285R variant was identified in a patient with an epileptic encephalopathy with severely refractory epilepsy (Table S2, patient 2). There is no family history of epilepsy, and the variant was inherited from his unaffected mother. Methionine 285 is located at the extracellular face of transmembrane segment 1 and is evolutionarily invariant in homologous channels from other mammals, birds, and fish. (Fig. S4B).
Both patients were also screened for variants in the voltage-gated sodium channel genes SCN1A and SCN2A. No pathogenic variants were identified (Table S3).
Functional Properties of Human KCNV2 Variants.
We determined the functional consequences of the R7K and M285R variants engineered in human Kv8.2 by coexpressing with human Kv2.1 in CHO cells. Coexpression of Kv2.1 with either R7K or M285R Kv8.2 variants resulted in significantly greater suppression of Kv2.1-mediated current compared with wild-type Kv8.2 for voltage steps from 0 mV to +60 mV (Fig. 4 A and B). Furthermore, M285R, but not R7K, exhibited a significant depolarizing shift in the voltage dependence of steady-state activation and significantly greater time constants of activation (τ) measured between 0 and +30 mV (Fig. 4 C and D). Neither variant differed from wild-type in the magnitude of cumulative inactivation evoked by moderate-frequency pulse trains (Fig. S2). Thus, both variants cause enhanced Kv2.1 suppression consistent with reduced neuronal delayed rectifier current. Furthermore, these effects are compounded by the slower kinetics and more depolarized voltage dependence of activation evoked by M285R.
Fig. 4.

Functional consequences of human KCNV2 variants. (A) Averaged whole-cell current traces normalized to membrane capacitance recorded from CHO cells coexpressing human Kv2.1 with wild-type or mutant Kv8.2 (n = 7 to 8 for each condition). (B) Current density–voltage relationships recorded from cells coexpressing Kv2.1 with wild-type or mutant Kv8.2. Current was measured at 1,990 ms after start of the test pulse, then normalized to membrane capacitance. (C) Voltage dependence of activation time constants for Kv2.1 coexpressed with wild-type or mutant Kv8.2 (*P < 0.05 compared with WT). Time constants were determined from monoexponential fits to the data. Currents recorded at voltages <0 mV were too small to determine time constants. Inset: Averaged current traces (black, hKv8.2-WT; blue, hKv8.2-R7K; red, hKv8.2-M285R) recorded from 10 to 500 ms after a test pulse to 0 mV and normalized to current amplitude measured at 500 ms. (D) Voltage dependence of steady-state activation. Tail-current amplitudes were measured at −30 mV after a 2,000-ms activating pulse from −60 to +60 mV. Currents were normalized to peak amplitude and fit with Boltzmann functions. Values for activation V1/2 were as follows: wild-type Kv8.2, 0.9 ± 1.2 mV; Kv8.2-R7K, 2.0 ± 1.6 mV (not significantly different from wild-type); and Kv8.2-M285R, 9.1 ± 1.1 mV (P < 0.01 compared with wild-type Kv8.2).
Discussion
In the present study we demonstrate important effects of Kcnv2 variants on seizure susceptibility in humans and mice. In transgenic mice, increased expression of Kcnv2 was sufficient to exacerbate the seizure phenotype in Scn2aQ54 mice. In human epilepsy patients, we identified two unique nonsynonymous variants that exerted increased suppression of delayed rectifier potassium currents. Although both human KCNV2 variants were inherited from an unaffected parent, they may act as susceptibility modifiers in the affected patients, as do the Kcnv2 variants in the Scn2aQ54 mouse model. Taken together, these results implicate Kcnv2 as an important genetic factor in epilepsy.
Kcnv2 encodes the voltage-gated potassium channel subunit Kv8.2, which is a silent subunit when expressed as a homotetramer. However, when coassembled as a heterotetramer with Kv2 family members, Kv8.2 influences membrane translocation and biophysical properties of these channels (10, 11). Kv2 channels are major contributors to delayed rectifier K+ current in hippocampal pyramidal neurons and act to dampen excitability during high-frequency stimulation (13, 19, 20). Inclusion of Kv8.2 in heteromeric channels results in suppression of Kv2-mediated delayed rectifier potassium currents, which could impair membrane repolarization and thus predispose neurons to increased excitability.
Direct interaction between Kv8.2 and Kv2.1 channels has been demonstrated by coimmunoprecipitation in HEK293 cells (11). The Kv8.2 and Kv2.1 subunits also show significant regional overlap in their central nervous system expression patterns. Within the hippocampus, transcripts for both Kcnv2 and Kcnb1, which encodes Kv2.1, are detected in the principal excitatory neurons of the pyramidal cell layers and the dentate gyrus (14, 21–24). Similarly, both Kcnv2 and Kcnb1 are expressed in the cortex, with high levels of transcript in Layers 2/3 and 5 (14, 21–24). This regional colocalization is consistent with an effect of Kv8.2 variants on Kv2.1 channels within cells of critical importance for seizure generation and propagation.
We had initially hypothesized that the amino acid differences between the resistant B6 and susceptible SJL strains were responsible for the divergent phenotypes (9). Functional characterization of the Kv8.2 amino acid differences in a heterologous expression system demonstrated that the B6 isoform was more effective at suppressing Kv2.1-mediated currents. Because these channels are localized in excitatory neurons and the B6 strain is more resistant, this result was counterintuitive and suggested that the in vivo physiology is more complex. Therefore, we sought to test the relative contribution of expression and amino acids differences in vivo. Relative expression of Kcnv2 in hippocampus was threefold greater in the susceptible SJL strain compared with B6. The Kcnv2 gene is located in a divergent haplotype block between B6 and SJL, with a high degree of noncoding sequence variation (four SNPs/kb) that may influence Kcnv2 transcript levels by altering the rate of transcription or mRNA stability (9). Increased transgenic expression in vivo increased the severity of the seizure phenotype in Scn2aQ54 mice, regardless of the amino acid sequence of the Kcnv2 transgene. This observation supports the view that higher expression of Kcnv2 in strain SJL is sufficient for increased phenotype severity. Increased expression of Kcnv2 would be predicted to decrease Kv2.1-mediated delayed rectifier potassium current, which could promote increased excitability under conditions of repetitive stimulation. In support of this mechanism, others have demonstrated that antisense knockdown of Kv2.1 in hippocampal slices resulted in increased CA1 pyramidal neuron excitability under conditions of high-frequency stimulation (19). Thus, increased expression of Kcnv2 alone could contribute to seizure susceptibility.
In further support of the importance of KCNV2 in seizure pathogenesis, we identified unique nonsynonymous variants in two unrelated children with epilepsy. Functional characterization of these KCNV2 variants, R7K and M285R, demonstrated that these variants also enhance Kv8.2-mediated suppression of Kv2.1 currents. These mutations would be predicted to decrease delayed rectifier potassium current in neurons, resulting in increased excitability under conditions of repetitive stimulation. The net effects would be similar to the effects of elevated expression of KCNV2. Consistent with the more severe clinical phenotype of patient 2, M285R exhibited additional kinetic defects, including a +10-mV shift in the voltage dependence of activation and slower activation kinetics. These defects are predicted to delay channel opening, which would likely further impair membrane repolarization and increase excitability. These results implicate enhanced Kv8.2 activity as a plausible genetically encoded factor in epilepsy susceptibility. Loss-of-function mutations in KCNV2 identified in autosomal recessive retinal cone dystrophy 3 (25–28) also implicate this channel as an important modulator of neuronal firing patterns.
Clinical severity in the two patients with KCNV2 variants differs, ranging from relatively benign febrile and afebrile seizures in patient 1 (R7K) to severe epileptic encephalopathy in patient 2 (M285R). However, it is not uncommon for missense mutations in the same gene to result in different epilepsy types of varying severity. For example, missense mutations in SCN1A and SCN2A have been reported in patients with the benign epilepsy syndrome GEFS+, as well as in patients with the severe epileptic encephalopathy Dravet syndrome (5, 6). Several factors may underlie the wide spectrum of phenotypes, including the location and nature of the amino acid substitution, divergent effects of the mutation on protein function, the genetic background of the individual, and environmental effects. Nonconservative substitution of arginine for methionine at the extracellular face of transmembrane segment 1 results in more profound biophysical defects than the conservative R7K substitution in the amino terminus. This difference in dysfunction is consistent with the difference in clinical severity. Additional resistance and susceptibility alleles at other loci are likely to contribute to the overall clinical phenotype. Although we found no evidence for interaction with mutant sodium channels in these patients, it is conceivable that the KCNV2 variants interact with other loci to influence epilepsy susceptibility. Patient 1 (R7K) has a positive family history of two paternal uncles with childhood epilepsy. Although the R7K variant was inherited from his unaffected mother, it is possible that there was paternal inheritance of additional genetic resistance or susceptibility factors.
Kcnv2 belongs to a group of potassium channel modulatory subunits that are electrically silent and cannot form functional homotetramers. These silent subunits form heterotetramers and modulate the properties of Kv2 and Kv3 channels, increasing the functional diversity of the channel subfamilies. This is particularly relevant for the Kv2 subfamily with only two members, Kv2.1 and Kv2.2. Targeting of modulatory subunits may offer a new therapeutic approach for fine-tuning neuronal excitability.
The interplay between ion channels contributes to overall neuronal excitability. Previous results from our laboratory and others have demonstrated genetic interactions between voltage-gated sodium, potassium, and calcium channels in mouse models of epilepsy (29–31). The present study implicates a unique potassium channel subtype in epilepsy and suggests that variants in ion channels may act to modify the clinical phenotype in human epilepsy. This may have implications for molecular diagnostic testing and suggests that expanded ion channel screening may improve the utility of molecular testing for clinical risk assessment and disease management in epilepsy.
In summary, our results implicate KCNV2 as a genetic modifier of epilepsy, and demonstrate that isolation of genetic modifiers in mice can lead to the identification of human epilepsy genes. Discovery of genes that influence susceptibility and disease progression will provide insight into the molecular events of epileptogenesis, improve molecular diagnostic utility, and identify novel therapeutic targets for improved treatment of human epilepsy.
Materials and Methods
Transgene Constructs.
The Kcnv2 open reading frame was amplified by RT-PCR from mouse brain RNA and cloned into an NSE transgene expression construct as previously described (7). The transgene contains the 4-kb promoter fragment from the rat NSE gene that includes 2.8 kb of flanking sequence, exon 1, intron 1, and 6 bp of noncoding sequence from exon 2, and the SV40 late polyadenylation sequence (17). Constructs were confirmed by sequencing before transgene microinjection.
Animals.
All experimental protocols were approved by the Vanderbilt University Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Scn2aQ54 transgenic mice congenic on the C57BL/6J background (B6.Q54) were established as described and are maintained by continued backcrossing of hemizygous transgenic males to C57BL/6J females. Transgenic mice carrying a B6 Kcnv2 cDNA [Tg(Kcnv2B)1Jak] or SJL/J Kcnv2 cDNA [Tg(Kcnv2S)1Jak] were generated by pronuclear injection into C57BL/6J fertilized oocytes in the University of Michigan Transgenic Animal Core or the Vanderbilt University Transgenic Mouse/ESC Shared Resource. Transgenic founders were identified by PCR genotyping as described below. Each founder was crossed to strain C57BL/6J to generate an independent transgenic line, and lines were maintained by continued backcrossing of transgenic hemizygotes to C57BL/6J. We generated two lines that carry the SJL Kcnv2 cDNA transgene (Kcnv2-S1, S2) and two lines that carry the B6 cDNA transgene (Kcnv2-B1, B2). We crossed B6.Kcnv2 transgenic mice to B6.Q54 mice to generate double-transgenic mice and single-transgenic littermate controls for experiments. Offspring of all genotypes were obtained at the expected Mendelian ratios.
Genotyping.
Genotyping of Scn2aQ54 mice was performed as previously described (8). Kcnv2 transgenics were identified by PCR genotyping of genomic DNA using primers located in intron 1 of NSE and in the Kcnv2 cDNA. These primers amplify a 935-bp band from the Kcnv2 cDNA transgene.
Phenotyping.
Mice were tail biopsied on postnatal day 14 and genotyped. We used the same phenotyping paradigm that was used for mapping of the Moe2 locus (8, 9). Briefly, Scn2aQ54;Kcnv2 double-transgenic mice and single-transgenic littermates were observed for visible seizures during 30-min observation periods at 3, 4.5, and 6 wk of age. Mice were transferred to a clean observation cage [7.75 in (width) × 12 in (depth) × 6.5 in (height)] just before the observation session. All observations occurred between 1300 hours and 1600 hours. Videotaped observation sessions were analyzed offline by a blinded observer using Observer XT behavioral coding software (Noldus). Assessment of visible partial motor seizures was based on prior extensive video-EEG monitoring that demonstrated coincident stereotyped behavioral and EEG abnormalities (7). Animals exhibiting one or more seizures during the observations were classified as having seizures (average frequency, three per 30 min). The proportion of mice with a seizure frequency of one or more per 30 min by 6 wk of age was compared between genotypes using Fisher's exact test (n ≥ 14 per group).
Qualitative RT-PCR.
For comparing relative Kcnv2 levels in strains B6 and SJL, hippocampi were dissected from 8-wk-old C57BL/6J and SJL/J mice (n = 7 per strain). For assessment of Kcnv2 transcript levels in transgenic mice, whole-brain RNA was isolated from 10- to 12-wk-old Kcnv2 transgenic mice and nontransgenic littermates (n = 5 per line). RNA was isolated using the RNeasy kit (Qiagen). Real-time RT-PCR was performed as previously described (9). Briefly, real-time PCR was performed in triplicate using QuantiTect SYBR Green PCR mix (Qiagen) on an ABI 7900HT system. All reported assays exhibited a single peak in dissociation analysis and no detectable signal in no-RT controls. Relative transcript levels were assessed using the 2−ΔΔCT method (16) with TATA binding protein as a reference gene (Qiagen QuantiTect Assay QT00198443). Statistical comparison between groups was made by the pairwise fixed reallocation randomization test (32).
Plasmids and Cell Transfection.
A full-length mouse Kv8.2 cDNA was generated by RT-PCR of mouse brain RNA and cloning into the pIRES2-smGFP mammalian expression vector, which contains the CMV promoter, an IRES-2 element, and soluble modified GFP (DF/HCC DNA Resource Core, EvNO00025960). A full-length mouse Kv2.1 cDNA clone was obtained from Open Biosystems (clone ID 5363843) and cloned into a modified pIRES2-DsRed vector (BD Biosciences-Clontech), in which we substituted the fluorescent protein cDNA with that of DsRed-MST (provided by Dr. Andras Nagy, University of Toronto, Toronto, Canada). A full-length human Kv8.2 cDNA clone was obtained from Open Biosystems (clone ID 40025518) and cloned into the pIRES2-smGFP vector. Mutations were introduced into Kv8.2 using the QuikChange Site-Directed Mutagenesis system (Agilent Technologies). Human Kv2.1 was amplified by RT-PCR from human brain total RNA (Clontech) and cloned into pIRES2-DsRed-MST. All clones were sequenced in their entirety to confirm their identity before transfection.
Expression of Kv8.2 and Kv2.1 constructs in CHO cells was achieved by transient plasmid transfection using FUGENE-6 (Roche) in which 1 μg of total cDNA was transfected with a mass ratio of 1:1. To study the pore-forming Kv2.1 subunit in the absence of Kv8.2, cells were transfected with pIRES2-DsRed-MST-Kv2.1 in combination with nonrecombinant pIRES2-smGFP vector. After transfection, cells were incubated for 48 h before use in electrophysiological experiments.
Electrophysiology.
Whole cell patch clamp recordings were performed as previously described (33), except the pipette solution was modified to contain lower K+ concentration to improve voltage-control (modified pipette solution, in mM: K+ aspartate 55, N-methyl-D-glucamine 55, aspartic acid 55, CaCl2 1, Hepes 10, EGTA 11, MgCl2 1, and K2ATP 5, pH 7.3). Whole-cell currents were measured from −80 to +60 mV (in 10-mV steps) from a holding potential of −80 mV. Data were collected for each experimental condition from at least three transient transfections and analyzed and plotted using a combination of Clampfit (Molecular Devices) and SigmaPlot 2000 (Systat Software). Statistical analyses were carried out using SigmaStat 2.03 (Systat Software), and P values are provided in the figures or listed in the figure legends. Statistical significance among two groups was determined using unpaired Student t test, and one-way ANOVA followed by Tukey post test was performed when comparing more than two groups. Whole-cell currents were normalized for membrane capacitance, and results are expressed as mean ± SEM. The number of cells used for each experimental condition is given in the figure legends.
Human KCNV2 Mutation Screening.
Research was approved by the local institutional review boards. Consent and DNA samples were obtained from study subjects. The two coding exons of KCNV2 were amplified from genomic DNA, and variants were identified by CSGE and sequencing using previously described methods (18). SCN1A and SCN2A were screened by PCR amplification and sequencing of the 26 coding exons. NINDS neurologically normal control panels NDPT079 and NDPT093 (Coriell Cell Repository) were screened for the R7K and M285 variants using custom Taqman assays on an ABI 7900HT system (Applied Biosystems).
Statistical Analysis.
Values are expressed as mean ± SEM. Comparisons of mean differences between groups were made by unpaired two-tailed Student t test or one-way ANOVA unless otherwise stated. Comparisons of proportions were made by Fisher's exact test. For qRT-PCR results, comparisons between groups were made by the pairwise fixed reallocation randomization test (32). P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank the patients and their families for their cooperation; Nicole Hawkins, Rebecca Somershoe, and Sarah Bergren for technical assistance; Miriam Meisler for valuable discussions; and the Vanderbilt Transgenic Mouse/Embryonic Stem Cell Shared Resource and the University of Michigan Transgenic Animal Model Core for generation of transgenic mice. This work was supported by National Institutes of Health Grants NS053792 (to J.A.K.), NS032387 (to A.L.G.), T32-MH64913 (to B.J.) and T32-GM007347 (to C.M.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1017539108/-/DCSupplemental.
References
- 1.Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935-1984. Epilepsia. 1993;34:453–468. doi: 10.1111/j.1528-1157.1993.tb02586.x. [DOI] [PubMed] [Google Scholar]
- 2.Leonardi M, Ustun TB. The global burden of epilepsy. Epilepsia. 2002;43(Suppl 6):21–25. doi: 10.1046/j.1528-1157.43.s.6.11.x. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. Atlas: Epilepsy Care in the World. Geneva: WHO Press; 2005. [Google Scholar]
- 4.Turnbull J, et al. Sacred disease secrets revealed: the genetics of human epilepsy. Hum Mol Genet. 2005;14(Spec No 2):2495–2500. [PubMed] [Google Scholar]
- 5.Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115:2010–2017. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meisler MH, O'Brien JE, Sharkey LM. Sodium channel gene family: Epilepsy mutations, gene interactions and modifier effects. J Physiol. 2010;588:1841–1848. doi: 10.1113/jphysiol.2010.188482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kearney JA, et al. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience. 2001;102:307–317. doi: 10.1016/s0306-4522(00)00479-6. [DOI] [PubMed] [Google Scholar]
- 8.Bergren SK, Chen S, Galecki A, Kearney JA. Genetic modifiers affecting severity of epilepsy caused by mutation of sodium channel Scn2a Mamm. Genome. 2005;16:683–690. doi: 10.1007/s00335-005-0049-4. [DOI] [PubMed] [Google Scholar]
- 9.Bergren SK, Rutter ED, Kearney JA. Fine mapping of an epilepsy modifier gene on mouse Chromosome 19. Mamm Genome. 2009;20:359–366. doi: 10.1007/s00335-009-9193-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Czirják G, Tóth ZE, Enyedi P. Characterization of the heteromeric potassium channel formed by kv2.1 and the retinal subunit kv8.2 in Xenopus oocytes. J Neurophysiol. 2007;98:1213–1222. doi: 10.1152/jn.00493.2007. [DOI] [PubMed] [Google Scholar]
- 11.Ottschytsch N, Raes A, Van Hoorick D, Snyders DJ. Obligatory heterotetramerization of three previously uncharacterized Kv channel alpha-subunits identified in the human genome. Proc Natl Acad Sci USA. 2002;99:7986–7991. doi: 10.1073/pnas.122617999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Misonou H, Mohapatra DP, Trimmer JS. Kv2.1: A voltage-gated k+ channel critical to dynamic control of neuronal excitability. Neurotoxicology. 2005;26:743–752. doi: 10.1016/j.neuro.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Murakoshi H, Trimmer JS. Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci. 1999;19:1728–1735. doi: 10.1523/JNEUROSCI.19-05-01728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen Institute for Brain Science Allen mouse brain atlas. 2009. Available at: http://mouse.brain-map.org. Accessed August 13, 2010.
- 15.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Forss-Petter S, et al. Transgenic mice expressing beta-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron. 1990;5:187–197. doi: 10.1016/0896-6273(90)90308-3. [DOI] [PubMed] [Google Scholar]
- 18.Plummer NW, et al. Exon organization, coding sequence, physical mapping, and polymorphic intragenic markers for the human neuronal sodium channel gene SCN8A. Genomics. 1998;54:287–296. doi: 10.1006/geno.1998.5550. [DOI] [PubMed] [Google Scholar]
- 19.Du J, Haak LL, Phillips-Tansey E, Russell JT, McBain CJ. Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv2.1. J Physiol. 2000;522:19–31. doi: 10.1111/j.1469-7793.2000.t01-2-00019.xm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohapatra DP, et al. Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels (Austin) 2009;3:46–56. doi: 10.4161/chan.3.1.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang PM, Glatt CE, Bredt DS, Yellen G, Snyder SH. A novel K+ channel with unique localizations in mammalian brain: Molecular cloning and characterization. Neuron. 1992;8:473–481. doi: 10.1016/0896-6273(92)90275-i. [DOI] [PubMed] [Google Scholar]
- 22.Hwang PM, Fotuhi M, Bredt DS, Cunningham AM, Snyder SH. Contrasting immunohistochemical localizations in rat brain of two novel K+ channels of the Shab subfamily. J Neurosci. 1993;13:1569–1576. doi: 10.1523/JNEUROSCI.13-04-01569.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 24.Maletic-Savatic M, Lenn NJ, Trimmer JS. Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J Neurosci. 1995;15:3840–3851. doi: 10.1523/JNEUROSCI.15-05-03840.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben Salah S, et al. Novel KCNV2 mutations in cone dystrophy with supernormal rod electroretinogram. Am J Ophthalmol. 2008;145:1099–1106. doi: 10.1016/j.ajo.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 26.Thiagalingam S, et al. Novel mutations in the KCNV2 gene in patients with cone dystrophy and a supernormal rod electroretinogram. Ophthalmic Genet. 2007;28:135–142. doi: 10.1080/13816810701503681. [DOI] [PubMed] [Google Scholar]
- 27.Wissinger B, et al. Cone dystrophy with supernormal rod response is strictly associated with mutations in KCNV2. Invest Ophthalmol Vis Sci. 2008;49:751–757. doi: 10.1167/iovs.07-0471. [DOI] [PubMed] [Google Scholar]
- 28.Wu H, et al. Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am J Hum Genet. 2006;79:574–579. doi: 10.1086/507568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glasscock E, Qian J, Yoo JW, Noebels JL. Masking epilepsy by combining two epilepsy genes. Nat Neurosci. 2007;10:1554–1558. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- 30.Kearney JA, et al. Severe epilepsy resulting from genetic interaction between Scn2a and Kcnq2. Hum Mol Genet. 2006;15:1043–1048. doi: 10.1093/hmg/ddl019. [DOI] [PubMed] [Google Scholar]
- 31.Martin MS, et al. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. 2007;16:2892–2899. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- 32.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanoye CG, et al. Distinct subdomains of the KCNQ1 S6 segment determine channel modulation by different KCNE subunits. J Gen Physiol. 2009;134:207–217. doi: 10.1085/jgp.200910234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.