

Abstract
Dislocation nucleation is essential to our understanding of plastic deformation, ductility, and mechanical strength of crystalline materials. Molecular dynamics simulation has played an important role in uncovering the fundamental mechanisms of dislocation nucleation, but its limited timescale remains a significant challenge for studying nucleation at experimentally relevant conditions. Here we show that dislocation nucleation rates can be accurately predicted over a wide range of conditions by determining the activation free energy from umbrella sampling. Our data reveal very large activation entropies, which contribute a multiplicative factor of many orders of magnitude to the nucleation rate. The activation entropy at constant strain is caused by thermal expansion, with negligible contribution from the vibrational entropy. The activation entropy at constant stress is significantly larger than that at constant strain, as a result of thermal softening. The large activation entropies are caused by anharmonic effects, showing the limitations of the harmonic approximation widely used for rate estimation in solids. Similar behaviors are expected to occur in other nucleation processes in solids.
Nucleation plays an important role in a wide range of physical, chemical, and biological processes (1–6). In the last two decades, the nucleation of dislocations in crystalline solids has attracted significant attention, not only for the reliability of microelectronic devices (7), but also as a responsible mechanism for incipient plasticity in nanomaterials (8–10) and nanoindentation (11–13). However, predicting the nucleation rate as a function of temperature and stress from fundamental physics is extremely difficult. Because the critical nucleus can be as small as a few lattice spacings, the applicability of continuum theory (14) becomes questionable. At the same time, the timescale of molecular dynamics (MD) simulations is about ten orders of magnitude smaller than the experimental timescale. Hence MD simulations of dislocation nucleation are limited to conditions at which the nucleation rate is extremely high (15, 16).
One way to predict the dislocation nucleation rate under common experimental loading rates (17) is to combine the transition state theory (TST) (5, 18) and the nudged elastic band (NEB) method (19). TST predicts that the nucleation rate per nucleation site in a crystal subjected to constant strain γ can be written as
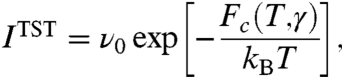 |
[1] |
where Fc is the activation free energy, T is temperature, and kB is Boltzmann’s constant. The frequency prefactor is ν0 = kBT/h, where h is Planck’s constant. Note that Fc(T,γ) = Ec(γ) - TSc(γ), where Ec and Sc are the activation energy and activation entropy, respectively. Here we assume the dependence of Ec and Sc on T is weak, which is later confirmed numerically for T ≤ 400 K. For a crystal subjected to constant stress σ, Fc(T,γ) in Eq. 1 should be replaced by the activation Gibbs free energy Gc(T,σ) = Hc(σ) - TSc(σ), where Hc is the activation enthalpy. Because the NEB method only computes the activation energy, the contribution of Sc is often ignored in rate estimates in solids. Recently, an approximation of Sc(σ) = Hc(σ)/Tm is used (17), where Tm is the surface disordering temperature. This approximation was questioned by subsequent MD simulations (20). The magnitude of Sc remains unknown because none of the existing methods for computing activation free energies (21–23) has been successfully applied to dislocation nucleation.
We successfully applied the umbrella sampling (21) method to compute the activation free energy for homogeneous and heterogeneous dislocation nucleation in copper. Based on this input, the nucleation rate is predicted using the Becker–Döring theory (24). Comparison with direct MD simulations at high stress confirms the accuracy of this approach. Both Fc(T,γ) and Gc(T,σ) show significant reduction with increasing T, corresponding to large activation entropies. For example, Sc(γ = 0.092) = 9kB and Sc(σ = 2 GPa) = 48kB are observed in homogeneous nucleation. We found that Sc(γ) is caused by the anharmonic effect of thermal expansion, with negligible contribution from the vibrational entropy. The large difference in the two activation entropies, ΔSc ≡ Sc(σ) - Sc(γ), is caused by thermal softening, which is another anharmonic effect. Similar behaviors are expected to occur in other nucleation processes in solids.
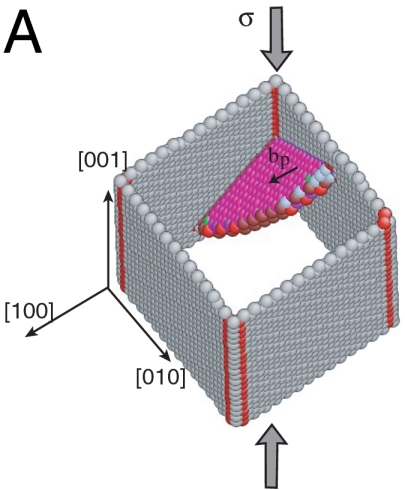
For simplicity, we begin with the case of homogeneous dislocation nucleation in the bulk. Even though dislocations often nucleate heterogeneously at surfaces or internal interfaces, homogeneous nucleation is believed to occur in nanoindentation (11) and in a model of brittle–ductile transition (25). It also provides an upper bound to the ideal strength of the crystal. Our model system is a copper single crystal described by the embedded atom method (EAM) potential (26). As shown in Fig. 1A, the simulation cell is subjected to a pure shear stress along 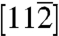 . The dislocation to be nucleated lies on the (111) plane and has the Burgers vector of a Shockley partial (27),
. The dislocation to be nucleated lies on the (111) plane and has the Burgers vector of a Shockley partial (27), 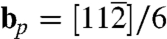 . Fig. 1B shows the shear stress strain relationship of the perfect crystal at different temperatures (before dislocation nucleation).
. Fig. 1B shows the shear stress strain relationship of the perfect crystal at different temperatures (before dislocation nucleation).
Fig. 1.


(A) Schematics of the simulation cell. The spheres represent atoms enclosed by the critical nucleus of a Shockley partial dislocation loop. (B) Shear stress–strain curves of the Cu perfect crystal (before dislocation nucleation) at different temperatures.
In this work, we predict the nucleation rate based on the Becker–Döring (BD) theory, which expresses the nucleation rate per nucleation site as
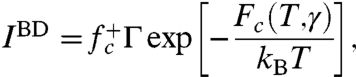 |
[2] |
where 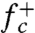 is the molecular attachment rate, and Γ is the Zeldovich factor (Materials and Methods). The BD theory and TST only differs in the frequency prefactor. Whereas TST neglects multiple recrossing over the saddle point by a single transition trajectory (5), the recrossing is accounted for in the BD theory through the Zeldovich factor.
is the molecular attachment rate, and Γ is the Zeldovich factor (Materials and Methods). The BD theory and TST only differs in the frequency prefactor. Whereas TST neglects multiple recrossing over the saddle point by a single transition trajectory (5), the recrossing is accounted for in the BD theory through the Zeldovich factor.
First, we establish the validity of the BD theory for dislocation nucleation by comparing it against direct MD simulations at a relatively high stress σ = 2.16 GPa (γ = 0.135) at T = 300 K, which predicts IMD = 2.5 × 108 s-1 (Materials and Methods). The key input to the BD theory is the activation Helmholtz free energy Fc(T,γ), which is computed by umbrella sampling. The umbrella sampling is performed in Monte Carlo simulations using a bias potential as a function of the order parameter n, which is chosen as the number of atoms inside the dislocation loop (Materials and Methods).
Fig. 2A shows the free energy function F(n) obtained from umbrella sampling for the specified (T,γ) condition. The maximum of F(n) gives the activation free energy Fc = 0.53 ± 0.01 eV and the critical nucleus size nc = 36. The Zeldovich factor (28), Γ = 0.055, is obtained from 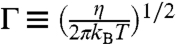 , where η = -∂2F(n)/∂n2|n=nc.
, where η = -∂2F(n)/∂n2|n=nc.
Fig. 2.


(A) Free energy of dislocation loop during homogeneous nucleation at T = 300 K, σxy = 2.16 GPa (γxy = 0.135) from umbrella sampling. (B) Size fluctuation of critical nuclei from MD simulations.
Using the configurations collected from umbrella sampling with n = nc as initial conditions, MD simulations give the attachment rate 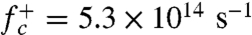 (Materials and Methods). Because the entire crystal is subjected to uniform stress, the number of nucleation sites is the total number of atoms, Natom = 14,976. Combining these data, the Becker–Döring theory predicts the total nucleation rate to be NatomIBD = 5.5 × 108 s-1, which is within a factor of three of the MD prediction. The difference between the two is comparable to our error bar. This agreement is noteworthy because no adjustable parameters (such as the frequency prefactor) is involved in this comparison. It shows that the Becker–Döring theory and our numerical approach are suitable for the calculation of the dislocation nucleation rate.
(Materials and Methods). Because the entire crystal is subjected to uniform stress, the number of nucleation sites is the total number of atoms, Natom = 14,976. Combining these data, the Becker–Döring theory predicts the total nucleation rate to be NatomIBD = 5.5 × 108 s-1, which is within a factor of three of the MD prediction. The difference between the two is comparable to our error bar. This agreement is noteworthy because no adjustable parameters (such as the frequency prefactor) is involved in this comparison. It shows that the Becker–Döring theory and our numerical approach are suitable for the calculation of the dislocation nucleation rate.
We now examine the dislocation nucleation rate under a wide range of temperature and strain (stress) conditions relevant for experiments and beyond the limited timescale of MD simulations. Fig. 3A shows the activation Helmholtz free energy Fc(T,γ) as a function of γ at different T. The zero temperature data are obtained with a minimum energy path (MEP) search using a modified version of the string method, similar to that used in refs. 29 and 17. The downward shift of Fc curves with increasing T and is the signature of the activation entropy Sc. Fig. 3C plots Fc as a function of T at γ = 0.092. For T ≤ 400 K, the data closely follow a straight line, whose slope gives Sc = 9kB. This activation entropy contributes a significant multiplicative factor, exp(Sc/kB) ≈ 104, to the absolute nucleation rate, and cannot be ignored.
Fig. 3.




Activation free energy for homogeneous dislocation nucleation in copper. (A) Fc as a function of shear strain γ at different T. The data for T = 400 K and T = 500 K are shown in the SI Appendix. (B) Gc as a function of shear stress σ at different T. Squares represent umbrella sampling data and lines represent zero temperature MEP search results using simulation cells equilibrated at different temperatures. (C) Fc as a function of T at γ = 0.092. Circles represent umbrella sampling data and the dashed line represents a polynomial fit. (D) Gc as a function of T at σ = 2.0 GPa from polynomial fit.
What causes this rapid drop of activation free energy with temperature? Thermal expansion and vibrational entropy are two candidate mechanisms. To examine the effect of thermal expansion, we performed a zero temperature MEP search at γ = 0.092, but with other strain components fixed at the equilibrated values at T = 300 K. This approach is similar to the quasi-harmonic approximation (QHA) (30, 31) often used in free energy calculations in solids, except that, unlike QHA, the vibrational entropy is completely excluded here. The resulting activation energy, 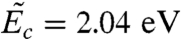 , is indistinguishable from the activation free energy Fc = 2.05 ± 0.01 eV at T = 300 K computed from umbrella sampling. Because atoms do not vibrate in the MEP search, this result shows that the dominant mechanism for the large Sc(γ) is thermal expansion, whereas the contribution from vibrational entropy is negligible. As temperature increases, thermal expansion pushes neighboring atoms further apart and weakens their mutual interaction. This expansion makes crystallographic planes easier to shear and significantly reduces the free energy barrier for dislocation nucleation. In the widely used harmonic approximation of TST, the activation entropy is often attributed to the vibrational degrees of freedom as
, is indistinguishable from the activation free energy Fc = 2.05 ± 0.01 eV at T = 300 K computed from umbrella sampling. Because atoms do not vibrate in the MEP search, this result shows that the dominant mechanism for the large Sc(γ) is thermal expansion, whereas the contribution from vibrational entropy is negligible. As temperature increases, thermal expansion pushes neighboring atoms further apart and weakens their mutual interaction. This expansion makes crystallographic planes easier to shear and significantly reduces the free energy barrier for dislocation nucleation. In the widely used harmonic approximation of TST, the activation entropy is often attributed to the vibrational degrees of freedom as 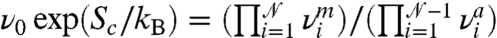 , where
, where 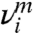 and
and 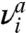 are the positive normal frequencies around the local energy minimum and activated state, respectively (5, 18, 32). However, here we see that Sc(γ) arises entirely from the anharmonic effect for dislocation nucleation. At T = 400 K and T = 500 K, we observe significant differences between Fc computed from umbrella sampling and
are the positive normal frequencies around the local energy minimum and activated state, respectively (5, 18, 32). However, here we see that Sc(γ) arises entirely from the anharmonic effect for dislocation nucleation. At T = 400 K and T = 500 K, we observe significant differences between Fc computed from umbrella sampling and 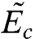 computed from MEP search in the expanded cell (SI Appendix). These differences must also be attributed to anharmonic effects.
computed from MEP search in the expanded cell (SI Appendix). These differences must also be attributed to anharmonic effects.
Although it is easier to control strain γ than stress σ in atomistic simulations, it is usually easier to apply stress in experiments, and experimental results are often expressed as a function of σ and T. To bridge between simulations and experiments, it is important to establish a connection between the constant stress and constant strain ensembles. In the constant strain ensemble, the system is described by the Helmholtz free energy F(n,T,γ), where n is the size of the dislocation loop and the activation Helmholtz free energy is defined as Fc(T,γ) ≡ F(nc,T,γ) - F(n = 0,T,γ). In the constant stress ensemble, the system is described by the Gibbs free energy G(n,T,σ), from the Legendre transform G = F - σγV, with σ ≡ V-1∂F/∂γ|n,T. Similarly, Gc(T,σ) ≡ G(nc,T,σ) - G(n = 0,T,σ). We have proved that Gc(T,σ) = Fc(T,γ) in the thermodynamic limit of V → ∞, when σ and γ satisfies the stress–strain relation of the perfect crystal, σ(γ,T). The difference between Fc and Gc when σ = σ(T,γ) is of the order 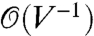 . The details of the proof will be published separately.
. The details of the proof will be published separately.
Combining the activation Helmholtz free energy Fc(T,γ) shown in Fig. 3A and the stress–strain relations shown in Fig. 1B, we obtain the activation Gibbs free energy Gc(T,σ), which is shown in Fig. 3B. We immediately notice that the curves at different temperatures are more widely apart in Gc(T,σ) than that in Fc(T,γ), indicating a much larger activation entropy in the constant stress ensemble. For example, Fig. 3D plots Gc as a function of T at σ = 2.0 GPa, from which we can obtain an averaged activation entropy of 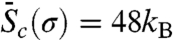 in the temperature range of [0,300 K]. This activation entropy contributes a multiplicative factor of
in the temperature range of [0,300 K]. This activation entropy contributes a multiplicative factor of 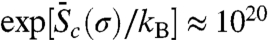 to the absolute nucleation rate.
to the absolute nucleation rate.
The dramatic difference between Sc(γ) and Sc(σ) may seem surprising. Indeed, they are sometimes used interchangeably (33, 34), although the conceptual difference between the two has been pointed out in the context of chemical reactions (35, 36). It is well-known that the entropy is independent of the ensemble of choice, i.e., S(n,T,γ) ≡ ∂F(n,T,γ)/∂T|n,γ and S(n,T,σ) ≡ ∂G(n,T,γ)/∂T|n,σ equal to each other as long as σ = V-1∂F/∂γ|n,T, which is true by definition. At the same time, the activation entropy is just the entropy difference between the activated state and the metastable state, i.e., Sc(T,γ) = S(nc,T,γ) - S(n = 0,T,γ) and Sc(T,σ) = S(nc,T,σ) - S(n = 0,T,σ). If the entropies in two ensembles can equal each other, it may seem puzzling how the activation entropies can be different.
The resolution of this apparent paradox is that under the constant applied stress, the nucleation of a dislocation loop causes a strain increase, i.e., σ(n = 0,T,γ) = σ(nc,T,γ+), with γ+ > γ. Based on this result, one can show that the difference in the activation entropies, ΔSc ≡ Sc(σ) - Sc(γ), equals S(n = 0,T,γ+) - S(n = 0,T,γ), which is the entropy difference of the perfect crystal at two slightly different strains. We can further show that ΔSc = -Ωc(σ)∂σ/∂T|γ, where Ωc ≡ -∂Gc/∂σ|T is the activation volume and -∂σ/∂T|γ describes the thermal softening effect. Hence ΔSc is always positive for nucleation processes in solids driven by shear stress. In the case of homogeneous dislocation nucleation ΔSc as large as 39kB is observed for homogeneous dislocation nucleation, which is mainly caused by its large activation volume Ωc. The numerical results enable us to examine the approximation (17) based on the so-called thermodynamic “compensation law” (37), which states that the activation entropy is proportional to the activation enthalpy (or energy). We find that Sc(γ) can be roughly approximated by Ec(γ)/T∗ with T∗ ≈ 3,000 K, whereas Sc(σ) is not proportional to Hc(σ) (SI Appendix).
To assess the applicability of these conclusions in heterogeneous nucleation, we studied dislocation nucleation from the corner of a [001]-oriented copper nanorod with {100} side surfaces under axial compression (Materials and Methods). Fig. 4B plots the activation free energy barrier as a function of axial compressive stress σ, which shows significant reduction of the activation free energy with temperature. For example, at the compressive elastic strain of ϵ = 0.03, the compressive stress is σ = 1.50 GPa at T = 0 K. The activation entropy Sc(ϵ) at this elastic strain equals 9kB, whereas the activation entropy Sc(σ) at this stress equals 17kB. Fig. 4C plots the contour lines of the predicted dislocation nucleation rate (per nucleation site) as a function of T and σ. To show the physical effect of the large activation entropies, the dashed lines plot the rate predictions if the effect of Sc(σ) were completely neglected. Significant deviations between the two sets of contour lines are observed, especially for T≥300 K and σ ≤ 1.5 GPa. For example, at T = 300 K and σ = 1.5 GPa (where a thick and a thin contour line cross), the neglect of activation entropy would cause an underestimate of the nucleation rate by 10 orders of magnitude.
Fig. 4.


(A) Heterogeneous dislocation nucleation in a copper nanorod under compression, visualized by AtomEye (38). (B) Gc as a function compressive stress σ at different T. (C) Contour lines of dislocation nucleation rate per site I as a function of T and σ. The predictions with and without accounting for activation entropy Sc(σ) are plotted in thick and thin lines, respectively. The nucleation rate of I ∼ 106 s-1 per site is accessible in typical MD timescales whereas the nucleation rate of I ∼ 10-4 - 10-9 is accessible in typical experimental timescales, depending on the number of nucleation sites.
In summary, we have shown that the Becker–Döring theory combined with free energy barriers determined by umbrella sampling can accurately predict the rate of homogeneous dislocation nucleation. In both homogeneous and heterogeneous dislocation nucleation, a large activation entropy at constant elastic strain is observed, and is attributed to the weakening of atomic bonds caused by thermal expansion. An even larger activation entropy is observed at constant stress, caused by thermal softening. Both effects are anharmonic in nature, and emphasize the need to go beyond harmonic approximation in the application of rate theories in solids. We believe our methods and the general conclusions are applicable to a wide range of nucleation processes in solids that are driven by shear stress, including cross slip, twinning, and martensitic phase transformation.
Materials and Methods
Molecular Dynamics.
The simulation cell for homogeneous dislocation nucleation has dimension 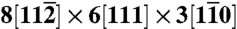 . Periodic boundary conditions (PBC) are applied to all three directions. To reduce artifacts from periodic image interactions, the applied stress is always large enough so that the diameter of the critical dislocation loop is smaller than half the width of the simulation cell.
. Periodic boundary conditions (PBC) are applied to all three directions. To reduce artifacts from periodic image interactions, the applied stress is always large enough so that the diameter of the critical dislocation loop is smaller than half the width of the simulation cell.
The shear strain γ is the x-y component of the engineering strain. The following procedure is used to obtain the pure shear stress–strain curve shown in Fig. 1B. At each temperature T and shear strain γxy, a series of 2 ps MD simulations under the canonical, constant temperature-constant volume (NVT) ensemble are performed. After each simulation, all strain components except γxy are adjusted according to the average virial stress until σxy is the only nonzero stress component. The shear strain is then increased by 0.01 and the process repeats until the crystal collapses spontaneously. The shear stress–strain data are fitted to a polynomial function, 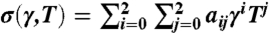 in the range of 0.09 ≤ γ ≤ 0.12 and 0 ≤ T ≤ 500 K.
in the range of 0.09 ≤ γ ≤ 0.12 and 0 ≤ T ≤ 500 K.
To obtain average nucleation time at σxy = 2.16 GPa (γ = 0.135) at 300 K, we performed 192 independent MD simulations using the NVT ensemble with random initial velocities. Each simulation ran for 4 ns. If dislocation nucleation occurred during this period, the nucleation time was recorded. This information is used to construct the function Ps(t), which is the fraction of MD simulation cells in which dislocation nucleation has not occurred at time t. Ps(t) can be well fitted to the form of exp(-IMDt) to extract the nucleation rate IMD.
To compute the attachment rate 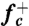 , we collected from umbrella sampling an ensemble of 500 atomic configurations for which n = nc, and ran MD simulations using each configuration as an initial condition. The initial velocities are randomized according to Boltzmann’s distribution. The mean square change of the loop size, 〈Δn2(t)〉, as shown in Fig. 2B, was fitted to a straight line,
, we collected from umbrella sampling an ensemble of 500 atomic configurations for which n = nc, and ran MD simulations using each configuration as an initial condition. The initial velocities are randomized according to Boltzmann’s distribution. The mean square change of the loop size, 〈Δn2(t)〉, as shown in Fig. 2B, was fitted to a straight line, 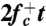 , to extract
, to extract 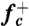 (39).
(39).
Free Energy Barrier Calculations.
The reaction coordinate n is defined for each atomic configurations in the following way. An atom is labeled as “slipped” if its distance from any of its original nearest neighbors has changed by more than the critical distance dc (40). We chose dc = 0.33, 0.38, and 0.43 A for T ≤ 400 K, T = 500 K, and T = 600 K, respectively. The slipped atoms were grouped into clusters; two atoms belong to the same cluster if their distance was less than the cutoff distance rc (3.4 A). The reaction coordinate n is the number of atoms in the largest cluster divided by two.
To perform umbrella sampling, a bias potential 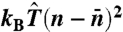 is superimposed on the EAM potential, where
is superimposed on the EAM potential, where 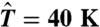 and
and 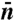 is the center of the sampling window. We chose
is the center of the sampling window. We chose 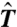 empirically so that the width of the sampling window on the n-axis would be about 10. The activation Helmholtz free energy data for homogeneous nucleation can be fitted very well by a polynomial function,
empirically so that the width of the sampling window on the n-axis would be about 10. The activation Helmholtz free energy data for homogeneous nucleation can be fitted very well by a polynomial function, 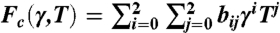 in the range of 0.09 ≤ γ ≤ 0.12 and 0 ≤ T ≤ 500 K.
in the range of 0.09 ≤ γ ≤ 0.12 and 0 ≤ T ≤ 500 K.
For heterogeneous dislocation nucleation, the size of the copper nanorod (17) is 15[100] × 15[010] × 20[001] with PBC along [001]. The activation Gibbs free energy data for heterogeneous nucleation is fitted to an empirical form Gc(σ,T) = A[(σ/σ0)p - 1]q - BσrT.
The error bar of activation free energies is about 0.5kB. Hence the error bar of activation entropies is about 0.5kB.
Supplementary Material
Acknowledgments.
This work is supported by National Science Foundation Grant CMS-0547681, a Department of Energy Scientific Discovery through Advanced Computing project on Quantum Simulation of Materials and Nanostructures, and the Army High Performance Computing Research Center at Stanford University. S.R. acknowledges support from the Weiland Family Stanford Graduate Fellowship..
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1017171108/-/DCSupplemental.
References
- 1.Walsh MR, Koh CA, Sloan ED, Sum AK, Wu DT. Microsecond simulations of spontaneous methane hydrate nucleation and growth. Science. 2009;326:1095–1098. doi: 10.1126/science.1174010. [DOI] [PubMed] [Google Scholar]
- 2.Anderson VJ, Lekkerkerker HNW. Insights into phase transition kinetics from colloid science. Nature. 2002;416:811–815. doi: 10.1038/416811a. [DOI] [PubMed] [Google Scholar]
- 3.Matsumoto M, Saito S, Ohmine I. Molecular dynamics simulation of the ice nucleation and growth process leading to water freezing. Nature. 2002;416:409–413. doi: 10.1038/416409a. [DOI] [PubMed] [Google Scholar]
- 4.Laaksonen A, Talanquer V, Oxtoby DW. Nucleation: Measurements, theory, and atmospheric applications. Annu Rev Phys Chem. 1995;46:489–524. doi: 10.1146/annurev.pc.46.100195.002421. [DOI] [PubMed] [Google Scholar]
- 5.Hänggi P, Talkner P, Borkovec M. Reaction-rate theory: Fifty years after Kramers. Rev Mod Phys. 1990;62:251–342. [Google Scholar]
- 6.ten Wolde PR, Frenkel D. Enhancement of protein crystal nucleation by critical density fluctuations. Science. 1997;277:1975–1978. doi: 10.1126/science.277.5334.1975. [DOI] [PubMed] [Google Scholar]
- 7.Izumi S, Ohta H, Takahashi C, Suzuki T, Saka H. Shuffle-set dislocation nucleation in semiconductor silicon device. Philos Mag Lett. 2010;90:707–714. [Google Scholar]
- 8.Li X, Wei Y, Lu L, Lu K, Gao H. Dislocation nucleation governed softening and maximum strength in nanotwinned metals. Nature. 2010;464:877–880. doi: 10.1038/nature08929. [DOI] [PubMed] [Google Scholar]
- 9.Li J. The mechanics and physics of defect nucleation. MRS Bull. 2007;32:151–159. [Google Scholar]
- 10.Zhu T, Li J, Ogata S, Yip S. Mechanics of ultra-strength materials. MRS Bull. 2009;34:167–172. [Google Scholar]
- 11.Li J, Van Vliet KJ, Zhu T, Yip S, Suresh S. Atomistic mechanisms governing elastic limit and incipient plasticity in crystals. Nature. 2002;418:307–310. doi: 10.1038/nature00865. [DOI] [PubMed] [Google Scholar]
- 12.Schuh CA, Mason JK, Lund AC. Quantitative insight into dislocation nucleation from high-temperature nanoindentation experiments. Nat Mater. 2005;4:617–621. doi: 10.1038/nmat1429. [DOI] [PubMed] [Google Scholar]
- 13.Schall P, Cohen I, Weitz DA, Spaepen F. Visualizing dislocation nucleation by indenting colloidal crystals. Nature. 2006;440:319–323. doi: 10.1038/nature04557. [DOI] [PubMed] [Google Scholar]
- 14.Xu G, Argon AS, Ortiz M. Critical configurations for dislocation nucleation from crack tips. Philos Mag A. 1997;75:341–367. [Google Scholar]
- 15.Bringa EM, et al. Shock deformation of face-centered-cubic metals on subnanosecond timescales. Nat Mater. 2006;5:805–810. doi: 10.1038/nmat1735. [DOI] [PubMed] [Google Scholar]
- 16.Tschopp MA, Spearot DE, McDowell DL. Atomistic simulations of homogeneous dislocation nucleation in single crystal copper. Modell Simul Mater Sci Eng. 15:693–709. [Google Scholar]
- 17.Zhu T, Li J, Samanta A, Leach A, Gall K. Temperature and strain-rate dependence of surface dislocation nucleation. Phys Rev Lett. 2008;100:025502. doi: 10.1103/PhysRevLett.100.025502. [DOI] [PubMed] [Google Scholar]
- 18.Vineyard GH. Frequency factors and isotope effects in solid state rate processes. J Phys Chem Solids. 1957;3:121–127. [Google Scholar]
- 19.Jónsson H, Mills G, Jacobsen KW. Nudged elastic band method for finding minimum energy paths of transitions. In: Berne BJ, Ciccotti G, Coker DF, editors. Classical and Quantum Dynamics in Condensed Phase Simulations. Singapore: World Scientific; 1998. pp. 385–404. [Google Scholar]
- 20.Warner DH, Curtin WA. Origins and implications of temperature-dependent activation energy barriers for dislocation nucleation in face-centered cubic metals. Acta Mater. 2009;57:4267–4277. [Google Scholar]
- 21.Frenkel D, Smit B. Understanding Molecular Simulation: From Algorithms to Applications. San Diego: Academic; 2002. [Google Scholar]
- 22.Ren WEW, Vanden-Eijnden E. Finite temperature string method for the study of rare events. J Phys Chem B. 2005;109:6688–6693. doi: 10.1021/jp0455430. [DOI] [PubMed] [Google Scholar]
- 23.Valeriani C, Allen RJ, Morelli MJ, Frankel D, ten Wolde PR. Computing stationary distributions in equilibrium and nonequilibrium systems with forward flux sampling. J Chem Phys. 2007;127:114109. doi: 10.1063/1.2767625. [DOI] [PubMed] [Google Scholar]
- 24.Becker R, Döring W. The kinetic treatment of nuclear formation in supersaturated vapors. Ann Phys (Weinheim, Ger) 1935;24:719–752. [Google Scholar]
- 25.Khantha M, Pope DP, Vitek V. Dislocation screening and the brittle-to-ductile transition: A Kosterlitz–Thouless type instability. Phys Rev Lett. 1994;74:684–687. doi: 10.1103/PhysRevLett.73.684. [DOI] [PubMed] [Google Scholar]
- 26.Mishin Y, Mehl MJ, Papaconstantopoulos DA, Voter AF, Kress JD. Structural stability and lattice defects in copper: Ab initio, tight-binding, and embedded-atom calculations. Phys Rev B: Condens Matter Mater Phys. 2001;63:224106. [Google Scholar]
- 27.Hirth JP, Lothe J. Theory of Dislocations. New York: Wiley; 1982. [Google Scholar]
- 28.Zeldovich YB. On the theory of new phase formation: Cavitation. Acta Physicochim URSS. 1943;18:1–22. [Google Scholar]
- 29.Zhu T, Li J, Samanta A, Kim HG, Suresh S. Interfacial plasticity governs strain rate sensitivity and ductility in nanostructured metals. Proc Natl Acad Sci USA. 2007;104:3031–3036. doi: 10.1073/pnas.0611097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foiles SM. Evaluation of harmonic methods for calculating the free energy of defects in solids. Phys Rev B: Condens Matter Mater Phys. 1994;49:14930–14938. doi: 10.1103/physrevb.49.14930. [DOI] [PubMed] [Google Scholar]
- 31.de Koning M, Miranda Caetano R, Antonelli A. Atomistic prediction of equilibrium vacancy concentraions in Ni3Al. Phys Rev B: Condens Matter Mater Phys. 2002;66:104110. [Google Scholar]
- 32.Gupta D, editor. Diffusion Processes in Advanced Technological Materials. New York: Williams Andrew Publishing; 2002. pp. 138–140. [Google Scholar]
- 33.Huntington HB, Shirn GA, Wajda ES. Calculation of the entropies of lattice defects. Phys Rev. 1955;99:1085–1091. [Google Scholar]
- 34.DiMelfi RJ, Nix WD, Barnett DM, Holbrook JH, Pound GM. The equivalence of two methods for computing the activation entropy for dislocation motion. Acta Mater. 1980;28:231–337. [Google Scholar]
- 35.Whalley E. Use of volumes of activation for determining reaction mechanisms. In: Gold V, editor. Advances in Physical Organic Chemistry. London: Academic; 1964. pp. 93–162. [Google Scholar]
- 36.Tonnet ML, Whalley E. Effect of pressure on the alkaline hydrolysis of ethyl acetate in acetone-water solutions. Parameters of activation at constant volume. Can J Chem. 1975;53:3414–3418. [Google Scholar]
- 37.Kemeny G, Rosenberg B. Compensation law in thermodynamics and thermal death. Nature. 1973;243:400–401. [Google Scholar]
- 38.Li J. AtomEye: An efficient atomistic configuration viewer. Modell Simul Mater Sci Eng. 2003;11:173–177. [Google Scholar]
- 39.Ryu S, Cai W. Validity of classical nucleation theory for Ising models. Phys Rev E: Stat, Nonlinear, Soft Matter Phys. 81:030601(R). doi: 10.1103/PhysRevE.81.030601. [DOI] [PubMed] [Google Scholar]
- 40.Ngan AHW, Zuo L, Wo PC. Size dependence and stochastic nature of yield strength of micron-sized crystals: A case study on Ni3Al. Proc R Soc A. 2006;462:1661–1681. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.